

Abstract
Synapses are highly specialized intercellular junctions organized by adhesive and scaffolding molecules that align presynaptic vesicular release with postsynaptic neurotransmitter receptors. The MALS/Veli–CASK–Mint-1 complex of PDZ proteins occurs on both sides of the synapse and has the potential to link transsynaptic adhesion molecules to the cytoskeleton. In this study, we purified the MALS protein complex from brain and found liprin-α as a major component. Liprin proteins organize the presynaptic active zone and regulate neurotransmitter release. Fittingly, mutant mice lacking all three MALS isoforms died perinatally with difficulty breathing and impaired excitatory synaptic transmission. Excitatory postsynaptic currents were dramatically reduced in autaptic cultures from MALS triple knockout mice due to a presynaptic deficit in vesicle cycling. These findings are consistent with a model whereby the MALS–CASK–liprin-α complex recruits components of the synaptic release machinery to adhesive proteins of the active zone.
Introduction
Synaptic transmission requires precise alignment of pre- and postsynaptic specializations. On the presynaptic side, synaptic vesicles containing neurotransmitters must be aligned and docked at active zones, where vesicles fuse with the presynaptic membrane for secretion (Südhof, 2004). On the postsynaptic side, neurotransmitter receptors must be clustered together with relevant signal transduction machinery to respond to released transmitters. Recent studies have begun to elucidate the molecular machinery responsible for the organization of synaptic junctions. Adhesion molecules that span the synaptic cleft function in both stabilization and definition of the presynaptic active zone and postsynaptic specialization (Ichtchenko et al., 1995; Fannon and Colman, 1996; Flanagan and Vanderhaeghen, 1998). Cytosolic molecules associated with these adhesive factors help position synaptic vesicles and neurotransmitter receptors on their respective sides of the synapse (Hata et al., 1996; Torres et al., 1998; Perego et al., 2000).
One such set of modular scaffolding proteins comprises a ternary complex of MALS/Veli (mammalian LIN-7/vertebrate homologue of LIN-7), CASK (peripheral plasma membrane protein), and Mint-1 (munc-18 interacting protein 1), which are vertebrate homologues of a complex first identified in Caenorhabditis elegans that mediates vulval development (Kaech et al., 1998). In mammalian brain, the MALS–CASK–Mint-1 complex occurs on both sides of synaptic junctions and is thought to serve distinct roles in these two locations. Presynaptically, this complex links to neurexin (Hata et al., 1996), an adhesion molecule that binds across the synapse to postsynaptic neuroligin (Ichtchenko et al., 1995). Furthermore, Mint-1 associates with Munc18-1, an essential component of the synaptic vesicle fusion machinery (Okamoto and Südhof, 1997). Postsynaptically, MALS binds to the N-methyl-d-aspartate (NMDA)–type of glutamate receptors (Jo et al., 1999) and is reported to transport NMDA receptor vesicles along microtubules (Setou et al., 2000).
Genetic studies have failed to establish the essential roles of the MALS–CASK–Mint-1 complex in brain. Three MALS genes exist in mammals (Borg et al., 1998; Butz et al., 1998; Jo et al., 1999), and targeted disruption of MALS-1 and MALS-2 leads to compensatory up-regulation of MALS-3 in the CNS (Misawa et al., 2001). Mint-1 mutant mice show no defects in excitatory synaptic transmission and only a subtle defect in inhibitory synaptic transmission (Ho et al., 2003). Also, no synaptic analysis has been reported for CASK knockouts that die at birth due to midline defects (Laverty and Wilson, 1998).
Several molecules that mediate synapse development have been identified through invertebrate genetic studies. For example, mutation of C. elegans syd-2 disperses presynaptic active zones (Zhen and Jin, 1999). A similar structural defect occurs in flies lacking the Drosophila melanogaster syd orthologue liprin-α, which exhibits a concomitant decrease in synaptic transmission (Kaufmann et al., 2002). Liprin-α binds to a receptor protein tyrosine phosphatase, Dlar (Serra-Pages et al., 1998), suggesting a model whereby liprin-α and Dlar cooperate to organize presynaptic active zones. How liprin-α links to the synaptic vesicle machinery remains uncertain.
To define the essential roles for the MALS complex in mammals, we purified the MALS complex from brain. Isolation of the MALS complex revealed an association with a family of cytoskeletal and presynaptic adhesion molecules. Importantly, we found liprin-α1, -α2, -α3, and -α4 in the MALS complex. Association with this complex is mediated through the SAM domains in liprin-α and an NH2-terminal region in CASK. Using the sterile α motif (SAM) domains of liprin-α as a dominant negative, we disrupted the MALS–liprin complex in dissociated neurons. To understand the function of the MALS complex, we generated mutant mice lacking all three MALS genes. Mice lacking any single gene were viable and fertile. However, mice lacking all three MALS genes died within one hour of birth. This perinatal lethality is associated with impaired presynaptic function, reflecting the presynaptic deficits of invertebrates lacking liprin-α orthologues. These studies establish a crucial role for the MALS complex in synaptic vesicle exocytosis and implicate liprin-α in this process.
Results
Proteomic characterization of the MALS complex in brain
To identify molecular roles for MALS, we assessed the composition of the MALS protein complex. We performed preparative immunoprecipitation of MALS-3 from brain homogenates and used MALS-3 knockout mice (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200503011/DC1) as a powerful control. A series of protein bands were present in the MALS-3 immunoprecipitation that were absent in precipitations from MALS-3 knockouts. Several known components of the MALS-3 complex were identified, including neurexin, CASKIN, NMDA receptor 2B, Mint-1, and PALS-1, which is a protein associated with lin-7 (Fig. 1 A). Silver staining of immunoprecipitates showed specific bands at 140, 120, and 105 kD (Fig. 1 A). Mass spectrometry indicated that the 105-kD band corresponds to CASK, the 120-kD band corresponds to SAP-97, and the 140-kD band contained Mint-1, as well as liprin-α2, -α3, and -α4 (Fig. 1 A). Western blotting confirmed the efficient coimmunoprecipitation of CASK, Mint-1, and liprin-α1 and -α2 (Fig. 1 B).
Figure 1.

Identification of a neuronal protein complex containing MALS and liprin-α. (A) Immunoprecipitation of MALS-3 from brain extracts showed a series of bands in heterozygote (H) that were absent from MALS-3 knockout (K). Bands were identified by MS/MS obtained using a micro-ionspray source attached to a mass spectrometer (red) and confirmed by Western blotting (black). Molecular weights are presented in blue and Mint-1 degradation products are shown with asterisks. (B) Western blotting of heterozygote and knockout brain extracts immunoprecipitated for MALS-3 shows specific association of CASK, Mint-1, liprin-α1, and -α2 with MALS-3. (C) Western blotting shows that MALS-3, CASK, and liprin-α2 are highly enriched with synaptophysin (Synphy) in the synaptosome (Syn) fraction and PSD-95 in PSD fractions. (D and E) Hippocampal cultures (28 DIV) were stained for MALS, liprin-α2, and synaptophysin. Immunostaining reveals that both liprin-α2 (D) and synaptophysin (E) partially colocalize with MALS (arrowheads). Bar, 20 μm.
Interaction of liprin-α with the MALS complex
Liprin-α mutants in D. melanogaster (and syd mutants in C. elegans) display impaired synaptic vesicle exocytosis. Our discovery that liprin-α binds to the MALS–CASK complex is novel. Consistent with this, MALS, CASK, and liprin-α2 were enriched in synaptic biochemical fractionations of brain extracts (Fig. 1 C). Furthermore, MALS partially colocalized with liprin-α2 and the presynaptic marker synaptophysin in cultured hippocampal neurons (Fig. 1, D and E).
Liprin-α proteins contain conserved coiled-coil regions, three SAM domains, and a COOH-terminal region that binds to certain PDZ domains (Fig. 2 B). Using immunoprecipitation analysis and the yeast two-hybrid system, we found that MALS-1 does not directly bind to liprin-α2 (Fig. 2, A [top left] and C). We therefore asked whether other core components of the MALS complex might directly associate with liprin-α2. Indeed, CASK, but not Mint-1, bound to liprin-α2 directly (Fig. 2 A, bottom left and top right). Furthermore, we found that CASK can link liprin-α2 to a MALS-1 complex (Fig. 2 A, bottom right). These biochemical associations also redirect protein distribution in transfected cells, and all three MALS isoforms can associate with CASK to form MALS–CASK–liprin-α complexes (Figs. 1 and 2; unpublished data).
Figure 2.

Domain mapping of the MALS–liprin-α interaction. (A) CASK, but not MALS-1 or Mint-1, coimmunoprecipitated liprin-α2 in transfected COS cells. In the presence of CASK, MALS coimmunoprecipitated liprin-α2. (B) Schematics representing the structural domains of MALS-3 (blue), CASK (orange), and liprin-α2 (yellow). (C) By yeast two-hybrid analysis, CASK, but not MALS-3, interacted with liprin-α2. For CASK–liprin-α binding, the SAM1 domain of liprin-α2 is sufficient for interaction with CASK. Both the CaMK-like domain and first L27 domain of CASK were necessary for binding to liprin-α2.
To define the site for interaction between liprin-α2 and CASK, we used the yeast two-hybrid system. We found that full-length CASK readily bound to full-length liprin-α2 (Fig. 2 C). This binding was mediated specifically by the first SAM domain in liprin-α2. Deletion analysis of CASK showed that the CAM kinase (CaMK) and first L27 domain of CASK were necessary for binding. We were unable to map this interaction further, suggesting that the binding domain may require large sequences for proper folding.
Targeted disruption of MALS-3 and breeding of MALS-deficient mice
To examine the essential roles for MALS in this complex, we targeted disruption of MALS-3. Our targeting vector replaced exons 3, 4, and 5 of MALS-3 with a neomycin cassette (Fig. S1, A–C). After targeted disruption in embryonic stem cells, we generated MALS-3–deficient mice. MALS-3 mutant mice were born at the expected Mendelian ratios and displayed no overt behavioral abnormalities. Western blotting showed a complete absence of MALS-3 protein in the knockout (Fig. S1 D). Histological inspection of brain showed no gross anatomical abnormalities. As previously reported (Misawa et al., 2001), MALS-3 occurs diffusely in numerous neuronal populations in the brain (Fig. S1 E). Furthermore, expression of MALS-3 is up-regulated, especially in the dentate gyrus region of the hippocampus, in MALS-1/2 double knockout mice (Fig. S1 E).
We interbred MALS-3 knockout mice with the previously generated MALS-1/2 mutants, which yielded 27 possible genotypes. These compound genotypes are presented in Fig. 3 A. We found that most mice lacking both MALS-1 and -3 died shortly after birth, whereas mice lacking MALS-1 and -2 were viable and fertile. Mice lacking MALS-2 and -3 and heterozygous for MALS-1 died during the second postnatal week. Finally, mice lacking all three MALS isoforms exhibited irregular, labored breathing and died within one hour of birth. The complete absence of MALS is not associated with embryonic lethality, as the predicted Mendelian ratio of fetuses was found when a caesarian section was performed at embryonic day 18 (Fig. 3 B).
Figure 3.

Generation of mice lacking all three MALS isoforms. (A) PCR genotyping of MALS mice. Single asterisk indicates mice that died within hours of birth and double asterisk indicates a line that died in their second postnatal week. (B) Statistics obtained from crossing MALS-1/2 K and MALS-3 H mice. Genotyping 2-wk-old mice showed no TKO mice. However, embryonic mice (E18) showed the predicted ratio of W, H, and K mice.
Disruption of the MALS–CASK–liprin-α complex
MALS triple knockout (TKO) mice appear anatomically normal at birth; however, their perinatal death and difficulty breathing suggest neurological deficits. To assess whether components of the MALS complex or other synaptic proteins show quantitative expression differences, we performed Western blotting in the MALS TKO mice as compared with MALS-1/2 knockout mice, which were phenotypically normal, or wild type (WT). We found that CASK levels are dramatically decreased in the MALS TKO, whereas levels of all other assayed synaptic proteins were normal in the TKO mice (Fig. 4, A and B). We also found that the synaptic localization of CASK, but not other synaptic markers, was partially disrupted in TKO neuronal cultures (Fig. 4, C and D).
Figure 4.
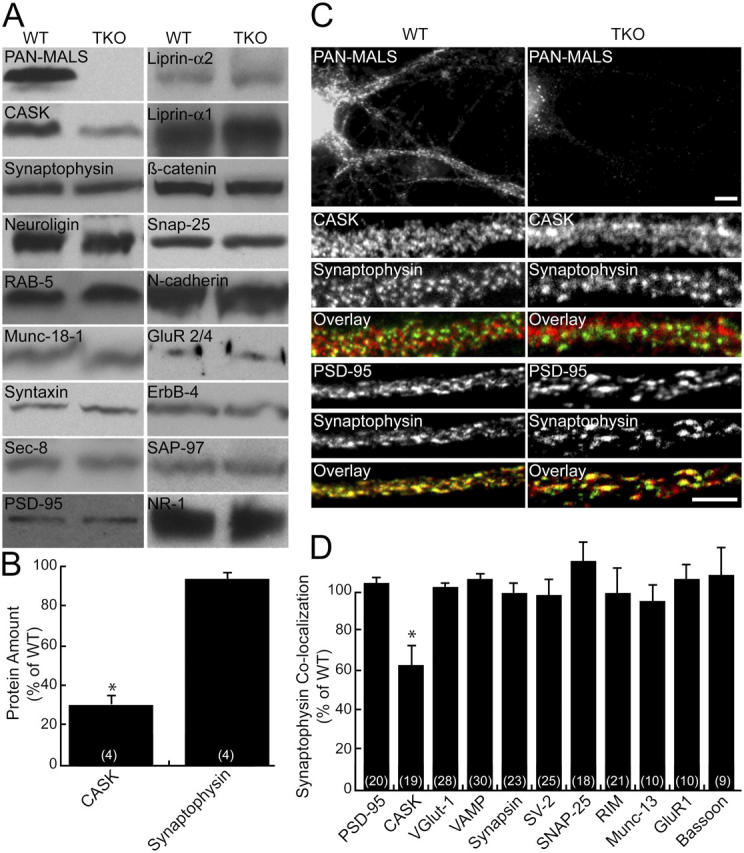
CASK expression is reduced in MALS-deficient mice. (A) Brains from E18 mice were immunoblotted for numerous synaptic proteins. (B) CASK was markedly reduced (31% of control ± 8; *, P < 0.01) in the TKOs, but no changes in other synaptic proteins were detected. (C and D) Similarly, cultured hippocampal neurons lacking MALS displayed normal localization of several pre- and postsynaptic markers but showed reduced colocalization of CASK (red) and synaptophysin (green), suggesting that CASK is partially lost from synapses (62% ± 12 of control; *, P < 0.01). Bars: (top) 10 μm; (bottom) 5 μm. All error bars represent SEM.
Liprins play important roles in synaptic development and function. Because we did not detect any change in liprin-α amount or localization (unpublished data), we suspected that the MALS–CASK complex may be a downstream effector for liprin-α's presynaptic function. To test this hypothesis, we generated dominant-negative liprin-α constructs consisting of the SAM domains fused to GFP in the PSCA1 Semliki Forest viral vector to disrupt the liprin-α–CASK interaction. Infection of neurons with Semliki virus expressing GFP alone showed that >90% of neurons were infected and that neither the virus nor GFP affected MALS distribution (Fig. 5 A). However, expression of the liprin dominant-negative (GFP-SAM) misdirected MALS to nonsynaptic sites and significantly disrupted synaptic localization of MALS (Fig. 5 B). These results suggest that liprin-αs are upstream of MALS and may function, at least partially, through their interaction with the MALS–CASK complex.
Figure 5.

A dominant-negative liprin-α disrupts presynaptic localization of MALS. Hippocampal cultures (35 DIV) were infected with Semliki Forest virus expressing either GFP or GFP fused to the SAM domains of liprin-α2. Whereas infection and expression of GFP had no effect on synaptic expression of MALS (A, arrowheads), expression of the dominant-negative (GFP-SAM) construct misdirected MALS to nonsynaptic sites (B, arrows) and resulted in a loss of presynaptic MALS (B, arrowheads). Quantification of immunofluorescence reveals that colocalization of MALS (red in overlayed images) with synaptophysin (green in overlayed images) is significantly reduced in neurons expressing GFP-SAM, from 77.3% ± 3.6 in uninfected neurons to 48.1% ± 2.1 and to 79.5% ± 2.0 in GFP-expressing neurons (P < 0.01). Bars: (top) 20 μm; (bottom) 10 μm.
MALS-deficient mice have deficient neurotransmitter release
Because of the perinatal death of MALS TKO mice, we were unable to assess electrophysiological parameters in forebrain neurons. Therefore, we generated microisland neuronal cultures from these mice to assess synaptic function. Individual neurons in such cultures are grown on isolated dots of substrate and form functional autaptic synapses. Because liprin-αs are necessary for proper synapse differentiation, we examined the morphology and number of synapses in autapses from TKO mice. We found that neurons from MALS TKO formed autaptic synapses in which presynaptic synaptophysin was juxtaposed to postsynaptic density (PSD)-95 (Fig. 6 A). The density of synapses in the TKO was comparable to WT (Fig. 6 B); however, the distribution of synaptic areas in the TKO was shifted to slightly larger sizes (Fig. 6 C).
Figure 6.
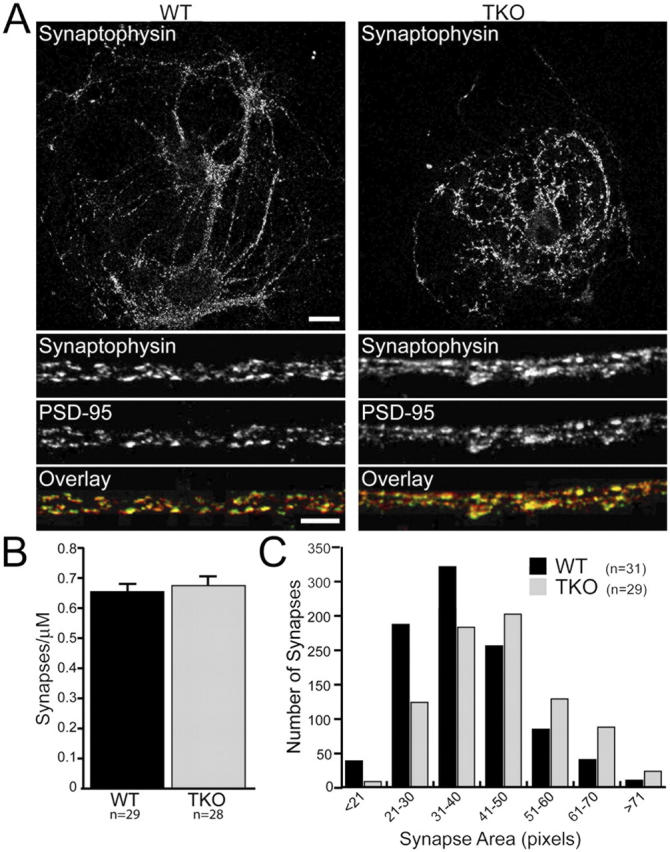
Synapse size, but not number, is altered in neurons lacking MALS. Autaptic cultures (14 DIV) from control and TKO mice were stained with antibodies to PSD-95 and synaptophysin. (A) Representative autaptic neurons from WT and TKO mice stained with synaptophysin. Bars: (top) 20 μm; (bottom) 5 μm. (B) PSD-95/synaptophysin staining revealed that the number of synapses was unaltered in TKO cultures (0.65 ± 0.15 and 0.67 ± 0.18 synapses/μm for WT and TKO, respectively). (C) As determined by synaptophysin staining, the distribution of presynaptic terminal size was shifted to the right in the TKO (P < 0.01). All error bars represent SEM.
Electrophysiological experiments demonstrated that excitatory postsynaptic currents (EPSCs) in the MALS TKO cultures were profoundly reduced relative to WT (Fig. 7 A). Furthermore, the rate and degree of EPSC depression during high frequency stimulation (10 Hz) was enhanced in the MALS TKO autapses (Fig. 7 B), suggesting that the MALS–liprin-α complex plays a role in presynaptic vesicle cycling. Because MALS mutant mice die around birth, structural analyses of mature synapses are not feasible. However, the magnitude and distribution of miniature EPSCs was comparable in MALS TKO and WT cultures (Fig. 7 C), arguing against a postsynaptic defect. Indeed, the ratio of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA)/NMDA components of the EPSC was normal (Fig. 7 D), suggesting that trafficking and clustering of these receptors are intact.
Figure 7.

Neurons from MALS-deficient mice display abnormal synaptic transmission. (A) EPSCs recorded in autaptic cultures prepared from E18 MALS TKO mice were profoundly reduced relative to EPSCs recorded in WT cultures (*, P = 0.01). Typical EPSCs recorded in WT and MALS TKO pyramidal cell autapses are shown above. (B) Normalized synaptic responses during a 10-Hz (4 s) stimulus train. Decay of EPSCs during the high frequency train was greater in the MALS TKO autapses than in WT autapses (*, P < 0.05). EPSCs returned to baseline values within 5 s of the end of the stimulus train in both populations (n = 18 and 7 for WT and MALS TKO, respectively, for the recovery). (C) The distribution and amplitude of miniature EPSCs was the same in autapses prepared from WT and MALS TKO mice (P > 0.1). (D) The ratio of AMPA EPSCs to NMDA EPSCs was also similar in WT and MALS TKO autapses (P > 0.1). AMPA EPSCs were measured at the peak (∼8–10 ms after the action potential), whereas NMDA EPSCs were measured at 40 ms after the action potential, a time point when the AMPA EPSC has decayed to baseline. Typical AMPA/NMDA EPSCs from the two populations are shown. All error bars represent SEM.
Discussion
MALS proteins are essential
This study establishes MALS as an essential protein family involved in neurotransmitter release. MALS are components of a large presynaptic protein complex that includes scaffolding proteins, adaptor proteins, and adhesion molecules (Butz et al., 1998). Importantly, we find that liprin-α isoforms are components of this complex. Because liprin-α orthologues define the dimensions of the active zone in invertebrate synapses (Zhen and Jin, 1999; Kaufmann et al., 2002), the MALS complex has the potential to link adhesion molecules to the exocytotic machinery.
Although each strain of individual MALS knockouts is viable and fertile, certain combinations are synthetically lethal. Mice lacking all three MALS isoforms die within one hour of birth and have a severe breathing defect. This defect resembles the phenotype of mice lacking all three neurexin isoforms (Missler et al., 2003), which are also part of the MALS complex (Hata et al., 1996). Previous studies showed that MALS can bind to the COOH termini of proteins typical for morphological development, including β-catenin and epidermal growth factor receptors (Garcia et al., 2000; Perego et al., 2000; Shelly et al., 2003). That MALS TKO mice show no gross external abnormalities suggests that MALS does not play general roles in tissue morphogenesis. Detailed histological evaluation may, however, reveal specific tissues whose development requires the MALS complex.
Presynaptic defect in MALS mutants
The perinatal lethality and labored breathing of MALS TKO are phenotypes often seen in mice with impaired synaptic transmission. Consistent with this, we found a profound presynaptic defect in these mutants. No change in the mEPSC amplitude distribution and no change in the ratio of AMPA/NMDA receptor–mediated currents were observed. In contrast to normal postsynaptic function, high-frequency stimulation produced an accelerated and more pronounced synaptic depression, suggesting greater depletion of vesicles in the readily releasable pool of MALS TKO mice. These results also imply that the MALS–CASK–liprin-α complex helps determine the size of the releasable pool and is important for replenishing this pool from the reserve pool.
Assembly of the MALS protein complex
The presynaptic defects in MALS TKO mice are paralleled by the association of MALS with a large presynaptic complex. Numerous modular protein interaction interfaces assemble this complex. The membrane-associated guanylate kinase CASK directly binds to many components and therefore constitutes the core. MALS uses its coiled-coil L27 domain to bind to the second of two L27 domains in CASK (Lee et al., 2002). The first L27 domain in CASK associates with the NH2-terminal L27 domain of SAP-97 (Lee et al., 2002). Neurexin binds to the PDZ domain of CASK (Hata et al., 1996) and Mint-1 binds to the CaMK domain of CASK (Butz et al., 1998; Borg et al., 1999). Furthermore, we find that liprin-α proteins associate with the NH2-terminal region of CASK, which includes the CaMK domain and the first L27 region. Previous studies showed that liprin-α proteins also bind to LAR-family receptor protein tyrosine phosphatase (Serra-Pages et al., 1998) and to the Rab3A binding protein RIM1α (Schoch et al., 2002). Our failure to detect LAR or RIM proteins in the MALS complex may suggest that interaction of CASK with the SAM domains of liprin-α occludes association with these other presynaptic molecules.
The liprin protein family comprises seven liprin isoforms that are subdivided into the α- and β-type. We identified only liprin-α family members as part of the MALS complex. The SAM1 domain of liprin-αs mediates their interaction with CASK. These domains are highly conserved between liprin-α proteins, sharing >90% identity. In contrast, liprin-α SAM1 domains share <45% identity with the most homologous liprin-β member. The SAM1 domain of liprin-α is evolutionarily conserved; mammalian liprin-αs share >90% identity with orthologues in D. melanogaster and C. elegans, which suggest that the liprin-α–CASK interaction is likely conserved.
Potential mechanisms for MALS complex regulating transmitter release
Our discovery that the MALS complex contains liprin-α proteins can explain how this complex participates in synaptic vesicle exocytosis. Genetic analysis of invertebrates shows that liprin-α in D. melanogaster and its homologue syd in C. elegans control presynaptic function and morphology (Zhen and Jin, 1999; Kaufmann et al., 2002). In addition to these developmental defects, these mutants show decreased synaptic transmission resembling that seen in the MALS TKO. Because the mammalian genome contains four liprin-α isoforms (Serra-Pages et al., 1998), functional analyses are difficult. Some biochemical work suggests that mammalian liprin-α proteins may be postsynaptic and regulate AMPA receptors through the glutamate receptor interacting protein GRIP (Wyszynski et al., 2002). lin-10 also has been shown to regulate GLR-1 in C. elegans (Rongo et al., 1998). MALS and liprin-αs are also enriched in PSD fractions from rat brain (Fig. 1 C) and partially colocalize with PSD-95 in cultured hippocampal neurons (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200503011/DC1). We cannot rule out the possibility that postsynaptic MALS may contribute to the synaptic defect in the MALS TKOs, but our failure to detect GRIP or AMPA receptors in the MALS complex suggests no role for postsynaptic liprin-α in our analysis. Because MALS mutant mice die around birth, conditional mutants of the MALS may be required to evaluate roles for this complex in synaptic morphogenesis.
This study provides the first evidence that the MALS complex plays an important role in controlling transmitter release at excitatory synapses in brain. The myriad of interactions for the core MALS–CASK complex has suggested diverse roles; however, decisive genetic evidence has been lacking. Our work is consistent with a model whereby this complex recruits components of the synaptic release machinery to adhesive proteins of the active zone. According to this proposal, the MALS–CASK complex would couple extracellular synaptic interactions to the intercellular organization of the presynaptic secretory machinery.
Materials and methods
Antibodies
Isoform-specific and pan antibodies against MALS-1, -2, and -3 were generated in rabbits as described previously (Misawa et al., 2001). Mouse anti–liprin-α1 (C3-77) and -α2 (245:2:1) antibodies have also been described previously (Serra-Pages et al., 1998). Mouse anti-CASK, MINT, ZO-2, Sec8, β-catenin, and N-cadherin were all purchased from Transduction Labs. Munc-18, Rab5, and neuroligin antibodies were purchased from Synaptic Systems GmbH. Rabbit anti-CASK and antisynaptophysin were obtained from Zymed Laboratories. The mouse anti–Erb-B4 antibody was purchased from Lab Vision/Neomarkers and anti–PSD-95 antibody was obtained from Affinity BioReagents, Inc. The mouse anti-GluR 2/4 antibody was purchased from Chemicon International and rabbit synaptophysin was obtained from Sigma-Aldrich.
Immunoprecipitations
Using a Potter homogenizer (Braun), adult mouse brains from either MALS-3 heterozygote or knockout mice were homogenized in three volumes of STE buffer (320 mM sucrose, 20 mM Tris, pH 8.0, and 2 mM EDTA) containing 10 μg/ml leupeptin and 200 μg/ml PMSF. Homogenates were spun at 20,000 g for 1 h and pellets were resuspended in TET buffer (20 mM Tris, pH 8.0, 1 mM EDTA, and 1.3% Triton X-100) containing 10 μg/ml leupeptin and 50 μg/ml PMSF. After rehomogenization with a Potter homogenizer and a 1-h incubation at 4°C, the lysates were spun at 100,000 g for 1 h. The supernatant was collected and precleared with protein A–Sepharose (GE Healthcare) for 1 h at 4°C. Precleared lysates were immunoprecipitated with 10 μg MALS-3 antibody or control rabbit IgG for 2 h at 4°C. To collect immunoprecipitated protein complexes, 80 μl of a 50% protein A–Sepharose slurry was added to the lysates and incubated for 1 h at 4°C. Immunoprecipitates were washed extensively and loaded onto SDS-PAGE to separate the proteins. Gels were either silver stained or transferred to nitrocellulose for Western blotting. Immunoprecipitations from COS cells were performed similarly, except that the cells were directly lysed in TET buffer and spun at 20,000 g for 10 min, and supernatants were collected.
Immunocytochemistry
Hippocampal cultures grown on poly-d-lysine–treated coverslips were fixed with 4% PFA on ice for 10 min followed by methanol at −20°C. After thorough washing with PBS containing 0.1% Triton X-100 (PBS-X), cells were blocked with PBS-X containing 3% normal goat serum (blocking solution). Primary antibodies were incubated for 2 h at RT or 4°C overnight in blocking solution. Cells were washed three times with PBS-X and incubated with secondary antibodies in blocking solution for 1 h at RT. Cells were washed with PBS-X and mounted onto slides with Fluoromount-G (Southern Biotechnology Associates, Inc.).
Colocalization experiments in neurons were performed using images acquired from a confocal microscope (model LSM510; Carl Zeiss MicroImaging, Inc.) and analyzed using image analysis software (MetaMorph). Neurons were chosen randomly for quantification. For all images, thresholds were set at a predetermined level and colocalization values were obtained using MetaMorph software. Statistical significance was determined by an unpaired t test.
Nano-LC-ESI-Qq-TOF tandem mass spectrometry (MS) analysis
Individual gel bands were reduced with 10 mM dithiothreitol at 56°C for 1 h followed by alkylation with 55 mM iodoacetamide for 45 min at RT. The proteins were digested overnight with 12 ng/μL trypsin at 37°C. Peptides were extracted with a 50% acetonitrile/5% formic acid solution. The peptides were dried down and resuspended in 0.1% formic acid, and then separated via HPLC using a 75 μM × 15 cm reverse phase C-18 column (LC Packings) at a flow rate of 350 nl/min running a 3–32% acetonitrile gradient in 0.1% formic acid on an 1100 series HPLC (Agilent Technologies). The LC eluent was coupled to a micro-ionspray source attached to a mass spectrometer (model QSTAR Pulsar; MDS Sciex). Peptides were analyzed in positive ion mode. MS spectra were acquired for 1 s, followed by 3-s MS/MS on the most intense multiply charged peak.
Subcellular fractionation
Subcellular fractions of rat brain were prepared by differential centrifugation. Brains were homogenized in buffer containing 320 mM sucrose and 10 mM Hepes-NaOH, pH 7.4. Homogenate (C, crude lysate) was centrifuged for 10 min at 1,000 g to produce a pellet. The supernatant was centrifuged at 13,800 g for 10 min to produce a pellet (P13.8) and supernatant. The pellet was resuspended in the original volume of homogenization buffer and centrifuged at 100,000 g to yield a pellet (P100, crude synaptosomal vesicle pellet) and supernatant (S100, crude cytosolic synaptosomal supernatant). Synaptosome and PSD fractions were isolated by a discontinuous sucrose gradient centrifugation using P13.8. The pellet was extracted twice with ice cold 0.5% Triton X-100 (synaptosome) and then centrifuged to obtain the PSD pellet.
Cell culture and transfections
Hippocampal cultures were prepared as described previously (Tomita et al., 2003). In brief, hippocampi were dissected from 17- and 18-d-old embryos, digested with papain solution and plated at a density of 5 × 104 neurons/well in 24-well dishes. Neurons were cultured in Neurobasal media (GIBCO BRL) supplemented with B27, penicillin, streptomycin, and l-glutamine according to the manufacturer's protocol (GIBCO BRL). COS cells were grown in DME supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Transfections were performed using LipofectAMINE Plus (Invitrogen) according to the manufacturer's protocols. Transfected cells were used for immunocytochemistry or immunoprecipitations 48 h after the transfection of plasmids.
Constructs
pcDNA3 MALS-1, -2, and -3 and HA-liprin-α2 constructs have been described previously (Serra-Pages et al., 1998; Misawa et al., 2001). For construction of EGFP-MALS and CASK, full-length clones were amplified by PCR and cloned downstream of EGFP. For construction of yeast two-hybrid clones, liprin-α2 constructs were amplified by PCR and cloned into the pGBKT7 vector. Liprin-α2 constructs were as follows: full-length (aa 2–1257), SAMS 1/2/3 (aa 705–1257), SAM 1 (aa 876–984), SAM 2 (aa 997–1105), and SAM 3 (aa 1086–1197). MALS and CASK clones were amplified and cloned into the pGADT7 vector as follows: full-length MALS-1 (aa 2–233), full-length CASK (aa 2–909), CASK STK/L27AB (aa 2–476), CASK L27/PDZ (aa 276–590), CASK PDZ/SH3GK (aa 476–909), CASK STK/L27A (aa 2–405), and CASK STK (aa 2–346). Viral constructs were subcloned from pGBKT7 into a modified pBK-CMV plasmid to produce an NH2-terminal GFP-fusion and then subcloned into PSCA1 plasmid.
Yeast two-hybrid
Yeast cotransformation was performed according to the manufacturer's protocol (CLONTECH Laboratories, Inc.) with the yeast strain AH109. Binding was assessed by quantification of colonies transformed with the indicated plasmids on –LWH plates. Interactions were scored as positive (+) if many (>50) individual colonies were observed or negative (−) if no colonies were present on the –LWH plate. Control plates (–LW) were used to verify transformation efficiency.
Isolation of MALS-3 genomic DNA and construction of targeting vector
A mouse MALS-3 cDNA probe was used to isolate bacterial artificial chromosome clones from a 129Sv/J mouse genomic library (Genome Systems, Inc.). The targeting vector was constructed using the pPNT replacement vector. A 1-kb region downstream from the targeted exons was PCR-amplified, digested with XhoI and NotI, and subcloned into the XhoI–NotI sites of pPNT. Similarly, a 5-kb genomic region upstream from the targeted exons was PCR-amplified, digested with BamHI and XbaI, and inserted into the BamHI–XbaI sites of the pPNT vector. In the targeting vector, the third, fourth, and fifth exons (as well as the third and fourth introns of MALS-3) were replaced with a neocassette.
Generation of MALS-3 null mice
The targeting vector was linearized with BamHI and electroporated into R-1 embryonic stem cells. Clones resistant to G418 and gancyclovir were analyzed for recombination by PCR. To ensure proper homologous recombination, PCR-positive clones were further analyzed by Southern blotting using probes containing genomic sequences outside of the targeting vector and with a neoprobe. Properly targeted clones were injected into blastocysts from C57BL6 (The Jackson Laboratory) mice and transferred to surrogate mothers. Male chimeras were mated with C57BL6 females for transmission of the mutated allele through the germ line. Heterozygous mice were interbred to generate MALS-3 null mice. The genotypes from these and subsequent matings were determined by Southern blotting or PCR using allele-specific primers as follows: GGAAGAAATGGAGTCCCGCTTTG and ACAGCAGGACAGAACTGTCC for the WT allele; GCTAAAGCGCATGCTCCAGACTG and ACAGCAGGACAGAACTGTCC for the targeted allele. The null phenotype was confirmed by Western blotting of brain homogenates with antibodies to MALS-3.
Generation of MALS TKO mice
Female MALS-1/2 null mice (Misawa et al., 2001) and male MALS-3 null mice were mated, generating MALS-1/2/3 triple heterozygous mice. Triple heterozygous mice were bred back to MALS-1/2 null mice, and MALS-1/2 double null and 3 heterozygous mice were selected. These mice were interbred to generate MALS-1/2/3 triple null mice. Genotyping for the MALS-1 and -2 targeted loci has been described previously (Misawa et al., 2001).
Electrophysiology
Microdot cultures were prepared as described previously (Bekkers and Stevens, 1991; Augustin et al., 1999). For whole cell voltage-clamp recordings in autaptic culture (10–18 days in vitro [DIV]), patch pipette solutions contained the following: 135 mM potassium gluconate, 10 mM Hepes, 1 mM EGTA, 4.6 mM MgCl2, 4 mM NaATP, 15 mM creatine phosphate, 50 U/ml phosphocreatine kinase, pH 7.3, and 300 mM mOsm. The extracellular solution contained 140 mM NaCl, 2.4 mM KCl, 10 mM Hepes, 10 mM glucose, 4 mM CaCl2, 4 mM MgCl2, pH 7.3, and 300 mM mOsm. To determine the ratio of AMPA/NMDA EPSCs, MgCl2 was omitted from the extracellular solution; the patch pipette and extracellular solutions were prepared as previously described (Tovar and Westbrook, 1999). Cells were held at −70 mV and stimulated at 0.1 Hz with a 1–4 ms 80 mV depolarizing current pulse. Pyramidal cells were distinguished based on the decay kinetics of the evoked current and by application of 10 μM CNQX at the end of the experiment.
Immunohistochemistry
Adult mice were anesthetized with pentobarbital and perfused with 4% PFA in 0.1 M phosphate buffer. The brain was removed and immersed in the same fixative for 4 h at 4°C and then cryoprotected in 20% sucrose in PBS overnight at 4°C. 35-μm free-floating sections were cut on a sliding microtome. Endogenous peroxidase activity was inactivated by incubating brain sections in 0.5% H2O2 for 10 min. Sections were blocked for 1 h in PBS containing 3% normal goat serum and then incubated in the same buffer containing diluted 0.1 μg/ml MALS-3 antibody for 2 d at 4°C. Immunohistochemical staining was performed with an avidin/biotin/peroxidase system (ABC Elite; Vector Laboratories) and DAB (Vector Laboratories).
Online supplemental material
Fig. S1 shows the targeted disruption of the MALS-3 gene. Fig. S2 shows the colocalization of MALS and liprin-α with PSD-95. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200503011/DC1.
Acknowledgments
The authors wish to thank David R.C. House for his technical help.
This work was supported by grants (to D.S. Bredt, R.A. Nicoll, O. Olsen, and K.A. Moore) from the National Institutes of Health and from the Christopher Reeves Paralysis Foundation (to D.S. Bredt).
The authors declare there is no financial conflict of interest related to this work.
Abbreviations used in this paper: AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate; CaMK, CAM kinase; DIV, days in vitro; EPSC, excitatory postsynaptic current; MS, mass spectrometry; NMDA, N-methyl-d-aspartate; PSD, postsynaptic density; SAM, sterile α motif; TKO, triple knockout; WT, wild type.
References
- Augustin, I., C. Rosenmund, T.C. Südhof, and N. Brose. 1999. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 400:457–461. [DOI] [PubMed] [Google Scholar]
- Bekkers, J.M., and C.F. Stevens. 1991. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc. Natl. Acad. Sci. USA. 88:7834–7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg, J.-P., Y. Yang, M. De Taddéo-Borg, B. Margolis, and R.S. Turner. 1998. The X11alpha protein slows cellular amyloid precursor protein processing and reduces Abeta40 and Abeta42 secretion. J. Biol. Chem. 273:14761–14766. [DOI] [PubMed] [Google Scholar]
- Borg, J.-P., M.O. Lõpez-Figueroa, M. de Taddèo-Borg, D.E. Kroon, R.S. Turner, S.J. Watson, and B. Ben Margolis. 1999. Molecular analysis of the X11-mLin-2/CASK complex in brain. J. Neurosci. 19:1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butz, S., M. Okamoto, and T.C. Südhof. 1998. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell. 94:773–782. [DOI] [PubMed] [Google Scholar]
- Fannon, A.M., and D.R. Colman. 1996. A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron. 17:423–434. [DOI] [PubMed] [Google Scholar]
- Flanagan, J.G., and P. Vanderhaeghen. 1998. The ephrins and Eph receptors in neural development. Annu. Rev. Neurosci. 21:309–345. [DOI] [PubMed] [Google Scholar]
- Garcia, R.A., K. Vasudevan, and A. Buonanno. 2000. The neuregulin receptor ErbB-4 interacts with PDZ-containing proteins at neuronal synapses. Proc. Natl. Acad. Sci. USA. 97:3596–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata, Y., S. Butz, and T.C. Südhof. 1996. CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J. Neurosci. 16:2488–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, A., W. Morishita, R.E. Hammer, R.C. Malenka, and T.C. Südhof. 2003. A role for Mints in transmitter release: Mint 1 knockout mice exhibit impaired GABAergic synaptic transmission. Proc. Natl. Acad. Sci. USA. 100:1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichtchenko, K., Y. Hata, T. Nguyen, B. Ullrich, M. Missler, C. Moomaw, and T.C. Südhof. 1995. Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell. 81:435–443. [DOI] [PubMed] [Google Scholar]
- Jo, K., R. Derin, M. Li, and D.S. Bredt. 1999. Characterization of MALS/Velis-1, -2, and -3: a family of mammalian LIN-7 homologs enriched at brain synapses in association with the postsynaptic density-95/NMDA receptor postsynaptic complex. J. Neurosci. 19:4189–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech, S.M., C.W. Whitfield, and S.K. Kim. 1998. The LIN-2/LIN-7/LIN-10 complex mediates basolateral membrane localization of the C. elegans EGF receptor LET-23 in vulval epithelial cells. Cell. 94:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann, N., J. DeProto, R. Ranjan, H. Wan, and D. Van Vactor. 2002. Drosophila liprin-alpha and the receptor phosphatase Dlar control synapse morphogenesis. Neuron. 34:27–38. [DOI] [PubMed] [Google Scholar]
- Laverty, H.G., and J.B. Wilson. 1998. Murine CASK is disrupted in a sex-linked cleft palate mouse mutant. Genomics. 53:29–41. [DOI] [PubMed] [Google Scholar]
- Lee, S., S. Fan, O. Makarova, S. Straight, and B. Margolis. 2002. A novel and conserved protein-protein interaction domain of mammalian Lin-2/CASK binds and recruits SAP97 to the lateral surface of epithelia. Mol. Cell. Biol. 22:1778–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa, H., Y. Kawasaki, J. Mellor, N. Sweeney, K. Jo, R.A. Nicoll, and D.S. Bredt. 2001. Contrasting localizations of MALS/LIN-7 PDZ proteins in brain and molecular compensation in knockout mice. J. Biol. Chem. 276:9264–9272. [DOI] [PubMed] [Google Scholar]
- Missler, M., W. Zhang, A. Rohlmann, G. Kattenstroth, R.E. Hammer, K. Gottmann, and T.C. Südhof. 2003. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 423:939–948. [DOI] [PubMed] [Google Scholar]
- Okamoto, M., and T.C. Südhof. 1997. Mints, Munc18-interacting proteins in synaptic vesicle exocytosis. J. Biol. Chem. 272:31459–31464. [DOI] [PubMed] [Google Scholar]
- Perego, C., C. Vanoni, S. Massari, R. Longhi, and G. Pietrini. 2000. Mammalian LIN-7 PDZ proteins associate with beta-catenin at the cell-cell junctions of epithelia and neurons. EMBO J. 19:3978–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongo, C., C.W. Whitfield, A. Rodal, S.K. Kim, and J.M. Kaplan. 1998. LIN-10 is a shared component of the polarized protein localization pathways in neurons and epithelia. Cell. 94:751–759. [DOI] [PubMed] [Google Scholar]
- Schoch, S., P.E. Castillo, T. Jo, K. Mukherjee, M. Geppert, Y. Wang, F. Schmitz, R.C. Malenka, and T.C. Südhof. 2002. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 415:321–326. [DOI] [PubMed] [Google Scholar]
- Serra-Pages, C., Q.G. Medley, M. Tang, A. Hart, and M. Streuli. 1998. Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J. Biol. Chem. 273:15611–15620. [DOI] [PubMed] [Google Scholar]
- Setou, M., T. Nakagawa, D.-H. Seog, and N. Hirokawa. 2000. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 288:1796–1802. [DOI] [PubMed] [Google Scholar]
- Shelly, M., Y. Mosesson, A. Citri, S. Lavi, Y. Zwang, N. Melamed-Book, B. Aroeti, and Y. Yarden. 2003. Polar expression of ErbB-2/HER2 in epithelia. Bimodal regulation by Lin-7. Dev. Cell. 5:475–486. [DOI] [PubMed] [Google Scholar]
- Südhof, T.C. 2004. The synaptic vesicle cycle. Annu. Rev. Neurosci. 27:509–547. [DOI] [PubMed] [Google Scholar]
- Tomita, S., L. Chen, Y. Kawasaki, R.S. Petralia, R.J. Wenthold, R.A. Nicoll, and D.S. Bredt. 2003. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J. Cell Biol. 161:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres, R., B.L. Firestein, J. Staudinger, H. Dong, E.N. Olson, R.L. Huganir, D.S. Bredt, N.W. Gale, and G.D. Yancopoulos. 1998. PDZ proteins bind, cluster and synaptically co-localize with Eph receptors and their ligands, the ephrins. Neuron. 21:1453–1463. [DOI] [PubMed] [Google Scholar]
- Tovar, K.R., and G.L. Westbrook. 1999. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J. Neurosci. 19:4180–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski, M., E. Kim, A.W. Dunah, M. Passafaro, J.G. Valtschanoff, C. Serra-Pages, M. Streuli, R.J. Weinberg, and M. Sheng. 2002. Interaction between GRIP and liprin-alpha/SYD2 is required for AMPA receptor targeting. Neuron. 34:39–52. [DOI] [PubMed] [Google Scholar]
- Zhen, M., and Y. Jin. 1999. The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature. 401:371–375. [DOI] [PubMed] [Google Scholar]