

Abstract
Palmitoylation of the vacuolar membrane protein Vac8p is essential for vacuole fusion in yeast (Veit, M., R. Laage, L. Dietrich, L. Wang, and C. Ungermann. 2001. EMBO J. 20:3145–3155; Wang, Y.X., E.J. Kauffman, J.E. Duex, and L.S. Weisman. 2001. J. Biol. Chem. 276:35133–35140). Proteins that contain an Asp-His-His-Cys (DHHC)–cysteine rich domain (CRD) are emerging as a family of protein acyltransferases, and are therefore candidates for mediators of Vac8p palmitoylation. Here we demonstrate that the DHHC-CRD proteins Pfa3p (protein fatty acyltransferase 3, encoded by YNL326c) and Swf1p are important for vacuole fusion. Cells lacking Pfa3p had fragmented vacuoles when stressed, and cells lacking both Pfa3p and Swf1p had fragmented vacuoles under normal growth conditions. Pfa3p promoted Vac8p membrane association and palmitoylation in vivo and partially purified Pfa3p palmitoylated Vac8p in vitro, establishing Vac8p as a substrate for palmitoylation by Pfa3p. Vac8p is the first N-myristoylated, palmitoylated protein identified as a substrate for a DHHC-CRD protein.
Introduction
Yeast vacuoles are organelles with roles in degradation and storage. In the budding yeast Saccharomyces cerevisiae, vacuoles undergo cycles of fragmentation and fusion (Jones et al., 1993). The fusion of vacuole fragments, termed homotypic vacuole fusion, has been a model system for studying membrane fusion. Vacuole fusion is a SNARE-mediated event that proceeds in three steps: disassembly of cis-SNARE complexes, docking of vacuoles, and membrane fusion (Wickner and Haas, 2000).
Modification of proteins with the fatty acid palmitate has an important role in membrane fusion events. Both intra-Golgi transport and vacuole fusion are stimulated by palmitoyl-Coenzyme A (palm-CoA) and are inhibited by the palmitoylation inhibitor 2-bromo-palmitate (Pfanner et al., 1990; Haas and Wickner, 1996; Veit et al., 2001). An important target of palmitoylation for homotypic vacuole fusion is Vac8p, a 63-kD protein composed primarily of 11 armadillo repeats. Vac8p is palmitoylated on one or more of three cysteine residues (Cys 4, 5, and 7) adjacent to the myristoylated NH2 terminus (Wang et al., 1998). Vac8p is essential for several membrane trafficking events in yeast, including vacuole fusion, vacuole inheritance, cytoplasm to vacuole targeting, and nuclear autophagy (Fleckenstein et al., 1998; Pan and Goldfarb, 1998; Wang et al., 1998, 2001; Pan et al., 2000). Of these processes, the NH2-terminal cysteine residues are required for vacuole fusion and inheritance, but are dispensable for cytoplasm to vacuole targeting (Wang et al., 1998, 2001).
Vac8p is present on vacuole membranes (Fleckenstein et al., 1998; Pan and Goldfarb, 1998; Wang et al., 1998) and both N-myristoylation and palmitoylation of Vac8p are important for this localization (Wang et al., 1998). During in vitro vacuole fusion assays, Vac8p is palmitoylated on purified vacuoles after disassembly of the cis-SNARE complexes (Veit et al., 2001). Although Vac8p is palmitoylated after priming, Vac8p does not appear to be required until later in the fusion reaction. Vacuoles lacking Vac8p proceed through docking and formation of trans-SNARE pairs, but do not fuse (Wang et al., 2001). In cells this translates to a fragmented vacuole phenotype (Pan and Goldfarb, 1998; Wang et al., 1998; Seeley et al., 2002). The specific role of Vac8p in fusion is unclear, but is likely to involve protein–protein interactions mediated by its many armadillo repeats. Because Vac8p still partially localizes to the vacuole membrane in the absence of palmitoylation, it has been proposed that palmitoylation may influence the function of Vac8p by directing it to subdomains within the vacuole membrane (Wang et al., 2001).
The importance of Vac8p palmitoylation for membrane fusion has generated interest in identifying the proteins that regulate this modification. Ykt6p is an essential SNARE protein involved in many membrane trafficking events including vacuole fusion (Ungermann et al., 1999). Inclusion of α-Ykt6p antibodies in vacuole fusion assays blocks both membrane fusion and palmitoylation of Vac8p (Dietrich et al., 2004). In vitro, the NH2-terminal longin domain of Ykt6p promotes Vac8p palmitoylation in a nonenzymatic manner (Dietrich et al., 2004). The role of Ykt6p in Vac8p palmitoylation in vivo has not been explored.
A family of proteins characterized by a conserved DHHC-CRD (Bohm et al., 1997; Putilina et al., 1999) has recently been linked to protein palmitoylation. Two yeast DHHC-CRD proteins, Erf2p (in complex with Erf4p) and Akr1p, palmitoylate COOH-terminal cysteine residues of the small GTPase Ras2p and the kinase Yck2p, respectively (Lobo et al., 2002; Roth et al., 2002). Both Erf2p and Akr1p require the DHHC-CRD for protein acyltransferase (PAT) activity suggesting that it is a PAT domain. We sought to determine whether the other yeast DHHC-CRD proteins also function as PATs. In this study we present genetic and biochemical evidence that the gene product of the uncharacterized open reading frame YNL326c is a PAT for Vac8p. Accordingly, we have assigned the name PFA3 (protein fatty acyltransferase 3), as this is the third member of the DHHC-CRD family found to have PAT activity. Pfa3p is the first DHHC-CRD protein found to palmitoylate a cysteine motif near a myristoylated NH2 terminus.
Results
DHHC-CRD proteins are required for vacuole fusion
As a first step toward identifying palmitoylation substrates of DHHC-CRD proteins, we generated yeast strains with the five small DHHC-CRD genes deleted (ERF2, PFA3/YNL326c, YOL003c, YDR459c, and SWF1) in various combinations. When examining the vacuole morphology of these strains by staining with the lipophilic dye FM 4–64, we found that pfa3Δswf1Δ cells had highly fragmented vacuoles (Fig. 1). The accumulation of small clustered vacuoles indicates that these cells have a defect in vacuole fusion. This is a synthetic phenotype, because pfa3Δ or swf1Δ cells have normal vacuole morphology when grown in rich media. The observed phenotype is a consequence of the gene deletions, because vacuole fusion is rescued by expression of either Pfa3p-2xFlag or Swf1p-myc (unpublished data).
Figure 1.

PFA3 and SWF1 promote vacuole fusion. Cells were grown in YPD or treated as indicated in minimal media (2 mM DTT for 2 h or 0.05% glucose for 4 h) and stained with FM 4–64 before microscopy. Bars, 2 μm.
Further examination of the pfa3Δ strain revealed that aberrant vacuole morphology was induced when the cells were stressed. In the presence of DTT, pfa3Δ vacuoles became highly fragmented whereas vacuole morphology in the wild-type (WT) strain did not change (Fig. 1). The same phenotype was detected when the cells were grown under conditions of glucose limitation (Fig. 1). We also observed DTT-induced vacuole fragmentation in the pfa3Δ strain from the Saccharomyces genome deletion project (Winzeler et al., 1999) and in pfa3Δ cells derived from YPH499 (unpublished data). Vacuole morphology of swf1Δ cells was not perturbed by DTT treatment (unpublished data). This suggests that both PFA3 and SWF1 are important for vacuole fusion, but PFA3 has a primary role that is apparent under stressful conditions in the single deletion strain.
As both Pfa3p and Swf1p contain DHHC-CRDs that are hypothesized to have PAT activity (Linder and Deschenes, 2003), the vacuole fragmentation in the deletion strains could be related to palmitoylation of one or more substrates. The fragmented vacuoles observed in pfa3Δswf1Δ or stressed pfa3Δ cells are strikingly similar to a reported phenotype of cells lacking the palmitoylated armadillo repeat-containing protein Vac8p. Interestingly, the vacuole fragmentation in vac8Δ cells increases when the cells are stressed by placement into hypotonic media (Wang et al., 2001). We created a vac8Δ strain and examined vacuole morphology. In rich media, some fragmentation was observed (Fig. 1). However, upon treatment with DTT, fragmentation increased dramatically, mimicking the shift observed in pfa3Δ cells. Given that Vac8p palmitoylation is required for vacuole fusion (Veit et al., 2001; Wang et al., 2001), we hypothesized that the phenotype observed in pfa3Δ or pfa3Δswf1Δ cells may be related to loss of Vac8p palmitoylation.
Pfa3p localizes to the vacuole membrane
To examine the localization of Pfa3p, a 3xGFP tag was integrated into the PFA3 locus, resulting in a COOH-terminally tagged protein. Fluorescence microscopy revealed association of Pfa3p-3xGFP with the vacuolar membrane, as indicated by codistribution with FM 4–64 (Fig. 2). This is consistent with the localization reported in the Yeast GFP Fusion Localization Database (yeastgfp.ucsf.edu; Huh et al., 2003). However, the images in the database show diffuse fluorescence in the vacuole lumen, whereas in our strain the fluorescence is concentrated on the limiting membrane. Although both proteins are COOH-terminally tagged, the database strain is the result of a single GFP integration that may account for the different localizations. The Pfa3p-3xGFP is functional because the vacuoles in this strain retained WT morphology upon DTT treatment (unpublished data). The vacuolar localization of both Pfa3p and Vac8p further supports the idea that these proteins are linked functionally. By contrast, Swf1p has been localized to the ER (Valdez-Taubas and Pelham, 2005).
Figure 2.

Pfa3p-3xGFP localizes to the vacuole membrane. Cells expressing Pfa3p-3xGFP (YML163) were stained with FM 4–64 and visualized by microscopy. Bars, represent 2 μm.
Vacuole fusion depends on PAT activity of Pfa3p
If Pfa3p promotes vacuole fusion through its predicted PAT activity then the DHHC-CRD is likely to play an important role in Pfa3p function. To examine the role of the Pfa3p DHHC-CRD in vacuole fusion, we sought to inactivate the DHHC-CRD within the context of the full-length protein. In Erf2p and Akr1p, mutation of the cysteine within the DHHC motif disrupts function in vivo by abolishing substrate palmitoylation (Lobo et al., 2002; Roth et al., 2002). Using this as a model for making an inactive Pfa3p, we mutated the DHHC cysteine, Cys134, to serine.
To explore the ability of PFA3 alleles to function in vacuole fusion, constructs using the endogenous PFA3 promoter and a COOH-terminal 2xFlag tag were created. WT PFA3 was placed in a centromeric plasmid to mimic endogenous expression levels. C134S Pfa3p-2xFlag could not be detected when expressed from a centromeric plasmid, so it was expressed from a high copy plasmid. Expression of both constructs was confirmed by Western blotting of whole cell lysates of pfa3Δ transformants (Fig. 3 B).
Figure 3.

An intact DHHC-CRD is required for PFA3 to function in vacuole fusion. (A) pfa3Δ cells transformed with vector (pRS313), PFA3-2xFlag (pML780), or C134S PFA3-2xFlag (pML782), were grown in selective media, treated with 2 mM DTT, and stained with FM 4–64 before microscopy. Pictures are representative of two independent transformants. Bars, 2 μm. (B) Whole cell lysates (50 μg) from pfa3Δ transformants used in A were probed with α-Flag antibody to detect expression of Pfa3p-2xFlag.
When expressed from a centromeric plasmid, Pfa3p-2xFlag rescued the vacuole fusion defect of DTT-treated pfa3Δ cells, as shown in Fig. 3 A. In contrast, C134S Pfa3p-2xFlag expression did not rescue the vacuole fusion defect. Thus we conclude that Cys134 of Pfa3p, and by extension a functional DHHC-CRD, is required for vacuole fusion.
Pfa3p promotes Vac8p palmitoylation and membrane association in vivo
Inactivation of the Pfa3p DHHC-CRD and loss of Vac8p palmitoylation have the same consequence: a defect in vacuole fusion. If Pfa3p palmitoylates Vac8p, we would expect to find a decrease in Vac8p palmitoylation in pfa3Δ cells. To evaluate the role of PFA3 in Vac8p palmitoylation in vivo, metabolic labeling with [3H]palmitate was performed. In pfa3Δ cells Vac8p-myc-GFP palmitoylation was decreased by 80% (Fig. 4 A), indicating that Pfa3p is required for a large portion of Vac8p palmitoylation. Surprisingly, Vac8p-myc-GFP palmitoylation was decreased to a similar level in pfa3Δ and pfa3Δswf1Δ cells. Considering SWF1 does not appear to influence Vac8p palmitoylation, it may promote vacuole fusion in some other manner.
Figure 4.
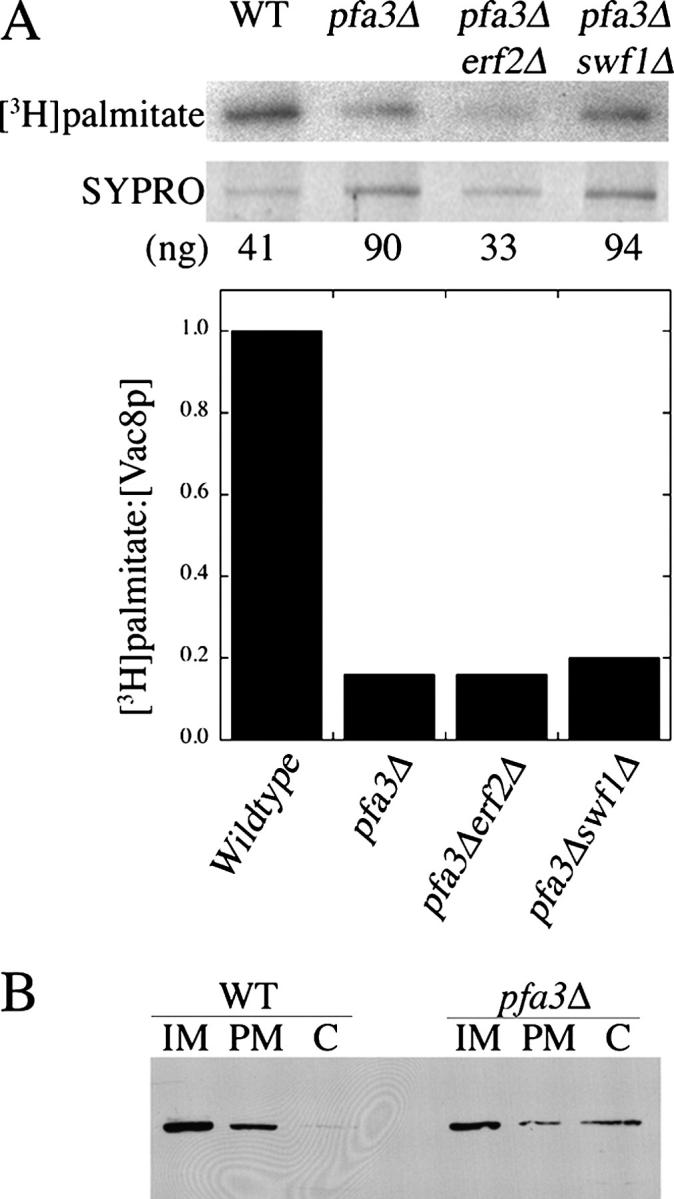
Pfa3p is required for Vac8p palmitoylation and membrane association in vivo. (A) WT, pfa3Δ, pfa3Δerf2Δ, and pfa3Δswf1Δ cells transformed with Vac8p-myc-GFP (pML735) were incubated with [3H]palmitate for 15 min before lysis and immunoprecipitation. Vac8p protein was quantitated from gels stained with SYPRO. Palmitoylation was detected by fluorography and quantitated by densitometry. Protein and [3H]palmitate values were normalized to wild type and expressed as ratios in the bar graph. (B) Whole cell lysates from WT or pfa3Δ cells transformed with Vac8p-myc-GFP underwent differential centrifugation to evaluate Vac8p association with internal membranes (IM, P13), plasma membrane (PM, P100), or cytosol (C, S100). Data shown in both A and B are representative of at least two experiments performed with independent transformants.
A reported consequence of the loss of Vac8p palmitoylation (through mutation of the three NH2-terminal cysteine residues) is an increase in cytosolic Vac8p (Wang et al., 1998). Because Vac8p palmitoylation was decreased in the absence of PFA3, we expected a corresponding increase in cytosolic Vac8p. This prediction was tested by examining the subcellular distribution of Vac8p in WT and pfa3Δ cells. Differential centrifugation was used to separate internal membranes (Fig. 4 B, IM), the plasma membrane (Fig. 4 B, PM), and cytosolic proteins (Fig. 4 B, C). As shown in Fig. 4 B, Vac8p-myc-GFP was concentrated in the internal membrane fraction in WT cells. In pfa3Δ cells, a substantial cytosolic fraction was detected, indicating that the decrease in palmitoylation we observed was sufficient to disrupt Vac8p membrane association.
Pfa3p autoacylates in vitro
Erf2p and Akr1p are palmitoylated in vitro when incubated with [3H]palm-CoA, and Akr1p is palmitoylated in vivo (Lobo et al., 2002; Roth et al., 2002). Mutations that block Erf2p and Akr1p palmitoylation also block substrate palmitoylation, indicating that the two events are linked (Lobo et al., 2002; Roth et al., 2002). To determine if Pfa3p shares this characteristic, Pfa3p-Flag was overexpressed in yeast. Membranes prepared from these yeast were evaluated for protein palmitoylation by incubation with [3H]palm-CoA followed by fluorography. As shown in Fig. 5, Pfa3p-Flag incorporated palmitate (lane 1). C134S Pfa3p-Flag was expressed at a similar level to WT Pfa3p-Flag, but did not autoacylate (Fig. 5, lane 2). The lack of autoacylation suggests that this mutant will also be deficient in PAT activity, and validates our use of this mutant as a loss of function allele in the rescue experiments described above.
Figure 5.

Pfa3p promotes Vac8p palmitoylation in membranes. Pfa3p-Flag (WT [pML395] or C134S[pML724]) and Vac8p-myc (WT [pML734] or Cys− [pML775]) constructs were coexpressed in yeast and membranes incubated with [3H]palm-CoA for 10 min and analyzed by fluorography or subjected to Western blot to evaluate protein expression. The result shown is representative of four experiments. The radiolabeled samples were exposed to film for 10 d.
Pfa3p promotes Vac8p palmitoylation in vitro
To determine whether Pfa3p palmitoylates Vac8p in vitro, we developed an assay based on the ability of Pfa3p-Flag to autoacylate in membranes. We hypothesized that overexpressed Pfa3p-Flag may be able to promote palmitoylation of a substrate that was also present in the membranes. To this end, Pfa3p-Flag and Vac8p-myc were coexpressed. When membranes were incubated with [3H]palm-CoA, incorporation of palmitate into Vac8p-myc was observed only in the presence of Pfa3p-Flag (Fig. 5, lane 4). Palmitoylation of Vac8p depended on Pfa3p autoacylation, as coexpression of C134S Pfa3p did not promote Vac8p palmitoylation (Fig. 5, lane 5). When coexpressed with Pfa3p-Flag, C4,5,7S Vac8p-myc (Cys−) was not palmitoylated (Fig. 5, lane 7), indicating that the reaction is occurring on the NH2-terminal cysteine residues. Both in vitro palmitoylation events occurred via the expected thioester linkage, as the incorporated palmitate was cleaved by treatment with hydroxylamine (unpublished data). In lanes 1 and 7, a weak palmitoylation signal is observed just below where the Vac8p-myc band is observed in lane 4. This likely represents palmitoylation of endogenous Vac8p present in the membranes.
The vacuole morphology phenotype and reduced palmitoylation of Vac8p in pfa3Δ cells suggest that Pfa3p is a PAT for Vac8p in yeast. To determine whether Vac8p palmitoylation is specific to Pfa3p, we tested Vac8p as a substrate for other DHHC-CRD proteins. The DHHC-CRD proteins were expressed in yeast as Flag-tagged constructs using a galactose-inducible promoter. Pfa3p, Akr1p, Erf2p (with its binding partner Erf4p), Yol003p, and Ydr459p expressed well, but Swf1p and Akr2p were expressed at low or undetectable levels and were not analyzed further (unpublished data). Membranes containing the overexpressed DHHC-CRD proteins were incubated with purified N-myristoylated Vac8p-myc-6xHis (myr-Vac8p) in a PAT assay. As shown in Fig. 6, autoacylation of the DHHC-CRD proteins was apparent in each case. However, Pfa3p-Flag membranes were the only ones to promote robust Vac8p palmitoylation. Low levels of Vac8p palmitoylation were observed with the Erf2p/Erf4p membranes. To test whether Erf2p/Erf4p contributes to palmitoylation of Vac8p in vivo, we performed radiolabeling studies in a pfa3Δerf2Δ strain. There was no further reduction in radioactive palmitate incorporation in Vac8p from that seen with pfa3Δ cells (Fig. 4 A), suggesting that Erf2p does not account for residual palmitoylation of Vac8p in these cells.
Figure 6.

Vac8p is selectively palmitoylated by Pfa3p. Membranes expressing Pfa3p-Flag, Yol003p-Flag, Ydr459p-Flag, Flag-Erf2p/GST-Erf4p, or Akr1p-Flag were incubated with 3.0 μg myr-Vac8p-myc-6xHis and [3H]palm-CoA for 10 min and analyzed by fluorography (5 d exposure). Data are representative of two independent experiments.
To further characterize the enzymology of Vac8p palmitoylation by Pfa3p, we sought to perform PAT assays with purified components. Pfa3p-Flag was expressed in yeast and purified by affinity-chromatography from membrane detergent extracts. A mock purification using detergent extracts from cells transformed with vector (pESC) was performed in parallel. PAT activity for Vac8p was enriched 36-fold in the Pfa3p preparation after Flag-immunoaffinity chromatography (Table I). No activity was detected in Flag peptide eluates from the vector control preparation. The silver stain shown in Fig. 7 A revealed a prominent 37-kD protein present only in the Pfa3p-Flag preparation. Except for an 82-kD protein, contaminating proteins were present in equivalent amounts in both the Pfa3p-Flag and the control preparations. The more abundant 82-kD protein was not reproducibly observed in silver stains of other Pfa3p preparations. Fig. 7 B shows that Flag immunoreactivity comigrated with the prominent 37-kD band in the silver stain. When incubated with [3H]palm-CoA in vitro, only the Pfa3p preparation exhibited a 37-kD protein that autoacylated (Fig. 7 C). Western blotting of a Pfa3p-Flag preparation demonstrated that Ykt6p did not copurify, although it was detected in the detergent extract (Fig. S1 http://www.jcb.org/cgi/content/full/jcb.200507048/DC1). These results establish that Vac8p palmitoylation by Pfa3p occurred independently of Ykt6p.
Table I.
Purification of Flag-Pfa3p
| Protein | Total Activity | Specific Activity | Purification | |
|---|---|---|---|---|
| mg | U | U/mg | fold | |
| Pfa3p | ||||
| Membranes | 10.0 | 88.0 | 8.8a | 1.0 |
| Extract | 6.0 | 64.0 | 10.7 | 1.2 |
| Elution | 0.005 | 1.6 | 320.0 | 36.0 |
| pESC | ||||
| Membranes | 10.0 | 1.2 | 0.12 | |
| Extract | 8.3 | 1.0 | 0.12 | |
| Elution | 0.001 | 0 | 0 |
One unit of activity is pmol of [3H]palm-Vac8p produced per minute.
Figure 7.

Partially purified Pfa3p-Flag autoacylates and palmitoylates Vac8p. Detergent extracts from Pfa3p-Flag–expressing cells or vector-transformed cells (pESC) were purified by Flag-immunoaffinity chromatography. Equal volumes of Pfa3p-Flag and pESC elutions were analyzed by (A) silver stain, (B) α-Flag Western blot, or (C) fluorography after incubation with [3H]palm-CoA. (D) Pfa3p-Flag (0.5 pmol) and myr-Vac8p-myc-6xHis (0.4 μg total protein/3 pmol Vac8p) were incubated with [3H]palm-CoA for the indicated time and analyzed by fluorography (inset; 4 d exposure) and scintillation counting (curve). Data are representative of two independent experiments.
The time course of Pfa3p-Flag autoacylation and myr-Vac8p-myc-6xHis palmitoylation is shown in Fig. 7 D. Autoacylation of Pfa3p-Flag occurred rapidly, reaching maximal levels within minutes (Fig. 7 D, inset). The amount of Pfa3p-Flag protein present in each reaction was ∼0.5 pmol. The amount of radiolabeled Pfa3p was not quantitated in these assays because the Pfa3p band was not visible on the gel to permit excision for scintillation counting. However, when filter-binding assays were used in parallel to quantitate the amount of palmitate incorporated into the Pfa3p-Flag, 15–20% of Pfa3p-Flag was palmitoylated after incubation with [3H]palm-CoA for 30 min. Palmitoylation of myr-Vac8p-myc-6xHis occurred with a slower time course; the assay was linear to 10 min and reached saturation at ∼60 min (Fig. 7 D, inset and curve). Palmitoylation of myr-Vac8p-myc-6xHis was dependent on substrate concentration (unpublished data). Maximal labeling of Vac8p occurred at a substoichiometric ratio of Pfa3p to Vac8p (0.5 pmol Pfa3p/15 pmol myr-Vac8p). Under these conditions, 1.1 pmol palmitate was incorporated into Vac8p, which is approximately twice the amount of Pfa3p-Flag protein in the assay. The ratio of pmol palmitate incorporated into Vac8p to pmol of PAT may be an underestimate if autoacylated Pfa3p-Flag represents the fraction of Pfa3p that can transfer palmitate to a substrate.
Discussion
We have identified a novel regulator of membrane fusion in yeast, the DHHC-CRD protein Pfa3p, and demonstrated that Pfa3p exerts its effects via palmitoylation of Vac8p. This is the first palmitoylation event assigned to Pfa3p, a vacuolar DHHC-CRD. In addition, we provide evidence that another DHHC-CRD protein, Swf1p, is involved in vacuole fusion through an unknown mechanism that may be independent of Vac8p. Pfa3p is the first DHHC-CRD protein reported to palmitoylate cysteine residues in the context of a NH2-terminal myristoylated motif. Our findings predict that members of the DHHC-CRD protein family will modify other N-myristoylated, palmitoylated proteins such as heterotrimeric G protein α subunits and nonreceptor tyrosine kinases.
Vacuoles in pfa3Δ cells are normal when the cells are grown in rich media, explaining why PFA3 was not identified in the genome-wide screen for vacuole morphology mutants (Seeley et al., 2002). In the absence of PFA3, there is still a low level of Vac8p palmitoylation that is apparently sufficient for the normal rate of vacuole fusion in a rapidly growing cell. Our results with pfa3Δ and vac8Δ cells suggest that cells increase vacuole fragmentation and fusion under stressful conditions. In pfa3Δ cells the low levels of palmitoylated Vac8p may not be able to support the increase in fusion, resulting in the accumulation of small vacuoles.
Whereas the fragmented vacuole phenotype of pfa3Δ cells is observed only under stressful conditions, Pfa3p is responsible for Vac8p palmitoylation under nonstressful conditions as well. Experiments performed under normal growth conditions show disruption of Vac8p palmitoylation and membrane association (Fig. 4). It is unknown if activity of DHHC-CRD proteins is regulated, but it would be of great interest to determine if Pfa3p activity is activated in response to stress to cope with a potentially increased demand for palmitoylated Vac8p.
Vacuole inheritance is another Vac8p function that has been reported to depend on palmitoylation (Wang et al., 1998). It is notable that pfa3Δ cells do not have a vacuole inheritance defect, although we did detect the defect in our vac8Δ strain (unpublished data). This suggests that a decreased amount of Vac8p palmitoylation is sufficient to support vacuole inheritance, or that the Vac8p cysteine residues have a palmitoylation-independent function. Alternatively, different cysteine residues may be important for the different functions of Vac8p. The Vac8p palmitoylation observed in pfa3Δ cells may be on a cysteine that is important for function in inheritance but not fusion.
The difference in vacuole morphology between pfa3Δ and pfa3Δswf1Δ cells indicates that Swf1p has a role in vacuole fusion. The similar level of Vac8p palmitoylation in pfa3Δ and pfa3Δswf1Δ cells suggests that Swf1p promotes vacuole fusion in a manner independent of Vac8p palmitoylation. The presence of a DHHC-CRD suggests that Swf1p is a PAT. It has recently been reported that deletion of SWF1 results in the loss of palmitoylation of the SNARE proteins Snc1, Syn8, and Tlg1 (Valdez-Taubas and Pelham, 2005). Swf1p is likely to be the enzyme responsible for palmitoylation of proteins modified at cysteine residues near their transmembrane domains like the aforementioned SNAREs as well as Vam3, a vacuolar t-SNARE (Valdez-Taubas and Pelham, 2005). Swf1p may exert its influence on vacuole fusion through palmitoylation of SNARE proteins that are involved in trafficking through the endosomal and Golgi compartments.
It has been reported that palmitoylation of Vac8p is mediated by the NH2-terminal longin domain of Ykt6p (Dietrich et al., 2004). Ykt6p is not present in our partially purified Pfa3p-Flag preparations that have activity toward Vac8p in vitro (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200507048/DC1). Therefore, we have discovered a PAT activity toward Vac8p that is independent of Ykt6p. A comparison of the properties of Ykt6p and Pfa3p suggest that they palmitoylate Vac8p by different mechanisms. The longin domain of Ykt6p binds palm-CoA (and CoA) through a noncovalent interaction and transfers it to Vac8p (Dietrich et al., 2004). The reaction does not appear to be enzymatic because it saturates at equimolar concentrations of longin domain and Vac8p (Dietrich et al., 2004). A direct comparison of the relative potencies of Ykt6p and Pfa3p is not possible because the amount of Vac8p palmitoylated by Ykt6p was not reported. In the case of Pfa3p, Vac8p palmitoylation is optimal at substoichiometric concentrations of Pfa3p to Vac8p. Maximal palmitoylation was observed at a 30:1 ratio of Vac8p/Pfa3p, suggesting that the reaction is catalytic. Similar to Erf2p and Akr1p, Pfa3p becomes acylated during the reaction and both autoacylation and palmitate transfer to Vac8p are dependent upon Cys134 in the DHHC motif. Future experiments will be directed at testing whether Pfa3p palmitoylation represents an acyl-intermediate.
Palmitoylation of Vac8p by Pfa3p-Flag occurs on a slow time scale, reaching saturation after 60 min. This may reflect suboptimal assay conditions. Pfa3p is an integral membrane protein and may function more effectively if reconstituted into liposomes. However, a slow turnover number for lipid-modifying enzymes is not unprecedented. Farnesyltransferase has a turnover number on the order of 1 min−1 (Reiss et al., 1990; Pompliano et al., 1993). It is also possible that the reaction we are observing is slow because we are missing another protein component. In the case of Erf2p, the Erf4p binding partner is required for both autoacylation and PAT activity (Lobo et al., 2002; and unpublished data). No proteins copurified with Pfa3p-Flag in stoichiometric quantities, however this could be because the Pfa3p-Flag was overexpressed. Erf4p is not likely to be a binding partner for Pfa3p because it is localized to the ER membrane (Zhao et al., 2002) whereas Pfa3p is on the vacuole.
Our genetic and biochemical data strongly argue that Pfa3p is a PAT for Vac8p. However, the residual Vac8p palmitoylation in pfa3Δ, pfa3Δswf1Δ, and pfa3Δerf2Δ cells argues for a second activity. The radiolabeling experiments were performed under conditions where Vac8p-myc-GFP was overexpressed. That in turn might allow Vac8p to encounter PATs that it would not associate with under physiological circumstances. However, vac8Δ cells have a more dramatic vacuole fragmentation phenotype under nonstressful conditions than pfa3Δ cells (Fig. 1), indicating that some endogenous Vac8p found in pfa3Δ cells is functional and therefore likely to be palmitoylated. Ykt6p and the DHHC-CRD proteins other than Pfa3p, Swf1p, and Erf2p are candidates for a second activity that palmitoylates Vac8p.
To date, a Vac8p homologue has not been identified in mammalian systems. However, the requirement for palm-CoA for mammalian Golgi fusion indicates that protein palmitoylation is important, even though the targets have not been identified (Glick and Rothman, 1987; Pfanner et al., 1990). Our results suggest that a DHHC-CRD protein may be involved in promoting Golgi fusion. Two mammalian DHHC-CRD proteins, HIP14 and GODZ, are present in the Golgi and are candidates for this role (Singaraja et al., 2002; Uemura et al., 2002). Continued efforts to localize and find substrates for DHHC-CRD proteins will further elucidate the roles of palmitoylation in membrane trafficking.
Materials and methods
Strains and constructs
Yeast culture and genetic manipulation were performed by standard methods (Adams et al., 1997). Yeast transformations were performed as described previously (Chen et al., 1992). SWY518 (MAT a) and SWY595 (MAT a/α) are ADE2-restored versions of the W303 background (Bucci and Wente, 1997). YPH499 is from Stratagene. Strains and plasmids are listed in Tables II and III.
Table II.
Plasmids used in this study
| Name | Description |
|---|---|
| pML395 | pESC-TRP, GAL promoter, PFA3-Flag |
| pML724 | pESC-TRP, GAL promoter, C134S pfa3-Flag |
| pML547 | pBS, PFA3-3xGFP-TRP1 |
| pML734 | pVT102U, ADH promoter, VAC8-myc |
| pML775 | pVT102U, ADH promoter, C4,5,7S vac8-myc |
| pML735 | pVT102U, ADH promoter, VAC8-myc-GFP |
| pML796 | pVT102U, ADH promoter, C4,5,7S vac8-myc-GFP |
| pML780 | pRS313, PFA3 promoter, PFA3-2xFlag |
| pML782 | pRS423, PFA3 promoter, C134S pfa3-2xFlag |
| pML810 | pVT102U, ADH promoter, SWF1-myc |
| pML658 | pQE60, VAC8-myc-6xHis |
| pBB131 | NMT1 (Duronio et al., 1990) |
| pML477 | pESC-TRP, GAL promoter, AKR1-Flag |
| pML393 | pESC-TRP, GAL promoter, YOL003c-Flag |
| pML394 | pESC-TRP, GAL promoter, YDR459c-Flag |
| Flag-ERF2 | pESC-TRP, GAL promoter, Flag-ERF2 (Lobo et al., 2002) |
| GST-ERF4 | pEG(KG), GAL promoter, GST-ERF4 (Lobo et al., 2002) |
Table III.
Strains used in this study
| Strain | Relevant genotype |
|---|---|
| SWY518 | WT |
| YML232 | pfa3Δ |
| YML236 | swf1Δ |
| YML237 | pfa3Δswf1Δ |
| YML249 | pfa3Δerf2Δ |
| YML216 | vac8Δ |
| YML163 | PFA3-3xGFP-TRP |
One copy of PFA3 was deleted in SWY595 with a PCR-generated kanamycin resistance cassette (pUG6; Guldener et al., 1996) to generate YML195. Dissection of YML195 generated a pfa3Δ haploid YML232. The haploid vac8Δ and swf1Δ strains (YML216 and YML236 respectively) were generated from SWY595 in the same manner. pfa3Δswf1Δ cells (YML237) and pfa3Δerf2Δ (YML249) were recovered from dissection of YML200 (PFA3/pfa3Δ SWF1/swf1Δ ERF2/erf2Δ YOL003c/yol003Δ YDR459c/ydr459Δ), which was created by repeated rounds of dissection and mating of strains carrying individual deletions. Integrations of deletion cassettes were confirmed by colony PCR. All haploids used in this study are MAT a.
All PCR products were ligated into pCR2.1-TOPO before subsequent cloning steps (Invitrogen). The PFA3, YOL003c, and YDR459c coding sequences were amplified from genomic DNA and cloned into pESC-TRP (Stratagene) as EcoRI-NotI fragments to generate pML395, pML393, and pML394 respectively. The AKR1 coding sequence was amplified from genomic DNA and cloned into pESC-TRP as a SpeI-SpeI fragment to generate pML477. Flag-Erf2p and GST-Erf4p expression constructs are described elsewhere (Lobo et al., 2002). For the plasmid rescue experiments, PFA3 plus 350 base pairs of the promoter region was amplified from genomic DNA and cloned into pRS313 (WT, pML780) or pRS423 (C134S, pML782) as a XhoI-BamHI fragment (Sikorski and Hieter, 1989). The 3′ primer used to amplify the region added a 2xFlag tag. Quikchange mutagenesis (Stratagene) was used to make the C134S pfa3 mutations in pML724 and pML782. The SWF1 coding region was amplified from genomic DNA and cloned into a modified Baculovirus vector (pBlueBac4.5B[−]) as a SpeI-SpeI fragment to add a COOH-terminal myc tag. SWF1-myc was then cut out of the pBlueBac construct (XhoI-SalI) and inserted into pVT102-U at the XhoI site to create pML810 (Vernet et al., 1987). VAC8 was amplified from genomic DNA with primers that added a 5′ BamHI site and a 3′ myc tag followed by a XhoI site, a stop codon, and a HindIII site. This product was cloned into pVT102U as a BamHI-HindIII fragment. GFP was then inserted into the COOH-terminal XhoI site to generate Vac8p-myc-GFP (pML735). C4,5,7S vac8 was made by amplifying VAC8 from pML734 with a mutagenic 5′ primer followed by cloning into pVT102U to generate pML775. Addition of GFP to pML775 resulted in pML796. All constructs and mutations were verified by sequencing.
Integration of a COOH-terminal 3xGFP tag into the PFA3 locus was made possible by modification of the pBS-3xGFP-TRP1 vector (Lee et al., 2003). A 3′ fragment of PFA3 (nucleotides 352–1005) was inserted into a BamHI site upstream of the 3xGFP to create pML547. This plasmid was digested within the PFA3 sequence at a unique restriction site, BsmI, and integrated into SWY595. Dissection yielded YML163 (MATa PFA3-3xGFP-TRP). Colony PCR and Western blotting confirmed proper integration.
The Vac8p-myc-6xHis bacterial expression plasmid was made by amplification of VAC8 from genomic DNA and cloning into pQE60 (QIAGEN) as an NcoI-BglII fragment. The 3′ primer added a COOH-terminal myc tag. The N-myristoyltransferase expression construct pBB131 is described elsewhere (Duronio et al., 1990).
Cell fractionation
WT and pfa3Δ cells were transformed with Vac8p-myc-GFP. Cells were grown in selective media and harvested in mid-log phase. Glass bead lysis and fractionation by differential centrifugation were performed as described (Wang et al., 1998). Whole cell lysates were adjusted to equal protein concentration before centrifugation. Equal percentages of the internal membranes (P13), plasma membrane (P100), and cytosol (S100) fractions were separated by SDS-PAGE and Western blotted with α-myc ascites.
Microscopy
Cells were grown and labeled in YPD unless treated with DTT or low glucose, in which case they were grown in minimal media. Rapidly growing cells were labeled with 20 μM FM 4–64 (Invitrogen) for 30 min and then collected and suspended in fresh media lacking FM 4–64 for 60 min (Vida and Emr, 1995). Cells were washed twice with nonfluorescent media (NF; 8 mM KPO4, pH 5.4, 2 mM MgSO4, 27 mM [NH4]2SO4, 2% glucose, 0.04× complete amino acids) before imaging. For DTT-treated samples, 2 mM DTT was added to cultures at the start of FM 4–64 labeling 2 h before microscopy. 2 mM DTT was maintained in the media after the labeling period and in the NF media washes. Low glucose samples were shifted to minimal media with 0.05% glucose 4 h before microscopy, and were washed and visualized in NF media containing 0.05% glucose.
Images were collected at room temperature with an Olympus IX-81 inverted microscope with a 100× objective, a camera (model CCD-300T-RC; Dage MTI) and NIH Image software. The only image processing was adjustment of brightness and/or contrast in Adobe Photoshop.
Metabolic palmitate labeling
Yeast cells were transformed with Vac8-myc-GFP constructs. Equal numbers of rapidly growing cells (25 OD600 units) were treated with 3 μg/ml cerulenin for 1 h and subsequently labeled with [3H]palmitate (45.0 Ci/mmol; NEN Life Science Products) at a final concentration of 0.2 mCi/ml for 15 min at 30°C. Cells were lysed as described (Wang et al., 1998). Whole cell lysates were extracted on ice for 1 h in lysis buffer containing 1% Triton X-100 (Roche), and then centrifuged at 15,000 g for 10 min. Vac8p-myc-GFP was immunoprecipitated from extracts with affinity-purified α-GFP antibody coupled to protein G–conjugated beads (GE Healthcare; Harlow and Lane, 1988). The GFP antibodies were generated and affinity purified as described (Seedorf et al., 1999). Immunoprecipitated proteins were resolved by SDS-PAGE and detected with SYPRO Ruby protein gel stain (Invitrogen). Vac8p was quantitated by extrapolation to a linear curve of known concentrations of bovine serum albumin. Gels were then soaked in fluor (15% methanol, 1 M sodium salicylate) and exposed to preflashed film. After fluorography, relative amounts of palmitoylation were determined by densitometry.
Expression of DHHC-CRD-Flag proteins
For purification of Pfa3p-Flag, pML395 or pESC were transformed independently into YPH499. Transformants were expanded in selective media with 2% ethanol and 2% glycerol as the carbon sources. Cells were induced with 1% galactose for 16 h at 23°C upon reaching OD600 = 0.4–0.6. For the in vitro membrane PAT assays presented in Fig. 5, WT (pML395) and C134S (pML724) Pfa3p-Flag constructs were cotransformed into YPH499 with Vac8p-myc constructs. For Fig. 6, YPH499 was independently transformed with DHHC-CRD-Flag expression constructs pML393, pML394, pML395, pML477, or Flag-ERF2 and GST-ERF4. DHHC-CRD-Flag expression was induced in the same manner as for the purifications, except that the galactose concentration was 4%. The different galactose concentration did not affect the level of Pfa3p-Flag expression.
Membrane preparation
Cells were lysed with a mini bead beater (Research Products International) in lysis buffer (LB: 50 mM Tris, pH 8.0, 150 mM NaCl, 10% glycerol, 1 mM EGTA, 1 mM EDTA, 7 μM pepstatin A, 10 μM leupeptin, 0.3 μM aprotinin, and 1 mM PMSF). After a low-speed spin to remove unbroken cells, lysates were centrifuged at 100,000 g for 20 min to collect the membrane fraction. Membranes were suspended in LB and protein concentration determined by Bradford assay.
In vitro PAT assays
[3H]Palm-CoA was synthesized as described previously (Dunphy et al., 1996). The in vitro assay contained 0.02% Triton X-100, 100 mM MES, pH 6.4, 1 mM DTT, and 0.4–0.8 μM [3H]palm-CoA. Assays were incubated at 30°C (Figs. 5 and 6 for 10 min, Fig. 7 C for 60 min) and stopped by addition of sample buffer containing 2 mM DTT. Samples were boiled for 1 min before SDS-PAGE. Gels were fixed and soaked in fluor before exposure to film at −80°C. For the quantification presented in Fig. 7 D, Vac8p was cut out of the gel after SDS-PAGE, Coomassie blue staining, and destaining. The gel slice was incubated in Soluene-350 (PE Corp.) at 50°C for 3 h before scintillation counting. Filter binding assays to evaluate Pfa3p-Flag autoacylation were performed as described (Dunphy et al., 1996) except for the addition of cytochrome c to a final concentration of 0.1 mg/ml before the addition of TCA/SDS to aid in protein precipitation.
Purification of Pfa3p-Flag and recombinant myristoylated Vac8p-myc-6xHis
Membranes expressing Pfa3p-Flag (5 mg/ml) were extracted with 1% Triton X-100 in LB for 1 h on ice. Insoluble material was pelleted at 100,000 g for 20 min. The extract was allowed to bind to α-Flag M2 agarose affinity gel (Sigma-Aldrich) for 1 h at 4°C. The beads were washed twice in wash 1 (LB: 0.3% Triton X-100, 0.1 mg/ml bovine liver lipids; Avanti Polar Lipids) and once with wash 2 (LB: 0.1% Triton X-100, 0.1 mg/ml bovine liver lipids). Bound proteins were eluted with wash 2 supplemented with 0.3 mg/ml Flag peptide (Sigma-Aldrich). A mock purification was performed in parallel with detergent extracts from cells transformed with the vector alone. Silver staining was performed essentially as described previously (Morrissey, 1981). Pfa3p-Flag concentration was determined by extrapolation to a linear curve of known concentrations of BSA using SYPRO Ruby protein gel stain (Molecular Probes) or estimated by comparison of Western blot signals to a Flag-tagged protein of known concentration. The extract and elutions were also probed with α-Ykt6p antibody, which was kindly provided by Dr. James McNew (Rice University, Houston, TX; McNew et al., 1997).
Recombinant myristoylated Vac8p-myc-6xHis was produced in Escherichia coli by coexpression with yeast N-myristoyltransferase (Duronio et al., 1990; Fig. S2). Details of the purification can be found in Online supplemental material. The protein concentration of the final preparation was determined by the method of Bradford. Approximately 50% of the final preparation was full-length myristoylated Vac8p-myc-6xhis, as estimated by examination of the Coomassie blue–stained gel (Fig. S2).
Online supplemental material
In Fig. S1, Ykt6p does not copurify with Pfa3p-Flag. Fig. S2 shows that recombinant Vac8-myc-6xhis is myristoylated. A description of the procedure for purification of myr-Vac8-myc-6xhis is available online. Online supplemental materials are available at http://www.jcb.org/cgi/content/full/jcb.200507048/DC1.
Acknowledgments
The authors would like to thank Drs. Robert Deschenes, John Swarthout, Heather True, and Kendall Blumer for helpful discussions and comments on the manuscript. We thank Dr. Kendall Blumer for the use of his microscope, Fanny Chang for assistance with microscopy, Wendy Greentree and Monica Croke for technical support, and Drs. Wei-Lih Lee, John Cooper, Robert Deschenes, and James McNew for reagents.
This study was supported by National Institutes of Health grant GM51466. J.E. Smotrys was supported by a Howard Hughes Medical Institute Predoctoral Fellowship and a Mr. and Mrs. Spencer T. Olin Fellowship for Women. M.J. Schoenfish is supported by the National Institutes of Health training grant T32GM07067 and the Lucille P. Markey Special Emphasis Pathway in Human Pathobiology. M.A. Stutz was supported by the Biomedical Research Apprenticeship Program summer program for undergraduate research. M.E. Linder is an Established Investigator of the American Heart Association.
J.E. Smotrys and M.J. Schoenfish contributed equally to this work.
Abbreviations used in this paper: DHHC-CRD, Asp-His-His-Cys cysteine-rich domain; LB, lysis buffer; myr-Vac8p, N-myristoylated Vac8p; NF, nonfluorescent media; palm-CoA, palmitoyl-CoenzymeA; PAT, protein acyltransferase; PFA, protein fatty acyltransferase; WT, wild type.
References
- Adams, A., D.E. Gottschling, C.A. Kaiser, and T. Stearns. 1997. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Plainview, NY. 177 pp.
- Bohm, S., D. Frishman, and H.W. Mewes. 1997. Variations of the C2H2 zinc finger motif in the yeast genome and classification of yeast zinc finger proteins. Nucleic Acids Res. 25:2464–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci, M., and S.R. Wente. 1997. In vivo dynamics of nuclear pore complexes in yeast. J. Cell Biol. 136:1185–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, D.C., B.C. Yang, and T.T. Kuo. 1992. One-step transformation of yeast in stationary phase. Curr. Genet. 21:83–84. [DOI] [PubMed] [Google Scholar]
- Dietrich, L.E., R. Gurezka, M. Veit, and C. Ungermann. 2004. The SNARE Ykt6 mediates protein palmitoylation during an early stage of homotypic vacuole fusion. EMBO J. 23:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy, J.T., W.K. Greentree, C.L. Manahan, and M.E. Linder. 1996. G-protein palmitoyltransferase activity is enriched in plasma membranes. J. Biol. Chem. 271:7154–7159. [DOI] [PubMed] [Google Scholar]
- Duronio, R.J., E. Jackson-Machelski, R.O. Heuckeroth, P.O. Olins, C.S. Devine, W. Yonemoto, L.W. Slice, S.S. Taylor, and J.I. Gordon. 1990. Protein N-myristoylation in Escherichia coli: reconstitution of a eukaryotic protein modification in bacteria. Proc. Natl. Acad. Sci. USA. 87:1506–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckenstein, D., M. Rohde, D.J. Klionsky, and M. Rudiger. 1998. Yel013p (Vac8p), an armadillo repeat protein related to plakoglobin and importin alpha is associated with the yeast vacuole membrane. J. Cell Sci. 111:3109–3118. [DOI] [PubMed] [Google Scholar]
- Glick, B.S., and J.E. Rothman. 1987. Possible role for fatty acyl-coenzyme A in intracellular protein transport. Nature. 326:309–312. [DOI] [PubMed] [Google Scholar]
- Guldener, U., S. Heck, T. Fielder, J. Beinhauer, and J.H. Hegemann. 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 24:2519–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, A., and W. Wickner. 1996. Homotypic vacuole fusion requires Sec17p (yeast alpha-SNAP) and Sec18p (yeast NSF). EMBO J. 15:3296–3305. [PMC free article] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 726 pp.
- Huh, W.K., J.V. Falvo, L.C. Gerke, A.S. Carroll, R.W. Howson, J.S. Weissman, and E.K. O'Shea. 2003. Global analysis of protein localization in budding yeast. Nature. 425:686–691. [DOI] [PubMed] [Google Scholar]
- Jones, H.D., M. Schliwa, and D.G. Drubin. 1993. Video microscopy of organelle inheritance and motility in budding yeast. Cell Motil. Cytoskeleton. 25:129–142. [DOI] [PubMed] [Google Scholar]
- Lee, W.L., J.R. Oberle, and J.A. Cooper. 2003. The role of the lissencephaly protein Pac1 during nuclear migration in budding yeast. J. Cell Biol. 160:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder, M.E., and R.J. Deschenes. 2003. New insights into the mechanisms of protein palmitoylation. Biochemistry. 42:4311–4320. [DOI] [PubMed] [Google Scholar]
- Lobo, S., W.K. Greentree, M.E. Linder, and R.J. Deschenes. 2002. Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J. Biol. Chem. 277:41268–41273. [DOI] [PubMed] [Google Scholar]
- McNew, J.A., M. Sogaard, N.M. Lampen, S. Machida, R.R. Ye, L. Lacomis, P. Tempst, J.E. Rothman, and T.H. Sollner. 1997. Ykt6p, a prenylated SNARE essential for endoplasmic reticulum-Golgi transport. J. Biol. Chem. 272:17776–17783. [DOI] [PubMed] [Google Scholar]
- Morrissey, J.H. 1981. Silver stain for proteins in polyacrylamide gels: a modified procedure with enhanced uniform sensitivity. Anal. Biochem. 117:307–310. [DOI] [PubMed] [Google Scholar]
- Pan, X., and D.S. Goldfarb. 1998. YEB3/VAC8 encodes a myristylated armadillo protein of the Saccharomyces cerevisiae vacuolar membrane that functions in vacuole fusion and inheritance. J. Cell Sci. 111:2137–2147. [DOI] [PubMed] [Google Scholar]
- Pan, X., P. Roberts, Y. Chen, E. Kvam, N. Shulga, K. Huang, S. Lemmon, and D.S. Goldfarb. 2000. Nucleus-vacuole junctions in Saccharomyces cerevisiae are formed through the direct interaction of Vac8p with Nvj1p. Mol. Biol. Cell. 11:2445–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanner, N., B.S. Glick, S.R. Arden, and J.E. Rothman. 1990. Fatty acylation promotes fusion of transport vesicles with Golgi cisternae. J. Cell Biol. 110:955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompliano, D.L., M.D. Schaber, S.D. Mosser, C.A. Omer, J.A. Shafer, and J.B. Gibbs. 1993. Isoprenoid diphosphate utilization by recombinant human farnesyl:protein transferase: interactive binding between substrates and a preferred kinetic pathway. Biochemistry. 32:8341–8347. [DOI] [PubMed] [Google Scholar]
- Putilina, T., P. Wong, and S. Gentleman. 1999. The DHHC domain: a new highly conserved cysteine-rich motif. Mol. Cell. Biochem. 195:219–226. [DOI] [PubMed] [Google Scholar]
- Reiss, Y., J.L. Goldstein, M.C. Seabra, P.J. Casey, and M.S. Brown. 1990. Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell. 62:81–88. [DOI] [PubMed] [Google Scholar]
- Roth, A.F., Y. Feng, L. Chen, and N.G. Davis. 2002. The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J. Cell Biol. 159:23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seedorf, M., M. Damelin, J. Kahana, T. Taura, and P.A. Silver. 1999. Interactions between a nuclear transporter and a subset of nuclear pore complex proteins depend on Ran GTPase. Mol. Cell. Biol. 19:1547–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley, E.S., M. Kato, N. Margolis, W. Wickner, and G. Eitzen. 2002. Genomic analysis of homotypic vacuole fusion. Mol. Biol. Cell. 13:782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski, R.S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singaraja, R.R., S. Hadano, M. Metzler, S. Givan, C.L. Wellington, S. Warby, A. Yanai, C.A. Gutekunst, B.R. Leavitt, H. Yi, et al. 2002. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum. Mol. Genet. 11:2815–2828. [DOI] [PubMed] [Google Scholar]
- Uemura, T., H. Mori, and M. Mishina. 2002. Isolation and characterization of Golgi apparatus-specific GODZ with the DHHC zinc finger domain. Biochem. Biophys. Res. Commun. 296:492–496. [DOI] [PubMed] [Google Scholar]
- Ungermann, C., G.F. von Mollard, O.N. Jensen, N. Margolis, T.H. Stevens, and W. Wickner. 1999. Three v-SNAREs and two t-SNAREs, present in a pentameric cis-SNARE complex on isolated vacuoles, are essential for homotypic fusion. J. Cell Biol. 145:1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez-Taubas, J., and H. Pelham. 2005. Swf1-dependent palmitoylation of the SNARE Tlg1 prevents its ubiquitination and degradation. EMBO J. 24:2524–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit, M., R. Laage, L. Dietrich, L. Wang, and C. Ungermann. 2001. Vac8p release from the SNARE complex and its palmitoylation are coupled and essential for vacuole fusion. EMBO J. 20:3145–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernet, T., D. Dignard, and D.Y. Thomas. 1987. A family of yeast expression vectors containing the phage f1 intergenic region. Gene. 52:225–233. [DOI] [PubMed] [Google Scholar]
- Vida, T.A., and S.D. Emr. 1995. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 128:779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y.X., N.L. Catlett, and L.S. Weisman. 1998. Vac8p, a vacuolar protein with armadillo repeats, functions in both vacuole inheritance and protein targeting from the cytoplasm to vacuole. J. Cell Biol. 140:1063–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y.X., E.J. Kauffman, J.E. Duex, and L.S. Weisman. 2001. Fusion of docked membranes requires the armadillo repeat protein Vac8p. J. Biol. Chem. 276:35133–35140. [DOI] [PubMed] [Google Scholar]
- Wickner, W., and A. Haas. 2000. Yeast homotypic vacuole fusion: a window on organelle trafficking mechanisms. Annu. Rev. Biochem. 69:247–275. [DOI] [PubMed] [Google Scholar]
- Winzeler, E.A., D.D. Shoemaker, A. Astromoff, H. Liang, K. Anderson, B. Andre, R. Bangham, R. Benito, J.D. Boeke, H. Bussey, et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 285:901–906. [DOI] [PubMed] [Google Scholar]
- Zhao, L., S. Lobo, X. Dong, A.D. Ault, and R.J. Deschenes. 2002. Erf4p and Erf2p form an endoplasmic reticulum-associated complex involved in the plasma membrane localization of yeast Ras proteins. J. Biol. Chem. 277:49352–49359. [DOI] [PubMed] [Google Scholar]