

Abstract
Drosophila transient receptor potential (TRP) serves dual roles as a cation channel and as a molecular anchor for the PDZ protein, INAD (inactivation no afterpotential D). Null mutations in trp cause impairment of visual transduction, mislocalization of INAD, and retinal degeneration. However, the impact of specifically altering TRP channel function is not known because existing loss-of-function alleles greatly reduce protein expression. In the current study we describe the isolation of a set of new trp alleles, including trp 14 with an amino acid substitution juxtaposed to the TRP domain. The trp 14 flies stably express TRP and display normal molecular anchoring, but defective channel function. Elimination of the anchoring function alone in trp Δ 1272, had minor effects on retinal morphology whereas disruption of channel function caused profound light-induced cell death. This retinal degeneration was greatly suppressed by elimination of the Na+/Ca2+ exchanger, CalX, indicating that the cell death was due primarily to deficient Ca2+ entry rather than disruption of the TRP-anchoring function.
Introduction
The transient receptor potential (TRP) superfamily is comprised of a large group of related cation channels that function in sensory processes ranging from phototransduction to touch, hearing, taste, olfaction, osmosensation, and thermosensation (for review see Montell, 2005b). As such, dissecting the specific contributions of TRP channels is central to understanding each of these sensory modalities. It is generally accepted that the roles of mammalian TRP proteins is to mediate influx of cations, such as Ca2+ and Na+. However, in the case of Drosophila TRP, which is the founding member of the superfamily that is required for phototransduction (Montell and Rubin, 1989), the protein functions both as a Ca2+-permeable channel and as a molecular anchor (Li and Montell, 2000; Tsunoda et al., 2001).
Many of the proteins that are essential for fly visual transduction are organized into a large macromolecular complex, referred to as the signalplex (for review see Montell, 2005a). The molecular scaffold that nucleates the signalplex, inactivation no afterpotential D (INAD; Shieh and Zhu, 1996), consists of multiple PDZ protein interaction modules and appears to be constantly associated with TRP as well as two other important signaling proteins, PLC (encoded by norpA [no receptor potential A, encodes PLC]) and PKC (encoded by inaC [inactivation no afterpotential C, encodes PKC]). These three core binding proteins depend on INAD for localization in the phototransducing compartment of the fly photoreceptor cells, the rhabdomeres (Chevesich et al., 1997; Tsunoda et al., 1997). In addition, at least five other signaling proteins appear to associate with INAD (Huber et al., 1996; Chevesich et al., 1997; Xu et al., 1998; Wes et al., 1999; Goel et al., 2001) and these latter proteins may interact dynamically with INAD. However, none of these noncore binding proteins requires interaction with INAD for normal localization.
A surprising finding is that there is a reciprocal requirement for association of TRP and INAD for concentration of these two proteins in the rhabdomeres (Li and Montell, 2000; Tsunoda et al., 2001). Deletion of the COOH-terminal four residues in TRP destroys the PDZ binding site and results in mislocalization of INAD (Li and Montell, 2000). In turn, the rhabdomeral distributions of PKC and PLC are also disrupted. The interaction between TRP and INAD is not necessary for targeting of these proteins, but rather for subsequent retention in the rhabdomeres. Also, unexpected was the finding that interference with the direct interactions between TRP and INAD had no major impact on activation of the TRP channels (Li and Montell, 2000), which in Drosophila photoreceptor cells is very rapid and occurs within milliseconds. These data demonstrate that the TRP channel functions as a molecular anchor, in addition to its more appreciated role as a cation channel.
The dual roles of TRP raise the question as to the impact of altering the channel activity independent of effects on the anchoring function. Null mutations in TRP result in light-dependent retinal degeneration, in addition to causing a transient response to bright light (Cosens and Perry, 1972; Chevesich et al., 1997). Retinal degeneration in fly photoreceptor cells is a common phenomenon that occurs as a result of mutations in nearly any protein important for phototransduction (for review see Pak, 1994; Montell, 1999). However, in most cases the mechanism underlying the retinal degeneration has not been clarified. In some mutants retinal degeneration occurs as a result of formation of stable rhodopsin–arrestin complexes, which in turn lead to endocytosis of rhodopsin (Alloway et al., 2000; Kiselev et al., 2000; Orem and Dolph, 2002). Ca2+ overload due to expression of a constitutively active TRP channel can also lead to rapid cell death in fly photoreceptor cells (Yoon et al., 2000; Wang et al., 2005). However, the mechanism underlying the retinal degeneration in trp-null mutant flies is not known. In particular, it is not clear whether the light-dependent retinal degeneration due to loss of trp function results from disruption of the anchoring role, or from lower Ca2+ influx during light stimulation. This question has not been possible to address because the existing loss-of-function mutations in trp have major impacts on protein levels and consequently disrupt both TRP functions (Montell and Rubin, 1989).
In addition to TRP, there are two related cation channels expressed in photoreceptors, transient receptor potential-like (TRPL), and transient receptor potential γ (TRPγ; Phillips et al., 1992; Xu et al., 2000). Currently, there are no loss-of-function mutations in TRPγ and elimination of TRPL has only subtle effects on the photoresponse (Niemeyer et al., 1996; Reuss et al., 1997; Leung et al., 2000). Nevertheless, TRPL contributes to phototransduction as flies that are missing both TRP and TRPL are blind (Niemeyer et al., 1996; Reuss et al., 1997).
In the current report, we describe the isolation of multiple new trp alleles, including one (trp 14) that specifically affected the channel function, but not the molecular anchoring role. In contrast to the wild-type light response, we found that in trp 14 photoreceptor cells, the light response was transient. This phenotype resulted from a missense mutation in TRP juxtaposed to the highly conserved TRP domain (for review see Montell, 2005b). In addition, we found that the light-induced retinal degeneration was as severe in trp 14 flies as in trp-null flies, trp P343. Conversely, elimination of the TRP–INAD interaction had relatively minor effects on the morphology of the photoreceptor cells. Finally, the retinal degeneration associated with either trp 14 or trp P343 was suppressed by a loss-of-function mutation in the Na+/Ca2+ exchanger, CalX. These results demonstrate that the cell death in trp mutant photoreceptor cells is due primarily to disruption of TRP channel activity and decreased light-dependent Ca2+ influx, rather than elimination of the TRP anchoring role.
Results
Identification of new trp alleles
To identify new alleles of the trp locus, we screened the recently isolated collection of chemically induced third chromosome mutations, which display defects in the electroretinogram (ERG) recording (for details see Wang et al., 2005). Exposure of wild-type flies to light results in two discriminable components in the ERG (Fig. 1 A). These include a sustained corneal negative maintained component arising from responses of all retinal cells (photoreceptor cells and pigment cells) and on- and off-transients emanating from activity in the second-order neurons in the optic lobes (for review see Montell, 1999). The classic trp phenotype is characterized by a transient response to light (Fig. 1 B), resulting from rapid light-dependent inactivation of the remaining TRPL cation channel.
Figure 1.

ERG phenotype and expression of TRP in trp14 flies. (A–F) ERG recordings. Flies <1 d after eclosion were dark-adapted for 2 min before exposure to a 10-s pulse of orange light (indicated by the event marker below the ERGs). (A) Wild type (wt); (B) trp P343; (C) trp 14; (D) trpl 302; (E) trpl 302;trp P343; (F) trpl 302;trp 14. (G) Time required for 60% return to the dark state. Shown are the mean times in seconds (s) based on ERGs obtained from ≥20 flies of the indicated genotypes. SDs are indicated. (H–J) Photoreceptor cell responses assayed by intracellular recordings. 1-d-old flies were dark adapted for 2 min before being exposed to a 10-s pulse of orange light. Scale bars are provided indicating the amplitude (mV) and timescale (s). (H) wt; (I) trp P343; (J) trp 14. (K) Expression of TRP in mutant alleles assayed on Western blots. Head extracts were prepared from flies <1 d after eclosion and a Western blot was first probed with anti-TRP antibodies and subsequently with anti-NORPA antibodies to provide a loading control. The positions of protein size markers (kD) are indicated. (L) Expression levels of core members of the signalplex in trp 14. The Western blot was first probed with anti-TRP antibodies and then reprobed with anti-INAD, anti-NORPA, anti-INAC, and anti-tubulin antibodies. (M) Relative TRP protein levels in trp 14 flies. The TRP protein level in trp 14 heads was compared with wt, trp P343, and trp 14 heterozygotes. The intensities of the bands were determined using a phosphoimager. SDs are indicated.
We crossed each of the third chromosome mutations to the strong trp P343 allele and identified five that failed to complement the recessive TRP phenotype and therefore represented new trp alleles. Four of the new trp alleles exhibited a transient ERG phenotype indistinguishable from trp P343 (unpublished data), whereas the phenotype of the fifth, trp 14 was distinct in that the decline in the light response was much slower than in trp P343 or other alleles isolated in this or previous studies (Fig. 1, C and G; 60% deactivation time: trp P343: 1.5 ± 0.2 s, trp 14: 5.2 ± 1.6 s). The trp 14 phenotype was due to an autonomous defect in the photoreceptor cells, rather than the pigment cells, as the slower decline in the receptor potential was evident in single photoreceptor cells assayed by performing intracellular recordings (Fig. 1, H–J).
Molecular defects and TRP expression in trp alleles
Currently, all of the existing loss-of-function mutations cause large reductions in protein levels, although the molecular lesions have not been defined. Among the extant trp alleles, the one with the strongest phenotype is trp P343. We sequenced the trp P343 genomic region and found that the TRP protein coding region was identical to wild type. Rather, there was a mutation in a conserved 5′ splice site that was essential for mRNA splicing (Table I). The mutation presumably results in instability of the mRNA, as no trp P343 mRNA is detected (Montell and Rubin, 1989). Given the strong phenotype and lack of mRNA or TRP protein expressed in trp P343 (Montell and Rubin, 1989), this allele would appear to represent a null.
Table I. Molecular defects in trp alleles.
| Allele | Type of mutation | Location | Specific change | TRP protein level |
|---|---|---|---|---|
| trpP343 | Splice site | Intron 7; first base of 5′ splice site |
GT to AT | ND |
| trp14 | Two missense | Pore loop; between TM6 and TRP box 1 | L612F; R671N | 60% wild- type level |
| trp38 | Promoter1 | ND | ||
| trp47 | Two missense | Adjacent to TRP box 1; TRP domain, between boxes 1 and 2 | T674A; W684R | 5% wild-type level |
| trp74 | Missense | Fourth ankyrin repeat | G167E | ND |
| trp92 | Deletion/ frameshift | Transmembrane domain 1 | Deletion of G in codon 336; stop after adding three residues | ND |
The trp genomic DNA encoding the transcribed region was sequenced for each trp allele. ND, not detected.
No mutation was found in trp 38, suggesting that there was a mutation affecting expression of the mRNA.
To examine the levels of TRP protein expressed in the new alleles described here, we performed Western blots. Among those alleles that displayed a phenotype typical of trp P343, three did not express any detectable TRP protein (Fig. 1 K, trp 38, trp 74, and trp 92), whereas a fourth expressed very low levels of TRP (Fig. 1 K, trp 47). This was in contrast to trp 14 flies in which TRP was expressed at ∼60% the level as in wild type (Fig. 1, K–M). Other rhabdomeral proteins including INAD, INAC, NORPA, and Rh1 (rhodopsin 1) were expressed at comparable levels in trp 14, trp P343, and wild-type flies (Fig. 1 L).
To determine the molecular defects associated with the five new trp alleles, we sequenced the genomic DNA and compared the sequences to that of the original isogenized stock used to conduct the mutagenesis. Among the four alleles that expressed very low or no detectable TRP, one had a frameshift mutation resulting in premature translation termination (trp 92), two had missense mutations (trp 47 and trp 74), and one had no mutation in the transcribed region and therefore may contain a mutation affecting the trp promoter (Table I, trp 38). The allele (trp 14) expressing TRP at 60% wild-type levels, and which exhibited a phenotype distinct from other trp alleles, had two missense mutations. One of these mutations changed an amino acid in the pore loop between transmembrane domains five and six (residue 612; leucine to phenylalanine), whereas the other was situated between the sixth transmembrane segment and a highly conserved sequence referred to as TRP box 1 (residue 671; arginine to glutamine).
Normal anchoring function in trp 14 mutant
Given that TRP has dual functions as a molecular anchor and as a cation channel (Li and Montell, 2000; Tsunoda et al., 2001), we considered whether the TRP phenotype in trp 14 was a consequence of perturbation of the anchoring role. Drosophila compound eyes consist of ∼800 repetitive units, referred to as ommatidia, each of which includes six outer photoreceptor cells (R1-6) and a central R7 or R8 cell in the distal region of the retina. Each photoreceptor cell contains a microvillar segment, the rhabdomere, where most of the proteins that function in phototransduction, such as the core members of the signalplex, are concentrated. These include TRP, protein kinase C (INAC), phospholipase C (NORPA), and INAD (Fig. 2 A). Mutations that eliminate or disrupt the anchoring role of TRP result in mislocalization of these core members such that they are present in both the rhabdomeres and cell bodies (Fig. 2 B; Chevesich et al., 1997; Tsunoda et al., 1997; Li and Montell, 2000; Tsunoda et al., 2001). However, other INAD binding partners, such as Rh1, do not depend on TRP for normal localization (Fig. 2 B; Li and Montell, 2000). We found that in trp 14 photoreceptor cells, each of the core and other rhabdomeral proteins examined displayed a rhabdomere localization pattern indistinguishable from wild type (Fig. 2 C). Consistent with these data, we found that INAD coimmunoprecipitated effectively with the TRP14 protein (Fig. 2 E). These data indicate that the TRP phenotype in trp 14 flies is not due to an alteration in the TRP anchoring function. Rather, they raise the possibility that the phenotype is due to a defect in TRP channel function.
Figure 2.

Normal anchoring role of TRP in trp14 flies. (A–D) Examination of the spatial distributions of INAD and the core INAD binding proteins. The cross sections of compound eyes were obtained from flies 5 d after eclosion maintained under a 12-h light–12-h dark cycle. Sections from flies of the following genotypes were probed with anti-NORPA, anti-INAC, anti-INAD, anti-TRP, and anti-Rh1 antibodies: (A) wild type; (B) trp P343; (C) trp 14; and (D) calx B,trp P343. (E) Normal interaction between INAD and TRP in trp 14. Fly head extracts were prepared and immunoprecipitations (IPs) were performed with anti-TRP antibodies. A Western blot containing the IPs was probed with anti-INAD antibodies. The positions of two protein size markers are indicated. (F) A Western blot containing 2% of the total extracts used for the coIPs was probed with anti-INAD antibodies.
Transient light response in trp 14 was due to mutation in trp rather than low levels of expression or mislocalization of TRP
In trp flies such as trp P343 the transient potential ERG phenotype is a consequence of inactivation of the TRPL channel during constant light stimulation. However, in trpl mutant flies, which express TRP but not TRPL, the ERG response is maintained during a typical 5–10-s light pulse (Fig. 1 D). To examine TRP14 channel function independent of TRPL, we introduced the trp 14 allele in a trpl-null mutant (trpl 302) background (Niemeyer et al., 1996). As previously shown, trpl 302;trp P343 flies are blind as they do not express TRPL or TRP (Niemeyer et al., 1996; Reuss et al., 1997). Flies harboring just the trpl 302 mutation show a response to 10 s of light similar to wild type, because these flies express wild-type TRP. In contrast, trpl 302;trp 14 flies displayed a TRP phenotype similar to trp 14 (Fig. 1, F and G; 60% deactivation time: trp P343 1.5 ± 0.2 s, trp 14: 5.2 ± 1.6 s, trpl 302;trp 14: 4.2 ± 0.8 s). These data indicate that the transient light response in trp 14 flies was due to disruption of TRP channel function.
To exclude that the trp 14 phenotype was a consequence of mislocalization of TRP14, we spatially localized the mutant protein. We performed immunostaining experiments and found that TRP14 was detected exclusively in the rhabdomeres, as was the case for wild-type TRP (Fig. 2, A and C). Therefore, the transient light response in trp 14 was not due to mislocalization of the TRP protein.
Given that trp 14 flies express a 40% lower concentration of TRP, we tested whether a transient light response could be induced by expression of low levels of TRP. Therefore, we generated transgenic flies that expressed varying levels of wild-type TRP under the control of the heat-shock promoter (hs-trp) and placed the transgene in a trpl 302;trp P343 genetic background. Even though use of the heat shock protein 70 (hsp70) promoter typically results in widespread expression, the TRP protein expressed under the control of the hsp70 promoter was found exclusively in the retina and not in the optic lobes or elsewhere in the adult head (Fig. 3 A). To induce different low levels of TRP, we exposed the hs-trp flies to 30, 60, and 120 min heat-shock treatments, which resulted in the production of ∼4%, 7%, and 10% of the wild-type levels of TRP, respectively (Fig. 3, B and C). Expression of only ∼10% the normal concentration of TRP restored an ERG in trpl 302;trp P343 flies, which was not transient (Fig. 3 G). Even 4% the normal levels of TRP did not cause a transient light response similar to trp 14, although the amplitude of the ERG was reduced (Fig. 3 E). These data indicate that the trp 14 mutant phenotype was due to the mutation of the TRP14 protein, rather than simply due to expression of low levels or mislocalized TRP protein.
Figure 3.

Expression of low levels of TRP rescues the trp ERG phenotype. (A) Expression of hs-trp results in expression primarily in the retina. Newly eclosed hs-trp,trp P343 flies were heat shocked for 2 h at 37°C and frozen sections, which were prepared 24 h later, were stained with anti-TRP antibodies. cb, central brain; la, lamina; me, medulla; re, retina. (B) Western blot showing TRP protein levels in hs-trp,trp P343 flies exposed to heat shock (hs) treatment of the indicated durations. The heat shocks were performed on newly eclosed flies and the head extracts were prepared 24 h later. The second lane from the left contains 10% the quantity of extracts (from wild-type fly heads) loaded in the other lanes. The same blot was reprobed with anti-tubulin antibodies. (C) Relative levels of TRP expressed in trpl;hs-trp,trp P343 flies exposed to different heat shock durations. The TRP protein levels after different heat shock times were compared with the band intensity obtained using 10% as much wild-type extracts. The quantification was performed using a phosphoimager. SDs are indicated. (D–G) ERG recordings obtained in trpl;hs-trp,trp P343 flies exposed to heat shock treatments of different lengths. Flies were heat shocked for either 0.5, 1, or 2 h at 37°C immediately after eclosion and the ERGs were performed 1 d later by dark adapting the flies for 2 min and exposing them to a 10-s pulse of orange light.
Channel activity critical to prevent light-dependent retinal degeneration
Strong loss-of-function mutations in trp, such as in trp P343, result in light dependent retinal degeneration (Fig. 4). Considering TRP has dual channel and nonchannel roles, disruption of either function could potentially cause retinal degeneration. To address which of these two roles is more critical to prevent retinal cell death, we examined the morphology of trp Δ 1272 (Li and Montell, 2000) and trp 14 ommatidia, which display specific defects in the scaffold and TRP channel functions, respectively. Wild-type ommatidia contain eight photoreceptor cell rhabdomeres, seven of which are present in any given plane regardless of their age or whether the flies were maintained in the dark or under a light–dark cycle (Fig. 4).
Figure 4.
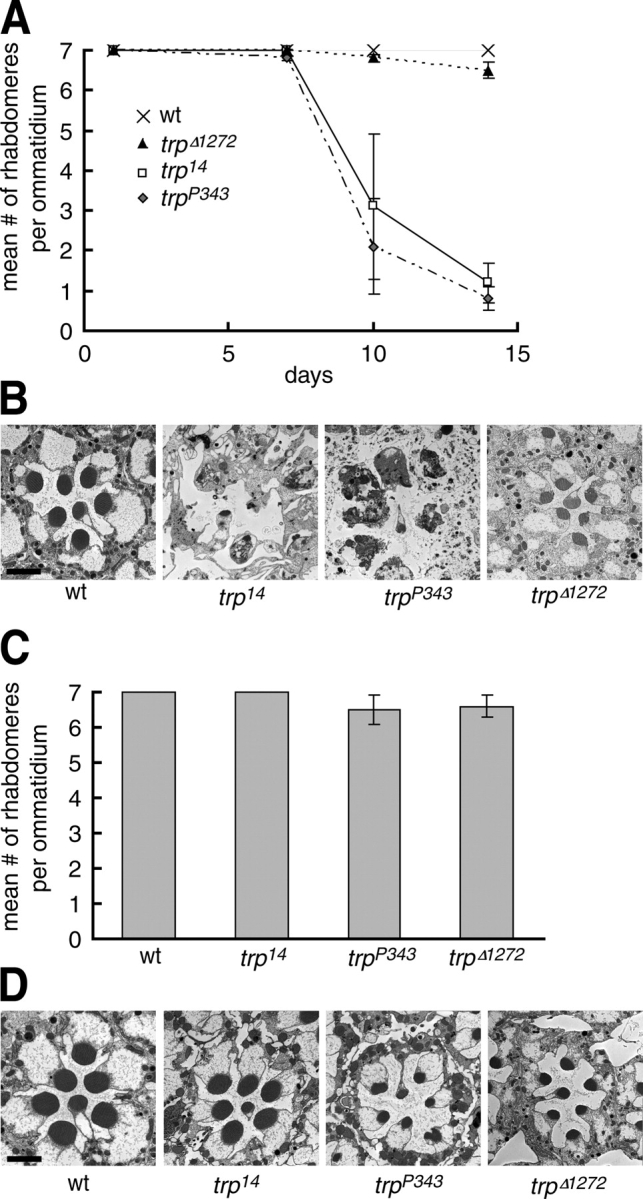
Light-dependent retinal degeneration in trp mutant flies. (A) The time-course of retinal degeneration of flies maintained under a 12-h light–12-h dark cycle. The mean numbers of rhabdomeres per ommatidium were determined by examining EM cross sections through the distal regions number of the compound eyes. Each point is based on ≥80 ommatidia from two separate flies each. Error bars indicate SDs. (B) Examination of the morphology of wt, trp 14, trp P343, and trp Δ 1272 by transmission EM. Cross sections were obtained from 14-d-old flies kept under a 12-h light–12-h dark cycle. (C) Mean number of rhabdomeres per ommatidium from 30-d-old dark-reared flies. SDs are indicated. (D) Morphology of 30-d-old dark-reared wt, trp 14, trp P343, and trp Δ 1272 flies examined by transmission EM. Bars, 2 μm.
We found that the retinal degeneration in trp 14 and trp P343 was much more severe than that in trp Δ 1272 flies. In both trp P343 and trp 14 flies, the rhabdomeres began to disappear between 7 and 10 d after eclosion and almost no rhabdomeres remained after 14 d of exposure to a 12-h light–12-h dark cycle (Fig. 4, A and B). By contrast most trp Δ 1272 flies maintained a full complement of seven rhabdomeres after 14 d of a light–dark cycle (Fig. 4 A). Nevertheless, the size of the rhabdomeres was typically smaller than in similarly aged wild-type and large intracellular vacuoles were present inside the cell bodies indicating that retinal degeneration had initiated (Fig. 4 B). By 30 d after eclosion, most of the rhabdomeres in these flies had degenerated (unpublished data). Thus, the retinal degeneration was much more severe in trp P343 and trp 14 than trp Δ 1272, indicating that the TRP channel function rather than the scaffold function was more critical to prevent the retinal cell death.
The retinal degeneration resulting from defects in TRP function was suppressed by maintaining the flies in the dark, which keeps the TRP channels in an inactive state. To assess the extent of suppression, we maintained the flies in the dark for 30 d, which was more than twice as long the 14-d light–dark cycle that caused elimination of almost all rhabdomeres in trp 14 or trp P343 flies. In dark-maintained trp Δ 1272 flies, seven rhabdomeres were present in ommatidia, although the size of the rhabdomeres was reduced (Fig. 4, C and D). Indistinguishable results were obtained with trp P343. The suppression of retinal degeneration was even more complete with trp 14, as all ommatidia contained a full set of seven rhabdomeres of normal size (Fig. 4, C and D). This result was striking as the retinal degeneration occurring under a light–dark cycle was significantly more rapid in trp 14 than in trp Δ 1272.
Rapid retinal degeneration of trp photoreceptors suppressed by mutations in calx and arr2
To explore the mechanism underlying the retinal degeneration in trp mutants, we used a genetic approach. The combination of results described above indicates that a defect in TRP channel function underlies the retinal degeneration in trp flies. If the basis of the retinal degeneration in the trp mutant was due to diminished Ca2+ influx during light stimulation, then the cell death might be reduced by mutations in the Na+/Ca2+ exchanger, CalX, which functions in Ca2+ extrusion in photoreceptor cells.
To test whether calx can suppress the retinal degeneration in trp P343 and trp 14, we examined the morphology of calx B,trp P343, and calx B,trp 14 compound eyes. As described above, trp 14 or trp P343 flies maintained under a light–dark cycle for 14 d displayed nearly complete loss of the rhabdomeres (Fig. 5, A–D). The retinal degeneration in calx B flies was even more severe as there were few rhabdomeres left after a 7 d light–dark cycle (unpublished data) and almost no rhabdomeres left after a 14-d light–dark cycle (Fig. 5, A and E). In contrast, most ommatidia from either calx B,trp P343 or calx B,trp 14 double mutant flies contained all the rhabdomeres after 14 d under a light–dark cycle (Fig. 5, A, F, and G). Moreover, the core signalplex proteins, NORPA, INAC, and INAD were mislocalized in calx B,trp P343 flies (Fig. 2 D), indicating that introducing the calx mutation did not prevent loss of the TRP scaffold function in trp P343. The effect of calx on trp was specific as the calx B mutation did not suppress the cell death resulting from mutations in other phototransduction genes such as inaC, which encodes an eye-enriched PKC (unpublished data). These results indicated that the retinal degeneration in trp 14 or trp P343 was a consequence of decreased intracellular Ca2+ levels, whereas the photoreceptor cell death in the calx mutant resulted from Ca2+ overload.
Figure 5.
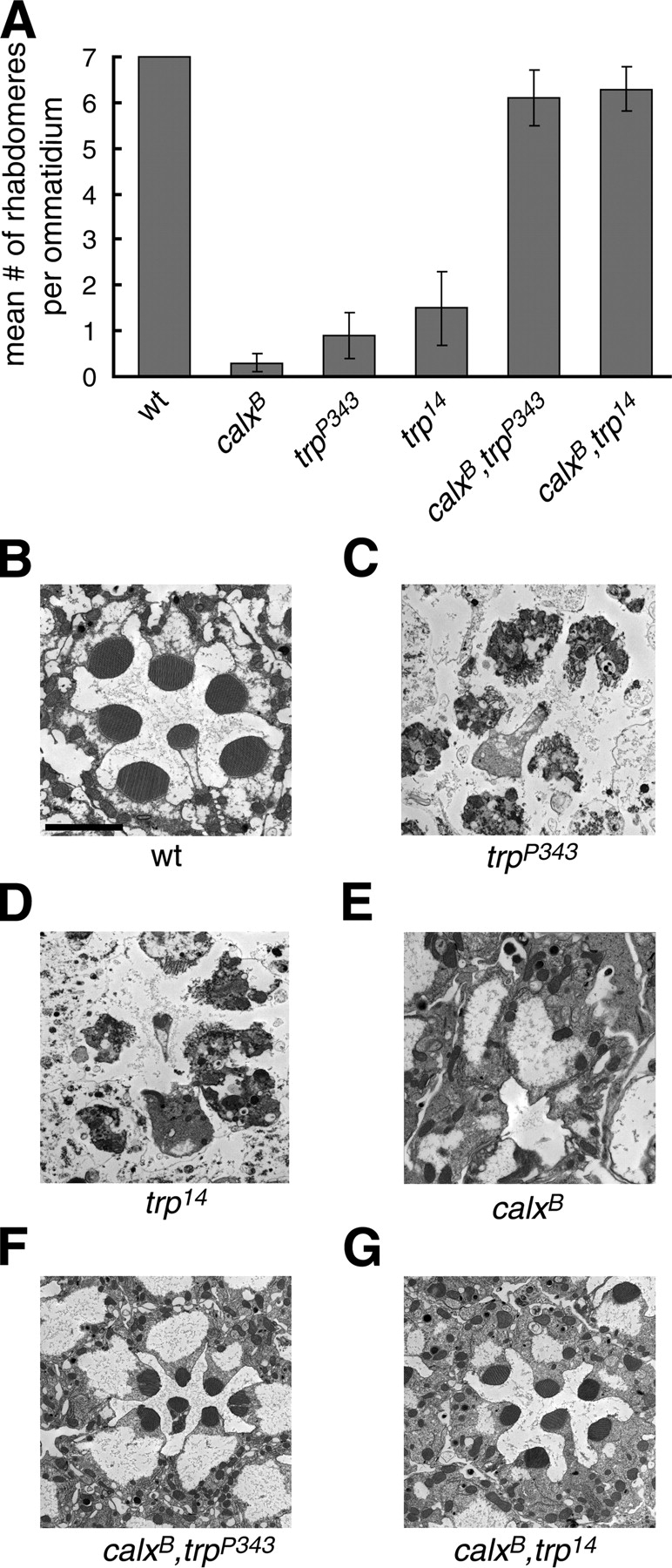
Suppression of retinal degeneration in trp mutant flies by the calx mutation. (A) Histogram of the mean number of rhabdomeres per ommatidium from 14-d-old flies held under a 12-h light–12-h dark cycle. The quantification is based on examination of EM cross sections from the distal region of the compound eyes from at least 80 ommatidia (two separate flies each). The error bars indicate SDs. (B–G) EM images of cross sections from: (B) wt; (C) trp P343; (D) trp 14; (E) calx B; (F) calx B,trp P343; and (G) calx B,trp 14 flies. Bar, 2 μm.
To explore further the mechanism of the retinal degeneration in trp 14 and trp P343, we considered whether it could be suppressed by mutations in the gene encoding the major arrestin (arrestin2 [arr2]; Dolph et al., 1993). Elimination of Arr2 reduces the retinal degeneration associated with certain mutations (Alloway et al., 2000; Kiselev et al., 2000), such as norpA (disrupts phospholipase C), which prevents light-dependent activation of TRP channels. The retinal degeneration in norpA results from formation of stable rhodopsin–arrestin complexes and subsequent endocytosis of rhodopsin (Orem and Dolph, 2002).
We found that strong mutations in arr2 partially suppressed the retinal degeneration in trp 14 and trp P343 flies. Whereas a 14-d exposure to a light–dark cycle resulted in extensive loss of rhabdomeres in arr2 5,trp P343 or trp 14 eyes (Fig. 6, B–D), most ommatidia in arr2 5,trp P343 or arr2 5,trp 14 double mutants contained seven rhabdomeres (Fig. 6, E and F). However, the sizes of the rhabdomeres were significantly reduced.
Figure 6.
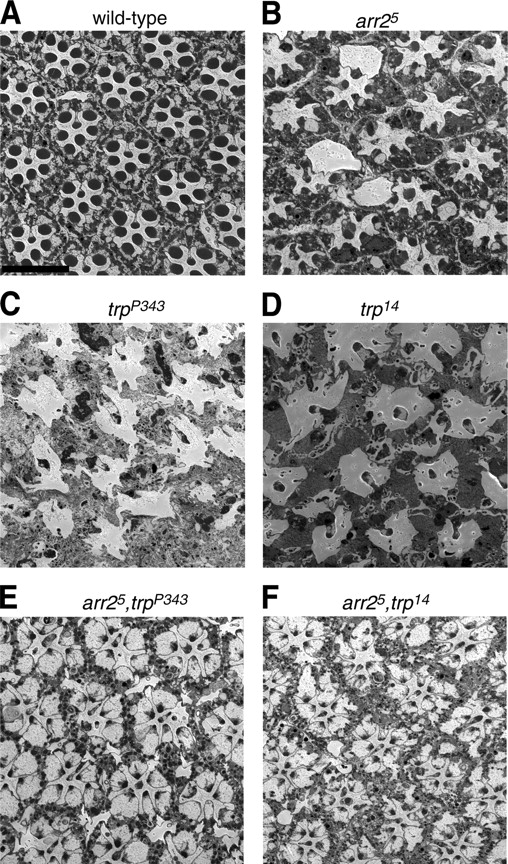
Suppression of retinal degeneration in trp flies by the arr2 mutation. EM images of cross sections from 14-d-old flies maintained under a 12-h light–12-h dark cycle. (A) Wild type; (B) arr2 5; (C) trp P343; (D) trp 14; (E) arr2 5,trp P343; (F) arr2 5,trp 14. Bar, 10 μm.
Substitution of a basic residue adjacent to the TRP domain causes a defect in channel activity
The two mutations in trp 14 alter residues in the pore loop or immediately NH2 terminal to TRP box 1 (residues 612 and 671, respectively; Fig. 7 A). Given the potential effects of a pore–loop mutation on ion selectivity and the highly conserved nature of the region between the sixth transmembrane segment (TM6) and the TRP domain (Montell, 2005b; Fig. 8 A), both of these mutations were in intriguing positions that could potentially be responsible for the trp 14 phenotype.
Figure 7.

R671Q mutation disrupts TRP channel function. (A) Locations of L612F and R671Q missense mutations in TRP14 indicated on a cartoon of the TRP protein. The gray rectangles represent the six transmembrane segments and the black box stands for TRP box 1. (B) Western blot showing expression of TRP in ninaE-trp, ninaE-trp 612F, and ninaE-trp 671Q transgenic flies. All three transgenes were in a trpl 302;trp P343 background. The fly head extracts used for the Western blot were prepared from flies <1 d after eclosion and probed with anti-TRP antibodies. The same blot was reprobed with anti-tubulin antibodies. (C–F) ERG recordings obtained from trpl 302;trp 14 (C) and flies containing the following transgenes in a trpl 302;trp P343 background: (D) ninaE-trp; (E) ninaE-trp 612F; and (F) ninaE-trp 671Q. To perform the ERGs, flies <1 d after eclosion were dark adapted for 2 min before being exposed to a 10-s pulse of orange light.
Figure 8.

TRPL channel function depends on a basic residue at position 678, which corresponds to arginine 671 in TRP. (A) Alignment of the TRP, TRPL, and TRPγ amino acid sequences between the sixth transmembrane domain (TM6) and TRP box 1. The amino acids in TRPL (residue 678) and TRPγ (residue 662) corresponding to arginine 671 in TRP are indicated by the box. (B) TRPL protein expression in flies containing the trpl transgenes. A wild-type trpl transgene and mutant transgenes containing the H678R and H678Q missense changes (trpl 678R and trpl 678Q, respectively) were fused to the ninaE promoter and introduced into a trpl 302;trp P343 background. The Western blot containing head extracts prepared from flies <1 d after eclosion was probed with anti-TRPL antibodies and reprobed with anti-tubulin antibodies. (C–G) ERG recordings were performed on flies <1 d after eclosion. The flies were dark adapted for 2 min before being exposed to a 10-s pulse of orange light. (C) trpl;trp P343; (D) trp P343. (E–G) The ERGs performed on the following transgenic flies were performed in a trpl 302;trp P343 background: (E) ninaE-trpl wt (trpl wt); (F) ninaE-trpl H678R (trpl 678R); and (G) ninaE-trpl H678Q (trpl 678Q). (H) Quantification of the ERG amplitudes obtained in the indicated trpl transgenic flies in a trpl 302;trp P343 background. SDs are indicated.
To determine whether one or both mutations were responsible for the transient light response phenotype in trp 14, we generated and tested transgenic flies expressing TRP isoforms with just the L612F or R671Q amino acid substitution (trp 612F and trp 671Q, respectively). The wild-type or mutant trp cDNAs were fused to the ninaE (neither inactivation nor afterpotential E [encodes Rh1]) promoter and introduced into the trpl 302;trp P343 double mutant background. We subsequently performed Western blots on the transgenic flies demonstrating that wild-type TRP, TRP612F, and TRP671Q were all expressed at similar levels (Fig. 7 B).
We found that the missense mutation juxtaposed to the TRP domain was responsible for the phenotype in trp 14. The trpl;ninaE-trp 612F,trp P343 flies displayed a wild-type ERG response (Fig. 7 E) indistinguishable from trpl;ninaE-trp wt,trp P343 (Fig. 7 D). Conversely, the trpl;ninaE-trp 671Q,trp P343 flies showed a transient ERG phenotype (Fig. 7 F). Moreover, the trp 671Q flies exhibited an ERG phenotype with a more rapid decline typical of trp P343 suggesting that the 612F mutation resulted in a slight suppression of the TRP phenotype. These data demonstrate that the missense mutation at residue 671 situated between the sixth transmembrane domain and TRP box 1 (Montell, 2005b) was the key mutation responsible for the trp 14 phenotype.
Mutation of TRPL in same position as TRP14 causes channel defect
The residues in TRPL and TRPγ, corresponding to the required arginine 671 in TRP, are also basic amino acids (histidine 678 and arginine 662, respectively; Fig. 8 A) suggesting that a basic residue at this position flanking the TRP domain is essential in the Drosophila TRPC channels. Therefore, we generated transgenic flies expressing derivatives of TRPL in which histidine 678 was replaced either with a conservative arginine substitution (trpl 678R) or with an uncharged glutamine (trpl 678Q). The mutant and wild-type trpl cDNAs were fused to the ninaE promoter and introduced into a trpl 302;trp P343 background. The transgenic TRPL proteins were all expressed at similar levels, though at an approximate sevenfold higher level than in wild-type due to the strong ninaE promoter (Fig. 8 B).
To determine the consequences of the mutations in TRPL, we performed ERGs after introducing the transgenes in a trpl 302;trp P343 background. Whereas the double trpl 302;trp P343 mutant was blind, overexpression of the wild-type TRPL in this genetic background (trpl wt) restored a transient response to light indistinguishable from trp P343 (Fig. 8, C–E). Furthermore, introduction of the trpl 678R transgene into the genome of trpl 302;trp P343 flies resulted in an ERG response similar to trpl wt (Fig. 8, E, F, and H). Thus, replacing histidine 678 with an arginine did not disrupt TRPL function. However, the amplitude of the ERG was significantly reduced in trpl 302;trp P343 flies expressing the trpl 678Q transgene with the histidine to glutamine substitution in residue 678 (Fig. 8, G and H). The combination of these results demonstrates a critical role of a basic residue at the corresponding positions in TRP and TRPL, flanking the highly conserved TRP box 1.
Discussion
Drosophila TRP is a multifunctional protein as it serves both as a cation channel and a molecular anchor required for the retention of the scaffold protein, INAD, in the rhabdomeres. The TRP scaffold function is critical because the consequent mislocalization of INAD in turn causes instability and mislocalization of TRP, PLC, and PKC. Thus, over time the core proteins in the signalplex are lost from the rhabdomeres and the visual response is reduced. In addition to TRP, other related proteins may also have dual roles as several vertebrate TRPs, such as TRPM2 (Perraud et al., 2001; Sano et al., 2001), TRPM6, and TRPM7 (Nadler et al., 2001; Runnels et al., 2001; Schlingmann et al., 2002; Walder et al., 2002), consist of channel domains fused to enzyme domains. In the case of Drosophila TRP, we have previously characterized the specific role of the anchoring function on the photoresponse, using transgenic flies expressing a derivative of TRP that is missing the INAD binding site (trp Δ 1272; Li and Montell, 2000). Surprisingly, young trp Δ 1272 flies display a normal photoresponse, although as the flies age, INAD and the core binding proteins are not retained in the rhabdomeres.
Null mutations in trp have at least three major consequences in photoreceptor cells. These include the inability to maintain a light response, mislocalization of INAD, PLC, and PKC, and light-induced retinal degeneration. However, it has not been possible to determine the physiological consequences resulting from specifically disrupting the TRP channel function independent of the anchoring role, as all of the previously described loss-of-function mutations (with the exception of trp Δ 1272) virtually eliminate the TRP protein. The trp 14 allele expressed relatively high levels of the TRP protein and exhibited a normal anchoring role as INAD coimmunoprecipitated with the TRP14 protein as effectively as with wild-type TRP. Furthermore, the spatial distributions of the core members of the signalplex were normal in trp 14 photoreceptor cells.
Rather than affecting the anchoring role, the mutation of the basic residue situated between the sixth transmembrane segment and the TRP domain, disrupted TRP channel function such that the response to light stimulation was transient. Though the molecular basis for the defect in TRP channel function is unclear, mutation of the corresponding basic residue in TRPL also disrupted the activity of this latter channel. Thus, this region would appear to play a critical role in TRPC channel function in vivo. The transient light response in trp 14 is not a simple consequence of the slightly lower expression of the mutant protein (60% of wild-type levels) as we found that expression of wild-type TRP at 4% the normal levels did not cause a transient light response, though the amplitude of the ERG was reduced. The TRP14 protein also displayed a wild-type rhabdomeral expression pattern, so that the phenotype was not due to mislocalization of the protein.
Of significance here, we found that the retinal degeneration associated with loss-of-function mutations in trp was due primarily to defects in channel function, rather than disruption of the anchoring role. This finding is surprising because elimination of the TRP scaffold function causes time-dependent instability and mislocalization of all four core proteins in the signalplex. Thus, low levels of TRP, INAD, PLC, and PKC result in less pronounced cell death than an amino acid substitution in TRP that disrupts channel function, but has no impact on the concentrations of the core proteins in the signalplex.
The basis for the retinal degeneration was decreased light-dependent Ca2+ influx because the cell death in either trp 14 or trp-null mutant flies (trp P343) was greatly reduced by strong loss-of-function mutations in the gene encoding the Na+/Ca2+ exchanger, CalX. This effect was not a consequence of suppression of the anchoring defect because the core signalplex proteins were still mislocalized in calx;trp P343 double mutant flies. Given that the strong light-dependent retinal degeneration in calx was reciprocally suppressed by the trp P343 or trp 14 mutations, these data also indicated that the cell death in calx resulted from Ca2+ overload.
The mechanism through which decreased Ca2+ influx causes cell death in trp appears to be due at least in part from increased rhodopsin–arrestin complexes. Stable rhodopsin–arrestin complexes and endocytosis of rhodopsin has been associated with degeneration resulting from mutations in the PLC and rhodopsin phosphatase (Alloway et al., 2000; Kiselev et al., 2000; Orem and Dolph, 2002). In the current study, we found that the trp-dependent retinal degeneration was partially suppressed by mutations in arr2. Because Ca2+/calmodulin-dependent phosphorylation of arrestin promotes the release of arrestin from rhodopsin (Alloway and Dolph, 1999), we suggest that a consequence of decreased light-dependent Ca2+ influx in trp 14 is reduced phosphorylation of arrestin, which in turn results in increased stability of arrestin–rhodopsin complexes. Alternatively, the reduced Ca2+ influx could result in increased arrestin–rhodopsin complexes due to affects on the rhodopsin phosphatase, RDGC (retinal degeneration C). The activity of RDGC is dependent on Ca2+/calmodulin (Lee and Montell, 2001) and loss of function mutations in rdgC result in stable rhodopsin–arrestin complexes and retinal degeneration (Kiselev et al., 2000).
The observation that decreased TRP-dependent Ca2+ influx underlies retinal degeneration in fly photoreceptor cells has potential implications in terms of the possible effects on cell survival resulting from loss-of-function mutations in vertebrate TRPs. It appears that constitutive activity of Drosophila and mammalian TRP leads to cell death due to Ca2+ overload (Yoon et al., 2000; Hara et al., 2002; Wehage et al., 2002; Aarts et al., 2003; Wang et al., 2005). Moreover, constitutive activity of TRPs by anoxic conditions has been proposed to underlie the massive cell death in the mammalian brain that can occur under anoxic conditions, such as occurs as a result of stroke (Agam et al., 2000; Yoon et al., 2000; Hara et al., 2002; Aarts et al., 2003; Wang et al., 2005).
The opposite of constitutive activation is elimination of TRP channel function and whether loss of vertebrate TRP-dependent Ca2+ influx leads to cell death has not been addressed. However, the results of the current analysis indicate that this is a likely possibility. Elimination TRPM7 from chicken DT40 cells results in cell death (Nadler et al., 2001), but the basis for the requirement for TRPM7 is not known. Given that TRPM7 consists of a TRP channel domain, fused to a COOH-terminal protein kinase domain, the cell death due to loss of TRPM7 could reflect a requirement for either the channel or kinase functions. Moreover, as TRPM7 is highly permeable to both Mg2+ and Ca2+ (Nadler et al., 2001), it is unclear the Mg2+ or Ca2+ influx is most important for viability. It will be of interest to determine whether the TRPM7-dependent cell death can be suppressed by inhibition of the Na+/Ca2+ exchanger, as we observed for Drosophila TRP and CalX.
Materials and methods
Fly stocks
The trp 14, trp 38, trp 47, trp 74, and trp 92 alleles were isolated by performing ethylmethylsulfonate mutagenesis and screening for third chromosome mutations affecting the ERG as we have described recently (Wang et al., 2005). The trp P343 (Montell and Rubin, 1989), calx B (Wang et al., 2005), trp Δ 1272 (Li and Montell, 2000), trpl 302 (Niemeyer et al., 1996), and arr2 5 (Alloway and Dolph, 1999) mutations were previously described.
Generation of transgenic flies
To express TRP under the control of the hsp70 promoter, the full-length trp cDNA was excised from pBluescriptKS+ (pBSNot-ctrp9) with NotI and XbaI and subcloned between the NotI and XbaI sites of pCaspeR-hs (Thummel and Pirrotta, 1992). To express the wild-type or mutated trp genes under the control of the major rhodopsin (ninaE) promoter, the corresponding cDNAs were subcloned between the NotI and XbaI sites of the pNX vector and the inserts were subsequently excised and introduced into the KpnI site of pCaspeR4. To express the wild-type or mutated TRPL under the control of ninaE promoter, the full-length wild-type or mutated trpl cDNAs were subcloned into the NotI site of the pNX vector and the inserts were subsequently excised and introduced into the KpnI site of pCaspeR4 (Thummel and Pirrotta, 1992). The constructs were injected into w 1118 embryos and transformants were identified on the basis of eye pigmentation. The third chromosome transgenes were selected and put into the trp P343 background by recombination.
ERG and intracellular recordings
ERG recordings were performed as previously described (Wes et al., 1999). In brief, two glass microelectrodes filled with Ringer's solution were inserted into small drops of electrode cream, which were placed on the surfaces of the compound eye and the thorax. A light projector (model 765; Newport Corp.) was used for stimulation. The ERGs were amplified with a electrometer (IE-210; Warner Instruments) and recorded with an analogue-to-digital converter (MacLab/4s) and the Chart v3.4/s program (A/D Instruments). All recordings were performed at room temperature. Intracellular recordings on photoreceptor cells were performed by placing a low-resistance microelectrode filled with 3 M KCl through a small hole on the compound eye and a reference electrode on the thorax as described (Xiong et al., 1994).
Western blots
Fly heads were homogenized in SDS sample buffer with a pellet pestle (Kimble-Kontes) and the proteins were fractionated by SDS-PAGE. The proteins were transferred overnight at 25V to Immobilon-P transfer membranes (Millipore) in Tris-glycine buffer. The blots were probed with the appropriate rabbit or mouse primary antibodies and subsequently with either IgG peroxidase conjugate (anti–rabbit or mouse, respectively; Sigma-Aldrich) or 125I-labeled protein A (NEN Life Science Products). In those cases in which mouse primary antibodies were used, the blots were probed with rabbit anti–mouse IgG (Sigma-Aldrich) before using the 125I-labeled protein A. The signals were detected using either ECL reagents (GE Healthcare) or a phosphoimager. Polyclonal rabbit anti-TRP (Chevesich et al., 1997), anti-INAD (Wes et al., 1999), and anti-NORPA antibodies (Wang et al., 2005) were previously described. Monoclonal anti-Rh1 and anti-tubulin antibodies were purchased from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Rabbit anti-TRPL antibodies were purchased from Chemicon.
Co-immunoprecipitations
Fly heads (2 mg) were homogenized in 200 μl of buffer A (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 10% sucrose, 1 mM EDTA, and complete protease inhibitor (Brakeman et al., 1997) containing 1% CHAPS with a pellet pestle. The extracts were centrifuged at 15,000 g for 5 min at 4°C to remove cellular debris. Homogenates were diluted with 800 μl buffer A to achieve a final concentration of 0.2% CHAPS. 1 μl of anti-TRP antibodies were added and incubated for 1 h at 4°C, followed by 10 min centrifugation at 4°C. To reduce nonspecific binding, the protein A–Sepharose beads (GE Healthcare) were first incubated with buffer A containing 0.2% CHAPS and 1% BSA for 1 h at 4°C. The immunocomplexes were then incubated for 30 min at 4°C with 50 μl of a 50% slurry of the blocked protein A–Sepharose beads in buffer A with 0.2% CHAPS, followed by 1 min centrifugation at 5,000 g to pellet the protein A–Sepharose beads and associated immunocomplexes. After washing twice with buffer A, the beads were resuspended in SDS sample buffer, fractionated by SDS-PAGE, and a Western blot was probed with anti-INAD antibodies.
Immunolocalizations
Hemisected fly heads were fixed with paraformaldehyde and embedded in LR White resin as described previously (Porter and Montell, 1993). Cross sections (0.5 μm) of compound eyes were cut through the distal region of the retina, which included the R7 cells, and stained with primary antibodies (1:200) and FITC-labeled secondary antibodies (1:200) as described previously (Porter and Montell, 1993). Samples were examined with a Axioplan microscope (Carl Zeiss MicroImaging, Inc.) using a Plan-Apochromat 63× objective and the images were acquired using a SensiCam camera (Cooke) and IPLab 3.6.5 software. The images were then transferred into Adobe Photoshop 7.0 for assembling the figures.
Transmission EM
Heads were dissected from flies reared under a 12-h light–12-h dark cycle or in constant darkness and embedded for transmission EM as described previously (Porter et al. 1992) except that 0.1 M sodium phosphate (pH 7.4) was used as the buffer. Thin sections (85 nm) prepared at a depth of 30 μm were examined by transmission EM using an electron microscope (FEI Tecnai 12; Carl Zeiss MicroImaging, Inc.). The images were acquired using a camera (model 794; Gatan) and Digital Micrograph software (Gatan, Inc.) and converted into tiff files.
Acknowledgments
We thank Mike Sepanski and Erin Black for preparing sections of compound eyes. We are indebted to Roger Hardie for many helpful discussions and comments on the manuscript.
This work was supported by a grant to C. Montell from the National Eye Institute (EY10852 and EY08117).
Abbreviations used in this paper: Arr2, arrestin2; CalX, Na+/Ca2+ exchanger; ERG, electroretinogram; hsp70, heat shock protein 70; INAC, inactivation no afterpotential C, encodes PKC; INAD, inactivation no afterpotential D; ninaE, neither inactivation nor afterpotential E (encodes Rh1); norpA, no receptor potential A, encodes PLC; RDGC, retinal degeneration C; Rh1, rhodopsin 1; TRP, transient receptor potential; TRPγ, transient receptor potential γ; TRPL, transient receptor potential-like.
References
- Aarts, M., K. Iihara, W.L. Wei, Z.G. Xiong, M. Arundine, W. Cerwinski, J.F. MacDonald, and M. Tymianski. 2003. A key role for TRPM7 channels in anoxic neuronal death. Cell. 115:863–877. [DOI] [PubMed] [Google Scholar]
- Agam, K., M. von Campenhausen, S. Levy, H.C. Ben-Ami, B. Cook, K. Kirschfeld, and B. Minke. 2000. Metabolic stress reversibly activates the Drosophila light-sensitive channels TRP and TRPL in vivo. J. Neurosci. 20:5748–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloway, P.G., and P.J. Dolph. 1999. A role for the light-dependent phosphorylation of visual arrestin. Proc. Natl. Acad. Sci. USA. 96:6072–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloway, P.G., L. Howard, and P.J. Dolph. 2000. The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron. 28:129–138. [DOI] [PubMed] [Google Scholar]
- Brakeman, P.R., A.A. Lanahan, R. Obrien, K. Roche, C.A. Barners, R.L. Huganir, and P.F. Worley. 1997. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature. 386:284–288. [DOI] [PubMed] [Google Scholar]
- Chevesich, J., A.J. Kreuz, and C. Montell. 1997. Requirement for the PDZ domain protein, INAD, for localization of the TRP store-operated channel to a signaling complex. Neuron. 18:95–105. [DOI] [PubMed] [Google Scholar]
- Cosens, D., and M.M. Perry. 1972. The fine structure of the eye of a visual mutant, A-type, of Drosophila melanogaster. J. Insect Physiol. 18:1773–1786. [DOI] [PubMed] [Google Scholar]
- Dolph, P.J., R. Ranganathan, N.J. Colley, R.W. Hardy, M. Socolich, and C.S. Zuker. 1993. Arrestin function in inactivation of G protein-coupled receptor rhodopsin in vivo. Science. 260:1910–1916. [DOI] [PubMed] [Google Scholar]
- Goel, M., R. Garcia, M. Estacion, and W.P. Schilling. 2001. Regulation of Drosophila TRPL channels by immunophilin FKBP59. J. Biol. Chem. 276:38762–38773. [DOI] [PubMed] [Google Scholar]
- Hara, Y., M. Wakamori, M. Ishii, E. Maeno, M. Nishida, T. Yoshida, H. Yamada, S. Shimizu, E. Mori, J. Kudoh, et al. 2002. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell. 9:163–173. [DOI] [PubMed] [Google Scholar]
- Huber, A., P. Sander, A. Gobert, M. Bähner, R. Hermann, and R. Paulsen. 1996. The transient receptor potential protein (Trp), a putative store-operated Ca2+ channel essential for phosphoinositide-mediated photoreception, forms a signaling complex with NorpA, InaC and InaD. EMBO J. 15:7036–7045. [PMC free article] [PubMed] [Google Scholar]
- Kiselev, A., M. Socolich, J. Vinos, R.W. Hardy, C.S. Zuker, and R. Ranganathan. 2000. A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron. 28:139–152. [DOI] [PubMed] [Google Scholar]
- Lee, S.J., and C. Montell. 2001. Regulation of the rhodopsin protein phosphatase, RDGC, through interaction with calmodulin. Neuron. 32:1097–1106. [DOI] [PubMed] [Google Scholar]
- Leung, H.T., C. Geng, and W.L. Pak. 2000. Phenotypes of trpl mutants and interactions between the transient receptor potential (TRP) and TRP-like channels in Drosophila. J. Neurosci. 20:6797–6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H.S., and C. Montell. 2000. TRP and the PDZ protein, INAD, form the core complex required for retention of the signalplex in Drosophila photoreceptor cells. J. Cell Biol. 150:1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell, C. 1999. Visual transduction in Drosophila. Annu. Rev. Cell Dev. Biol. 15:231–268. [DOI] [PubMed] [Google Scholar]
- Montell, C. 2005. a. TRP channels in Drosophila photoreceptor cells. J. Physiol. 567.1:45-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell, C. 2005b. The TRP superfamily of cation channels. Sci. STKE. 2005:re3. [DOI] [PubMed]
- Montell, C., and G.M. Rubin. 1989. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 2:1313–1323. [DOI] [PubMed] [Google Scholar]
- Nadler, M.J., M.C. Hermosura, K. Inabe, A.L. Perraud, Q. Zhu, A.J. Stokes, T. Kurosaki, J.P. Kinet, R. Penner, A.M. Scharenberg, and A. Fleig. 2001. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 411:590–595. [DOI] [PubMed] [Google Scholar]
- Niemeyer, B.A., E. Suzuki, K. Scott, K. Jalink, and C.S. Zuker. 1996. The Drosophila light-activated conductance is composed of the two channels TRP and TRPL. Cell. 85:651–659. [DOI] [PubMed] [Google Scholar]
- Orem, N.R., and P.J. Dolph. 2002. Loss of the phospholipase C gene product induces massive endocytosis of rhodopsin and arrestin in Drosophila photoreceptors. Vision Res. 42:497–505. [DOI] [PubMed] [Google Scholar]
- Pak, W.L. 1994. Retinal degeneration mutants of Drosophila. In Molecular Genetics of Inherited Eye Disorders. A.F. Wright and B. Jay, editors. Harwood Academic Publishers, Chur, Switzerland. 29–52.
- Perraud, A.L., A. Fleig, C.A. Dunn, L.A. Bagley, P. Launay, C. Schmitz, A.J. Stokes, Q. Zhu, M.J. Bessman, R. Penner, et al. 2001. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 411:595–599. [DOI] [PubMed] [Google Scholar]
- Phillips, A.M., A. Bull, and L.E. Kelly. 1992. Identification of a Drosophila gene encoding a calmodulin-binding protein with homology to the trp phototransduction gene. Neuron. 8:631–642. [DOI] [PubMed] [Google Scholar]
- Porter, J.A., J.L. Hicks, D.S. Williams, and C. Montell. 1992. Differential localizations of and requirements for the two Drosophila ninaC kinase/myosins in photoreceptor cells. J. Cell Biol. 116:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, J.A., and C. Montell. 1993. Distinct roles of the Drosophila ninaC kinase and myosin domains revealed by systematic mutagenesis. J. Cell Biol. 122:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuss, H., M.H. Mojet, S. Chyb, and R.C. Hardie. 1997. In vivo analysis of the Drosophila light-sensitive channels, TRP and TRPL. Neuron. 19:1249–1259. [DOI] [PubMed] [Google Scholar]
- Runnels, L.W., L. Yue, and D.E. Clapham. 2001. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 291:1043–1047. [DOI] [PubMed] [Google Scholar]
- Sano, Y., K. Inamura, A. Miyake, S. Mochizuki, H. Yokoi, H. Matsushime, and K. Furuichi. 2001. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 293:1327–1330. [DOI] [PubMed] [Google Scholar]
- Schlingmann, K.P., S. Weber, M. Peters, L. Niemann Nejsum, H. Vitzthum, K. Klingel, M. Kratz, E. Haddad, E. Ristoff, D. Dinour, et al. 2002. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat. Genet. 31:166–170. [DOI] [PubMed] [Google Scholar]
- Shieh, B.-H., and M.-Y. Zhu. 1996. Regulation of the TRP Ca2+ channel by INAD in Drosophila photoreceptors. Neuron. 16:991–998. [DOI] [PubMed] [Google Scholar]
- Thummel, C.S., and V. Pirrotta. 1992. New pCaSpeR P element vectors. Drosoph. Inf. Serv. 71:150. [Google Scholar]
- Tsunoda, S., J. Sierralta, Y. Sun, R. Bodner, E. Suzuki, A. Becker, M. Socolich, and C.S. Zuker. 1997. A multivalent PDZ-domain protein assembles signalling complexes in a G-protein-coupled cascade. Nature. 388:243–249. [DOI] [PubMed] [Google Scholar]
- Tsunoda, S., Y. Sun, E. Suzuki, and C. Zuker. 2001. Independent anchoring and assembly mechanisms of INAD signaling complexes in Drosophila photoreceptors. J. Neurosci. 21:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walder, R.Y., D. Landau, P. Meyer, H. Shalev, M. Tsolia, Z. Borochowitz, M.B. Boettger, G.E. Beck, R.K. Englehardt, R. Carmi, and V.C. Sheffield. 2002. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat. Genet. 31:171–174. [DOI] [PubMed] [Google Scholar]
- Wang, T., H. Xu, J. Oberwinkler, Y. Gu, R.C. Hardie, and C. Montell. 2005. Light activation, adaptation, and cell survival functions of the Na+/Ca2+ exchanger CalX. Neuron. 45:367–378. [DOI] [PubMed] [Google Scholar]
- Wehage, E., J. Eisfeld, I. Heiner, E. Jungling, C. Zitt, and A. Lückhoff. 2002. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J. Biol. Chem. 277:23150–23156. [DOI] [PubMed] [Google Scholar]
- Wes, P.D., X.-Z.S. Xu, H.-S. Li, F. Chien, S.K. Doberstein, and C. Montell. 1999. Termination of phototransduction requires binding of the NINAC myosin III and the PDZ protein INAD. Nat. Neurosci. 2:447–453. [DOI] [PubMed] [Google Scholar]
- Xiong, W.-C., H. Okano, N.H. Patel, J.A. Blendy, and C. Montell. 1994. repo encodes a glial-specific homeo domain protein required in the Drosophila nervous system. Genes Dev. 8:981–994. [DOI] [PubMed] [Google Scholar]
- Xu, X.Z., F. Chien, A. Butler, L. Salkoff, and C. Montell. 2000. TRPγ, a Drosophila TRP-related subunit, forms a regulated cation channel with TRPL. Neuron. 26:647–657. [DOI] [PubMed] [Google Scholar]
- Xu, X.Z., A. Choudhury, X. Li, and C. Montell. 1998. Coordination of an array of signaling proteins through homo- and heteromeric interactions between PDZ domains and target proteins. J. Cell Biol. 142:545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, J., H.C. Ben-Ami, Y.S. Hong, S. Park, L.L. Strong, J. Bowman, C. Geng, K. Baek, B. Minke, and W.L. Pak. 2000. Novel mechanism of massive photoreceptor degeneration caused by mutations in the trp gene of Drosophila. J. Neurosci. 20:649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]