

Abstract
Members of the fibroblast growth factor (FGF) family and the FGF receptors (FGFRs) have been implicated in mediating various aspects of mammary gland development and transformation. To elucidate the molecular mechanisms of FGFR1 action in a context that mimics polarized epithelial cells, we have developed an in vitro three-dimensional HC11 mouse mammary epithelial cell culture model expressing a drug-inducible FGFR1 (iFGFR1). Using this conditional model, iFGFR1 activation in these growth-arrested and polarized mammary acini initially led to reinitiation of cell proliferation, increased survival of luminal cells, and loss of cell polarity, resulting in the disruption of acinar structures characterized by the absence of an empty lumen. iFGFR1 activation also resulted in a gain of invasive properties and the induction of matrix metalloproteinase 3 (MMP-3), causing the cleavage of E-cadherin and increased expression of smooth muscle actin and vimentin. The addition of a pan MMP inhibitor abolished these phenotypes but did not prevent the effects of iFGFR1 on cell proliferation or survival.
Introduction
The pathways through which FGFs and their receptors signal appear to play important roles not only in normal development and wound healing but also in tumor formation and progression (Powers et al., 2000). To date, 22 distinct FGFs have been discovered that can act in an autocrine or paracrine fashion through a class of cell-surface tyrosine kinase receptors (RTKs; Grose and Dickson, 2005). The ligand binding capacity and tissue distribution of each receptor is highly variable (Dailey et al., 2005).
Several studies have now linked inappropriate expression of FGFs and their receptors to the development of human breast cancer (Dickson et al., 2000). For example, in some breast cancers, FGF receptors (FGFRs) 1, 2, and 4 are amplified (Penault-Llorca et al., 1995). Thus, a better understanding of the role of FGF signaling during breast cancer progression may facilitate the development of cancer therapies to target this signaling pathway.
The diversity and complexity of FGF ligand and receptor interactions makes it difficult to delineate the molecular mechanisms of action of individual FGF ligands or receptors, which determine a specific cellular response. Thus, an inducible FGFR (iFGFR) was developed as a ligand-independent dimerization and activation system to study the early events in FGFR signaling and transformation (Welm et al., 2002). In mouse mammary tumor virus–iFGFR1 transgenic mice, acute induction of iFGFR1 results in increased lateral budding of the mammary ductal epithelium and sustained activation induces alveolar hyperplasia and invasive lesions, which are associated with ECM remodeling and vascular branching in the stroma adjacent to these lesions. These data suggest a potential role for FGFs in initiating branching in the mammary gland and in restructuring the stroma to provide support for the developing ductal network. In addition, these results indicate that FGFR1 signaling may play a critical role in early mammary tumor progression.
To date, many of the studies of FGF signaling have been performed in 3T3 fibroblasts in two-dimensional (2D) culture systems, and it was not possible to study the effects of this pathway in a polarized architecture that mimics acinar structures of mammary epithelial cells in vivo. Unlike monolayer cultures, mammary epithelial cells grown in three dimensions recapitulate several features of mammary epithelium in vivo (Schmeichel and Bissell, 2003; Shaw et al., 2004). Therefore, the three-dimensional (3D) culture system has been used as a unique approach to investigate the signaling pathways involved in the early progression of cancer (Muthuswamy et al., 2001; Radisky et al., 2001; Debnath et al., 2003b).
Several human and mouse mammary epithelial cell lines have been shown to form acinar structures when grown in a basement membrane matrix (Shaw et al., 2004). In this paper, we show for the first time that another extensively used immortalized murine mammary epithelial cell line, HC11, can form acinus-like structures containing a single layer of polarized, growth-arrested cells when grown within a growth factor–reduced matrix. HC11 cells are a clonal mouse mammary epithelial cell line derived from a midpregnant mouse mammary gland. Upon hormonal stimulation, these cells differentiate and express milk proteins in vitro (Ball et al., 1988). We therefore combined the well-defined synthetic chemical ligand–induced dimerization strategy and HC11 3D cell cultures to establish a new model system to help dissect the molecular mechanisms through which iFGFR1 activation disrupts the polarized architecture of acinar units in the mammary gland and promotes an invasive phenotype.
Using this model system, we demonstrate that iFGFR1 activation rapidly disrupted cell polarity, induced proliferation, and promoted cell survival. In addition, matrix metalloproteinase 3 (MMP-3) expression was induced and appears to play a critical role during an epithelial-to-mesenchymal transition (EMT) characterized by cleavage of E-cadherin, expression of mesenchymal markers, and cell invasion. Importantly, our data suggest that increased MMP expression by iFGFR1 was critical for EMT but not for regulation of cell survival or proliferation. Furthermore, small interfering RNA (siRNA) targeted against MMP-3, but not -9, blocked cleavage of E-cadherin. These studies describe the first-time use of an inducible dimerization model to study FGFR1 signaling in an HC11 3D culture system.
Results
3D. culture of HC11 mammary epithelial cells
We generated 3D acinar structures by plating single HC11 cells on the matrix. At day 6, immunostaining and confocal imaging for phosphorylated histone H3 (p-histone H3), a marker of mitosis; α6 integrin, a basal surface marker (Fig. 1 A); and phospho-ezrin/radixin/moesin (p-ERM), an apical marker (Fig. 1 B), indicated that the structures consisted of unpolarized proliferative cells. We did not observe the staining of cleaved caspase 3, an apoptosis marker, at day 6 (unpublished data). At day 8, apical–basal polarization appeared within the cell clusters before lumen formation. The absence of p-histone H3 staining (Fig. 1 C) and the lack of increase in the size of the acini after day 8 suggested that the acini had reached a growth-arrested state. Immunostaining for α6 integrin and cleaved caspase 3 (Fig. 1 E) suggested that the acini consisted of two populations of cells: a polarized outer layer of cells that directly contacted the matrix and survived and an inner cluster of apoptotic and poorly polarized cells that did not contact the matrix. Additionally, HC11 acini displayed other aspects of polarity, such as basal deposition of laminin V, a basement membrane component that was detected by immunostaining with an anti–laminin V antibody (Fig. 1 F), and enrichment of p-ERM at the apical surface (Fig. 1 D). After 10–12 d in culture, the majority of the acini contained 15–40 polarized cells surrounding an empty lumen. Polarization of the cells was confirmed by immunostaining for α6 integrin and p-ERM (Fig. 1 G) and laminin V (Fig. 1 H). These results suggest that HC11 cells grown in three dimensions recapitulate several features of mammary epithelial cells in vivo, providing the structural and functional context necessary for examining the biological roles of genes during early-stage tumorigenesis.
Figure 1.
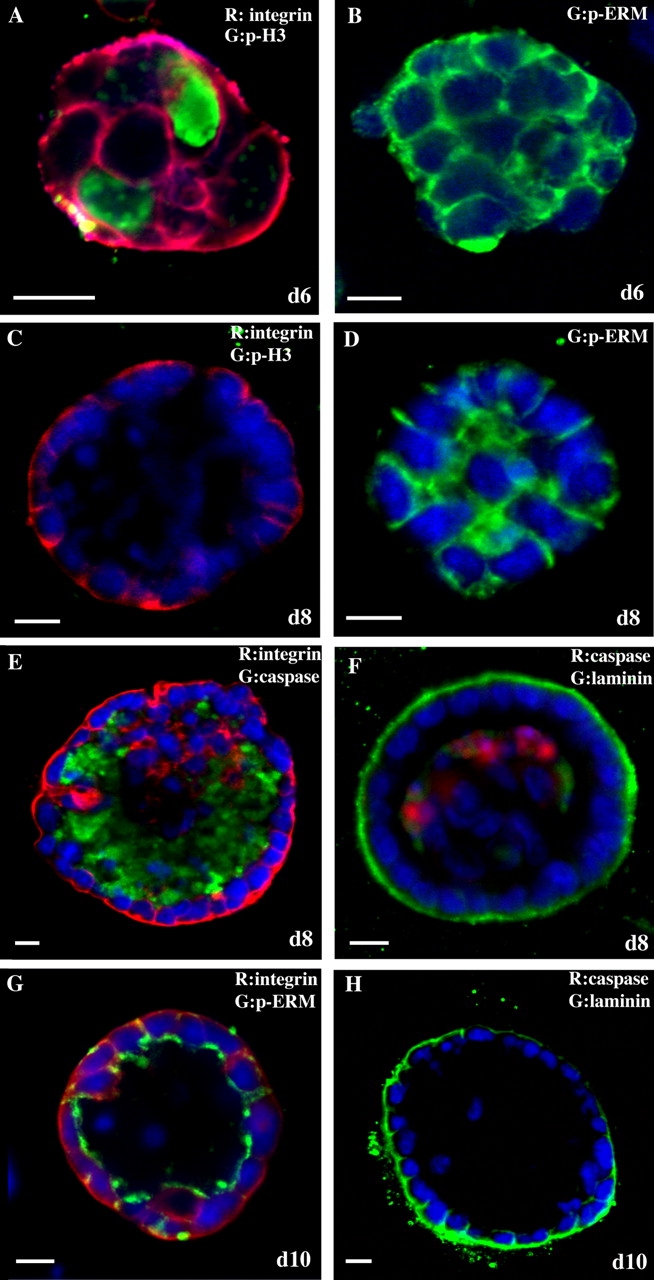
HC11 cells form polarized, growth-arrested acini when cultured on a basement membrane matrix. Representative confocal microscopic images are shown. Day 6 HC11 acini were immunostained with antiserum to p-histone H3 (A, green) and α6 integrin (A, red) or p-ERM (B, green). Day 8 HC11 acini were immunostained with antiserum to p-histone H3 (C, green) and α6 integrin (C, red), p-ERM (D, green), cleaved caspase 3 (E, green) and α6 integrin (E, red), and laminin V (F, green) and cleaved caspase 3 (F, red). Day 10 HC11 acini were immunostained with antiserum to α6 integrin (G, red) and p-ERM (G, green) and to laminin V (H, green). Bars, 10 μm.
iFGFR1 activation disrupts HC11 acinar morphology
To examine the downstream effects of iFGFR1 activation in HC11 3D cultures, we generated stable cell lines expressing iFGFR1. Two clones expressing different levels of iFGFR1 as determined by immunoblotting for expression of HA-tagged iFGFR1 (R1-high [R1-H] and R1-low [RI-L]) were chosen for further analysis (Fig. 2 A). To determine whether dimerization of iFGFR1 in the two clones activates downstream signaling pathways, we analyzed whether iFGFR1 induction could activate extracellular regulated kinase (ERK) in 2D cultures. On AP20187 induction, both clones showed sustained activation of ERK, which correlated with the level of iFGFR1 expression (Fig. 2, B and C). This result indicated that both clones transduce signals downstream of iFGFR1 to activate ERK. Cell cycle analysis was performed on the clones grown in two dimensions to determine whether the AP20187-induced iFGFR1 signaling pathway could also stimulate a biological response, such as proliferation. For this experiment, the clones were treated with AP20187 in serum-free medium, and cellular DNA content was determined by flow cytometry of propidium iodide–stained cells. Both clones showed a greater than twofold increase in the number of cells in S phase in response to AP20187 (unpublished data), which was consistent with previous studies using HC11 cells transiently transduced with iFGFR1 (Welm et al., 2002). These results suggested that dimerization of iFGFR1 in the two clones was sufficient to stimulate a biological response.
Figure 2.
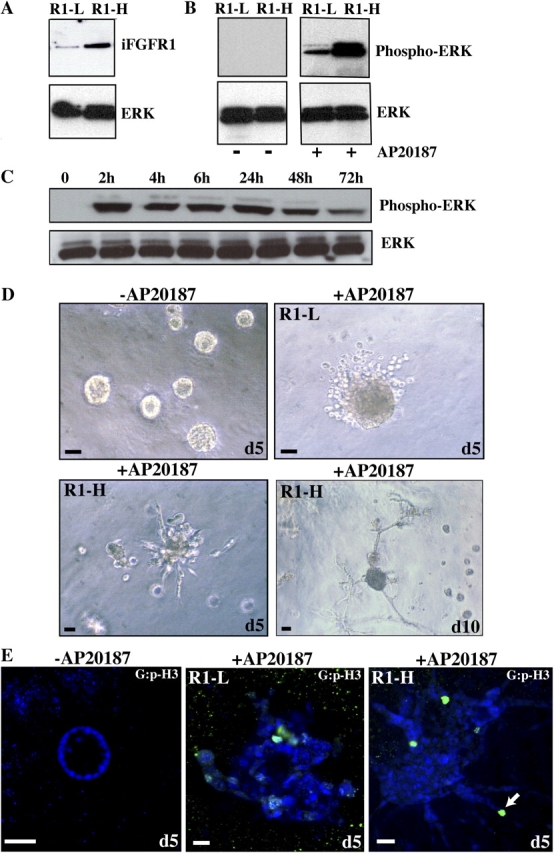
iFGFR1 activation alters HC11 acinar morphogenesis. (A) Two stable clones of HC11 cells expressing different levels of iFGFR1 (R1-L and -H) were examined by immunoblotting with anti–HA epitope antibody. (B) ERK activation of two clones was assessed by immunoblotting with anti–p-ERK after treatment with AP20187 for 30 min. Equal loading of protein on the blot was determined by reprobing the membrane with anti-ERK antibody. (C) Time course of ERK activation after iFGFR1 induction was examined by immunoblotting using anti–p-ERK antibody. ERK is shown as a control for equal protein loading. The two clones gave similar results, so only R1-L is shown here. (D) 10-d-old iFGFR1 acini were treated with (+) or without (−) AP20187 for 5 or 10 d. Brightfield images are shown. Bars, 50 μm. (E) Confocal images of acinar cultures treated for 5 d and immunostained by anti–p-histone H3 (green) are shown. TOPRO-3 (blue) labeled the nuclei. The arrow indicates where cells continued to proliferate and invade the matrix, which was observed after 5 d of treatment. Bars, 25 μm.
To examine the effects of iFGFR1 activation in preformed polarized HC11 acini, single cells of each clone were seeded on the matrix and allowed to grow for 10 d to form growth-arrested acini. When grown on the matrix in the absence of AP20187, the HC11 clones expressing iFGFR1 displayed the same growth and morphogenesis properties as nontransduced HC11 cells (unpublished data). After 10 d, growth-arrested polarized acini had formed and AP20187 were added to the medium for 5 d. Activation of iFGFR1 induced striking changes in the morphology of the acinar structures (Fig. 2 D). In contrast to the untreated iFGFR1 acini, >80% of the treated acini displayed dramatically increased acinar size (∼10-fold larger) and an invasive phenotype. Immunostaining with the anti–p-histone H3 antibody and confocal analyses of these structures demonstrated that untreated iFGFR1 acini remained growth arrested, whereas proliferation was reinitiated in the treated iFGFR1 acini that now contained lumens that were completely filled with cells (Fig. 2 E). Although the R1-H and -L clones had similar proliferation properties (not depicted), their invasive behaviors correlated with their iFGFR1 expression level. In a few acini (5% or less), cells dissociated and invaded the matrix after 24 h of AP20187 treatment. In 80–90% of the treated acini, a progressive architectural disruption occurred, and the acini displayed an invasive phenotype after 5 d of treatment. 5 d after stimulation, the R1-L clone had individual cells that dissociated from the acini and invaded the matrix, whereas the R1-H clone had long projections containing multiple cells that invaded the matrix (Fig. 2 D). The cells in these projections continued to proliferate (as determined by anti–p-histone H3 immunostaining) and invade the matrix, which was observed after 5 d of treatment (Fig. 2 E, arrow). Together, these results demonstrate that although proliferation in response to iFGFR activation was similar in the two clones, the severity of the invasive phenotype correlated with the level of FGFR1 expression.
iFGFR1 activation disrupts polarity, prevents luminal apoptosis, reinitiates proliferation, and promotes invasion in HC11 acini
Our previous study of iFGFR1 transgenic mice showed that iFGFR1 activation prevented apoptosis, induced proliferation, and altered cell polarity in mammary epithelial cells. Furthermore, iFGFR1-induced lesions have invasive characteristics (Welm et al., 2002). To examine whether iFGFR1 homodimers were able to activate downstream signaling and recapitulate some phenotypes of transgenic mice in polarized, growth-arrested 3D acini, iFGFR1 acini were stimulated with AP20187 for 24 h or 5 d, and cell polarity, proliferation, and apoptosis were monitored by immunostaining with the appropriate markers.
The serine/threonine protein kinase Akt (protein kinase B), a regulator of cellular proliferation and apoptosis (Scheid and Woodgett, 2001), is a downstream target of FGF signaling. To examine whether Akt signaling was activated on iFGFR1 induction in 3D cultures, we performed immunostaining on treated and untreated iFGFR1 acini using a phosphospecific Akt antibody that recognized an activation-specific phosphorylation site (anti–p-Akt Ser 473; Fig. 3 A, a and b). The untreated acini exhibited a random pattern of Akt activation only in the outer layer cells that directly contacted the matrix, whereas the treated structures displayed significantly increased levels of activated Akt present in both the luminal cells and the outer layer cells. This result suggests that the iFGFR1 homodimer is able to activate a physiological FGF signaling pathway in 3D cultures.
Figure 3.

iFGFR1 activation inhibits luminal apoptosis, induces proliferation, promotes cell invasion, and disrupts cell polarity. Two clones gave similar results, so only representative images of R1-L are shown here. For each antibody staining, ∼70 structures from three independent experiments were examined, and ∼85% of them displayed the similar phenotypes. (A) Day 10 iFGFR1 acini were either not treated (−) or treated (+) with AP20187 for 24 h. Representative confocal images of structures immunostained with anti–p-Akt (Ser 473; a and b, green) or anti–p-histone H3 (c and d, green) and anti–α6 integrin (c and d, red) are shown. (B) Day 10 iFGFR1 acini either not treated or treated with AP20187 for 5 d. Representative confocal images of structures immunostained with antibodies against p-ERM (a and b, green), α6 integrin (c and d, red) and p-histone H3 (c and d, green), or cleaved caspase 3 (e and f, green) are shown. TOPRO-3 stained the nuclei (blue). Bars, 25 μm. (C) Quantitation of p-histone H3 staining of untreated (UT), 24-h treated (T1D), and 5-d treated (T5D) acini. The total number of nuclei and the number of nuclei staining positive for p-histone H3 were counted in 74 untreated acini, 83 24-h–treated acini, and 76 5-d–treated acini. Using these numbers, the percentage of p-histone H3–positive cells was calculated. The data are presented as the mean and SEM of fold change in the percentage of p-histone H3 positively staining cells among these three groups from three independent experiments.
To determine whether iFGFR1 activation could reinitiate proliferation in 3D cultures, we stimulated day 10 iFGFR1 acini with AP20187 for 24 h or 5 d and then immunostained them with anti–p-histone H3 antibody to detect mitotic cells. The untreated iFGFR1 acini rarely displayed positive staining of p-histone H3 (Fig. 3, A [c] and B [c]). 24 h after stimulation, an approximately eightfold increase in the number of cells staining positive for p-histone H3 was observed in the treated iFGFR1 acini compared with untreated acini (Fig. 3, A [d] and C). This increase in proliferation was still detectable after 5 d of treatment, when we observed an approximately fourfold increase compared with untreated acini (Fig. 3, B [d] and C). These data suggest that iFGFR1 activation has the ability to reinitiate cell proliferation in the growth-arrested HC11 acini. Negative feedback mechanisms involving Sprouty, Sef, and Cbl (Hanafusa et al., 2002; Wong et al., 2002; Kovalenko et al., 2003) may attenuate iFGFR1 signaling after 5 d of treatment, leading to reduced cell proliferation.
To test whether iFGFR1 activation was able to disrupt cell polarity in the polarized acini, we performed immunostaining on iFGFR1 acini with antibodies against the polarity markers α6 integrin (Fig. 3, A [c and d] and B [c and d]) and p-ERM (Fig. 3 B, a and b). After 24 h of treatment, cell polarity within the acini was rapidly disrupted because α6 integrin staining was no longer limited to the basal-lateral region as seen in untreated acini. In addition, the enriched apical localization of p-ERM was also disrupted and was now detected around the entire periphery of the cells (unpublished data). This disruption of cell polarity persisted after 5 d of treatment. Thus, iFGFR1 activation resulted in a sustained loss of cell polarity.
To determine whether iFGFR1 activation could promote cell survival in the acini, apoptosis was analyzed by immunostaining with an anti–cleaved caspase 3 antibody. In comparison with the untreated acini, after 5 d of treatment, a dramatic decrease in the number of cells staining positive for cleaved caspase 3 was detected in the luminal cells (Fig. 3 B, e and f). The increased cell proliferation and inhibition of cell death appears to account for the increased size and filled lumens of treated iFGFR1 acini. Therefore, iFGFR1 activation has the ability to reinitiate cell proliferation and disrupt cell polarity in these growth-arrested polarized HC11 acini, suggesting that it can be used to investigate the early stages of tumor progression induced by iFGFR1 signaling.
iFGFR1 activation leads to dissociation of E-cadherin and β-catenin from the cell membrane and expression of mesenchymal markers
The invasive behavior of the cells upon iFGFR1 dimerization suggested that iFGFR1 signaling might affect molecules involved in cell–cell adhesion. To examine whether iFGFR1 activation disrupts membrane localization of E-cadherin and β-catenin, day 10 iFGFR1 acini were stimulated with AP20187 for 5 d and immunostained with anti–E-cadherin and anti–β-catenin antibodies (Fig. 4, A and B). Untreated iFGFR1 acini exhibited well-defined cell–cell junctions as indicated by membrane localization of E-cadherin and β-catenin, which were concentrated toward the basal-lateral region of the membrane. In the acini with iFGFR1 activation, diffusely localized E-cadherin and β-catenin were detected as indicated by diminished and punctate staining of both proteins. This finding demonstrated that localization of these adhesive molecules to the cell–cell junctions was lost in the architecturally disrupted acini.
Figure 4.

iFGFR1 activation promotes EMT. 10-d-old iFGFR1 acini were cultured in the media with (+) or without (−) AP20187 for 5 d. Two clones displayed similar EMT phenotype after treatment, so only representative images of R1-L are shown here. Approximately 90 structures from three independent experiments were examined, and ∼80% of them displayed the similar phenotypes. Structures were immunostained with antibodies against E-cadherin (A, green), β-catenin (B, green), SMA (C [a and b], green) and p-histone H3 (C [a and b], red), or vimentin (C, c). TOPRO-3 (blue) marked the nuclei. Arrows indicate where E-cadherin and β-catenin have dissociated from the cell membrane. Bars, 25 μm.
FGFR1 has been suggested to be involved in EMT during mouse mesodermal development (Ciruna et al., 1997; Xu et al., 1999), and mislocalization and loss of E-cadherin at cell–cell junctions is an early event that is essential for cell invasion and EMT (Bracke et al., 1996; Bates and Mercurio, 2005). To investigate whether iFGFR1 activation can promote EMT in the HC11 3D culture system, day 10 iFGFR1 acini treated for 5 d with AP20187 were immunostained with antibodies to detect expression of mesenchymal markers such as smooth muscle actin (SMA) and vimentin (Fig. 4 C, b and c). In contrast to the untreated acini that did not express these markers (Fig. 4 C, a), SMA and vimentin expression were readily detectable in the iFGFR1-activated acini. This result, together with the acquisition of the invasive phenotype and loss of E-cadherin expression at cell–cell junctions, suggested that iFGFR1 activation results in EMT.
iFGFR1 activation alters the motility of monolayer HC11 cells
Disruption of cadherin-mediated adhesion has been suggested to affect the movement of epithelial monolayers (Owens et al., 2000; Wrobel et al., 2004). Because we also observed loss of membrane localization of E-cadherin upon iFGFR1 activation in HC11 cells in 2D cultures (unpublished data), we investigated whether activation of iFGFR1 affected the motility of HC11 monolayer cells. For this experiment, we examined the ability of iFGFR1-expressing HC11 cells in monolayer to break their intercellular contacts and move freely out of the epithelial sheet to repair a wound in the monolayer in response to AP20187 treatment (Fig. 5 A). After 20 h, cells expressing iFGFR1 did not migrate without AP20187 treatment. The R1-L cells displayed a slower level of migration into the wound space, whereas the R1-H cells migrated into the wound space rapidly after stimulation and nearly closed the wound in 20 h. To further confirm this result, a quantitative transwell migration assay was performed. Although both R1-L and -H cells migrated better than untreated cells, ∼2.5 times more migrating cells were detected in the R1-H clone than in the R1-L clone (Fig. 5 B). Therefore, the level of the motility of iFGFR1 stable clones correlated with the expression level of the iFGFR1 gene.
Figure 5.
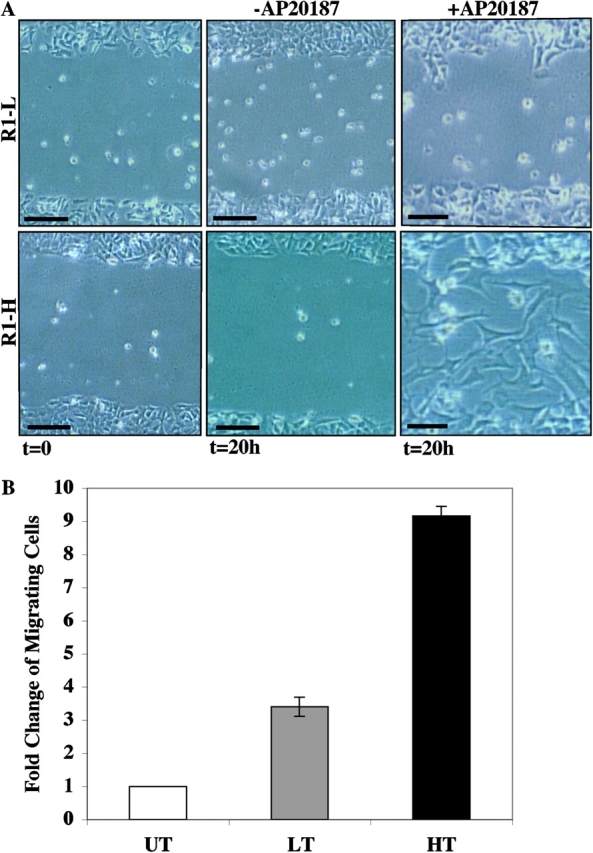
iFGFR1 activation in HC11 cells induces migration. (A) Phase-contrast images of wound closure of R1-L and -H cells that were incubated in medium with (+) or without (−) AP20187 for 20 h. These images are representative of three separate experiments. Bars, 20 μm. (B) R1-L and -H cells were induced to migrate toward serum. The migration ability was calculated by dividing the total number of migrating cells of treated R1-L (LT) and -H cells (HT) by the number of untreated cells (UT) that migrated. Each bar represents the mean and SEM of samples measured in triplicate.
iFGFR1 activation induces MMP-3 expression and cleavage of E-cadherin
FGF signaling has been reported to induce MMP-3 expression (Pintucci et al., 2003), and MMP-3 overexpression has been reported to result in E-cadherin cleavage and EMT (Lochter et al., 1997). We therefore examined the expression level of MMP-3 after iFGFR1 activation by quantitative RT-PCR (Fig. 6 A). A significant increase in MMP-3 expression was observed after 24 h of iFGFR1 activation in HC11 monolayer cultures. A similar increase in MMP-9 expression was also detected (unpublished data). Interestingly, although induction of MMP-3 and -9 expression was observed in both the R1-H and -L clones, the level of the induction correlated with the iFGFR1 gene expression level (unpublished data).
Figure 6.
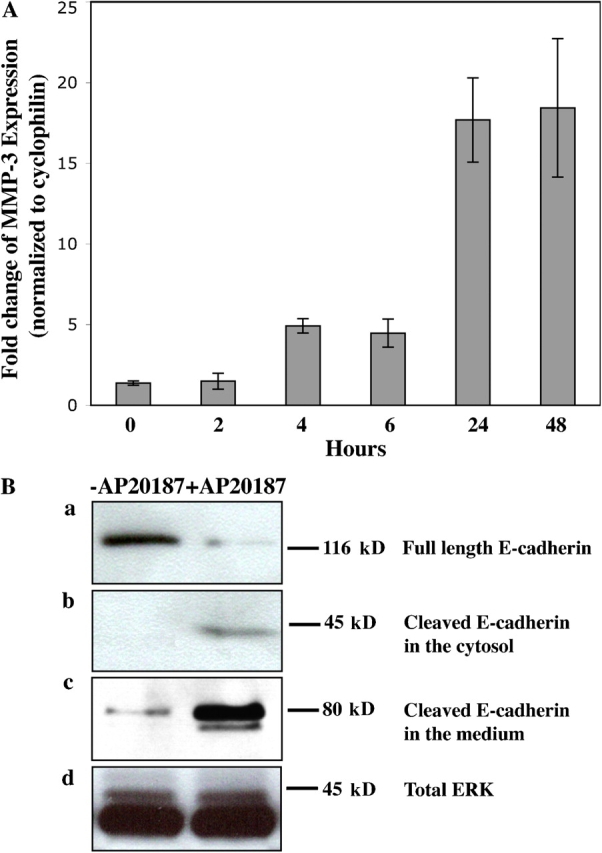
iFGFR1 activation induces MMP-3 expression and triggers cleavage of E-cadherin. (A) Quantitative RT-PCR demonstrates MMP3 induction after iFGFR1 activation in R1-L cells. The mean and SEM from four independent experiments are shown. (B) iFGFR1 cells cultured on tissue culture plastic were serum starved for 16 h and then treated with AP20187 or ethanol solvent for 4 d. Two clones showed similar results, so only R1-L results are shown here. (a) Total levels of cytoskeleton-associated E-cadherin were detected by immunoblotting of lysates using DECMA-1 antibody. (b) Cleaved E-cadherin in the cytosol was detected by immunoblotting using an anti–E-cadherin antibody recognizing the cytoplasmic tail of E-cadherin. (c) The extracellular fragment of E-cadherin found in the conditioned medium was detected using the DECMA-1 antibody. (d) ERK is shown as a control for equal protein loading.
To test whether iFGFR1 activation resulted in cleavage of E-cadherin, cell lysates and conditioned medium were collected from iFGFR1 clones in monolayer culture after 4 d of AP20187 treatment. Immunoblotting using antibodies against E-cadherin on the cell lysates revealed a reduction in the level of full-length E-cadherin (120 kD) expression after treatment (Fig. 6 B, a). A 40-kD fragment of E-cadherin was observed in the cell extract (Fig. 6 B, b), whereas an 80-kD cleaved product of E-cadherin was detected in the conditioned medium after treatment (Fig. 6 B, c). Total ERK was shown to indicate equal protein loading from the cell extracts (Fig. 6 B, d), and Ponceau S staining confirmed equal loading of the proteins from conditioned medium (not depicted). Because we did not detect down-regulation of E-cadherin expression in response to iFGFR1 activation by quantitative RT-PCR (unpublished data), rapid loss of full-length E-cadherin was probably attributable to proteolytic cleavage.
The iFGFR1-induced EMT phenotype can be blocked by a pan MMP inhibitor and is irreversible
MMPs are known to promote cellular invasion both in vitro and in vivo (Egeblad and Werb, 2002). To examine whether the invasive phenotypes induced by iFGFR1 activation are dependent on MMP activity, 10-d-old iFGFR1 acini were treated with AP20187 in the absence or presence of a pan MMP inhibitor, GM6001, for 5 d. In the AP20187-treated iFGFR1 acini, invasion of cells into the matrix was abolished in the presence of the MMP inhibitor (Fig. 7, A and B). Additionally, immunostaining for E-cadherin revealed that it was maintained at the cell–cell junctions in the GM6001-treated cultures but not in control cultures (Fig. 7 C). To determine whether the EMT phenotype was also prevented, AP20187-treated or AP20187/GM6001-treated iFGFR1 acini were immunostained with the anti-SMA antibody (Fig. 7 D). Inhibition of MMP activity abolished SMA expression in the HC11 acini with activated iFGFR1, suggesting that MMP activity was required for these cells to undergo EMT.
Figure 7.
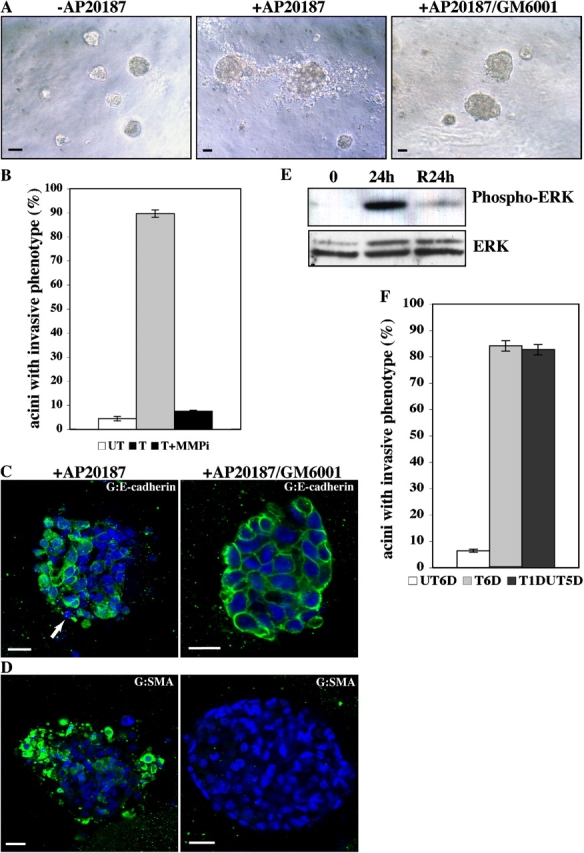
EMT resulting from iFGFR1 activation in HC11 acini is dependent on MMP activity and is not reversible. Two clones showed similar results, so only R1-L results are shown here. (A) 10-d-old iFGFR1 acini were treated (+) or not treated (−) with AP20187 and the MMP inhibitor GM6001 for 5 d. Brightfield images of acini are shown. Bars, 25 μm. (B) Quantitation of invasive phenotypes of iFGFR1 acini in the absence of AP20187 (UT), in the presence of AP20187 (T), or in the presence of AP20187 and GM6001 (T+MMPi). The mean and SEM from three independent experiments are shown. (C and D) Confocal images of day 10 iFGFR1 acini treated with AP20187 (+AP20187) or with AP20187 and GM6001 (+AP20187/GM6001) for 5 d. Structures were stained with TOPRO-3 (C, blue) and anti–E-cadherin (C, green) or with anti-SMA (D, green). Arrow indicates where E-cadherin expression became punctate or lost. Bars, 25 μm. (E) ERK activation of R1 cells induced with AP20187 for 24 h and then withdrawn (R24h) or not (24h) was assessed by immunoblotting with anti–p-ERK. Equal loading of protein on the blot was determined by reprobing the membrane with anti-ERK antibody. (F) Quantitation of invasive phenotypes of iFGFR1 acini that were untreated (UT6D), treated for 6 d (T6D), or withdrawn from AP20187 for 5 d after a 24-h induction (T1DUT5D). The means and SEMs from three independent experiments are shown.
Interestingly, in the presence of AP20187 and GM6001, iFGFR1 acini still displayed increased acinar size and filled lumens. Additionally, immunostaining of AP20187/GM6001-treated iFGFR1 acini with antibodies to detect cell proliferation, apoptosis, and polarity showed results similar to those found in the iFGFR1 acini treated with only AP20187 (unpublished data). These data suggest that the induction of MMP activity is only required for iFGFR1 to induce cell invasion and EMT.
To test whether the EMT-like effect induced by iFGFR1 is reversible, the iFGFR1 acini were treated with AP20187 for 24 h and then AP20187 was removed and the phenotype was evaluated after 5 d. No reduction in the number of acini displaying the invasive phenotype was observed (Fig. 7 F). These acini also showed similar levels of vimentin and SMA staining (unpublished data). This result suggests that the EMT-like effect is not reversible. Interestingly, although dramatically reduced p-ERK was observed by immunoblotting after withdrawal of AP20187 for 24 h (Fig. 7 E), similar elevated levels of MMP-3 and -9 expression were observed by quantitative RT-PCR compared with the cells from which AP20187 was not withdrawn (not depicted). This suggests that there may be a positive feedback loop downstream of iFGFR1 signaling to maintain the expression of target genes even in the absence of continued iFGFR1 activation. The slightly elevated level of p-ERK observed after AP20187 withdrawal as compared with untreated cells may also be sufficient to maintain the induction of downstream target genes accounting for the lack of reversibility of the EMT phenotype.
MMP-3 is essential for E-cadherin cleavage induced by iFGFR1 activation
To determine whether MMP-3 plays an essential role during iFGFR1-induced EMT, we performed a test to see whether inhibition of MMP-3 expression in the iFGFR1 cells affects a critical step of EMT, loss of E-cadherin from cell–cell junctions. To do so, predesigned MMP-3 and -9 siRNA pools of four siRNA duplexes were used. Because a similar EMT effect was observed in both 2D and 3D cultures after iFGFR1 activation, the effects of MMP-3 and -9 siRNA pools were examined in 2D cultures. After siRNA transfection, a significant reduction in MMP-3 and -9 expression was detected in untreated and treated cells, respectively, by quantitative RT-PCR. After AP20187 treatment, MMP-3 siRNA–transfected cells displayed induction of MMP-9 expression but little MMP-3 induction (Fig. 8 A). Similarly, MMP-9 siRNA–transfected cells displayed induction of MMP-3 expression but little MMP-9 induction (unpublished data). This suggests that the MMP siRNAs are specific for their appropriate targets. siGLO, an RNA-induced silencing complex (RISC)–free siRNA, which was used as control, indicated that the transfection efficiency of the siRNA reached almost 90%. Additionally, siGLO-transfected cells displayed induction of both MMP-3 and -9 similar to nontransfected cells.
Figure 8.
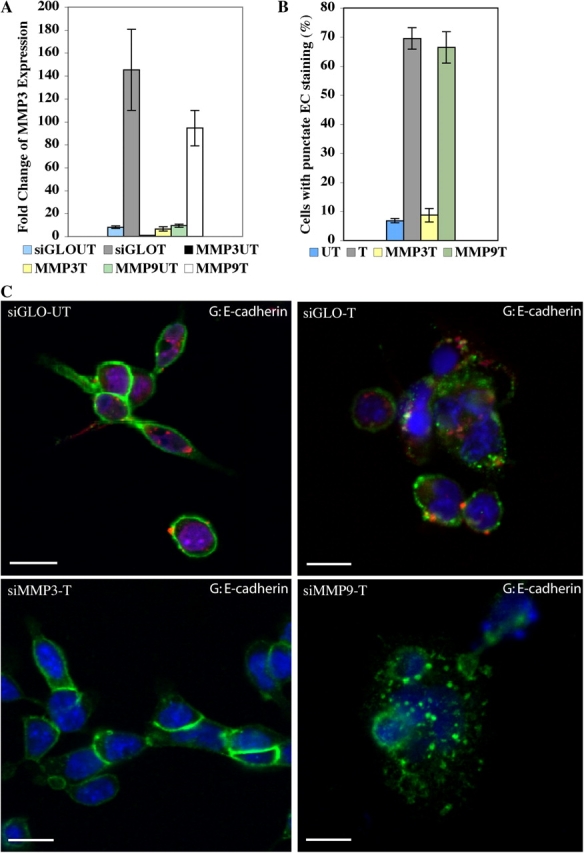
Suppression of MMP-3 expression inhibits E-cadherin cleavage. (A) Quantitative RT-PCR demonstrates MMP-3 expression and induction after iFGFR1 activation (in hours) were significantly reduced in R1 cells transfected with MMP-3 siRNA pool but not with MMP-9 siRNA pool or siGLO control siRNA. The mean and SEM from three independent experiments are shown. (B) Quantitation of E-cadherin punctate staining of untreated iFGFR1 cells transfected with siGLO and AP20187-treated iFGFR1 cells transfected with siGLO, MMP-3 siRNA, or MMP-9 siRNA. The mean and SEM from three independent experiments are shown. (C) Confocal images of the cells immunostained with anti–E-cadherin (green). TOPRO-3 marked the nuclei (blue). siGLO control siRNA displays red fluorescence signal. Bars, 10 μm.
Interestingly, a significant reduction in the percentage of cells showing punctate staining of E-cadherin was observed in AP20187-treated MMP-3 siRNA–transfected iFGFR1 cells but not in siGLO control or MMP-9 siRNA–transfected R1 cells (Fig. 8, B and C). This suggests that MMP-3, but not -9, is involved in cleavage of E-cadherin downstream of iFGFR1 activation. These data are consistent with the reported function of MMP-3 in the other model systems (Lochter et al., 1997; Noe et al., 2001). More important, they suggest that MMP-3 is the critical downstream target of iFGFR1 activation involved in EMT.
Discussion
We have previously described the AP20187-dependent iFGFR1 dimerization system that was developed and applied to a transgenic mouse model (Welm et al., 2002). Although a transgenic mouse model is critical for understanding certain aspects of epithelial tumorigenesis, such as the roles of the inflammatory response and angiogenesis, the complexity of the in vivo environment makes it difficult to delineate the molecular mechanisms involved in the early stages of oncogenesis. Therefore, we analyzed the molecular mechanisms of iFGFR1 in 3D cultures of HC11 mouse mammary epithelial cells to mimic the conditions under which iFGFR1 is activated in vivo.
There are two distinct stages of phenotypic progression observed in this 3D culture model: the first stage includes rapid loss of polarity, reinitiation of proliferation, and reduction of luminal cell apoptosis (observed 24 h after iFGFR1 activation). The second stage involves invasion of cells into the surrounding matrix and EMT (observed 4–5 d after iFGFR1 activation; Fig. 9 A).
Figure 9.
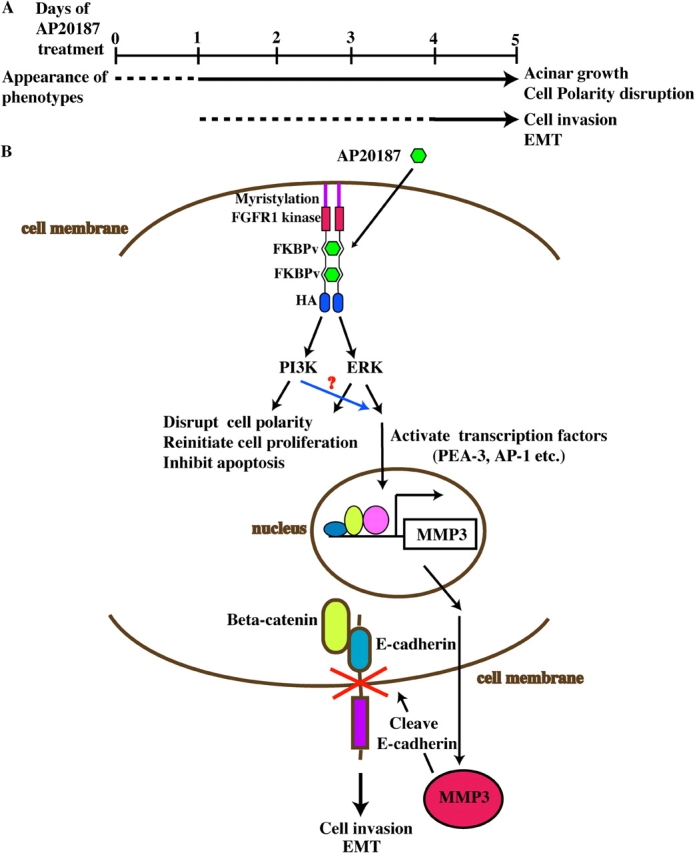
Model for iFGFR1-induced cell invasion and EMT in HC11 3D culture system. (A) Two distinct stages of phenotypic progression were observed in an HC11 3D culture system after iFGFR1 activation. The dashed line suggests that the phenotypes are still progressing, and the solid line indicates that the phenotypes have been established. (B) Conditional dimerization of the iFGFR1 fusion protein by AP20187 activated several downstream signaling pathways, which may eventually lead to cell polarity disruption, acinar growth, invasive phenotypes, and EMT.
Although several RTKs, including colony-stimulating factor receptor (CSF-1R), ErbB2, and Met, have been studied in MCF10A 3D cultures (Shaw et al., 2004), the present study is the first time that FGFR1 signaling has been examined in a 3D culture system. In the HC11 model system, iFGFR1 activation is inducible and allows us to study the consequences of iFGFR1 signaling with time on cell proliferation and invasion in the preformed and growth-arrested HC11 acinar structures. The only other example of using this AP20187-inducible system in a 3D culture system is the activation of ErbB2 in MCF10A mammary acini, which resulted in the formation of structures containing multiple acinar units with filled lumens but without invasive properties. Cooperation of the ErbB2 and TGFβ signaling pathways was required for the sustained, elevated activation of ERK and the induction of migration and invasion of MCF10A cells (Muthuswamy et al., 2001; Seton-Rogers et al., 2004). This result suggests that coordination of these two signal transduction pathways is required to promote cell invasion. Thus, it is surprising that FGFR1 signaling can promote both proliferation and invasion in the absence of other oncogenic events, and this may be a unique property of this RTK.
Another RTK, CSF-1R, when co-overexpressed with its ligand in MCF10A 3D cultures, resulted in a cell invasion phenotype similar to that observed after FGFR1 activation characterized by loss of E-cadherin from plasma membrane and release of individual cells into the matrix. However, in this case no reduction in total E-cadherin was observed. Thus, it was suggested that other mechanisms, such as an increased rate of endocytosis or a defect in the recycling of internalized molecules, may be involved in the loss of E-cadherin from the plasma membrane (Wrobel et al., 2004). Therefore, FGFR1 and CSF-1R appear to regulate cell invasion through distinct mechanisms. Although RTKs can activate a similar array of downstream signaling effectors, including ERK and phosphatidylinositol-3 kinase, their phenotypes as determined in 3D cultures appear to be quite distinct (Shaw et al., 2004). The current study of FGFR1 signaling in 3D cultures supports this observation.
To understand how iFGFR1 regulates cell invasion, we first demonstrated that iFGFR1 activation markedly disrupted cell–cell contacts characterized by the loss of E-cadherin and β-catenin at the cell membrane. Adherens junctions represent a powerful invasion suppressor complex in normal mammary as well as other epithelial cells (Herrenknecht et al., 1991; McCrea and Gumbiner, 1991).
Multiple mechanisms of inactivation of the E-cadherin–catenin complex in normal development and tumor formation have been reported, including genetic mutation, gene inactivation through methylation, repression of expression by Snail and other family members, and cleavage by MMPs (Berx et al., 1996; Bracke et al., 1996; Lochter et al., 1997; Ciruna and Rossant, 2001). Although it has been proposed that Snail expression downstream of FGFR1 is required for the normal repression of E-cadherin expression during early embryonic development (Ciruna and Rossant, 2001), we did not detect induction of Snail expression or inhibition of E-cadherin expression by quantitative RT-PCR in our system (unpublished data). This finding suggests that in the HC11 model during cell invasion, loss of E-cadherin is regulated through iFGFR1 by other mechanisms. Interestingly, after iFGFR1 activation, we observed a rapid increase in Twist expression, a known transcriptional repressor of E-cadherin during EMT (Yang et al., 2004). Because Twist has also been suggested to function as a transcriptional activator (Yang et al., 2004), it may be involved in inducing vimentin expression downstream of iFGFR1 signaling.
MMP-3 is one MMP that has been suggested to lead directly or indirectly to the cleavage of E-cadherin (Lochter et al., 1997; Noe et al., 2001) and is involved in promoting EMT and invasion in mammary tumor cells (Sternlicht et al., 1999). It has also been suggested to function through Rac1b and reactive oxygen species to induce expression of Snail and EMT (Radisky et al., 2005). That the effects of iFGFR1 signaling on cell invasion are dependent on MMP-3 activity is supported by several observations. First, quantitative RT-PCR revealed a rapid and dramatic increase in MMP-3 expression on FGFR1 activation, which was followed by E-cadherin cleavage, detected by immunoblotting. Therefore, in the HC11 model system, the decrease in E-cadherin after iFGFR1 activation is not regulated at the transcriptional level, which is dependent on increased Snail expression but is likely the result of cleavage by MMP-3. Second, the introduction of MMP-3 siRNA, not MMP-9 siRNA, into iFGFR1 cells inhibited the cleavage of E-cadherin, thus demonstrating the important role of MMP-3 during iFGFR1-induced EMT.
In iFGFR1-activated acini, the induction of SMA and vimentin expression was also detected. Expression of these mesenchymal markers together with the loss of E-cadherin and β-catenin from cell–cell contacts as well as the invasive behavior of the cells suggest that the HC11 mammary epithelial cells were converted from an epithelial to a mesenchymal phenotype after the activation of iFGFR1. Although it has been known that FGFR1 can stimulate EMT in mesoderm during gastrulation, it is intriguing to find that FGFR1 activation can also induce EMT in mammary epithelial cells. EMT is a hallmark of tumor progression associated with the acquisition of invasive and metastatic features of breast carcinomas (Thompson et al., 1994; Birchmeier and Birchmeier, 1995). Abolishment of these phenotypes by the pan MMP inhibitor GM6001 further suggests that MMP activity is required for iFGFR1 signaling to promote EMT and invasive ability of mammary epithelial cells.
In addition to MMP-3, we also observed induction of MMP-9 expression and activity after iFGFR1 activation (Welm et al., 2002). Thus, MMP-9 may function to activate local growth factors, stimulate angiogenesis in vivo, and degrade the ECM during cell invasion, whereas MMP-3 is critical for the cleavage of E-cadherin. This is another potential mechanism through which FGFR1 may act to promote cell invasion.
It is still not clear how cell polarity, proliferation, and survival are regulated by iFGFR1. Because the changes in these cellular processes upon iFGFR1 activation were not blocked by the addition of the MMP inhibitor (unpublished data), it is possible that they are regulated by independent signaling cascades downstream of iFGFR1. Therefore, these data suggest that MMP-3 induction may play an important role downstream of iFGFR1 during invasion and cell migration but was not required for iFGFR1-mediated effects on cell proliferation, cell polarity, and cell survival.
Based on these and previous studies that have reported the ability of MMP-3 to cleave E-cadherin and promote EMT (Lochter et al., 1997; Noe et al., 2001), we propose a model whereby iFGFR1 dimerization activates several downstream signaling pathways that lead to cell polarity disruption, acinar growth, and an invasive phenotype (Fig. 9 B). Multiple transcription factors can be activated and bind to the MMP-3 promoter to induce its expression, eventually resulting in increased MMP-3 activity in mammary epithelial cells. Subsequently, MMP-3 cleaves E-cadherin at the cell membrane, resulting in cell invasion and EMT. This may also provide one potential mechanism by which FGFR1 functions during ductal morphogenesis to regulate the invasion of mammary epithelium into the stromal fat pad. Aberrant regulation of this pathway may lead to early tumor progression. Thus, these results provide new insight into the possible mechanisms through which FGFR1 may contribute to breast cancer progression.
Although the iFGFR1 HC11 3D culture system has been proven to be an excellent system for studying some aspects of the FGFR1 signaling pathway (e.g., cell invasion and proliferation), inflammation and angiogenesis, two processes that are involved in tumor initiation and progression within the iFGFR1 transgenic mouse model, cannot be studied in this monotypic HC11 3D culture system. Although it may be possible for some of these processes to be modeled in organotypic 3D cultures, further analysis of iFGFR1 in transgenic mice will most likely be required to characterize the molecular mechanisms that regulate these processes.
Materials and methods
Plasmids and cell culture
iFGFR1 was cloned into the EcoRI sites of the pBabe-puro using an MfeI fragment of pMMP-iFGFR1 including the FGFR1 intracellular kinase domain, two tandem FKBPv (FK506 binding protein) domains, the NH2-terminal myristoylation, and HA epitope sequences (Welm et al., 2002). HC11 cells were grown in growth media containing RPMI 1640 medium (JRH Biosciences) supplemented with 10% FBS (JRH Biosciences), 2 mM l-glutamine (JRH Biosciences), 10 ng/ml EGF (Life Technologies), 5 μg/ml insulin (Sigma-Aldrich), and 50 μg/ml gentamicin (Sigma-Aldrich). The 293T cell growth media contained DMEM (GIBCO BRL), 10% FBS, and 50 μg/ml gentamicin.
Generation of HC11s expressing iFGFR1
Cell transduction was done as described previously (Welm et al., 2002). Stable populations were obtained by selection with 2 μg/ml puromycin (Sigma-Aldrich). iFGFR1 expression was confirmed by immunoblotting. The selected clonal cell lines R1-H and -L were routinely grown in the medium with 2 μg/ml to maintain selection pressure. Serum starvation media for HC11 cells contained only RPMI supplemented with either 30 nM AP20187 (Ariad Pharmaceuticals) in ethanol or an equal volume of ethanol diluent alone as control.
Morphogenesis assay
HC11 cells were treated with trypsin and resuspended in growth medium. The cells were pelleted and resuspended in assay medium (RPMI supplemented with 2% FBS, 2 mM l-glutamine, 2.5 ng/ml EGF, 5 μg/ml insulin, and 50 μg/ml gentamicin) at a concentration of 25,000 cells/ml. Eight-chambered RS glass slides (Nalgene) were coated with 40 μl Matrigel basement membrane matrix (CLONTECH Laboratories, Inc.) per well and left to solidify for 15 min. The cells were mixed 1:1 with assay medium containing 4% Matrigel, and 400 μl was added to each chamber of the Matrigel-coated eight-chambered slide. Assay medium was replaced every 4 d. For stimulation with AP20187, the assay medium was replaced with the assay medium containing 100 μM AP20187 at day 10. For stimulation with AP20187 and GM6001 (Sigma-Aldrich), the assay medium was replaced with the assay medium containing 100 μM AP20187 and 10 μM GM6001 at day 10.
Wound healing assay
HC11 cells were seeded on glass coverslips at 5 × 104 cells per well and grown to confluence before overnight starvation in RPMI. A single wound was introduced in the monolayer using a micropipette tip, and the medium was replaced with RPMI supplemented with either 30 μM AP20187 in ethanol or an equal volume of ethanol diluent. Wound closure was monitored for 20 h.
Migration assay
0.5 ml of serum was added to the wells of a 12-well plate (Falcon). 5,000 R1-L and -H cells were plated in 0.5 ml of serum-free RPMI in 12-well format cell culture inserts containing 8-μm pores (Falcon). After a 20-h incubation, cells that had not migrated through the filters were removed using a cotton swab and the filters were stained with hematoxylin.
Immunoblot analysis
The following antibodies were used for immunoblot analysis: phospho-ERK and ERK (Cell Signaling); HA epitope (Santa Cruz Biotechnology, Inc.); DECMA-1, a rat monoclonal antibody that recognizes the extracellular domain of murine E-cadherin (Sigma-Aldrich); and E-cadherin, which recognizes the cytoplasmic tail of E-cadherin (Transduction Laboratories). Protein extracts from cultured cells were isolated by lysing the monolayer cell with RIPA buffer (1% Triton X-100, 1% NaDOC, 0.1% SDS, 20 mM Tris HCl, pH 7.5, 150 mM NaCl, and 1 mM EDTA) supplemented with Protease cocktail inhibitor (Roche) and phosphatase inhibitor (1 mM NaVO4 and 1 mM NaF) for 15–30 min at 4°C. Lysates were cleared for 15 min at 13,000 rpm, and supernatants were collected. Conditioned medium from monolayer cell cultures were harvested, centrifuged at 2,000 g for 15 min, and passed through a 0.22-μm filter, concentrated by Centricon (Millipore). Protein extracts and concentrated condition medium were quantitated by Bradford assay (Bio-Rad Laboratories). Proteins (30 μg) were resolved by SDS-PAGE and transferred onto polyvinylidene fluoride membranes (Millipore). For immunostaining, blots were quenched with 5% nonfat dry milk in TBS containing 0.5% Tween 20. The membranes were incubated with primary antibody at a dilution of 1:1,000, followed by three 5-min washes and incubation with horseradish peroxidase–conjugated secondary antibodies at a dilution of 1:5,000. Chemiluminescence was developed based on the manufacturer's protocol (Pierce Chemical Co.).
Indirect immunofluorescence and image acquisition
The following primary antibodies were used for indirect immunofluorescent detection of specific antigens: anti–p-histone H3 (Ser10; Upstate Biotechnology); anti–cleaved caspase 3 (Asp175; Cell Signaling); anti–phospho-ezrin (Thr567)/radixin (Thr564)/moesin (Thr558) (Cell Signaling); anti-CD49f (Cell Signaling); anti–laminin V (Chemicon); anti–E-cadherin (Transduction Laboratories); anti–β-catenin (nonphosphorylated; Upstate Biotechnology); anti–phospho-Akt (Ser473; Cell Signaling). All first antibodies were used at a dilution of 1:200. Secondary antibodies were as follows: anti-mouse or anti-rabbit Texas red; anti-mouse or anti-rabbit coupled with Alexa Fluor dyes; anti-rat Texas red (Invitrogen). All secondary antibodies were used at a dilution of 1:500. Nuclei were stained with TOPRO-3 (Invitrogen). Immunostaining was performed as previously described (Debnath et al., 2003a). Confocal analyses were performed using a laser-scanning confocal microscope (model 510; Carl Zeiss MicroImaging, Inc.). The acquisition software used was LSM image browser (Carl Zeiss MicroImaging, Inc.). Phase-contrast images were captured using an inverted microscope (CK40-SLP; Olympus) and video camera (CCD-IRIS/RGB; Sony). The acquisition software used was Photoshop 5.0 (Adobe).
Quantitative RT-PCR
RNA was extracted from monolayer cells using Trizol as recommended by the manufacturer (Invitrogen). 5-μg samples of RNA were treated with DNase I as recommended by the manufacturer (Invitrogen) followed by generation of cDNA using Superscript II (Invitrogen). One tenth of the final reaction volume was used in quantitative PCR reactions as described previously (Yuen et al., 2002) containing 1 mM MgCl2, platinum Taq (Invitrogen), and 1× SYBR green (Invitrogen). PCR was performed using a thermocycler (ABI PRISM 7500; Applied Biosystems) with the following cycles: 94°C for 30 s, 55°C for 30 s, and 72°C for 34 s for 40 cycles. Relative quantitation of MMP-3 gene expression was calculated and normalized to cyclophilin expression levels using the 2−ΔΔCt method (Livak and Schmittgen, 2001). The following primer sequences were used: for MMP-3, 5′-TCCATGGAGCCAGGATTTC-3′ and 5′-TGCGAAGATCCACTGAAGAAGT-3′ (designed by F. Behbod, Baylor College of Medicine, Houston, TX), and for cyclophilin, 5′-TGAGCACTGGGGAGAAAGG-3′ and 5′-TTGCCATCCAGCCACTCAG-3′. MMP-9 primers were purchased from Superarray.
siRNA
SMARTpool siRNA for MMP-3 and -9 and siGLO RISC-free siRNA as the control were introduced into subconfluent (75–80%) HC11 cells using DharmaFECT transfection reagent (Dharmacon) according to the manufacturer's instructions. After 24 h, the media were changed to RPMI, and the cells were starved overnight. The media were replaced with RPMI supplemented with either 30 μM AP20187 in ethanol or an equal volume of ethanol diluent the following day. After the induction for 48 h, the cells were collected for RNA analysis and immunostaining.
Online supplemental material
Fig. S1 shows untreated or treated R1-L and -H acini at low magnification. Online supplemental material is available at www.jcb.org/cgi/content/full/jcb.200505098/DC1.
Acknowledgments
We are grateful to Dr. F. Behbod for designing MMP-3 primers. We would also like to thank Dr. S. Muthuswamy, Dr. P.D. McCrea, and Dr. S. Grimm for helpful discussions.
W. Xian was supported by a postdoctoral fellowship from the Department of Defense Breast Cancer Research Program (DAMD 17-02-1-0285). K.L. Schwertfeger was supported by a Ruth L. Kirschstein National Research Service Awards fellowship (CA 97676-01). T.C. Vargo-Gogola was supported by a postdoctoral fellowship from the Department of Defense Breast Cancer Research Program (DAMD 17-03-1-0325). This research was supported by a National Institutes of Health grant (CA16303).
Abbreviations used in this paper: 2D, two-dimensional; 3D, three-dimensional; CSF-1R, colony-stimulating factor receptor; EMT, epithelial-to-mesenchymal transition; ERK, extracellular regulated kinase; FGFR, FGF receptor; iFGFR, inducible FGFR; MMP, matrix metalloproteinase; p-ERM, phospho-ezrin/radixin/moesin; p-histone H3, phosphorylated histone H3; RTK, tyrosine kinase receptor; siRNA, small interfering RNA; SMA, smooth muscle actin.
References
- Ball, R.K., R.R. Friis, C.A. Schoenenberger, W. Doppler, and B. Groner. 1988. Prolactin regulation of beta-casein gene expression and of a cytosolic 120-kd protein in a cloned mouse mammary epithelial cell line. EMBO J. 7:2089–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates, R.C., and A.M. Mercurio. 2005. The epithelial-mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol. Ther. 4:365–370. [DOI] [PubMed] [Google Scholar]
- Berx, G., A.M. Cleton-Jansen, K. Strumane, W.J. de Leeuw, F. Nollet, F. van Roy, and C. Cornelisse. 1996. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene. 13:1919–1925. [PubMed] [Google Scholar]
- Birchmeier, W., and C. Birchmeier. 1995. Epithelial-mesenchymal transitions in development and tumor progression. EXS. 74:1–15. [DOI] [PubMed] [Google Scholar]
- Bracke, M.E., F.M. Van Roy, and M.M. Mareel. 1996. The E-cadherin/catenin complex in invasion and metastasis. Curr. Top. Microbiol. Immunol. 213:123–161. [DOI] [PubMed] [Google Scholar]
- Ciruna, B., and J. Rossant. 2001. FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev. Cell. 1:37–49. [DOI] [PubMed] [Google Scholar]
- Ciruna, B.G., L. Schwartz, K. Harpal, T.P. Yamaguchi, and J. Rossant. 1997. Chimeric analysis of fibroblast growth factor receptor-1 (Fgfr1) function: a role for FGFR1 in morphogenetic movement through the primitive streak. Development. 124:2829–2841. [DOI] [PubMed] [Google Scholar]
- Dailey, L., D. Ambrosetti, A. Mansukhani, and C. Basilico. 2005. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 16:233–247. [DOI] [PubMed] [Google Scholar]
- Debnath, J., S.K. Muthuswamy, and J.S. Brugge. 2003. a. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 30:256–268. [DOI] [PubMed] [Google Scholar]
- Debnath, J., S.J. Walker, and J.S. Brugge. 2003. b. Akt activation disrupts mammary acinar architecture and enhances proliferation in an mTOR-dependent manner. J. Cell Biol. 163:315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson, C., B. Spencer-Dene, C. Dillon, and V. Fantl. 2000. Tyrosine kinase signalling in breast cancer: fibroblast growth factors and their receptors. Breast Cancer Res. 2:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad, M., and Z. Werb. 2002. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2:161–174. [DOI] [PubMed] [Google Scholar]
- Grose, R., and C. Dickson. 2005. Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev. 16:179–186. [DOI] [PubMed] [Google Scholar]
- Hanafusa, H., S. Torii, T. Yasunaga, and E. Nishida. 2002. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat. Cell Biol. 4:850–858. [DOI] [PubMed] [Google Scholar]
- Herrenknecht, K., M. Ozawa, C. Eckerskorn, F. Lottspeich, M. Lenter, and R. Kemler. 1991. The uvomorulin-anchorage protein alpha catenin is a vinculin homologue. Proc. Natl. Acad. Sci. USA. 88:9156–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko, D., X. Yang, R.J. Nadeau, L.K. Harkins, and R. Friesel. 2003. Sef inhibits fibroblast growth factor signaling by inhibiting FGFR1 tyrosine phosphorylation and subsequent ERK activation. J. Biol. Chem. 278:14087–14091. [DOI] [PubMed] [Google Scholar]
- Livak, K.J., and T.D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. [DOI] [PubMed] [Google Scholar]
- Lochter, A., S. Galosy, J. Muschler, N. Freedman, Z. Werb, and M.J. Bissell. 1997. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 139:1861–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea, P.D., and B.M. Gumbiner. 1991. Purification of a 92-kDa cytoplasmic protein tightly associated with the cell-cell adhesion molecule E-cadherin (uvomorulin). Characterization and extractability of the protein complex from the cell cytostructure. J. Biol. Chem. 266:4514–4520. [PubMed] [Google Scholar]
- Muthuswamy, S.K., D. Li, S. Lelievre, M.J. Bissell, and J.S. Brugge. 2001. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol. 3:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noe, V., B. Fingleton, K. Jacobs, H.C. Crawford, S. Vermeulen, W. Steelant, E. Bruyneel, L.M. Matrisian, and M. Mareel. 2001. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J. Cell Sci. 114:111–118. [DOI] [PubMed] [Google Scholar]
- Owens, D.W., V.G. Brunton, E.K. Parkinson, and M.C. Frame. 2000. E-cadherin at the cell periphery is a determinant of keratinocyte differentiation in vitro. Biochem. Biophys. Res. Commun. 269:369–376. [DOI] [PubMed] [Google Scholar]
- Penault-Llorca, F., F. Bertucci, J. Adelaide, P. Parc, F. Coulier, J. Jacquemier, D. Birnbaum, and O. deLapeyriere. 1995. Expression of FGF and FGF receptor genes in human breast cancer. Int. J. Cancer. 61:170–176. [DOI] [PubMed] [Google Scholar]
- Pintucci, G., P.J. Yu, R. Sharony, F.G. Baumann, F. Saponara, A. Frasca, A.C. Galloway, D. Moscatelli, and P. Mignatti. 2003. Induction of stromelysin-1 (MMP-3) by fibroblast growth factor-2 (FGF-2) in FGF-2−/− microvascular endothelial cells requires prolonged activation of extracellular signal-regulated kinases-1 and -2 (ERK-1/2). J. Cell. Biochem. 90:1015–1025. [DOI] [PubMed] [Google Scholar]
- Powers, C.J., S.W. McLeskey, and A. Wellstein. 2000. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer. 7:165–197. [DOI] [PubMed] [Google Scholar]
- Radisky, D., C. Hagios, and M.J. Bissell. 2001. Tumors are unique organs defined by abnormal signaling and context. Semin. Cancer Biol. 11:87–95. [DOI] [PubMed] [Google Scholar]
- Radisky, D.C., D.D. Lery, L.E. Littlepape, H. Liu, C.M. Nelson, J.E. Fata, D. Leake, E.L. Godden, D.G. Albertson, M.A. Nieto, et al. 2005. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 436:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid, M.P., and J.R. Woodgett. 2001. Phosphatidylinositol 3′ kinase signaling in mammary tumorigenesis. J. Mammary Gland Biol. Neoplasia. 6:83–99. [DOI] [PubMed] [Google Scholar]
- Schmeichel, K.L., and M.J. Bissell. 2003. Modeling tissue-specific signaling and organ function in three dimensions. J. Cell Sci. 116:2377–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seton-Rogers, S.E., Y. Lu, L.M. Hines, M. Koundinya, J. LaBaer, S.K. Muthuswamy, and J.S. Brugge. 2004. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc. Natl. Acad. Sci. USA. 101:1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, K.R., C.N. Wrobel, and J.S. Brugge. 2004. Use of three-dimensional basement membrane cultures to model oncogene-induced changes in mammary epithelial morphogenesis. J. Mammary Gland Biol. Neoplasia. 9:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht, M.D., A. Lochter, C.J. Sympson, B. Huey, J.P. Rougier, J.W. Gray, D. Pinkel, M.J. Bissell, and Z. Werb. 1999. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 98:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, E.W., J. Torri, M. Sabol, C.L. Sommers, S. Byers, E.M. Valverius, G.R. Martin, M.E. Lippman, M.R. Stampfer, and R.B. Dickson. 1994. Oncogene-induced basement membrane invasiveness in human mammary epithelial cells. Clin. Exp. Metastasis. 12:181–194. [DOI] [PubMed] [Google Scholar]
- Welm, B.E., K.W. Freeman, M. Chen, A. Contreras, D.M. Spencer, and J.M. Rosen. 2002. Inducible dimerization of FGFR1: development of a mouse model to analyze progressive transformation of the mammary gland. J. Cell Biol. 157:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, A., B. Lamothe, A. Lee, J. Schlessinger, and I. Lax. 2002. FRS2α attenuates FGF receptor signaling by Grb2-mediated recruitment of the ubiquitin ligase cbl. Proc. Natl. Acad. Sci. USA. 99:6684–6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel, C.N., J. Debnath, E. Lin, S. Beausoleil, M.F. Roussel, and J.S. Brugge. 2004. Autocrine CSF-1R activation promotes Src-dependent disruption of mammary epithelial architecture. J. Cell Biol. 165:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X., C. Li, K. Takahashi, H.C. Slavkin, L. Shum, and C.X. Deng. 1999. Murine fibroblast growth factor receptor 1alpha isoforms mediate node regression and are essential for posterior mesoderm development. Dev. Biol. 208:293–306. [DOI] [PubMed] [Google Scholar]
- Yang, J., S.A. Mani, J.L. Donaher, S. Ramaswamy, R.A. Itzykson, C. Come, P. Savagner, I. Gitelman, A. Richardson, and R.A. Weinberg. 2004. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 117:927–939. [DOI] [PubMed] [Google Scholar]
- Yuen, T., W. Zhang, B.J. Ebersole, and S.C. Sealfon. 2002. Monitoring G-protein-coupled receptor signaling with DNA microarrays and real-time polymerase chain reaction. Methods Enzymol. 345:556–569. [DOI] [PubMed] [Google Scholar]