

Abstract
Autophagic degradation of ubiquitinated protein aggregates is important for cell survival, but it is not known how the autophagic machinery recognizes such aggregates. In this study, we report that polymerization of the polyubiquitin-binding protein p62/SQSTM1 yields protein bodies that either reside free in the cytosol and nucleus or occur within autophagosomes and lysosomal structures. Inhibition of autophagy led to an increase in the size and number of p62 bodies and p62 protein levels. The autophagic marker light chain 3 (LC3) colocalized with p62 bodies and coimmunoprecipitated with p62, suggesting that these two proteins participate in the same complexes. The depletion of p62 inhibited recruitment of LC3 to autophagosomes under starvation conditions. Strikingly, p62 and LC3 formed a shell surrounding aggregates of mutant huntingtin. Reduction of p62 protein levels or interference with p62 function significantly increased cell death that was induced by the expression of mutant huntingtin. We suggest that p62 may, via LC3, be involved in linking polyubiquitinated protein aggregates to the autophagy machinery.
Introduction
Several reports have described p62, which is also named sequestosome 1 (SQSTM1), as a common component of protein aggregates that are found in protein aggregation diseases affecting both the brain and the liver. These include Lewy bodies in Parkinsons disease, neurofibrillary tangles in Alzheimer's disease, and huntingtin aggregates (Kuusisto et al., 2001a, 2002; Zatloukal et al., 2002; Nagaoka et al., 2004). In the liver Mallory bodies, hyaline bodies in hepatocellular carcinoma and α1 antitrypsin aggregates contain p62 (Zatloukal et al., 2002). All of these aggregates contain polyubiquitinated proteins. The 440–amino acid–long p62 protein has an NH2-terminal Phox and Bem1p (PB1) domain followed by a ZZ type zinc finger domain, a PEST region containing putative phosphorylation sites, and a COOH-terminal ubiquitin-associated (UBA) domain (Geetha and Wooten, 2002). The latter domain binds ubiquitin noncovalently (Vadlamudi et al., 1996). This raises the possibility that p62 could be recruited to ubiquitinated protein aggregates as a result of its ability to bind polyubiquitin via the UBA domain (Donaldson et al., 2003). The NH2-terminal PB1 domain is used both for the polymerization of p62 and for binding to other proteins containing PB1 domains (Gong et al., 1999; Sanz et al., 1999, 2000; Avila et al., 2002; Cariou et al., 2002; Lamark et al., 2003).
The p62 protein level increases after oxygen radical stress. Both mRNA and protein levels increase, suggesting an induced transcription of the gene (Ishii et al., 1997). The transcription factor Nrf2 is activated after oxidative stress, and induction of p62 is severely inhibited in cells from Nrf2 knockout mice (Ishii et al., 2000). Inhibition of proteasomal activity also causes induction of p62 (Ishii et al., 1997; Kuusisto et al., 2001b; Thompson et al., 2003). Interestingly, p62 was recently identified as a protein that is induced as a response to the expression of mutant huntingtin (Nagaoka et al., 2004). Huntington's disease is a late onset progressive autosomal dominant neurodegenerative disorder caused by the expression of mutant forms of the huntingtin (Htt) protein containing a polyglutamine expansion encoded by CAG repeats in exon 1 of the huntingtin gene (Vonsattel and DiFiglia, 1998). The disease causes selective neuronal cell death in the striatum. Cells expressing the mutant form of huntingtin display both diffuse and aggregated localization of the protein. The mutant protein has cytotoxic properties, and aggregation seems to be a mechanism for cell survival (Arrasate et al., 2004). Protein inclusions formed by aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by macroautophagy (hereafter referred to as autophagy; Kegel et al., 2000; Ravikumar et al., 2002, 2004), which is a bulk degradation pathway in which a double or multimembrane-bound structure called the autophagosome forms to sequester cytoplasm. Subsequently, the autophagosome fuses with the lysosome, and its content and internal membranes are degraded as it recycles the macromolecules (Levine and Klionsky, 2004; Yoshimori, 2004). Most long-lived proteins and some organelles are degraded by autophagy, and autophagy, in addition to cellular homeostasis, has also been implicated in cellular differentiation, tissue remodelling, growth control, bacterial and viral infections, cell defense, adaptation to adverse environments, neurodegenerative diseases, cardiomyopathies, apoptosis, and cancer (Cuervo, 2004). Among the autophagosomal marker proteins are Atg8 in yeast and light chain 3 (LC3) in mammals (Kabeya et al., 2000). After synthesis, LC3 is cleaved at its COOH terminus to produce the cytosolic LC3-I form. LC3-I is converted to LC3-II, which is tightly associated with the autophagosomal membrane probably via conjugation to phosphatidylethanolamine (Kabeya et al., 2000, 2004).
In this study, we report that the polyubiquitin-binding and homopolymerizing p62 protein may, via LC3, be involved in linking polyubiquitinated protein aggregates to the autophagic machinery, facilitating the clearance of such aggregates and, thereby, contributing to reduced toxicity of mutant huntingtin expression.
Results
The p62 bodies in the cytoplasm of HeLa cells are ubiquitin-containing protein aggregates
The p62 protein has been reported to be present in several types of cytoplasmic inclusions in neurodegenerative diseases and other protein aggregation diseases (Zatloukal et al., 2002). In a previous study, we noted that endogenous as well as ectopically expressed p62 was present in numerous round bodies in the perinuclear area of human HeLa cells (Lamark et al., 2003). By studying this further using both a monoclonal and polyclonal antibody, the p62 protein was found to be located in the cytoplasm, with weak staining in the nucleus in ∼85% of the cells (Fig. 1 A). In the cytoplasm, the protein is enriched in round bodies with a diameter ranging from the detection limit of the confocal microscope (0.1 μm) to 2 μm. These structures seemed to be of two distinct types: a high number of faint, 0.1–0.2-μm-diameter bodies and 5–10 times more intense bodies ranging from 0.5 to 1 μm in diameter. Interestingly, ∼5% of the cell population contained large p62 bodies that were 1–2 μm in diameter. In ∼15% of the cells, fluorescence intensities in the cytoplasm and nucleus were similar. Nuclear p62 was enriched in speckles or bodies, with a minority of the cells (∼0.5%) displaying a nuclear accumulation of p62, and 0.5–1-μm-diameter p62 bodies were stained 5–10 times more intensely than the diffuse nuclear p62. In broad terms, this pattern of p62 localization was observed in a wide range of cell lines, including HeLa, HEK293, MDCK II, U2OS, A431, HT1080, TERT fibroblasts, NIH3T3 fibroblasts, and several human neuroblastoma cell lines. Although the reason for heterogeneous p62 distribution is not known, it is evident that p62 bodies in one form or another are widespread among various cell types.
Figure 1.
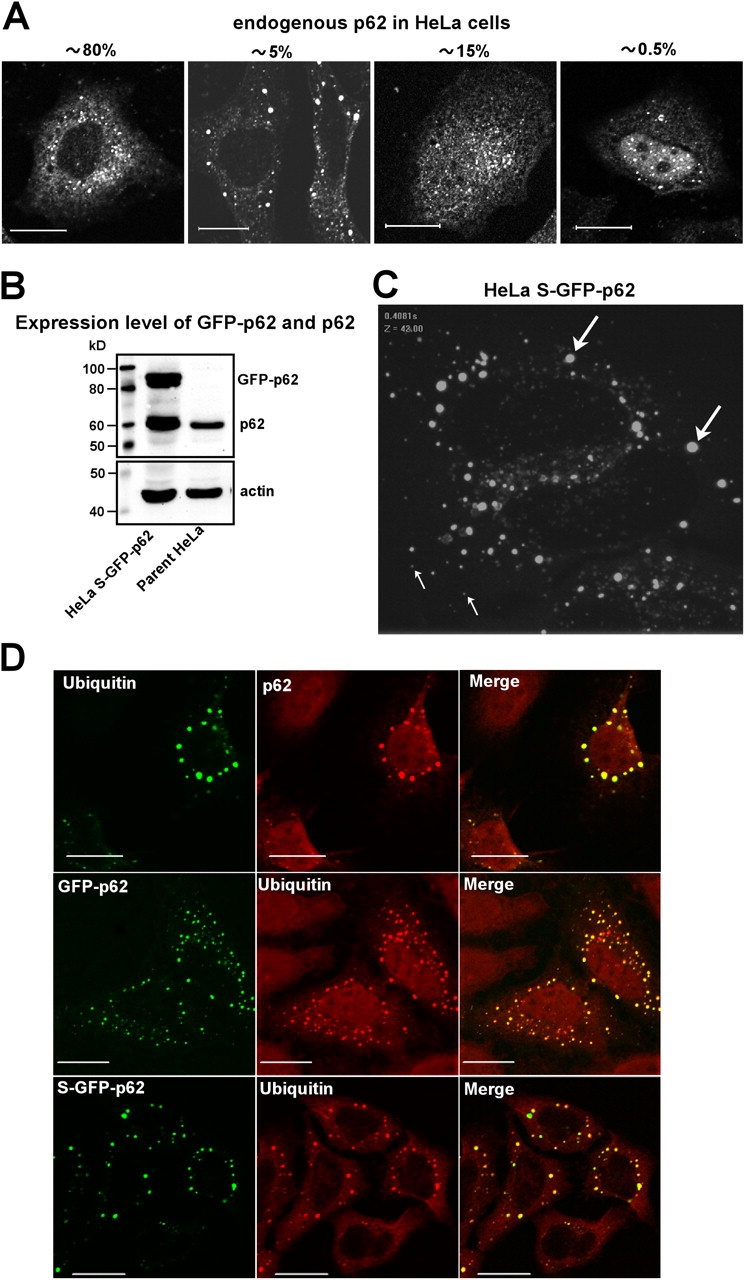
The p62 bodies in the cytoplasm of HeLa cells are ubiquitin-containing protein aggregates. (A) HeLa cells were fixed and stained for p62. The different patterns of p62 distribution were scored in >200 cells as cytoplasmic with small bodies, cytoplasmic with large and intense bodies, equal nuclear and cytoplasmic staining, and nuclear enriched. The percentage of cells in the respective groups is indicated. (B) Expression level of GFP-p62 in HeLa cells stably expressing GFP-p62 (S–GFP-p62) compared with the level of endogenous p62 in the parent HeLa cells. (C) Cells stably expressing GFP-p62 display a cytoplasmic punctuate localization of GFP-tagged p62 similar to endogenous p62. Video microscopy of live cells demonstrated that the small, faint bodies (thin arrows) displayed directed migration, whereas the majority of the larger, intense bodies (thick arrows) were nonmigratory (Videos 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200507002/DC1). A still image from Video 1 is shown here. (D) Bodies containing either endogenous p62 or transiently or stably transfected GFP-p62 all contain polyubiquitin. HeLa cells were fixed and stained with p62 and polyubiquitin (clone FK1) mAbs directly coupled with AlexaFluor555 (red) and AlexaFluor488 (green), respectively. Cells expressing GFP-p62 were only stained for polyubiquitin (red). Bars, 20 μm.
We generated a HeLa cell line (stable [S] GFP-p62) that stably expressed a moderate level of the GFP-p62 fusion protein at a level that was about fourfold higher than in the parent cell line (Fig. 1 B). The S–GFP-p62 cell line displayed all four distributions depicted in Fig. 1 A, but the population of large, round bodies was increased from 5 to 40%. Video confocal microscopy of the GFP-tagged p62 fusion protein also suggested that the p62 bodies could be divided into two populations (Fig. 1 C and Videos 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200507002/DC1): a high number of faint, mobile bodies (most of them smaller than 0.3 μm) and larger bodies (>0.5 μm) with low or no mobility. Notably, these larger and more intensely labeled structures were equally fluorescent throughout the whole structure, suggesting that they are not vesicles. These bodies seemed to grow by fusion, and their size and intensity increased with increasing expression levels. Altogether, these results suggest that p62 bodies represent two different populations differing in size, intensity, and mobility. Importantly, as far as we could observe by confocal fluorescence microscopy, all p62 bodies contained ubiquitin (Fig. 1 D). This is consistent with the ability of p62 to bind to polyubiquitin via its UBA domain.
Both the PB1 and UBA domains are needed for p62 to form cytoplasmic bodies
To map the domains of p62 involved in the formation of cytoplasmic bodies, different GFP-p62 cDNA constructs were transiently transfected into NIH3T3 cells, and the fusion proteins were analyzed by confocal fluorescence microscopy (Fig. 2 A). NIH3T3 cells were used because they have a low level of endogenous p62, but similar data were obtained with HeLa cells (not depicted). All deletion constructs lacking the PB1 domain (i.e., p62(124–440), p62(256–440), and p62(385–440)) have a completely diffuse distribution in NIH3T3 cells (Fig. 2 A and not depicted). This is also the case with the D69A mutant, which abrogates PB1 domain–mediated polymerization of p62 (Fig. 2 A; Lamark et al., 2003). This strongly indicates that the PB1 domain–mediated polymerization of p62 is essential for the formation of cytoplasmic bodies.
Figure 2.
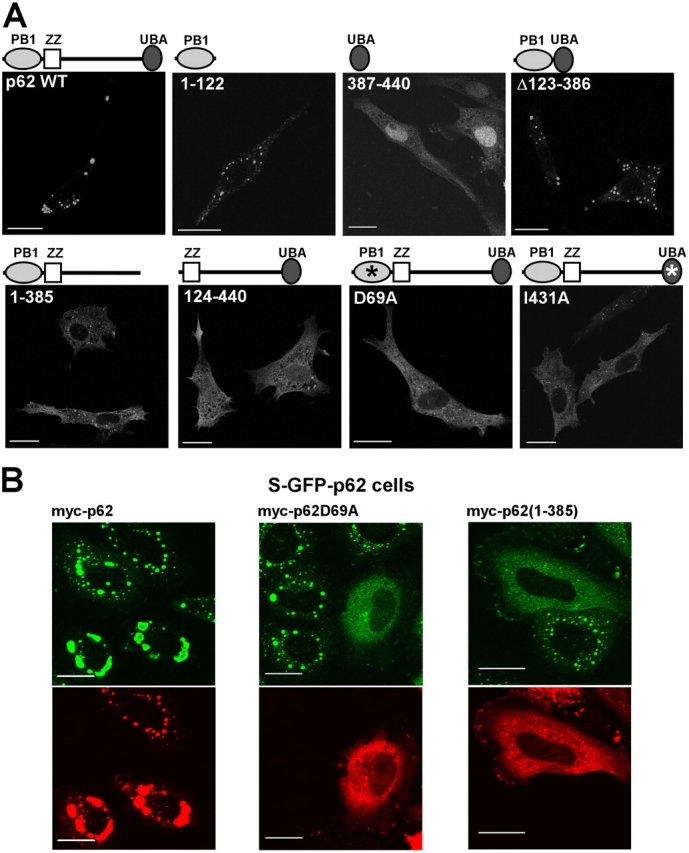
Both the PB1 and UBA domains are needed for p62 to form cytoplasmic bodies. (A) The indicated deletion constructs were fused COOH terminal to GFP and expressed in NIH3T3 fibroblasts. Asterisks indicate point mutations in the PB1 (D69A) and UBA (I431A) domains, respectively. (B) S–GFP-p62 cells were transiently transfected with the indicated myc-tagged p62 constructs, fixed, and stained for the myc tag. Bars, 20 μm.
Given its ability to bind to polyubiquitin, the UBA domain may be needed for the formation of cytoplasmic bodies. In support of this, we found that a construct lacking the UBA domain (p62(1–385)) and the I431A mutant, which affects the folding of the UBA domain, both showed a diffuse localization in transfected NIH3T3 cells (Fig. 2 A). The isolated PB1 domain of p62(1–122) formed large bodies (Fig. 2 A), as did a construct encompassing amino acids 1–256 (not depicted). However, we consistently observed that the structures formed by these truncated p62 constructs were distinct, with a less regular shape than the round bodies formed by full-length p62. In contrast, a construct containing only the PB1 and UBA domains (p62Δ123–385) formed bodies that were indistinguishable from those formed by full-length p62. These data suggest that both the PB1 and UBA domains are necessary and sufficient for the localization of full-length p62 into the ubiquitin-containing bodies.
In vivo, the length of p62 polymers may be reduced as a result of the binding of p62 to other PB1 domain–interacting partners. In line with this, overexpression of p62 D69A (Fig. 2 B) or p62 R21A (not depicted) counteracted the formation of GFP-p62 bodies in S–GFP-p62 cells. These PB1 domain mutants presumably compete with wild-type p62 for binding to a growing chain of p62 molecules acting as chain terminators. However, overexpression of the p62ΔUBA construct myc-p62(1–385) also prevented the formation of cytoplasmic dots in S–GFP-p62 cells (Fig. 2 B). In contrast to PB1 mutants, deletion of the UBA domain had no effect on the ability of p62 to interact with itself or to form polymers in vitro (unpublished data). Only the PB1 domain is needed for polymerization of p62 in vitro. Therefore, the role of the UBA domain may be to cross-link p62 polymers, presumably by interacting with polyubiquitinated proteins.
p62 bodies are found both as membrane-free protein aggregates (sequestosomes) and as membrane-confined autophagosomal and lysosomal structures
To characterize the two populations of p62 bodies, we performed a series of colocalization experiments in both fixed and live cells. We looked at both endogenous p62 and ectopically expressed GFP-p62 or epitope-tagged p62. There was no colocalization of early endosomal markers with p62 bodies. Early endosome antigen 1 (EEA1) did not colocalize with endogenous p62 or ectopically expressed GFP-p62 (Fig. 3 A). In line with this, there was no colocalization between p62 and EGF receptor (fixed cells) or fluorescent-labeled EGF (live cells) at time points up to 15 min of internalization (unpublished data). However, at 30 and 60 min, there was a minor fraction of the smaller p62 bodies that colocalized with EGF receptor or its ligand (unpublished data). Moreover, there was no association of GFP-p62 with the recycling compartment that was visualized by internalization of fluorescently labeled transferrin (unpublished data). Thus, these data suggested that the weakly stained smaller p62 bodies could be late endosomes or lysosomes. CD63 is often used as a marker for the late endosomes/lysosomes and is highly enriched in multivesicular endosomes (Escola et al., 1998). In S–GFP-p62 cells, immunostaining with an anti-CD63 antibody revealed colocalization with a fraction of the smaller GFP-p62 structures (Fig. 3 B). The large p62 bodies were consistently negative for CD63. By performing immuno-EM with antibodies against CD63 and GFP, vesicles containing both CD63 and GFP-p62 were identified, although CD63 and GFP-p62 were also found in separate compartments within the same vesicular structures (Fig. 3 C).
Figure 3.
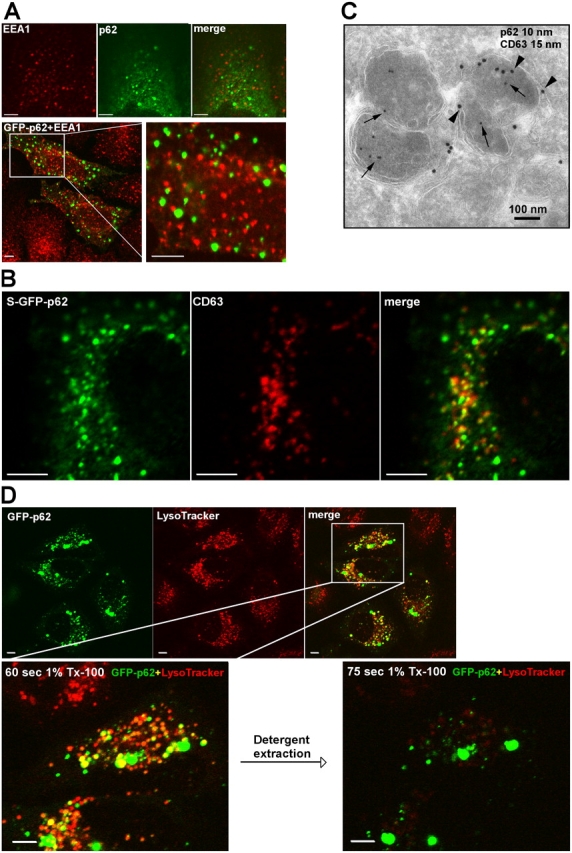
p62 bodies are found both as membrane-free protein aggregates and as membrane-confined autophagosomal and lysosomal structures. (A) Localization of bodies containing endogenous p62 or transiently expressed GFP-p62 relative to EEA1-positive early endosomes. Endogenous p62 were stained green using p62 antibodies directly labeled with AlexaFluor488, and EEA1 was stained red with EEA1 mAbs directly labeled with AlexaFluor555. Alternatively, transiently expressed GFP-p62 was expressed in HeLa cells, and EEA1 was stained red with EEA1 mAb (bottom). The boxed area is shown to the right at a higher magnification. (B) Colocalization of GFP-p62 and CD63 (stained red using a mAb) in HeLa cells stably expressing GFP-p62. (C) Immunoelectron micrograph of S–GFP-p62 stained with a GFP pAb (10-nm gold particle, arrows) and monoclonal CD63 (15-nm gold particle, arrowheads). (D) Rapid detergent extraction of GFP-p62 from LysoTracker-positive acidic organelles. HeLa cells transiently expressing GFP-p62 were labeled with LysoTracker for 60 min. The detergent extractions were imaged in a time series with 15-s time intervals after adding 1% Triton X-100. The boxed area is shown in the bottom two images at a higher magnification before and after detergent extraction. Bars (A, B, and D), 20 μm.
To further analyze whether p62 partly localizes to the late endosome/lysosomal compartment, we took advantage of the fluorescent dye LysoTracker, which displays intense fluorescence when it faces the acidic environment in the late endosomal/lysosomal compartment. As shown in Fig. 3 D, LysoTracker labeled a subfraction of the p62 bodies, and these structures were rapidly lost by detergent extraction, whereas the intense bodies were detergent resistant. As a control, we did the same experiment with GFP-tagged p40phox, which, similar to p62, contains a PB1 domain but is located in endosomes. As expected, p40phox was extracted from the endosomes at the same time as the LysoTracker fluorescence was lost (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200507002/DC1).
We were not able to detect endogenous p62 by immuno-EM. However, by performing immuno-EM on S–GFP-p62 cells, we observed both membrane-free protein aggregates (sequestosomes) and membrane-surrounded p62 bodies (Fig. 4 A). To further characterize the sequestosomes on the ultrastructural level, we performed correlative immunofluorescence and EM. Proteasomal inhibitors such as PSI induce a prominent increase in the amount of p62 protein in cells (Kuusisto et al., 2001b; Thompson et al., 2003). We took advantage of this to increase the size of endogenous p62 bodies in HeLa cells to facilitate EM/immunofluorescence studies. The results show that the large and intensely fluorescent p62 structures are membrane-free protein aggregates that are not related to endocytic vesicles (Fig. 4 B). The aggregates had a filamentous appearance and seemed to exclude any cytosolic material. Both in the stably transfected cell line and in transiently transfected HeLa cells, p62 was found within double membrane structures that are indicative of autophagosomes (Fig. 4 C). Autophagic structures and autolysosomes represented the dominant fraction of p62 bodies in S–GFP-p62 cells.
Figure 4.

p62 is found both in autophagosomes and in cytosolic aggregates/sequestosomes. (A) Immuno-EM of GFP-p62. S–GFP-p62 cells were labeled with rabbit anti-GFP (Abcam) followed by protein A–gold (15 nm). We observed labeling in membrane-free cytosolic structures and sequestosomes (arrows) as well as in endosomes (Endo). (B) Correlative immunofluorescence/EM of HeLa cells treated with 10 μM PSI for 5 h displaying typical sequestosomes. The insets show two magnifications of the sequestosome, which is labeled 1. (C) Representative image of a p62-containing autophagosome. HeLa cells transfected with GFP-p62 were immunogold labeled as in A. Note the cisternal-like membrane (arrowheads) surrounding the GFP-positive material. The arrow indicates a fused endosome.
p62 bodies are degraded by autophagy
The presence of GFP-p62 within autophagosomes suggests that p62 bodies are degraded by autophagy. Pulse-chase experiments in which cells were added to 35S-labeled methionine indicate a half-life of p62 in HeLa cells of ∼6 h, and almost all of the radiolabeled protein was lost after 24 h (Fig. 5 A). The protein synthesis inhibitor cycloheximide is known to cause a drastic reduction in autophagy-induced protein degradation (Lawrence and Brown, 1993; Abeliovich et al., 2000). Consistently, the level of p62 protein was stable in cells treated with cycloheximide for as long as 24 h (Fig. 5 A).
Figure 5.
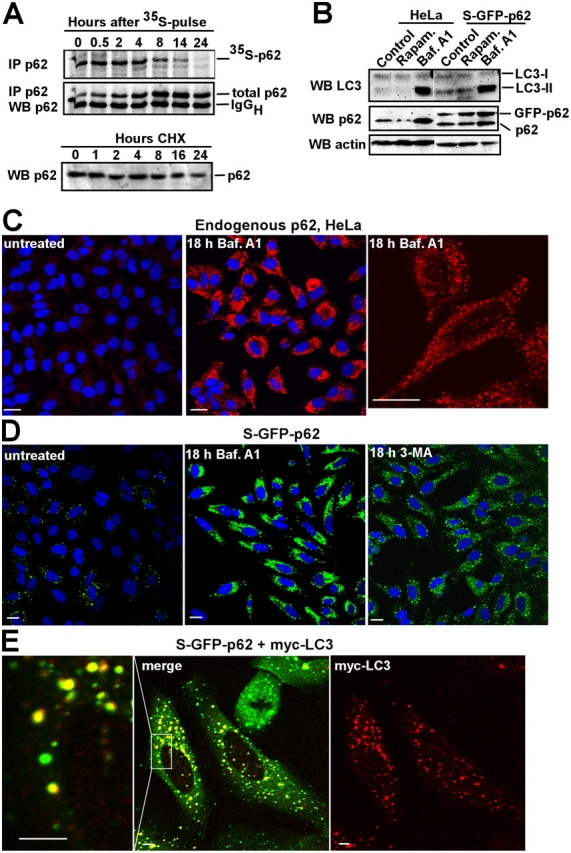
p62 bodies are degraded by autophagy. (A) Comparison of p62 degradation using pulse-chase labeling with 35S-methionine and immunoblotting after cycloheximide (CHX) treatment. HeLa cells were pulsed with 35S-methionine and incubated for the indicated times in nonradioactive medium. After immunopurification, the amount of radioactive p62 was determined by autoradiography, and the total amount of p62 was determined by an immunoblot of the same membrane. The p62 level in total cellular lysates after different times of 10 μg/ml cycloheximide treatment determined by immunoblotting (bottom). (B) The levels of p62 and LC3-II change with autophagic activity. HeLa or S–GFP-p62 cells were either left untreated or rapamycin (10 μg/ml) or bafilomycin A1 (Baf. A1; 10 μg/ml) was added for 18 h. Immunoblots were sequentially probed using LC3, p62, and actin antibodies. (C and D) The amount of p62 located to cytoplasmic bodies increases upon inhibition of autophagy. HeLa cells or S–GFP-p62 HeLa cells were left untreated or bafilomycin A1 was added for 18 h, the cells were fixed, and p62 was either stained red using a p62 mAb (C) or imaged directly (D). Nuclei were visualized using the Draq5 DNA stain. The settings for imaging were identical for the treated cells and the untreated control. (E) The majority of S–GFP-p62 cytoplasmic bodies are stained with antibodies recognizing transiently expressed LC3. S–GFP-p62 cells were transiently transfected with myc-LC3. Myc-LC3 was stained red using an anti-myc tag mAb. The boxed area indicates the part of the cell that is shown to the left at a higher magnification. Bars, 5 μm.
To further explore the relationship between autophagy and p62 degradation, cells were treated with rapamycin, which is an inducer of autophagy (Noda and Ohsumi, 1998), or bafilomycin A1. Bafilomycin A1 is an inhibitor of the vacuolar ATPase, which blocks the fusion of autophagosomes with lysosomes, leading to an accumulation of autophagosomal structures (Yamamoto et al., 1998). Rapamycin treatment caused a slight decrease of endogenous p62, whereas treatment with bafilomycin A1 for 18 h resulted in an accumulation of endogenous p62 in HeLa cells (Fig. 5 B). It also resulted in an increase of both endogenous and GFP-tagged p62 in S–GFP-p62 cells (Fig. 5 B). LC3 is involved during the late steps of autophagy after the isolation membrane has formed (Klionsky, 2005). The unmodified LC3-I form is cytosolic, whereas LC3-II is presumably covalently attached to phosphatidylethanolamine at its COOH terminus and is tightly bound to autophagosomal membranes, serving as an important marker for autophagy (Kabeya et al., 2000, 2004; Mizushima, 2004). A Western blot with an antibody against LC3 and the highest affinity for the LC3-II form (Kabeya et al., 2000) revealed a strong induction of LC3-II upon treatment of HeLa cells and S–GFP-p62 cells with bafilomycin A1 (Fig. 5 B). Interestingly, the background level of LC3-II is higher in S–GFP-p62 cells than in the parent HeLa cells, suggesting a higher autophagic activity in these cells (Fig. 5 B). Consistent with the Western blot data (Fig. 5 B), immunostaining revealed an extensive accumulation of small p62 bodies (0.3–0.8 μm) in HeLa cells that were treated with bafilomycin A1 (Fig. 5 C). Similarly, GFP-p62–containing dots accumulated in S–GFP-p62 cells upon treatment with bafilomycin A1 (Fig. 5 D). Treatment of cells with 3-methyladenine, which inhibits the sequestration step during autophagy, also resulted in an accumulation of p62 in cytoplasmic dots, although the effect was less pronounced than that of bafilomycin A1 (Fig. 5 D).
Presently, there are no antibodies available that allow immunostaining of endogenous LC3 in mammalian cells, and overexpressed myc-LC3 could only be detected using anti-myc antibodies. After transient transfection, myc-LC3 was present in a large fraction of the cytoplasmic bodies formed by endogenous p62 in HeLa cells (not depicted), and it also strongly colocalized with GFP-p62 in S–GFP-p62 cells (Fig. 5 E). Generally, it was difficult to find myc-LC3–positive dots that did not also contain p62. It should be noted that in HeLa cells, autophagosomes are small and are commonly visualized as dots by fluorescence microscopy (Mizushima, 2004). In cells treated with bafilomycin A1, there was an extensive accumulation of cytoplasmic bodies containing both p62 and myc-LC3 (unpublished data). Coexpression of GFP-LC3 with p62 fused to a novel, very bright, red fluorescent protein, tdTomato (Shaner et al., 2004), enabled the visualization of p62-LC3–positive bodies in living cells. Video confocal microscopy of HeLa cells coexpressing tdTomato-p62 and GFP-LC3 showed that many of the punctuate structures containing p62 and LC3 had a high mobility (Video 3, available at http://www.jcb.org/cgi/content/full/jcb.200507002/DC1).
LC3 is associated with the isolation membrane during its formation and remains on the membrane after a spherical autophagosome has formed. Transiently overexpressed GFP-LC3 has, therefore, been shown to be a very good marker for autophagy, as its localization changes from diffuse to a punctuate or dotted pattern when autophagy is induced (Mizushima, 2004). GFP-LC3 dots represent isolation membranes and autophagosomes (Mizushima, 2004). Amino acid starvation induces autophagy and results in a transient increase in the number of autophagosomes. In line with this, we found that the fraction of HeLa cells with GFP-LC3 in punctuated structures increased from 27 to 53% after amino acid starvation in Hanks medium for 60 min. Almost all of the LC3-positive bodies stained positive for endogenous p62 (Fig. 6 A). Interestingly, the redistribution of overexpressed LC3 into punctuated structures appeared to depend on the presence of p62. In cells transfected twice with small interfering RNA (siRNA) to deplete endogenous p62, very few cells contained punctuated GFP-LC3 structures (Fig. 6 A). In contrast, cooverexpression of HA-p62 strongly increased the frequency of cells with punctuated GFP-LC3 (Fig. 6 A). Cooverexpression of the D69A mutant, which inhibits PB1 domain–mediated polymerization, resulting in a diffuse localization of p62 (Lamark et al., 2003), also resulted in a diffuse localization of GFP-LC3 (Fig. 6 A). Consistent with a direct or indirect association between GFP-LC3 and p62, both endogenous p62 and overexpressed HA-p62 coimmunoprecipitated with GFP-LC3 from HeLa cell extracts (Fig. 6 B). When the D69A mutant was overexpressed, less p62 was coimmunoprecipitated because polymers of p62 were not formed. However, when a p62 mutant lacking the UBA domain was coexpressed with GFP-LC3, both endogenous p62 and p62ΔUBA were efficiently coimmunoprecipitated (Fig. 6 B). Together, the aforementioned results suggest a close association between LC3 and p62 bodies and that a large fraction of p62 bodies are degraded by autophagy.
Figure 6.
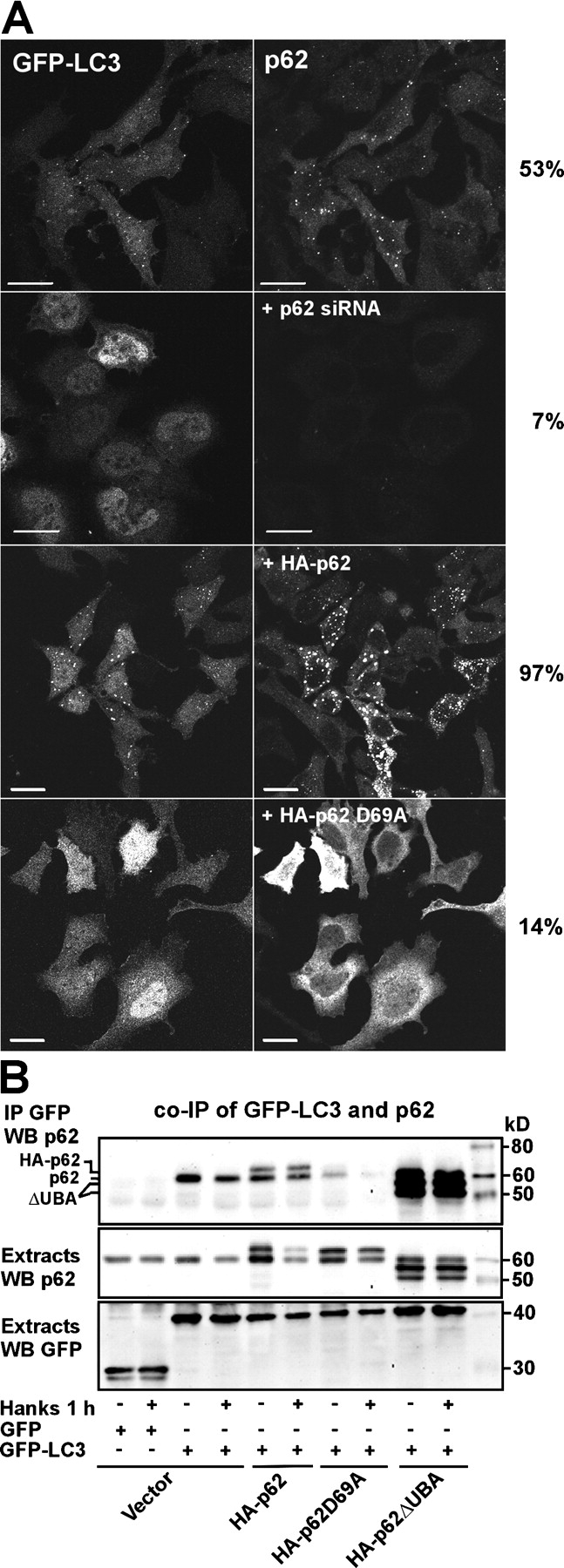
p62 is required for the formation of GFP-LC3–positive punctuated structures in HeLa cells. (A) HeLa cells transiently transfected with GFP-LC3 alone or cotransfected with siRNA against p62, HA-p62, or HA-p62 D69A were either left in normal medium or starved for amino acids for 1 h. The cells were fixed, and p62 was stained using a p62 mAb. More than 200 randomly selected cells for each condition were scored for the cytoplasmic pattern of GFP-LC3 as either diffuse or punctuate. The frequency of GFP-LC3–positive cells with punctuate localization are indicated to the right. (B) Endogenous p62 as well as coexpressed myc-tagged wild-type p62 or a UBA deletion mutant of p62 coimmunoprecipitated with GFP-LC3 from HeLa cell extracts. GFP or GFP-LC3 was immunoprecipitated from total cellular extracts after cotransfecting the indicated constructs. The cell cultures were either left untreated or starved for amino acids for 1 h as indicated. Copurified endogenous or ectopically expressed myc-tagged p62 constructs were detected using the p62 mAb. Bars, 20 μm.
p62 forms a shell around huntingtin protein aggregates
The p62 protein is associated with protein aggregates in a number of aggregation diseases (Kuusisto et al., 2001a; Zatloukal et al., 2002; Nagaoka et al., 2004). However, the role of p62 in the handling of such aggregates is unknown. To start analyzing the possible functional role of p62, we chose an established model in which we expressed an NH2-terminal fragment (amino acids 1–171) of huntingtin containing a 68–amino acid polyglutamine expansion (N-HttQ68) that was found to be causative for disease development (Saudou et al., 1998). Expression of Flag-tagged N-HttQ68 caused the formation of cytoplasmic aggregates in ∼30% of the expressing cells, with a strong colocalization of p62 and N-HttQ68 in these aggregated structures (Fig. 7 A). These structures also contained ubiquitin (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200507002/DC1). In the rest of the cells, the N-HttQ68 protein was diffusely distributed in the cytosol. The cells containing huntingtin aggregates also seemed to express high levels of p62, whereas the cells containing diffuse huntingtin did not show an increase in immunostained p62. Confocal microscopy at higher resolution revealed that p62 apparently formed a shell surrounding GFP–N-HttQ68–containing aggregates (Fig. 7 B). However, a possibility could be that antibodies may not penetrate to the core of the aggregate, giving an illusion of a p62-containing shell. Thus, to test this more rigorously, we expressed DsRed2-tagged p62 together with GFP–N-HttQ68 and observed live cells in the confocal microscope. With this strategy, it was clear that DsRed-p62 formed a shell surrounding the huntingtin aggregate (Fig. 7 C). The DsRed2 protein alone did not show any association with the huntingtin aggregates (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200507002/DC1). Similarly, when p62 was fused to the nonaggregating, very brightly fluorescing tdTomato, many structures with p62 surrounding GFP–N-HttQ68 aggregates were observed (Fig. 7 D). Stably expressed GFP-tagged p62 could also be seen to enclose aggregates of transiently expressed Flag-HttQ68 (not depicted). It has previously been shown that huntingtin aggregates are degraded by autophagy (Ravikumar et al., 2002, 2004). Interestingly, we found that myc-LC3 protein localized to structures containing GFP-p62 and Flag–N-HttQ68 in S–GFP-p62 cells (Fig. 7 E, top). By coexpressing Flag–N-HttQ68 and tdTomato-LC3, we were able to show that LC3 was part of the shell surrounding the huntingtin aggregates (Fig. 7 E, bottom). We speculate that a p62/LC3-containing shell around huntingtin aggregates might serve to mark these for autophagic degradation.
Figure 7.
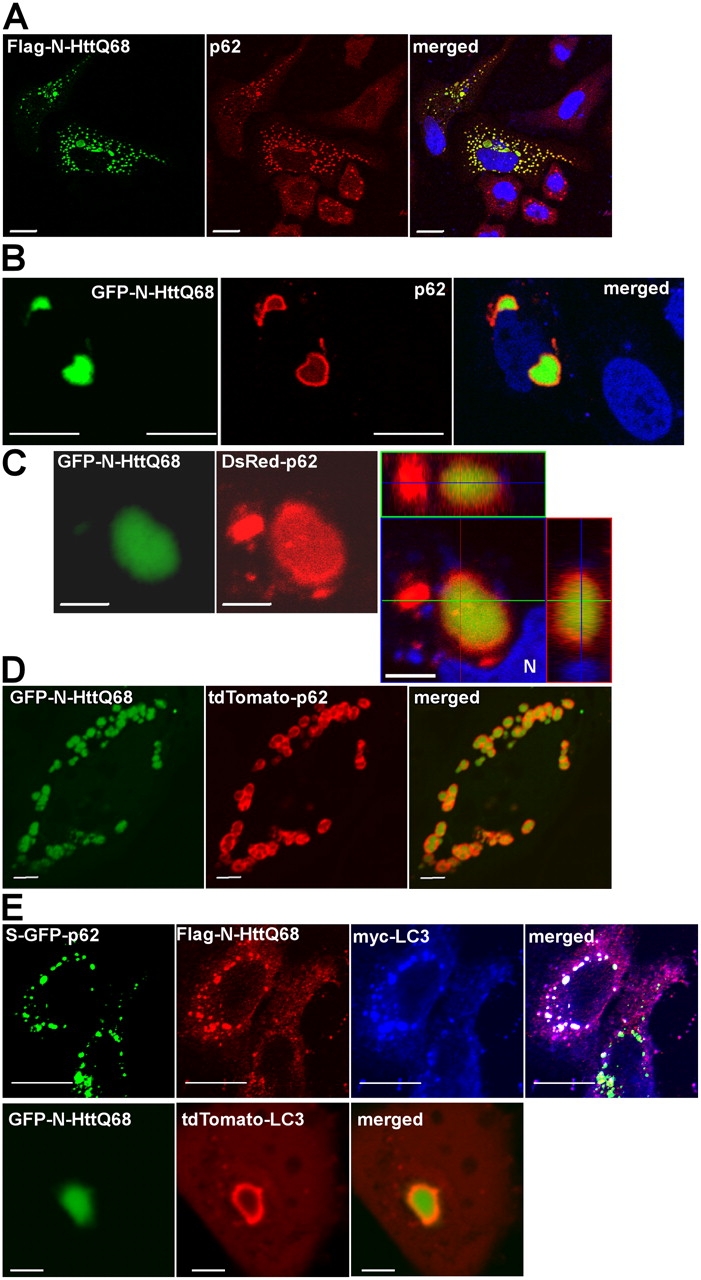
p62 forms a shell around huntingtin protein aggregates. (A) HeLa cells transiently expressing a Flag-tagged NH2-terminal fragment (amino acids 1–171) of huntingtin containing a 68-polyglutamine expansion, Flag–N-HttQ68. After 48 h, the cells were fixed, and Flag–N-HttQ68 was stained green using a Flag mAb. Endogenous p62 was stained red using the p62 pAb. (B) Endogenous p62 forms a shell around aggregated GFP–N-HttQ68. Transiently transfected HeLa cells were fixed 48 h after transfection, and endogenous p62 was detected using a p62 mAb (red). (C and D) The p62 shell surrounding GFP–N-HttQ68 was detected independently of the use of antibodies. (C) GFP–N-HttQ68 and DsRed-tagged p62 were cotransfected into HeLa cells, and images of live cells were obtained after 48 h of expression. The left panel shows one confocal plane of a representative GFP–N-HttQ68 and DsRed-p62 double-positive structure, and the right panel shows the Z-stack of planes with side views of the aggregate at the side and at the top. N, nucleus. (A–C) Nuclei were detected using Draq5. (D) GFP–N-HttQ68 and tdTomato-tagged p62 were cotransfected into HeLa cells, and images of live cells were obtained after 24 h of expression. (E) Transiently expressed Flag–N-HttQ68 and myc-LC3 colocalize in GFP-p62–positive structures. S–GFP-p62 cells were fixed 48 h after transfection, Flag–N-HttQ68 was stained red using a Flag tag mAb, and myc-LC3 was stained far red (blue) using a chicken myc tag pAb (top). The bottom panel shows a live cell image of a GFP–N-HttQ68 aggregate surrounded by a shell containing tdTomato-LC3. Bars, 5 μm.
p62 shows a protective effect against cell death that is induced by overexpression of polyglutamine-expanded huntingtin
Expression of N-HttQ68 caused the induction of apoptosis, which was scored as GFP-positive, rounded, and detached cells that displayed condensed or fragmented nuclei (Fig. 8 A). Expression of wild-type N-HttQ17 gave the same low level of apoptosis as the GFP control (not depicted). The percentage of HttQ68 that induced cell death after 48 h of expression varied from experiment to experiment, ranging from 17 to 46% of the GFP-HttQ68–transfected HeLa cells with a mean of 29% in five independent experiments. We consistently observed that reduction of endogenous p62 levels by expression of p62 antisense RNA increased apoptosis of GFP–N-HttQ68–expressing cells. There was also a less pronounced but significant increase of apoptosis in control cells expressing GFP alone (Fig. 8 B). Also, expressing siRNA against p62 caused a reduced level of endogenous p62 and a corresponding increase in apoptosis after expression of mutant huntingtin (unpublished data). Interestingly, coexpression of a deletion construct of p62 lacking the UBA domain (p62ΔUBA) gave a potent increase in the frequency of huntingtin-expressing apoptotic cells (Fig. 8 B). These findings were observed in both HeLa cells and the neuroblastoma cell line SHSY-5Y. Because the depletion of p62 by antisense RNA or siRNA and interference with the formation of p62 bodies by p62ΔUBA overexpression increased huntingtin-induced cell death, we reasoned that increasing p62 levels by overexpressing p62 might have a protective effect. We could indeed observe a small protective effect in every experiment we performed (Fig. 8 B), but HeLa cells already have a reasonably high level of p62 expression, and mere overexpression could simply direct p62 into sequestosomes. Altogether, these results indicate that p62 protects against huntingtin-induced cell death.
Figure 8.
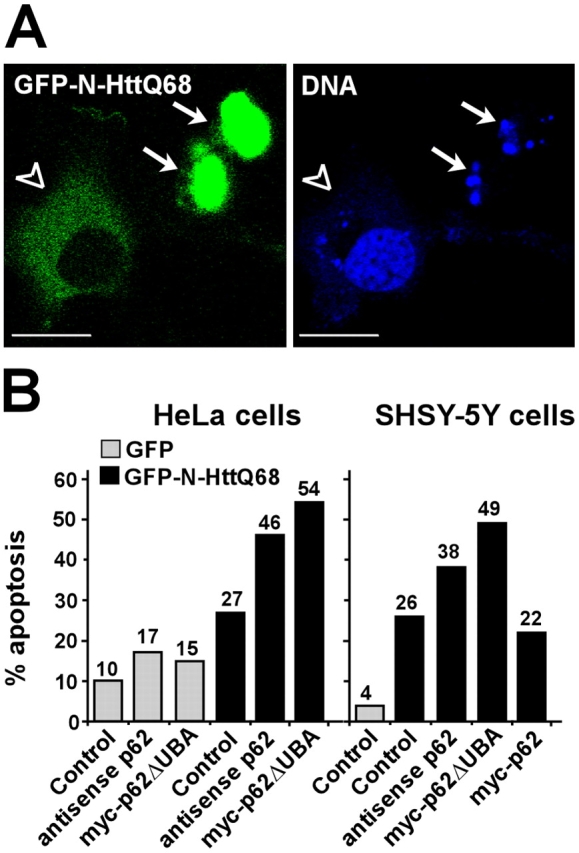
Interfering with p62 protein level or function increases cell death induced by mutant huntingtin. (A) GFP-positive cells were scored as live or apoptotic by the appearance of nuclear Draq5 staining. After 48 h of expression, GFP-positive cells attached to the surface with a normal nuclear appearance (open arrowheads) and were scored as live, whereas rounded, partially detached cells with a condensed or fragmented nuclei (arrows) were scored as dead. More than 200 GFP-positive cells in randomly selected microscope views were scored. Bars, 20 μm. (B) The effect of coexpressing either a p62 antisense construct or a deletion construct of p62 lacking the UBA domain on N-HttQ68–induced cell death in HeLa and SHSY-5Y neuroblastoma cells.
Discussion
Proteins are degraded via two main pathways in eukaryotic cells. Short-lived proteins are degraded by the proteasome, whereas long-lived proteins are degraded by autophagy. Protein aggregates that form during oxidative stress and other conditions, leading to misfolding and aggregation of proteins such as polyglutamine and polyalanine expansion mutations, are degraded by autophagy (Ravikumar et al., 2002, 2004). Autophagy is generally thought of as a nonspecific bulk degradation mechanism. It is presently unclear whether there is a specific recognition or targeting of polyubiquitinated protein aggregates by the autophagic machinery.
Our data suggest that p62 may link polyubiquitinated proteins to the autophagic machinery. This function seems dependent on both the polymerization of p62 via the NH2-terminal PB1 domain and polyubiquitin binding via the COOH-terminal UBA domain of p62. Both endogenous and ectopically expressed p62 could be copurified with the autophagy marker LC3. Both p62 and LC3 colocalized with mutant huntingtin aggregates. Such aggregates were recently shown to be degraded by autophagy (Ravikumar et al., 2002, 2004). Very recently, studies of conditional knockout mice of Atg7 demonstrated that autophagy is needed for clearance of ubiquitin-positive aggregates (Komatsu et al., 2005). We found that p62 formed a shell surrounding huntingtin aggregates. Cell death induced by the expression of aggregation-prone mutant huntingtin was increased both in HeLa and SHSY-5Y neuroblastoma cells after antisense RNA–mediated depletion of p62 levels or by interfering with p62 function by expressing a p62 deletion mutant lacking the UBA domain.
We found that p62 was located in two different types of bodies in the cytosol. The first type of structure appears as large protein aggregates (sequestosomes) that are not surrounded by a membrane and have very low mobility in living cells. However, the majority of p62 bodies in S–GFP-p62 cells were generally smaller structures with a much higher mobility that colocalized poorly with early endosomal markers but strongly with coexpressed myc- or GFP-tagged LC3. The finding that a high number of p62 bodies colocalized with LysoTracker and the lysosomal marker CD63 is consistent with p62 being localized to autophagosomes. By detergent extraction of live cells, we observed two populations of p62 bodies. LysoTracker-positive p62 bodies were rapidly lost after extraction, whereas LysoTracker-negative structures were not dissolved by 1% Triton X-100. This is consistent with p62 being partly located to membrane-enclosed autophagosomes and partly in cytosolic sequestosomes. Induction of autophagy by amino acid starvation led to a clear increase in the number of GFP-LC3–labeled autophagosomes, and all of these were positive for endogenous p62. The idea that a large fraction of p62 bodies are autophagosomes was also supported by the extensive accumulation of p62-LC3–positive structures observed in cells upon treatment with bafilomycin A1. Bafilomycin A1 inhibits the autophagosome–lysosome fusion step leading to accumulation of autophagosomal vacuoles. EM experiments confirmed that p62 is found both in autophagosomes and in sequestosomes and that p62 is colocalized with CD63 in cytoplasmic membrane-enclosed autophagosomal structures. Interestingly, we found p62 and LC3 to be components of the same protein complex by coimmunoprecipitation. It should be noted that coexpression of p62 with GFP-LC3 did not increase the total amount of p62 that was copurified with LC3. This suggests that the interaction might not be direct but may depend on a limiting third cellular factor. This notion is consistent with our failure to detect any interaction between GST-p62 and in vitro translated LC3 in a GST pull-down assay (unpublished data). Furthermore, the p62 D69A mutant that inhibits polymerization of p62 was very inefficiently coimmunoprecipitated with GFP-LC3, suggesting that polymeric p62 is important for interaction with LC3. Our data suggest that p62 is needed for the accumulation of GFP-LC3 in dots in HeLa cells in response to amino acid starvation. No GFP-LC3 dots were formed in response to amino acid starvation in cells depleted for endogenous p62 after transfection with siRNA. Similarly, no GFP-LC3 dots were formed in cells coexpressing mutants of p62, resulting in diffuse localization of endogenous p62. These results indicate that p62 polymerization is important for autophagosome formation in HeLa cells.
Previous studies have established p62 as a stress response protein induced by oxidative stress (Ishii et al., 1997, 2000). The protein has also been identified as a common component in protein aggregates that was found in a wide range of protein aggregation diseases (Zatloukal et al., 2002). Recently, expression of mutant huntingtin was shown to induce p62 (Nagaoka et al., 2004). Interestingly, reactive oxygen species are produced in response to proteasomal inhibition (Ling et al., 2003), and aggregation-prone mutant proteins with expanded polyglutamine stretches inhibit proteasomal activity (Bence et al., 2001). Thus, the induction of p62 in aggregation diseases might also be caused by reactive oxygen species. Prostaglandin J2 is known to induce oxidative stress by causing decreases in glutathione, glutathione peroxidase, and mitochondrial membrane potential as well as increases in the production of protein-bound lipid peroxidation products (Kondo et al., 2001). In human neuroblastoma cells, p62 is needed for the sequestration of ubiquitinated proteins into bodies in response to treatment with the inflammatory agent prostaglandin J2 (Wang and Figueiredo-Pereira, 2005). Our present results provide a molecular mechanism of how p62 might recognize ubiquitinated protein bodies and present these to the autophagic machinery.
Mutant huntingtin is found to be both diffuse in some cells and aggregated in others. It seems clear that aggregation is a mechanism for cell survival (Arrasate et al., 2004). In line with this, Steffan et al. (2004) found that ubiquitination of mutant huntingtin is an important way to detoxify the protein, whereas sumoylation of the same residues prevents aggregation and leads to cell death. Autophagy is important for the clearance of huntingtin aggregates, and induced autophagy leads to increased cell survival of cells expressing mutant huntingtin (Ravikumar et al., 2002, 2004). Similar to Nagaoka et al. (2004), we found that mutant huntingtin could also form aggregates in the absence of p62. Thus, we believe that the protective role of p62 may be to recruit autophagosomal components to the polyubiquitinylated protein aggregates rather than to help or facilitate the formation of these aggregates. Given the essential role of autophagy in preventing protein aggregate–induced neurodegeneration, p62 and proteins with related functions could be attractive targets for the development of neuroprotective drugs.
Materials and methods
Cell transfections
Subconfluent HeLa and SHSY-5Y neuroblastoma cells were transfected using LipofectAMINE Plus (Invitrogen). Stable transfectants were selected using G418, and GFP-positive colonies were isolated and subcloned twice using minimal dilution. A siGENOME SMARTpool SQSTM1 siRNA oligonucleotide mixture against p62 (Dharmacon) was transfected twice, with a 24-h interval at 100 nM using LipofectAMINE Plus. siRNA (Dharmacon) against human tissue factor was used as an unrelated control. Knockdown of p62 by siRNA was verified by immunoblotting.
Antibodies and reagents
The following antibodies were used: anti-p62 and EEA1 mAbs (BD Transduction Laboratories); p62 pAb (Progen Biotechnik); polyubiquitin mAb (clone FK1; Affinity BioReagents, Inc.); CD63 mAbs (Developmental Studies Hybridoma Bank, University of Iowa); anti-GFP antibody (Ab290; Abcam Ltd.); anti-EGF receptor and myc-tag antibodies (9E10; Santa Cruz Biotechnology, Inc.); antiactin antibody (Sigma-Aldrich); and LC3 antibody (gift from T. Yoshimori, National Institute of Genetics, Shizuoka, Japan; Kabeya et al., 2000). All fluorescent Alexa-labeled secondary antibodies, LysoTracker, fluorescent-labeled dextran, and EGF as well as a Zenon kit for direct Alexa labeling of mAbs were obtained from Invitrogen. Bafilomycin A1, epoximicin, 3-methyladenine, and rapamycin were all purchased from Sigma-Aldrich. Proteasome inhibitor I (PSI) was obtained from Calbiochem. Draq5 was obtained from Biostatus Ltd. Redivue Pro-mix 35S-methionine was obtained from GE Healthcare.
Plasmids
The following vectors have been described previously (Lamark et al., 2003): pCW7-mycUbi (a gift from J. Lukas, Danish Cancer Society, Copenhagen, Denmark; Thullberg et al., 2000); pcDNA3-myc-LC3 and pEGFP-C1-LC3 (Simonsen et al., 2004); the Gateway entry clones pENTR-p62 and pENTR-p40phox; the Gateway destination vectors pDestEGFP-C1 and pDestmyc; and the Gateway expression vectors pDestHA-p62, pDestmyc-p62, pDestHA-p62 D69A, pDestmyc-p62 D69A, pDestEGFP-p62 D69A, and pEGFP-p62. pENTR-p62 I431A was constructed by the mutagenesis of pENTR-p62 using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The p62 deletion constructs pENTR-p62Δ123–386, pENTR-p62(387–440), pENTR-p62(1–122), pENTR-p62(124–440), and pENTR-p62(1–385) were made by subcloning of the indicated p62 fragments from pENTR-p62 into Gateway entry vectors (pENTR1A or pENTR3C; Invitrogen). pENTR-Htt(1–171)68Q-Flag was made by the subcloning of human huntingtin(1–171)68Q-FLAG cDNA from plasmid pcDNA1-Htt(1–171)68Q-Flag (obtained from U. Moens, University of Tromsø, Tromsø, Norway; Saudou et al., 1998) into pENTR3C. The mammalian tdTomato fusion expression vector ptdTomato-C1 was made by exchanging the EGFP gene of pEGFP-C1 (CLONTECH Laboratories, Inc.) with tdTomato from the bacterial expression vector pRSET-B-tdTomato (obtained from R. Tsien, University of California, San Diego, San Diego, CA; Shaner et al., 2004). ptdTomato-LC3 was made by inserting a 400-bp EcoRI–BamHI fragment from pEGFP-C1-LC3 into ptdTomato-C1 cut with the same enzymes. The Gateway destination vector pDest-tdTomato-C1 was made by exchanging the EGFP gene of pDestEGFP-C1 with tdTomato, and Gateway destination vector pDestDsRED2-C1 was constructed by the insertion of Gateway cassette B (Gateway vector conversion system; Invitrogen) into pDsRED2-C1 (CLONTECH Laboratories, Inc.). The Gateway expression vectors pDestDsRED2-p62, pDest-tdTomato-p62, pDestEGFP-p62(1–122), pDestEGFP-p62(387–440), pDestEGFP-p62Δ123–386, pDestEGFP-p62(1–385), pDestmyc-p62(1–385), pDestEGFP-p62 (124–440), pDestEGFP-p62 I431A, pDestEGFP-Htt(1–171)68Q-Flag, and pDestEGFP-p40phox were made using Gateway recombination reactions (Invitrogen). The antisense p62 construct pAS-p62-c was made by religation of pAS-p62 (Lamark et al., 2003) after digestion with EcoRV. All constructs were verified by DNA sequencing (BigDye; Applied Biosystems). Oligonucleotides for mutagenesis, PCR, and DNA sequencing reactions were obtained from Eurogentec.
Confocal microscopy analyses
The cell cultures were directly examined under the microscope or fixed in 4% PFA and stained as previously described (Lamark et al., 2003). Live cells were imaged at 37°C in Hanks medium containing 10% serum, whereas fixed cells were imaged at RT in PBS. Images were collected using a microscope (Axiovert 200; Carl Zeiss MicroImaging, Inc.) equipped with a 40× 1.2W C-Apochroma objective and a confocal module (LSM510 META; Carl Zeiss MicroImaging, Inc.) and using the LSM5 software version 3.2 (Carl Zeiss MicroImaging, Inc.). Images were processed using Photoshop (Adobe). Z-stack video images were acquired using an imager (Ultraview RS Live Cell; PerkinElmer) on the Axiovert microscope with a 63× NA 1.4 plan-Apochromat objective. In total, 8–10 confocal 1-μm planes covering the whole height of the cells were superimposed for each time point. The video was acquired with Imaging Suite software (PerkinElmer) and compressed using Premiere Pro software (Adobe). Two-color live cell videos were obtained using the LSM510 META confocal module imaging a single confocal plane. Videos were compressed using QuickTime Pro.
EM
Cells for immuno-EM were fixed and embedded as described previously (Peters et al., 1991). Small blocks were cut and infused with 2.3 M sucrose for 1 h, mounted on silver pins, and frozen in liquid nitrogen. Ultrathin cryosections were cut at −110°C on an ultramicrotome (Ultracut; Leica) and collected with a 1:1 mixture of 2% methyl cellulose and 2.3 M sucrose. Sections were transferred to formvar/carbon-coated grids and labeled with primary antibodies followed by a bridging secondary antibody and protein A–gold conjugates essentially as described previously (Slot et al., 1991). For double-labeling experiments, we included a blocking step between the first protein A–gold and the second primary antibody (15-min incubation in 0.1% glutaraldehyde and 0.1 M PBS). After embedding in 2% methyl cellulose/0.4% uranyl acetate, we observed sections at 80 kV in an electron microscope (CM10; Philips).
Correlative immunofluorescence/EM was performed on HeLa cells that were grown on gridded coverslips (Eppendorf). Cells were treated with PSI for 3 h, fixed in 3% PFA/PBS (30 min), permeabilized with 0.05% saponin/PBS, and labeled with mouse anti-p62 (1:2,000) followed by donkey anti–mouse Cy2 (1:500). After embedding in moviol, the samples were observed on a microscope (LSM510 META; Carl Zeiss MicroImaging, Inc.), and the localization of interesting cells was recorded. For EM observation, the same coverslips were then fixed with 2% glutaraldehyde in 0.1 M phosphate buffer and postfixed with 2% OsO4 and 1.5% KFeCN in 0.1 M phosphate buffer. Thereafter, the coverslips were stained en bloc with 4% uranyl acetate for 60 min, dehydrated in ethanol, and embedded in Epon. After polymerization, the coverslips were removed with 40% hydrofluoric acid, and the flat specimens were glued onto Epon stubs. The block was thereafter trimmed down to the regions observed on the gridded coverslips in the fluorescence microscope and sectioned parallel to the substratum at 50–70-nm section thickness. The sections were poststained with lead citrate (2 min). Electron micrographs were taken and overlaid with the confocal images in Photoshop.
Immunoblot and immunoprecipitations
All expression constructs were controlled by the immunoblotting of total cell extracts that was made after stable transfection or 24 h after transient transfection as described previously (Lamark et al., 2003). Cells were labeled with 0.14 μCi/ml 35S-methionine by a 30-min pulse in methionine-free medium, washed in PBS, and incubated for different times in normal medium. Endogenous p62 was immunoprecipitated using the p62 mAb from total cell lysates from 35S-methionine cells as described previously (Lamark et al., 2003). GFP-LC3 was immunoprecipitated from total cellular extracts using anti-GFP antibody.
Online supplemental material
Video 1 shows the migration of S–GFP-p62 expressed in HeLa cells over 2 min. In total, 130 image stacks were collected with a time interval of 1 s. Video 2 shows the migration of S–GFP-p62 expressed in HeLa cells over 1 h. 60 image stacks were collected with a time interval of 60 s. Video 3 shows dynamic movements of pDest-tdTomato-LC3 and GFP-p62 transiently coexpressed in HeLa cells. One confocal plane was imaged for 3 min with a time interval of 4 s. Videos 1, 2, and 3 are displayed as 15, 6, and 10 frames per second, respectively. Fig. S1 shows rapid detergent extraction of both GFPp40phox- and LysoTracker-positive vesicles in HeLa cells. Fig. S2 shows a lack of association between DsRed fluorescent protein and a GFP–N-HttQ68 aggregate. Fig. S3 shows the accumulation of endogenous p62 and polyubiquitin in mutant huntingtin aggregates. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200507002/DC1.
Acknowledgments
We thank Hege Avsnes Dale and Endy Spriet at the Molecular Imaging Centre in Bergen for help with video images from the live cell imager. We are grateful to R. Tsien for the gift of pRSET-B-tdTomato, J. Lukas for pCW7-mycUbi, U. Moens and F. Saudou for pcDNA1-Htt(1–171)68Q-Flag, and T. Yoshimori for LC3 antibodies.
This work was supported by grants to T. Johansen from the Norwegian Research Council, the Norwegian Cancer Society, the Aakre Foundation, Simon Fougner Hartmanns Familiefond, and the Blix Foundation. A. Brech is the recipient of a career fellowship from The National Programme for Research in Functional Genomics in Norway of the Norwegian Research Council.
Abbreviations used in this paper: EEA1, early endosome antigen 1; LC3, light chain 3; PB1, Phox and Bem1p; siRNA, small interfering RNA; UBA; ubiquitin associated.
References
- Abeliovich, H., W.A. Dunn Jr., J. Kim, and D.J. Klionsky. 2000. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J. Cell Biol. 151:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate, M., S. Mitra, E.S. Schweitzer, M.R. Segal, and S. Finkbeiner. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 431:805–810. [DOI] [PubMed] [Google Scholar]
- Avila, A., N. Silverman, M.T. Diaz-Meco, and J. Moscat. 2002. The Drosophila atypical protein kinase C-ref(2)p complex constitutes a conserved module for signaling in the toll pathway. Mol. Cell. Biol. 22:8787–8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence, N.F., R.M. Sampat, and R.R. Kopito. 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 292:1552–1555. [DOI] [PubMed] [Google Scholar]
- Cariou, B., D. Perdereau, K. Cailliau, E. Browaeys-Poly, V. Bereziat, M. Vasseur-Cognet, J. Girard, and A.F. Burnol. 2002. The adapter protein ZIP binds Grb14 and regulates its inhibitory action on insulin signaling by recruiting protein kinase Czeta. Mol. Cell. Biol. 22:6959–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo, A.M. 2004. Autophagy: in sickness and in health. Trends Cell Biol. 14:70–77. [DOI] [PubMed] [Google Scholar]
- Donaldson, K.M., W. Li, K.A. Ching, S. Batalov, C.C. Tsai, and C.A. Joazeiro. 2003. Ubiquitin-mediated sequestration of normal cellular proteins into polyglutamine aggregates. Proc. Natl. Acad. Sci. USA. 100:8892–8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escola, J.M., M.J. Kleijmeer, W. Stoorvogel, J.M. Griffith, O. Yoshie, and H.J. Geuze. 1998. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 273:20121–20127. [DOI] [PubMed] [Google Scholar]
- Geetha, T., and M.W. Wooten. 2002. Structure and functional properties of the ubiquitin binding protein p62. FEBS Lett. 512:19–24. [DOI] [PubMed] [Google Scholar]
- Gong, J., J. Xu, M. Bezanilla, R. van Huizen, R. Derin, and M. Li. 1999. Differential stimulation of PKC phosphorylation of potassium channels by ZIP1 and ZIP2. Science. 285:1565–1569. [DOI] [PubMed] [Google Scholar]
- Ishii, T., T. Yanagawa, K. Yuki, T. Kawane, H. Yoshida, and S. Bannai. 1997. Low micromolar levels of hydrogen peroxide and proteasome inhibitors induce the 60-kDa A170 stress protein in murine peritoneal macrophages. Biochem. Biophys. Res. Commun. 232:33–37. [DOI] [PubMed] [Google Scholar]
- Ishii, T., K. Itoh, S. Takahashi, H. Sato, T. Yanagawa, Y. Katoh, S. Bannai, and M. Yamamoto. 2000. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 275:16023–16029. [DOI] [PubMed] [Google Scholar]
- Kabeya, Y., N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi, and T. Yoshimori. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya, Y., N. Mizushima, A. Yamamoto, S. Oshitani-Okamoto, Y. Ohsumi, and T. Yoshimori. 2004. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117:2805–2812. [DOI] [PubMed] [Google Scholar]
- Kegel, K.B., M. Kim, E. Sapp, C. McIntyre, J.G. Castano, N. Aronin, and M. DiFiglia. 2000. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 20:7268–7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D.J. 2005. The molecular machinery of autophagy: unanswered questions. J. Cell Sci. 118:7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu, M., S. Waguri, T. Ueno, J. Iwata, S. Murata, I. Tanida, J. Ezaki, N. Mizushima, Y. Ohsumi, Y. Uchiyama, et al. 2005. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, M., T. Oya-Ito, T. Kumagai, T. Osawa, and K. Uchida. 2001. Cyclopentenone prostaglandins as potential inducers of intracellular oxidative stress. J. Biol. Chem. 276:12076–12083. [DOI] [PubMed] [Google Scholar]
- Kuusisto, E., A. Salminen, and I. Alafuzoff. 2001. a. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 12:2085–2090. [DOI] [PubMed] [Google Scholar]
- Kuusisto, E., T. Suuronen, and A. Salminen. 2001. b. Ubiquitin-binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochem. Biophys. Res. Commun. 280:223–228. [DOI] [PubMed] [Google Scholar]
- Kuusisto, E., A. Salminen, and I. Alafuzoff. 2002. Early accumulation of p62 in neurofibrillary tangles in Alzheimer's disease: possible role in tangle formation. Neuropathol. Appl. Neurobiol. 28:228–237. [DOI] [PubMed] [Google Scholar]
- Lamark, T., M. Perander, H. Outzen, K. Kristiansen, A. Overvatn, E. Michaelsen, G. Bjorkoy, and T. Johansen. 2003. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J. Biol. Chem. 278:34568–34581. [DOI] [PubMed] [Google Scholar]
- Lawrence, B.P., and W.J. Brown. 1993. Inhibition of protein synthesis separates autophagic sequestration from the delivery of lysosomal enzymes. J. Cell Sci. 105:473–480. [DOI] [PubMed] [Google Scholar]
- Levine, B., and D.J. Klionsky. 2004. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell. 6:463–477. [DOI] [PubMed] [Google Scholar]
- Ling, Y.H., L. Liebes, Y. Zou, and R. Perez-Soler. 2003. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J. Biol. Chem. 278:33714–33723. [DOI] [PubMed] [Google Scholar]
- Mizushima, N. 2004. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 36:2491–2502. [DOI] [PubMed] [Google Scholar]
- Nagaoka, U., K. Kim, N.R. Jana, H. Doi, M. Maruyama, K. Mitsui, F. Oyama, and N. Nukina. 2004. Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J. Neurochem. 91:57–68. [DOI] [PubMed] [Google Scholar]
- Noda, T., and Y. Ohsumi. 1998. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 273:3963–3966. [DOI] [PubMed] [Google Scholar]
- Peters, P.J., J.J. Neefjes, V. Oorschot, H.L. Ploegh, and H.J. Geuze. 1991. Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for transport to lysosomal compartments. Nature. 349:669–676. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B., R. Duden, and D.C. Rubinsztein. 2002. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11:1107–1117. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B., C. Vacher, Z. Berger, J.E. Davies, S. Luo, L.G. Oroz, F. Scaravilli, D.F. Easton, R. Duden, C.J. O'Kane, and D.C. Rubinsztein. 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36:585–595. [DOI] [PubMed] [Google Scholar]
- Sanz, L., P. Sanchez, M.J. Lallena, M.T. Diaz-Meco, and J. Moscat. 1999. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 18:3044–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz, L., M.T. Diaz-Meco, H. Nakano, and J. Moscat. 2000. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 19:1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou, F., S. Finkbeiner, D. Devys, and M.E. Greenberg. 1998. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 95:55–66. [DOI] [PubMed] [Google Scholar]
- Shaner, N.C., R.E. Campbell, P.A. Steinbach, B.N. Giepmans, A.E. Palmer, and R.Y. Tsien. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22:1567–1572. [DOI] [PubMed] [Google Scholar]
- Simonsen, A., H.C. Birkeland, D.J. Gillooly, N. Mizushima, A. Kuma, T. Yoshimori, T. Slagsvold, A. Brech, and H. Stenmark. 2004. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 117:4239–4251. [DOI] [PubMed] [Google Scholar]
- Slot, J.W., H.J. Geuze, S. Gigengack, G.E. Lienhard, and D.E. James. 1991. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol. 113:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan, J.S., N. Agrawal, J. Pallos, E. Rockabrand, L.C. Trotman, N. Slepko, K. Illes, T. Lukacsovich, Y.Z. Zhu, E. Cattaneo, et al. 2004. SUMO modification of Huntingtin and Huntington's disease pathology. Science. 304:100–104. [DOI] [PubMed] [Google Scholar]
- Thompson, H.G., J.W. Harris, B.J. Wold, F. Lin, and J.P. Brody. 2003. p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene. 22:2322–2333. [DOI] [PubMed] [Google Scholar]
- Thullberg, M., J. Bartek, and J. Lukas. 2000. Ubiquitin/proteasome-mediated degradation of p19INK4d determines its periodic expression during the cell cycle. Oncogene. 19:2870–2876. [DOI] [PubMed] [Google Scholar]
- Vadlamudi, R.K., I. Joung, J.L. Strominger, and J. Shin. 1996. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J. Biol. Chem. 271:20235–20237. [DOI] [PubMed] [Google Scholar]
- Vonsattel, J.P., and M. DiFiglia. 1998. Huntington disease. J. Neuropathol. Exp. Neurol. 57:369–384. [DOI] [PubMed] [Google Scholar]
- Wang, Z., and M.E. Figueiredo-Pereira. 2005. Inhibition of sequestosome 1/p62 up-regulation prevents aggregation of ubiquitinated proteins induced by prostaglandin J2 without reducing its neurotoxicity. Mol. Cell. Neurosci. 29:222–231. [DOI] [PubMed] [Google Scholar]
- Yamamoto, A., Y. Tagawa, T. Yoshimori, Y. Moriyama, R. Masaki, and Y. Tashiro. 1998. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 23:33–42. [DOI] [PubMed] [Google Scholar]
- Yoshimori, T. 2004. Autophagy: a regulated bulk degradation process inside cells. Biochem. Biophys. Res. Commun. 313:453–458. [DOI] [PubMed] [Google Scholar]
- Zatloukal, K., C. Stumptner, A. Fuchsbichler, H. Heid, M. Schnoelzer, L. Kenner, R. Kleinert, M. Prinz, A. Aguzzi, and H. Denk. 2002. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 160:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]