

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by a selective degeneration of motor neurons, atrophy, and paralysis of skeletal muscle. Although a significant proportion of familial ALS results from a toxic gain of function associated with dominant SOD1 mutations, the etiology of the disease and its specific cellular origins have remained difficult to define. Here, we show that muscle-restricted expression of a localized insulin-like growth factor (Igf) -1 isoform maintained muscle integrity and enhanced satellite cell activity in SOD1G93A transgenic mice, inducing calcineurin-mediated regenerative pathways. Muscle-specific expression of local Igf-1 (mIgf-1) isoform also stabilized neuromuscular junctions, reduced inflammation in the spinal cord, and enhanced motor neuronal survival in SOD1G93A mice, delaying the onset and progression of the disease. These studies establish skeletal muscle as a primary target for the dominant action of inherited SOD1 mutation and suggest that muscle fibers provide appropriate factors, such as mIgf-1, for neuron survival.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neuromuscular disorder involving the degeneration of motor neurons accompanied by skeletal muscle atrophy and paralysis. Transgenic mice ubiquitously overexpressing human SOD1 mutants develop motor neuron disease resembling ALS (Gurney et al., 1994) and provide useful model to test potential therapeutic strategies in disease treatment.
Notably, restriction of SOD1 mutant expression selectively to post-natal motor neurons failed to produce detectable sign of pathology or motor-neuron disease (Lino et al., 2002), suggesting that other cell types may be involved in ALS-associated neurodegeneration.
Indeed, analysis of chimeras generated between wild-type and SOD1 mutant mouse embryonic cells revealed that wild-type non neuronal cells in adult chimeric animals extended the survival of SOD1 mutant motor neurons, suggesting that the neurodegenerative action of mutant SOD1 may operate through a dominant paracrine activity emanating from nonneuronal cells (Clement et al., 2003).
Skeletal muscle is an untested component in the motor neurodegenerative effects of SOD1 mutations. Adult muscle fibers are a source of signals that influence neuron survival, axonal growth, and maintenance of synaptic connections (Funakoshi et al., 1995). Among these, insulin-like growth factor 1 (Igf-1) has been implicated in anabolism of muscle and nerve tissues, inducing muscle hypertrophy and promoting neuronal survival (Musarò and Rosenthal, 2002).
In a recent study, injection of SOD1 mutant mouse muscle with an adeno-associated virus carrying an Igf-1 gene prolonged life and delayed disease progression (Kaspar et al., 2003). Retrograde transport of the AAV–Igf-1 vector to the spinal cord was reported necessary to achieve therapeutic effects (Kaspar et al., 2003). However, it is not clear from that study which Igf-1 isoform was used, or whether the effects of Igf-1 expressed in motor neurons were cell autonomous.
Two major isoforms of Igf-1, originating from alternative splicing, have been described, differing in structure and functions (Musarò and Rosenthal, 2002). These isoforms are referred as circulating (class 2) and local (class 1) Igf-1. Igf-1 class 2 transcripts predominate in the liver, are highly growth hormone (GH) responsive and as such are major endocrine effectors of GH. By contrast, class 1 transcripts are widely expressed in all tissues, but remain preferentially confined to the tissue of expression. The contribution to more localized accumulation of Igf-1 class 1 seems due to the combination of exon 1, Ea peptide, and a lower binding affinity to Igf binding proteins, compared with circulating class 2 isoform (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200407021/DC1).
To assess the effects of supplemental Igf-1 directly on atrophic SOD1 skeletal muscle, we exploited a transgenic mouse expressing a full-length precursor of the local isoform of Igf-1 (mIgf-1) that is normally induced transiently in response to muscle damage, but does not enter the circulation (Musarò et al., 2001; Musarò and Rosenthal, 2002). Muscle restricted mIgf-1 transgene (MLC/mIgf-1) exerts its effects in an autocrine/paracrine manner, circumventing the adverse side effects of systemic Igf-1 administration. Expression of the MLC/mIgf-1, delivered either as an inherited transgene or somatically on an AAV vector, induces muscle hypertrophy and strength, and preserves regenerative capacity in senescent and dystrophic mice (Barton-Davis et al., 1998; Musarò et al., 2001; Barton et al., 2002) through enhanced stem cell recruitment (Musarò et al., 2004).
In the present study, we establish skeletal muscle as a primary target in inherited forms of ALS by showing that localized expression of the coinherited MLC/mIgf-1 transgene exclusively in the skeletal muscle of SOD1G93A mice counteracted the symptoms of ALS, induced satellite cell activity, stabilized neuromuscular junctions, and led to a reduction in astrocytosis in the SOD1G93A spinal cord. These observations suggest novel approaches to the attenuation of motor neuronal degradation and underscore the importance of Igf-1 isoform selection when designing therapeutic strategies for ALS.
Results and discussion
Muscle-specific mIgf-1 delays the progression of the disease and prolongs the life span of SOD1G93A mice
To evaluate the effects of mIgf-1 on the SOD1G93A neurodegenerative phenotype, we compared double transgenic SOD1G93A and MLC/mIgf-1 transgenic mice to their SOD1G93A littermates. The SODG93A and SOD1G93A × MLC/mIgf-1 (SOD1G93A/mIgf-1) transgenic mice were selected for same expression level of the human transgenic protein (Fig. 1 a). Notably, the mIgf-1 transgene was selectively expressed in skeletal muscle of both MLC/mIgf-1 and SOD1G93A/mIgf-1 mice (Fig. 1 b, lanes 2 and 4), whereas it was not expressed in the brain and spinal cord (Fig. 1 b, lanes 5–8), or in skeletal muscle of wild-type and SODG93A mice (Fig. 1 b, lanes 1 and 3).
Figure 1.

mIgf-1 expression delays the progression of the disease and enhances the survival of SOD1G93A mutant mice. (a) Western blot analysis of human SOD transgenic protein in wild-type (lane 1), MLC/mIgf-1 (lane 2), SOD1G93A (lane 3) and SOD1G93A/mIgf-1 (lane 4) transgenic muscle. (b) Northern blot analysis for mIgf-1 transgene expression in skeletal muscle of wild-type (wt; lane 1), MLC/mIgf-1 (lane 2), SOD1G93A (lane 3) and SOD1G93A/mIgf-1 (lane 4) transgenic mice; in brain and spinal cord of MLC/mIgf-1 (lanes 5 and 7) and SOD1G93A/mIgf-1 (lanes 6 and 8) mice. (c) Age of onset of disease symptoms; average onset: SOD1G93A (n = 30) = 111 ± 1.8; SOD1G93A/mIgf-1 (n = 30) = 120.8 ± 1.0 (d) analysis of the progression of the disease; average of disease duration: SOD1G93A (n = 30) =12 ± 0.6; SOD1G93A/mIgf-1 (n = 30) = 32 ± 0.8 (e) survival analysis; average survival: SOD1G93A (n = 30) = 123 ± 1.4; SOD1G93A/mIgf-1 (n = 30) = 152.8 ± 1.4.
At 111 ± 1.8 d old, disease onset was observed in the mutant SOD1G93A transgenic mice (n = 30; Fig. 1 c). Notably, the SOD1G93A mice died within 10 d ± 0.6 of clinical disease onset (Fig. 1 d), with a maximal life span of 145 d (Fig. 1 e). By contrast, localized expression of mIgf-1 delayed the onset (Fig. 1 c) and in particular the progression (Fig. 1 d) of disease, increasing the survival of SOD1G93A/mIgf-1 mice (n = 30) by ∼30 d to a maximal life span of 175 d (Fig. 1 e). Differences between SOD1G93A and SOD1G93A/mIgf-1 were significantly relevant for onset (χ2 LR = 18.7, P < 0.0001), progression (χ2 LR = 67.0, P < 0.0001), and survival (χ2 LR = 63.2, P < 0.0001).
mIgf-1 expression attenuates muscle atrophy, increasing satellite cell activation in SOD1G93A mice
SOD1G93A (n = 7) and SOD1G93A/mIgf-1 (n = 7) transgenic mice were analyzed at different stages of disease. At 123 d, motor neuronal degeneration of SOD1G93A mice was accompanied by severe muscle atrophy (Fig. 2 a, E), and complete muscle paralysis (hereafter this stage is indicated as paralysis stage). In contrast, at the same age, SOD1G93A/mIgf-1 transgenic mice did not show evident signs of muscle disease, but only difficulty in extending the limbs when suspended (hereafter this stage is indicated as symptom onset). Moreover, muscle atrophy was substantially attenuated in SOD1G93A/mIgf-1 offspring even after onset of denervation and paralysis stage (153 d old; Fig. 2 a, F; Fig. 2 b). In addition, markers of satellite cell activity, such as Pax-7 and desmin, were increased to varying extents in affected SODG93A mice (Fig. 2 c), whereas hallmarks of satellite cell activity and fiber maturation, including centralized nuclei (Fig. 2 a, yellow arrows), Pax-7 isoforms, desmin, myogenin, and neonatal myosin heavy chain (MyHC) expression were present exclusively in the SOD1G93A/mIgf-1 muscles at all stages of disease, including at paralysis stage (Fig. 2 c and not depicted), Thus, satellite cell activation is likely to contribute to the maintenance of muscle phenotype induced by mIgf-1 expression (Barton-Davis et al., 1999).
Figure 2.
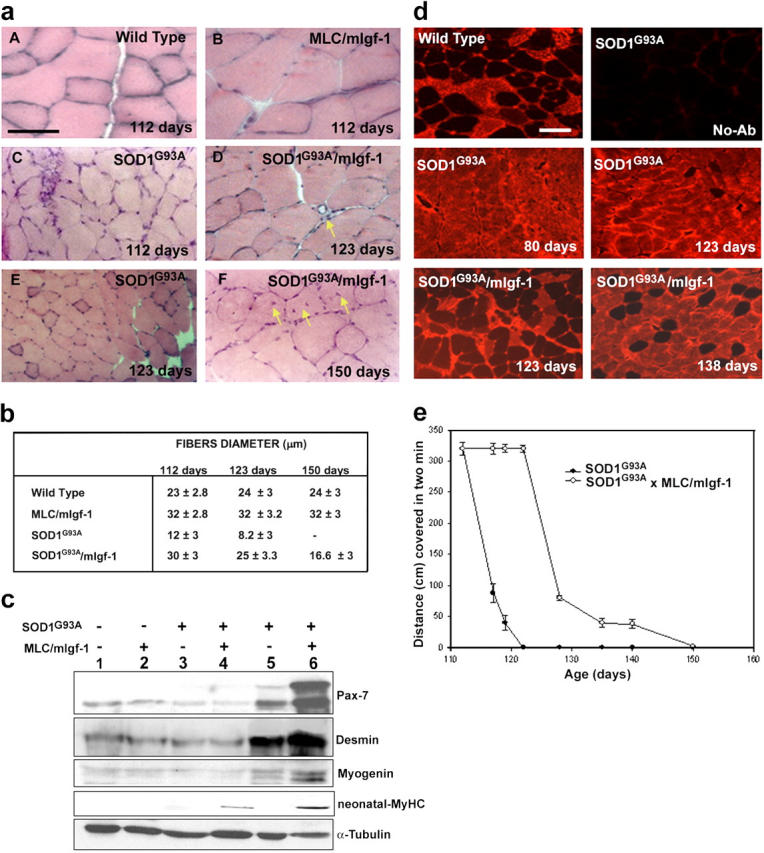
mIgf-1 expression attenuates muscle wasting and promotes regenerative pathways in SOD1G93A mice. (a) Histological analysis of wt, MLC/mIgf-1, SOD1G93A and SOD1G93A/mIgf-1 muscle at different age and stage of disease. Bar, 20 μm (b) Analysis of fiber size differences in the quadriceps underscores the relative attenuation of muscle atrophy in SOD1G93A/mIgf-1 compared with SOD1G93A mice. (c) Western blot analysis of molecular markers of muscle regeneration, activated satellite cells, and maturation. Muscle protein lysates were obtained from quadriceps of wt (lane 1), MLC/mIgf-1 (lane 2), SOD1G93A (lanes 3 and 5) and SOD1G93A/mIgf-1 (lanes 4 and 6) transgenic mice at different ages of the clinical disease (lanes 3 and 4 at 28 d old; lanes 5 and 6 at 123 d old). Immunoblotting for α-tubulin served as a control for protein loading. (d) Immunofluorescence analysis of MyHC-fast performed on soleus muscles of wt, SOD1G93A and SOD1G93A/mIgf-1 before (80 d) and after symptom onset (123, 138 d old). Bar, 50 μm. (e) Walk test of SOD1G93A (closed circles) and SOD1G93A/mIgf-1 (open circles) transgenic mice.
Motor neurons are known to regulate the properties of the myofibers they innervate by selective activation of fiber specific gene expression. Immunohistochemical analysis (Fig. 2 d) revealed that fiber type composition was altered in SOD1G93A soleus muscle even before overt disease (80 d), with a shift toward a fast fiber type. In contrast, normal heterogeneity of muscle fibers was maintained for a more extended period in SOD1G93A/mIgf-1 mice, which showed shifts in fiber composition only at later stage of disease (Fig. 2 d, 138 d). By paralysis stage (153 d) there was not significant difference in fiber type composition between SOD1G93A and SOD1G93A/mIgf-1 mice (not depicted). The alteration in the heterogeneity of SOD1G93A muscle fibers indicate an alteration in motor neuron activity even before overt disease and support the hypothesis that delaying the progression and severity of ALS disease may depend on the maintenance of muscle integrity.
The walk test analysis (Fig. 2 e), performed at different ages, revealed that at 112 d SOD1G93A mice (n = 7) showed symptom onset without evident alterations in functional parameters. The condition of SOD1G93A mice rapidly deteriorated at 117 d, as shown by the shortening of their walk (Fig. 2 e). In contrast, the pathological sign of disease were delayed in SOD1G93A/mIgf-1 transgenic mice (n = 7), as shown by the capacity of the mice to walk 233 ± 5.6 cm further when analyzed at same age as SOD1G93A mice, and by their ability to move for a more extended period of time (Fig. 2 e).
These data suggest that promotion of muscle satellite cell activity and hypertrophy through mIgf-1 can be considered as an alternate therapeutic approach to counteract muscle wasting associated with ALS disease.
An activated calcineurin isoform is induced in SOD1G93A/mIgf-1 muscle
One of the signal transduction pathway involved in muscle remodeling is the protein phosphatase calcineurin-A (CnA). CnA overexpression has been implicated in myocyte hypertrophy (Musarò et al., 1999), fiber type conversion (Chin et al., 1998), and tissue remodeling after injury (Sakuma et al., 2003). In a study of selective CnA subunit isoform expression, we recently established that the alternatively spliced variant CnA-β1 is up-regulated in regenerating skeletal muscle fibers (unpublished data).
In this study, we verified whether activation of satellite cells and maintenance of muscle phenotype involved the induction of CnA-β1 expression. Although the normally low levels of CnA-β1 expression were not raised in SOD1G93A muscles (Fig. 3 a lane 3; Fig. 3 b lane 5, Fig. 3 c), or in uninjured wild-type and MLC/mIgf-1 muscle (Fig. 3, a and b, lanes 1 and 2), SOD1G93A/mIgf-1 regenerating muscle dramatically increased CnA-β1 transcripts (73 ± 1.3%; Fig. 3 a lane 4) and protein (52 ± 2%; Fig. 3 b lane 6, Fig. 3 c) after onset of clinical symptoms. Thus, specific signaling elicited by CnA-β1 may contribute to the prolongation of muscle integrity even after denervation.
Figure 3.
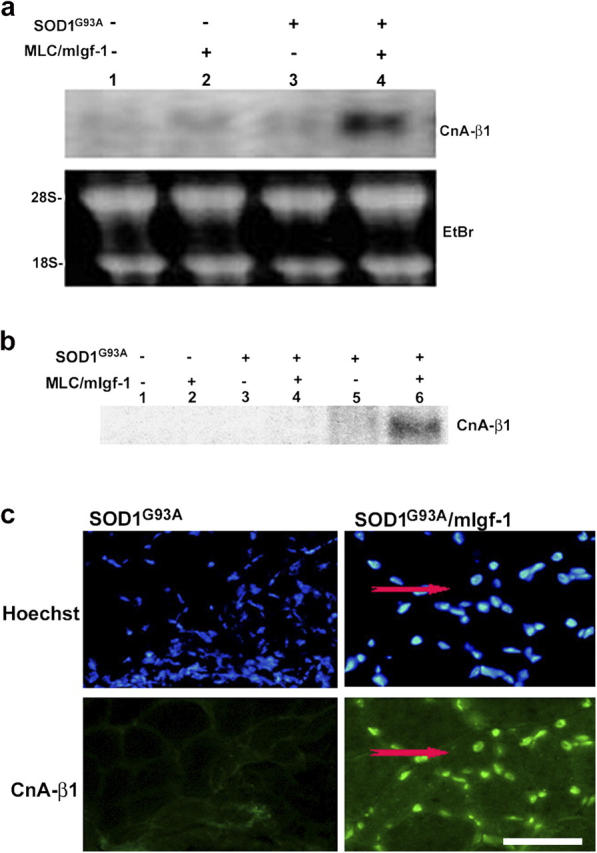
Transgenic mIgf-1 expression induces chronic CnA-β1 expression in SOD1G93A mice. (a) Northern blot analysis of CnA-β1 expression in wt (lane 1), MLC/mIgf-1 (lane 2), SOD1G93A (lane 3) and SOD1G93A/mIgf-1 (lane 4) transgenic mice. Ethidium bromide staining was used to verify equal loading of the RNA sample. (b) Lysates of the same muscle tissues used in Fig. 2 c were tested by Western blotting using a CnA-β1–specific antibody. (c) Immunofluorescence of transverse sections from quadriceps muscles of SOD1G93A and SODG93A/mIgf-1 at paralysis stage. CnA-β1 shows a nuclear localization. A regenerating fiber is indicated by the presence of central nucleus (red arrows). Nuclei were visualized by Hoechst dye (blue). Bar, 20 μm.
Preservation of neuromuscular junctions in SOD1G93A/mIgf-1 mice
Alterations in motor neuronal activity typical of denervated muscle and motor neuron diseases also affect the configuration of neuromuscular junctions in SOD1G93A mice, characterized by the diffusion of acetylcholine receptor (AChR) postsynaptic clusters (Fig. 4 a, yellow arrow). At 123 d, SOD1G93A paralyzed muscle showed 56 ± 0.2% of diffuse AChR expression, whereas AChR cluster aggregates (Fig. 4 a) were preserved in muscles of age-matched SOD1G93A/mIgf-1 mice, which showed only 3.3 ± 0.4% of diffuse AChR expression. At comparable end-stage disease, SOD1G93A/mIgf-1 muscle displayed only 18 ± 0.4% of diffuse AChR expression. These results were confirmed by Northern blot analysis (Fig. 4 b); high AChR expression levels in SOD1G93A muscle were reduced in SOD1G93A/mIgf-1 mice at all stages observed. Densitometric analysis (n = 6) revealed that AChR mRNA expression in SOD1G93A paralyzed muscle (123 d) was 68 ± 2.4% higher than that observed in age matched SOD1G93A/mIgf-1 mice; whereas the increase in mRNA expression in SOD1G93A mice was of 32 ± 1.9% when SOD1G93A and SOD1G93A/mIgf-1 mice where analyzed at comparable end-stage disease. This suggests that mIgf-1 delays the progression of the disease, stabilizing the innervation of muscle fibers.
Figure 4.
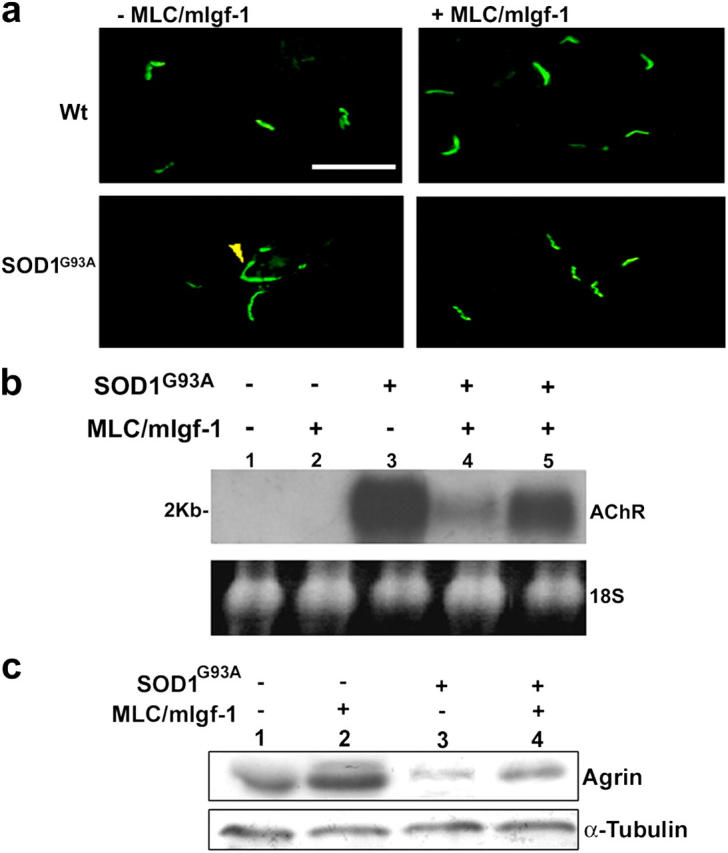
Maintenance of the neuromuscular junction configuration in SOD1G93A/mIgf-1 transgenic mice. (a) Immunofluorescent analysis of transverse sections from muscles of wt, MLC/mIgf-1, SOD1G93A and SOD1G93A/mIgf-1 transgenic mice at 123 d old. α-bungarotoxin antibody identified diffusion of AChR expression in SOD1G93A muscle (yellow arrow); whereas SODG93A/mIgf-1 muscle maintained AChR clusters. Bar, 20 μm. (b) Northern blot of total RNA samples from quadriceps of wt (lane 1), MLC/mIgf-1 (lane 2), SOD1G93A (lane 3) and SOD1G93A/mIgf-1 (lanes 4 and 5) transgenic muscles at 123 (lanes 1–4) and 150 (lane 5) days old, hybridized with AChR 32P-labeled probe. Ethidium bromide staining was used to verify equal loading of the RNA sample. (c) Western blot of agrin from quadriceps of wt (lane 1), MLC/mIgf-1 (lane 2), SOD1G93A (lane 3) and SOD1G93A/mIgf-1 (lane 4) transgenic muscles. SOD1G93A and SOD1G93A/mIgf-1 mice were analyzed at comparable end-stage disease.
The localization of AChR clusters at the end plate requires the expression of agrin, a large proteoglycan in the synaptic cleft that plays an important role in the maintenance of the molecular architecture of the postsynaptic membrane (McConville and Vincent, 2002). Agrin expression was significantly down-regulated in paralyzed SOD1G93A compared with SODG93A/mIgf-1 muscle (Fig. 4 c) analyzed at comparable end-stage disease, further underscoring a role for local expression of mIgf-1 in the maintenance of muscle innervation.
Muscle-restricted mIgf-1 prolongs motor neuronal function in SOD1G93A mice
Histological analysis of the ventral spinal cord revealed that SOD1G93A mice (n = 7) presented a progressive reduction in the number of motor neuron from clinical onset to end-stage disease. Specifically, SOD1G93A mice showed a reduction of 37 and 55% in the number of motor neurons at clinical onset (112 d) and at end-stage disease (123 d), respectively (Fig. 5 a). In contrast, mIgf-1 expression induced motor neuron survival in SOD1G93A/mIgf-1 mice (n = 7) at all ages and stages observed, with significant differences at 112 and 123 d old (Fig. 5 a), demonstrating that mIgf-1 expression preserves the motor neuron during the progression of ALS.
Figure 5.
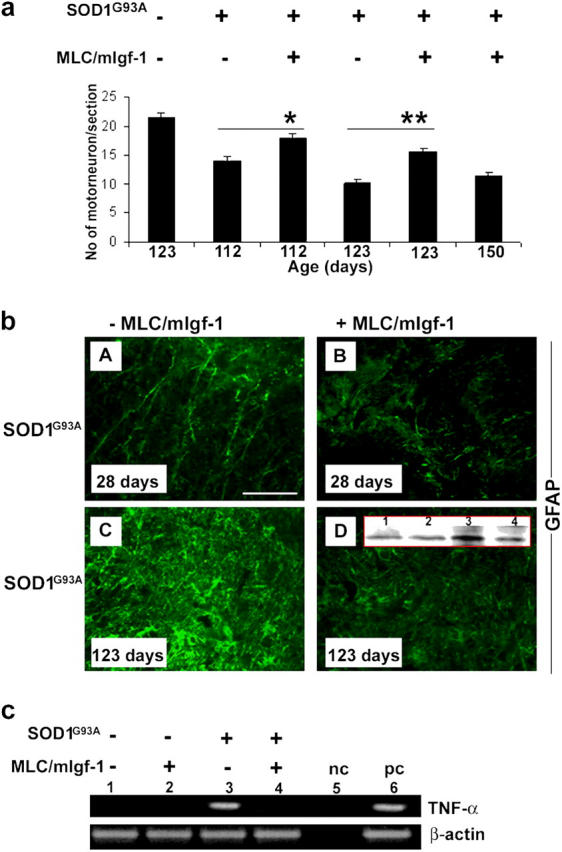
Transgenic mIgf-1 expression protects motor neuron from degeneration. (a) Quantification of surviving motor neurons in the ventral spinal cord of wild-type, SOD1G93A and SOD1G93A/mIgf-1 mice at different ages. *P < 0.001; **P < 0.001. (b) Immunofluorescence analysis identify GFAP positive astrocytes in ventral horn of SOD1G93A and SOD1G93A/mIgf-1 mice at different ages: A and B, 28 d old; C and D, 123 d old. The intensity of the GFAP signal revealed progressive astrocytosis. Bar, 20 μm. Insert in D shows Western blot for GFAP in the spinal cord of SOD1G93A (lanes 1 and 3) and SOD1G93A/mIgf-1 (lanes 2 and 4) mice at 28 (lanes 1 and 2) and 123 (lanes 3 and 4) d old. (c) RT-PCR analysis of TNF-α and β-actin of wt (lane 1), MLC/mIgf-1 (lane 2), SOD1G93A (lane 3) and SOD1G93A/mIgf-1 (lane 4) transgenic mice at 123 d old. Lane 5 shows a negative control (nc) consisting of RT-PCR mix without cDNA template. Lane 6 identifies the RNA positive control (pc) for TNF-α obtained from spleen.
One of the prominent markers of motor neuron dysfunction in ALS mice is the activation of astrocytes, leading to motor weakness and neural loss (Hall et al., 1998). Comparable patterns of glial fibrillary acidic protein (GFAP) immunoreactivity were found in spinal cords of SOD1G93A (n = 6) and SODG93A/mIgf-1 (n = 6) transgenic mice before the symptom onset (28 d; Fig. 5 b, A, B, and D, insert, lanes 1 and 2). However, at paralysis stage (123 d) the spinal cord of SOD1G93A mice demonstrated a marked increase in astroglial activation (Fig. 5 b C), with an increase in GFAP expression of ∼55 ± 0.2% (Fig. 5 b, D, insert, lane 3) compared with the GFAP expression levels displayed in the spinal cord of age matched SOD1G93A/mIgf-1 transgenic mice (Fig. 5 b, D and insert, lane 4). At comparable end-stage disease, there were no significant differences in GFAP expression between SOD1G93A and SOD1G93A/mIgf-1 mice, although SOD1G93A mice continued to express 13% more GFAP as compared with SOD1G93A/mIgf-1 mice (unpublished data).
The activation of astroglia can be correlated with the expression of certain cytokines, such as TNF-α, which enhance the response to inflammatory states and contribute to the progression of neurological dysfunction in SOD1G93A mice (Elliott, 2001). Although TNF-α expression was normally undetectable in the CNS of healthy mice (Fig. 5 c, lanes 1 and 2), it accumulated in the spinal cord of SOD1G93A mice at paralysis stage (123 d; Fig. 5 c, lane 3). In contrast, TNF-α expression was not apparent in the spinal cord of SOD1G93A/mIgf-1 transgenic mice (Fig. 5 c, lane 4). mIgf-1 hypertrophic muscle therefore functions as a protective tissue for the CNS, modulating reactive astrocytosis and inflammatory cytokines that normally exacerbate the pathogenesis of ALS disease.
ALS is emerging as a “multi-systemic” disease in which the alteration in structural, physiological, and metabolic parameters in different cell types (muscle, motor neuron, glia) may act synergistically to exacerbate the disease and evidences a functional cross-talk between neuronal and not neuronal cells (Rothstein et al., 1996; Ferri et al., 2004). The present study serves to refocus therapeutic strategies to attenuate motor neuronal degradation toward skeletal muscle. It remains to be determined whether the dramatic prolongation of CNS tissue integrity in SOD1G93A/mIgf-1 mice derives from the direct retrograde transport of transgenic mIgf-1, or indirect action either through distal activation of endogenous Igf-1 expression, or through other trophic factors secreted by SODG93A/mIgf-1 muscle.
In a previous study, retrograde transport of the AAV–Igf-1 vector from muscle to motor neuron was deemed necessary to achieve the therapeutic effects, because the same gene delivered to the muscle by a lentiviral vector, which cannot retrotransport, proved ineffective.
The results of the present study suggest other explanations. The Igf-1 cDNA used by Kaspar et al. (2003) may have encoded the more prevalent circulating gene product, which once secreted from the muscle disperses in the circulation (Musarò and Rosenthal, 2002). In contrast, the mIgf-1 isoform used in our present and previous studies (Barton-Davis et al., 1998; Musarò et al., 2001; Barton et al., 2002; Musarò et al., 2004) does not enter the circulation, but accumulates in the tissue of synthesis where its autocrine/paracrine function is concentrated. The importance of Igf-1 isoform choice in designing therapeutic strategies cannot be overstressed, because their diverse biological activities lead to radically different outcomes (for review see Musarò and Rosenthal, 2002). Whatever its mode of action, the feasibility of localized synthesis of the mIgf-1 isoform to activate survival mechanisms in distal damaged tissues represents a powerful approach to counteract the degeneration of both muscle and motor neuron in ALS disease.
Materials and methods
Mice
SODG93A transgenic mice (Jackson Laboratory) express a transgenic human mutant SOD1G93A allele containing the Gly93 →Ala (G93A) substitution, driven by its endogenous human promoter (Gurney et al., 1994). The SOD1G93A B6J mice were crossed with MLC/mIgf-1 FVB mice (Musarò et al., 2001) for seven different generations to obtain SOD1G93A/mIgf-1 B6J inbred transgenic mice.
Walk test
The walk test was performed in a scaled ramp, accordingly to the method reported by Gurney et al. (1994). The mice were allowed to explore the cage for 1 min and then they were left to walk for 2 min. The hind feet of the mice were painted with ink and the track left by the mice were recorded on a paper tape. The test was performed in horizontal, laminar flow hood to maintain barrier conditions.
Histological and immunofluorescence analysis
For histological analysis, 7-μm tissue cryosections were fixed in 4% PFA and stained with hematoxylin/eosin. For immunofluorescence analysis, sections were fixed with 4% PFA, washed in PBS with 1% BSA and 0.2% Triton X-100, preincubated for 1 h in 10% goat serum at RT, and incubated overnight at 4°C with primary antibodies: neonatal MyHC (neo-MyHC), MyHC-fast, Alexa Fluor 488–conjugated α-bungarotoxin, CnA-β1 (from C. Klee, National Institutes of Health, Bethesda, MD), GFAP. Nuclei were visualized by Hoechst staining.
For motor neuron quantification, fixed spinal cord slices were immunostained with SMI-32 monoclonal antibody
Inverted microscope (Axioskop 2 plus; Carl Zeiss MicroImaging, Inc.) using 20 or 40× lenses was used, and images were processed using Axiovision 3.1.
RNA preparation and Northern analysis
RNA was extracted from muscles by RNA-TRIzol–kit (GIBCO BRL). Total RNA was separated in 1.3% agarose gel and hybridized as described previously (Musarò and Rosenthal, 1999).
RT-PCR analysis
RNA was isolated from spinal cord of wild-type, MLC/mIgf-1, and SODG93A and SOD1G93A/mIgf-1 transgenic mice. The following oligonucleotides were used: TNF-α sense, 5′-CCCAGACCCTCACACACTCAGAT-3′ and anti-sense, 5′-TTGTCCCTTGAAGAGAACCTG-3′; β-actin sense, 5′-GTGGGCCGCTCTAGGCACAA-3′ and anti-sense, 5′-CTCTTTGATGTCACGCACGATTTC-3′.
Protein extraction and Western blot analysis
Protein extraction was performed in lysis buffer (50 mM Tris-HCl, pH 7.4, 1% wt/vol Triton X-100, 0.25% sodium deoxycholate, 150 mM sodium chloride, 1 mM PMSF, 1 μg/ml of each aprotinin, leupeptin, pepstatin, 1 mM sodium orthovanadate, 1 mM sodium fluoride). Equal amounts of protein from each muscle lysate were separated in SDS polyacrylamide gel and transferred onto a hybond C extra nitrocellulose membrane. Filters were blotted with antibodies against hSOD, Pax7, myogenin, desmin, neo-MyHC (from S. Schiaffino, University of Padova, Padova, Italy), Agrin, GFAP.
Online supplemental material
Organization of the Igf-1 gene
As its name implies, Igf-1 is similar to insulin in structure. The mature Igf-1 is a single-chain protein of 70 aa and differs from insulin by retention of the C-domain, by a short extension of the A-domain to include a novel domain D, and by the presence of variable COOH-terminal extension peptides (E-peptides; Fig. S1). The rodent Igf-1 gene contains six exons, separated by five introns (Fig. S1). Exons 1 and 2 encode distinct 5′UTRs, as well as different parts of the signal peptide, and are therefore termed leader exons. Separate promoters drive exons 1 and 2 transcription. Exon 3 encodes 27 aa that are part of the signal peptide and common to all isoforms, as well as part of the mature Igf-1 peptide. Exon 4 encodes the rest of the mature peptide and 16 aa of the NH2-terminal region of the E-peptide, which is also common to all Igf-1 mRNAs. Exons 5 and 6 encode two distinct COOH-terminal E-peptides (Ea, Eb) and the 3′UTR. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200407021/DC1.
Acknowledgments
We thank F. Palombi, E. Paolucci, B.M. Scicchitano, and S. De Grossi for help at various steps of the work. We thank S. Orlando for statistical analysis.
This work was supported by grants to A. Musarò from Telethon-Italy (grant GP0098Y01), Min. Salute; to M. Molinaro from Ministero dell'Università e Ricerca Scientifica (MIUR), and to N. Rosenthal from the Muscular Dystrophy Association.
The authors do not have financial interest in relation to this submission.
Abbreviations used in this paper: AChR, acetylcholine receptor; ALS, amyotrophic lateral sclerosis; CnA, calcineurin; GFAP, glial fibrillary acidic protein; Igf, insulin-like growth factor; mIgf-1, local isoform of Igf-1; MyHC, myosin heavy chain; SOD1, superoxide dismutase1; wt, wild-type.
References
- Barton, E.R., L. Morris, A. Musarò, N. Rosenthal, and H.L. Sweeney. 2002. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J. Cell Biol. 157:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton-Davis, E.R., D.I. Shoturma, A. Musarò, N. Rosenthal, and H.L. Sweeney. 1998. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc. Natl. Acad. Sci. USA. 95:15603–15607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton-Davis, E.R., D.I. Shoturma, and H.L. Sweeney. 1999. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta. Physiol. Scand. 167:301–305. [DOI] [PubMed] [Google Scholar]
- Chin, E.R., E.N. Olson, J.A. Richardson, Q. Yang, C. Humphries, J.M. Shelton, H. Wu, W. Zhu, R. Bassel-Duby, and R.S. Williams. 1998. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 12:2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement, A.M., M.D. Nguyen, E.A. Roberts, M.L. Garcia, S. Boillee, M. Rule, A.P. McMahon, W. Doucette, D. Siwek, R.J. Ferrante, et al. 2003. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 302:113–117. [DOI] [PubMed] [Google Scholar]
- Elliott, J.L. 2001. Cytokine upregulation in a murine model of familial amyotrophic lateral sclerosis. Brain Res. Mol. Brain Res. 95:172–178. [DOI] [PubMed] [Google Scholar]
- Ferri, A., M. Nencini, A. Casciati, M. Cozzolino, D.F. Angelini, P. Longone, A. Spalloni, G. Rotilio, and M.T. Carri. 2004. Cell death in amyotrophic lateral sclerosis: interplay between neuronal and glial cells. FASEB J. 18:1261–1263. [DOI] [PubMed] [Google Scholar]
- Funakoshi, H., N. Belluardo, E. Arenas, Y. Yamamoto, A. Casabona, H. Persson, and C.F. Ibanez. 1995. Muscle-derived neurotrophin-4 as an activity-dependent trophic signal for adult motor neurons. Science. 268:1495–1499. [DOI] [PubMed] [Google Scholar]
- Gurney, M.E., H. Pu, A.Y. Chiu, M.C. Dal Canto, C.Y. Polchow, D.D. Alexander, J. Caliendo, A. Hentati, Y.W. Kwon, H.X. Deng, et al. 1994. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 264:1772–1775. [DOI] [PubMed] [Google Scholar]
- Hall, E.D., J.A. Oostveen, and M.E. Gurney. 1998. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 23:249–256. [DOI] [PubMed] [Google Scholar]
- Kaspar, B.K., J. Llado, N. Sherkat, J.D. Rothstein, and F.H. Gage. 2003. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 301:839–842. [DOI] [PubMed] [Google Scholar]
- Lino, M.M., C. Schneider, and P. Caroni. 2002. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 22:4825–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConville, J., and A. Vincent. 2002. Diseases of the neuromuscular junction. Curr. Opin. Pharmacol. 2:296–301. [DOI] [PubMed] [Google Scholar]
- Musarò, A., and N. Rosenthal. 1999. Maturation of the myogenic program is induced by postmitotic expression of insulin-like growth factor I. Mol. Cell. Biol. 19:3115–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musarò, A., and N. Rosenthal. 2002. The role of local insulin-like growth factor-1 isoforms in the pathophysiology of skeletal muscle. Curr. Genomics. 3:149–162. [Google Scholar]
- Musarò, A., K.J. McCullagh, F.J. Naya, E.N. Olson, and N. Rosenthal. 1999. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 400:581–585. [DOI] [PubMed] [Google Scholar]
- Musarò, A., K. McCullagh, A. Paul, L. Houghton, G. Dobrowolny, M. Molinaro, E.R. Barton, H.L. Sweeney, and N. Rosenthal. 2001. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 27:195–200. [DOI] [PubMed] [Google Scholar]
- Musarò, A., C. Giacinti, G. Borsellino, G. Dobrowolny, L. Pelosi, L. Cairns, S. Ottolenghi, G. Bernardi, G. Cossu, L. Battistini, et al. 2004. Muscle restricted expression of mIGF-1 enhances the recruitment of stem cells during muscle regeneration. Proc. Natl. Acad. Sci. USA. 101:1206–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein, J.D., M. Dykes-Hoberg, C.A. Pardo, L.A. Bristol, L. Jin, R.W. Kuncl, Y. Kanai, M.A. Hediger, Y. Wang, J.P. Schielke, and D.F. Welty. 1996. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 16:675–686. [DOI] [PubMed] [Google Scholar]
- Sakuma, K., J. Nishikawa, R. Nakao, K. Watanabe, T. Totsuka, H. Nakano, M. Sano, and M. Yasuhara. 2003. Calcineurin is a potent regulator for skeletal muscle regeneration by association with NFATc1 and GATA-2. Acta Neuropathol. (Berl.). 105:271–280. [DOI] [PubMed] [Google Scholar]