

Abstract
Although Rho regulates cytokinesis, little was known about the functions in mitosis of Cdc42 and Rac. We recently suggested that Cdc42 works in metaphase by regulating bi-orient attachment of spindle microtubules to kinetochores. We now confirm the role of Cdc42 by RNA interference and identify the mechanisms for activation and down-regulation of Cdc42. Using a pull-down assay, we found that the level of GTP-Cdc42 elevates in metaphase, whereas the level of GTP-Rac does not change significantly in mitosis. Overexpression of dominant-negative mutants of Ect2 and MgcRacGAP, a Rho GTPase guanine nucleotide exchange factor and GTPase activating protein, respectively, or depletion of Ect2 by RNA interference suppresses this change of GTP-Cdc42 in mitosis. Depletion of Ect2 also impairs microtubule attachment to kinetochores and causes prometaphase delay and abnormal chromosomal segregation, as does depletion of Cdc42 or expression of the Ect2 and MgcRacGAP mutants. These results suggest that Ect2 and MgcRacGAP regulate the activation and function of Cdc42 in mitosis.
Introduction
During mitosis cells segregate their duplicated DNA equally to each pole and divide into two daughter cells. The cytoskeleton exerts different mechanical forces and governs this important biological process. For example, at the onset of mitosis, the instability of microtubules (MTs) increases. In prometaphase to metaphase, MTs emanating from centrosomes bind to the kinetochores of each sister chromatid. Once stabilized, the bipolar attachment of MTs to chromosomes ensures that chromosomes congress correctly at the metaphase plate. Sister chromatids are then segregated to each pole in anaphase by the combined actions of MT shrinkage and MT-associated motor proteins (Wittmann et al., 2001). In anaphase to telophase, F-actin and myosin accumulate in the cell cortex in the middle of the dividing cell to form the contractile ring, which contracts and cleaves the cell into two (Wang, 2001). Although the overall progression through mitosis is regulated by cyclin and cyclin-dependent kinase signaling, much remains to be learned about the signaling mechanism that determines the aforementioned spatio-temporal actions of the cytoskeleton.
Rho GTPases control a variety of cellular processes such as cell adhesion, cell motility, and cell contraction through induction of specific types of actin cytoskeleton and by local regulation of MT dynamics (Etienne-Manneville and Hall, 2002). Although the role of Rho in induction and maintenance of the contractile ring during cytokinesis has been well documented (Kishi et al., 1993; Mabuchi et al., 1993), the role for other Rho GTPases in the regulation of mitotic events was, until recently, unknown. Clostridium difficile toxin B is known to inactivate all members of Rho GTPases by selective glucosylation of the critical threonine residue (Aktories et al., 2000). We have recently found that treatment of HeLa cells with toxin B delays the progression of mitosis in prometaphase and that expression of Cdc42 mutants but not those of Rac1 or RhoA mimics this phenotype (Yasuda et al., 2004). In these cells, spindle MTs fail to bind to chromosomes in the correct bipolar manner, and chromosomes fail to congress at the metaphase plate. These findings suggest that Cdc42 plays an important role in regulating the attachment of spindle MTs to the kinetochore.
Here, we extend this study to confirm the role of Cdc42 in mitosis by RNA interference (RNAi) and to further identify the regulatory mechanisms for activation and down-regulation of this Rho GTPase during cell division. Like other small GTPases, Rho GTPases cycle between the GDP-bound inactive state and the GTP-bound active state. The exchange of bound GDP for GTP is catalyzed by guanine nucleotide exchange factors (GEFs), whereas the hydrolysis of GTP by the GTPases is accelerated by GTPase activating proteins (GAPs; Zheng, 2001; Moon and Zheng, 2003). Previous studies showed that activation of Rho in mitosis is regulated by the action of a Rho exchanger, Ect2, and by the action of a Rho GAP, MgcRacGAP.
Ect2 is a GEF for Rho GTPases, containing the hallmark Dbl homology (DH) domain and pleckstrin homology (PH) domain tandem motif (Miki et al., 1993). Prokopenko et al. (1999) showed in Drosophila melanogaster that Pebble, the Ect2 orthologue, interacts genetically with RhoA and is required for the formation of the contractile ring and initiation of cytokinesis. Tatsumoto et al. (1999) found in mammalian cells that the inhibition of Ect2 function, either by overexpression of deletion mutants lacking the DH and PH domains or by injection of anti-Ect2 antibodies, generated multinucleated cells, indicating that Ect2 is involved in cytokinesis. They further reported that Ect2, which is localized exclusively in the nucleus of interphase cells, is released into the cytoplasm upon nuclear membrane breakdown, is localized on the spindles in metaphase, and accumulates in the cleavage furrow in telophase and then in the midbody at the end of cytokinesis. Using the pull-down assay, we previously verified the importance of Ect2 in mitotic activation of RhoA in HeLa cells (Kimura et al., 2000). We showed that GTP-RhoA accumulates in telophase, and that this accumulation can be abolished by expression of dominant-negative forms of Ect2 in synchronized HeLa cells. Although these studies have established a role for Ect2 in Rho activation and cytokinesis, little is known about its action on other Rho GTPases in mitosis. Ect2 catalyzes in vitro GDP-GTP exchange for Rac1 and Cdc42 as potently as for Rho (Tatsumoto et al., 1999).
MgcRacGAP is a Rho GAP conserved from Caenorhabditis elegans and Drosophila to mammals (Agnel et al., 1992; Toure et al., 1998; Jantsch-Plunger et al., 2000; Kawashima et al., 2000). Hirose et al. (2001) expressed a catalytically inactive mutant of MgcRacGAP, MgcRacGAPR386A, and found production of multinucleate cells by this expression and suggested that this GAP is required for cytokinesis. MgcRacGAP exhibits a similar spatio-temporal localization to that of Ect2: it is present in the nucleus of interphase cells, localizes to the spindle in metaphase and anaphase, and accumulates in the midbody at the end of cytokinesis (Hirose et al., 2001). Jantsch-Plunger et al. (2000) found that a mutation in cyk-4, a MgcRacGAP orthologue in C. elegans, caused disassembly of the central spindle and failure of cytokinesis. Together these results suggest that MgcRacGAP/CYK-4 acts on RhoA and regulates cytokinesis. However, CYK-4 stimulates GTP hydrolysis of not only RhoA but also Rac1 and Cdc42 in vitro. Furthermore, in vitro MgcRacGAP exhibits greater activity toward Rac1 and Cdc42 than toward RhoA (Kawashima et al., 2000). These results raise the possibility that this molecule may also function to regulate Cdc42 and Rac1 in mitosis.
In the present work, we have adopted a variety of strategies to address the aforementioned questions. We have first synchronized HeLa cells and performed pull-down assays for the GTP-bound form of Cdc42 and Rac-1 to identify mitosis-associated changes in the activation of these two GTPases. We have examined the effects of the expression of the dominant-negative forms of Ect2 and MgcRacGAP, or RNAi of Ect2, on the level of GTP-bound Cdc42 and Rac1 during mitosis. We have also investigated mitotic phenotypes of cells either subjected to RNAi for Cdc42 and Ect2, or overexpressing the aforementioned Ect2 and MgcRacGAP mutants. Our results indicate that Ect2 and MgcRacGAP regulate the activation and function of Cdc42 in mitosis.
Results
Changes in the levels of GTP-bound Cdc42 and Rac during mitosis
We used thymidine and nocodazole either alone or in combination to enrich HeLa S3 cells in G2 phase, prometaphase, metaphase, telophase, and G1 phase, as described in Materials and methods. To clarify the activation of Cdc42 and Rac in cell division, we used a GST fusion of the Cdc42-Rac–interacting binding domain (CRIB) of Pak1 (GST-CRIB-Pak; Matsuo et al., 2002) to pull-down the GTP-bound forms of Rac1 or Cdc42 from the lysates of these cells. We then subjected fractions of the precipitates to immunoblot analysis for Cdc42 and Rac, using antibodies specific to each GTPase. The amounts of the GTP-bound form of each GTPase were then quantified. GTP-bound Cdc42 accumulated to some extent during G2 phase. After nocodazole removal, the level of GTP-Cdc42 decreased in prometaphase, elevated transiently in metaphase, reaching a level similar to that found for G2/G1 phases, and then decreased again in telophase (Fig. 1 A). However, the level of GTP-bound Rac1 did not change significantly during mitosis (Fig. 1 A). As a reference, we also monitored the change in GTP-Rho, which, in agreement with previous findings (Kimura et al., 2000), peaked in telophase (Fig. 1 A).
Figure 1.
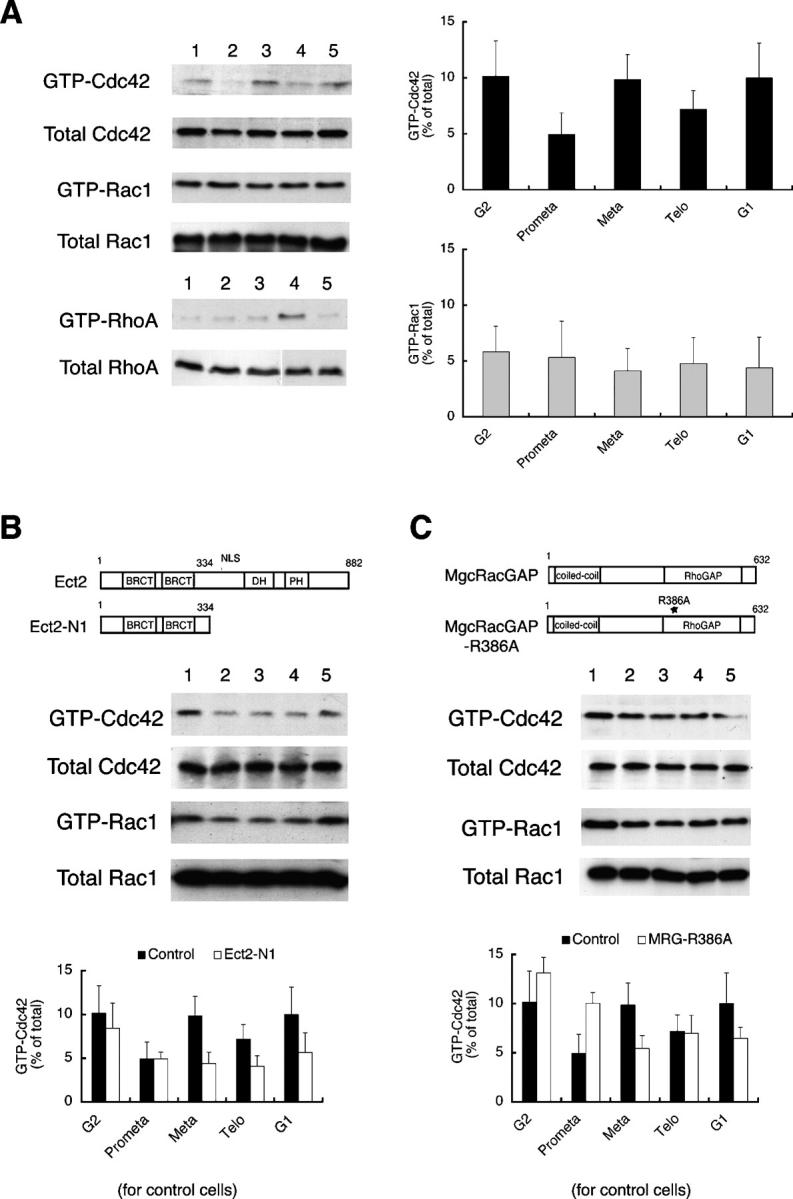
Regulation of Cdc42 activation in mitosis by Ect2 and MgcRacGAP. (A) Cell cycle–associated changes in the level of the GTP-bound form of each Rho GTPase. HeLa S3 cells were synchronized, collected in G2 phase (lane 1), prometaphase (lane 2), metaphase (lane 3), telophase (lane 4), and G1 phase (lane 5), respectively, and subjected to the pull-down assay to detect GTP-Cdc42 or GTP-Rac, as described in Materials and methods. Typical immunoblots are shown on the left, and the results of the quantitative analysis are on the right. Values are shown as means ± SEM (n = 4 and 3 for Cdc42 and Rac, respectively). (B) Suppression of Cdc42 activation in metaphase by expression of a dominant-negative mutant of Ect2. Synchronized HeLa cells were transfected with pCEV32F-Ect2-N1, collected at various phases of the cell cycle, and subjected to the pull-down assay as described in Materials and methods. The top diagram shows the domain structures of full-length Ect2 and Ect2-N1 (BRCT, BRCA carboxyl terminus domain; DH, Dbl homology domain; PH, pleckstrin homology domain; NLS, nuclear localization signal). The middle panel is representative of a typical experiment, and the bottom panel represents the results of quantitative analysis of three independent experiments (open bars, experiments with Ect2-N1; closed bars, the control experiment shown in A). (C) Prevention of down-regulation of Cdc42 by expression of a catalytically inactive mutant of MgcRacGAP. Synchronized HeLa cells were transfected with pME18S-MgcRacGAPR386A, collected at various phases of the cell cycle, and subjected to the pull-down assay as described in Materials and methods. Top diagrams show the structure of MgcRacGAP and MgcRacGAPR386A (coiled-coil, coiled-coil domain; RhoGAP, GTPase activating homology domain). The middle panel is representative of a typical experiment, and the bottom panel represents the results of quantitative analysis of three independent experiments (open bars, experiments with MgcRacGAPR386A; closed bars, the control experiment shown in A).
Ect2 and MgcRacGAP regulate activation of Cdc42 during mitosis
The NH2-terminal fragment of Ect2 lacking the DH-PH domain (Ect2-N1) functions as a dominant-negative mutant and suppresses Rho activation in telophase (Tatsumoto et al., 1999; Kimura et al., 2000). To test if Ect2 is involved in the activation of Cdc42 in metaphase, we overexpressed this Ect2 mutant in mitotic HeLa cells and performed pull-down assays with lysates from these cells. Expression of Ect2-N1 suppressed the accumulation of GTP-bound Cdc42 in metaphase to the level found in prometaphase. As a consequence, the level of GTP-Cdc42 remained low from prometaphase to telophase in the Ect2-N1–overexpressing cells (Fig. 1 B). However, Ect2-N1 expression had no effect on the level of GTP-bound Rac1 (Fig. 1, A and B).
Next, we investigated the involvement of MgcRacGAP in the regulation of Cdc42 activation in mitosis. To this end, we used MgcRacGAPR386A, a mutant that is devoid of GAP activity and that is known to produce multinucleate cells when overexpressed (Hirose et al., 2001). We transfected thymidine-synchronized HeLa cells with this GAP mutant and overexpressed the mutant protein in mitotic cells. The lysates of the transfected cells were then subjected to the pull-down assay. Overexpression of MgcRacGAPR386A elevated the amount of GTP-Cdc42 in prometaphase to a level comparable to that found in metaphase in control cells (Fig. 1 C), resulting in premature accumulation of GTP-Cdc42. Curiously, MgcRacGAPR386A expression tended to suppress the Cdc42 activation thereafter (see Discussion). In contrast, the MgcRacGAPR386A expression had little effect on the activation of Rac1 (Fig. 1 C).
We confirmed the aforementioned findings by depletion of endogenous Ect2 with RNAi. As shown in Fig. 2 A, the treatment of HeLa cells with RNAi for Ect2 potently suppressed the expression of the endogenous protein to <20% of that found in control cells after 48 h. We then examined the accumulation of GTP-Cdc42 in cells subjected to RNAi. Although cells subjected to control RNAi exhibited an accumulation of GTP-Cdc42 in metaphase, no accumulation was found in cells subjected to Ect2 RNAi (Fig. 2 B). We also wished to verify the role of MgcRacGAP by RNAi, but we were unable to obtain a significant decrease of this protein by the RNAi strategy we used for Ect2. These results suggest that Ect2 and MgcRacGAP regulate the level of GTP-Cdc42 in mitosis, the former activating Cdc42 in metaphase and the latter suppressing Cdc42 activation in prometaphase. Interestingly, neither Ect2 nor MgcRacGAP appears to regulate Rac activation in mitosis, although they show catalytic activity toward Rac in vitro.
Figure 2.

RNAi for Ect2 and its effects on accumulation of GTP-Cdc42 in metaphase. (A) Depletion of Cdc42 or Ect2 by RNAi. HeLa cells were subjected to RNAi for Ect2 (Ect2 RNAi), Cdc42 (Cdc42 RNAi), or E. coli LacZ (control RNAi). After 24 and 48 h, the cells were lysed and subjected to immunoblot with the respective antibody. (B) Effects of Ect2 depletion on accumulation of GTP-Cdc42 in metaphase. HeLa S3 cells were transfected with control or Ect2 siRNA after the first thymidine block. The cells then underwent a second thymidine block and, after release from this block, were collected at different phases of the cell cycle and subjected to the pull-down assay. One sixth of the precipitates and one twentieth of the supernatant were used to show the amount of GTP-Cdc42 and total Cdc42, respectively. The top panel represents typical immunoblots and the bottom represents the results of quantitative analysis of three independent experiments (closed bars, experiments with control RNAi; open bars, Ect2 RNAi experiment). Values are shown as means ± SEM (n = 3).
RNAi for Cdc42 or Ect2 causes a delay in metaphase progression
We have recently suggested that Cdc42 regulates the bi-oriented attachment of spindle MTs to kinetochores during metaphase (Yasuda et al., 2004). This suggestion was based on experimental findings using toxin B and Cdc42 mutants. Here, we confirmed these findings by depletion of Cdc42 with RNAi, and also by depletion of Ect2. As with RNAi for Ect2, RNAi for Cdc42 in HeLa cells effectively suppressed expression of the endogenous protein to <10% of that found in control cells (Fig. 2 A). Therefore, we transfected HeLa cells with the respective RNAi and cultured the cells for 50 h, during which time they were subjected to a double thymidine block to synchronize the cell cycle progression. Cells were collected at different time points after release from the second thymidine block and then subjected to flow-cytometry analysis. We found that whereas cells subjected to control RNAi or RNAi for Cdc42 or Ect2 progressed through S phase at comparable rates, exit from mitosis was consistently delayed in the cells subjected to Cdc42 or Ect2 RNAi when compared with control RNAi cells; percentages of cells with 4N DNA content 11 h after thymidine release were 33, 32, and 33 for control, Cdc42, or Ect2 RNAi cells, respectively; by 12 h, percentages were 26, 32, and 33; and by 15 h, they were 18, 24, and 26. The cells were then fixed 12 h after release, and the mitotic index of each population was determined by staining for MTs and chromosomes (Fig. 3 A). The mitotic index of control RNAi cells was <9%, whereas the indexes for Cdc42 and Ect2 RNAi cells were significantly higher and were 22 and 19%, respectively. Interestingly, combined RNAi for Cdc42 and Ect2 together increased the mitotic index (17.5%) to nearly the values observed with RNAi for either alone. This lack of synergy is consistent with our hypothesis that Ect2 and Cdc42 work in the same pathway. Immunofluorescence analysis of mitotic cells in these populations revealed that although the majority of cells had formed a bipolar spindle, chromosomal congression was often impaired; in some cells, chromosomes were widely scattered and in others some chromosomes remained around either pole (Fig. 3 B). Quantitative analysis revealed that the proportion of cells that showed such misaligned chromosomes was significantly higher in the Cdc42- or Ect2-depleted cell population when compared with the control cell population (Fig. 3 C). Consequently, the proportion of cells in metaphase was significantly lower in the Cdc42- or Ect2-depleted cell populations. Staining of these cells both for CENP-A, a kinetochore protein, and Mad2 revealed the presence of Mad2 at a significant number of kinetochores (Fig. 3 D). Mad2 associates with the kinetochore in the absence of spindle MT binding where it functions to arrest cell cycle progression, thus ensuring that the cell progresses to anaphase only when all kinetochores have attached with spindle MTs (Cleveland et al., 2003). These results indicate the loss of MT attachment to kinetochores in cells where either Cdc42 or Ect2 or both of these proteins have been depleted. Interestingly, however, significant proportions of the cells subjected to RNAi for Ect2 or Cdc42 nonetheless progressed to anaphase and subsequently to telophase. Consistently, most of these cells progressed to G1, resulting in cells with micro- and macronuclei (Fig. 3, C and E). These results suggest that the spindle checkpoint mechanism was impaired in these cell populations. Our previous work showed that Cdc42 acts on its effector, mDia3, which localizes to kinetochores in mitosis, and that toxin B treatment disrupts this localization (Yasuda et al., 2004). We wondered if depletion of Ect2 or Cdc42 has similar effects on mDia3 localization. We found that, although the mDia3 signals were seen as small punctate spots next to those of CENP-A in control RNAi cells, these signals became diffuse and blurred upon Ect2/Cdc42 depletion (Fig. 3 F).
Figure 3.

Prometaphase delay by RNAi for Cdc42 or Ect2 in HeLa cells. (A) Mitotic index. Cells subjected to RNAi were synchronized in S phase by double thymidine block, and were fixed at 12 h after release from the second block. The RNAi cells were identified by coexpression of pEGFP-histone H2Bk. The cells were stained for MTs and chromosomes and the mitotic index was calculated by examining more than 250 cells in each of four independent experiments. Results are the mean ± SEM (n = 4). *, P < 0.05 versus control RNAi cells. (B) Immunofluorescence. Cells treated as in A and expressing EGFP-H2Bk (green) were stained for chromosomes (DAPI, blue) and β-tubulin (red). Arrowheads indicate misaligned chromosomes. (C) Prometaphase delay. Mitotic cells in each population were examined and the percentages of cells in prometaphase (cells with misaligned chromosomes), metaphase (cells with chromosomes aligned at the metaphase plate), or ana/telophase (cells showing chromosome segregation) were determined. Results are mean ± SEM (n = 3). (D) Mad2 staining. Cells treated as in A were subjected to staining for Mad2 (green), CENP-A (red), and chromosomes (DAPI, blue). Insets are enlargements of the indicated regions of interest. (E) Production of aberrant multinucleate cells. Cells subjected to respective RNAi were fixed at 14 h after the release and stained for β-tubulin (red) and chromosomes (DAPI, blue and right column). Arrowheads show micronuclei. (F) mDia3 localization. HeLa cells subjected to control RNAi or RNAi for Ect2 or Cdc42 were subjected to staining for mDia3 (green), CENP-A (red), and with DAPI (blue). Bars: (B, E, and F) 10 μm; (D) 2 μm.
To gain a greater insight into this process, we used video microscopy to monitor the mitotic progression of cells subjected to Ect2 or Cdc42 depletion. Whereas the control cells progressed from prometaphase to telophase within 50 min (Fig. 4 A and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200408085/DC1), chromosomes in cells subjected to Ect2 RNAi took longer to assemble at the metaphase plate, and this assembly was never quite complete. In many cells, some chromosomes remained adjacent to either pole, whereas the majority appeared to assemble at the metaphase plate (Fig. 4 B and Video 2, available at http://www.jcb.org/cgi/content/full/jcb.200408085/DC1). A similar phenotype was observed in cells subjected to RNAi for Cdc42 alone (Fig. 4 C and Video 3, available at http://www.jcb.org/cgi/content/full/jcb.200408085/DC1) or both Ect2 and Cdc42 together (not depicted). In addition, depletion of Ect2 resulted in failure of cytokinesis in 40% of the cells observed (n = 19), thus confirming the role of Ect2 in the regulation of cytokinesis. Together, these results confirm our previous suggestion that Cdc42 regulates the bipolar attachment of spindle MTs to kinetochores before chromosome congression in metaphase and further suggest that Ect2 regulates the activation of Cdc42 during this process.
Figure 4.

Ect2 and Cdc42 depletion induce abnormalities in chromosome alignment and segregation in HeLa cells. HeLa cells were cotransfected with pEGFP-EB1 and pDsRed2-histone H2Bk together with control siRNA (A), Ect2 siRNA (B), or Cdc42 siRNA (C) and time-lapse imaging was taken after 48 h. The bottom panels of each set show the EGFP-EB1 images only. The movies of these time-lapse acquisitions are available online as Videos 1–3 (available at http://www.jcb.org/cgi/content/full/jcb.200408085/DC1).
Expression of dominant-negative Ect2 and MgcRacGAP mutants causes prometaphase delay
The aforementioned role of Ect2 was further confirmed by expression of the dominant-negative mutant Ect2-N1. HeLa cells were synchronized with thymidine in early S phase and transfected with Ect2-N1. The cells were released from the thymidine block and enriched in mitosis by the use of nocodazole. The cells were then allowed to enter and progress through mitosis upon nocodazole removal. At 120 min after nocodazole removal, the majority of control cells expressing GFP had reached the end stage of telophase (Fig. 5 A, a and b). When we expressed Ect2-N1 in mitotic cells, we observed a significant increase in the number of binucleate cells containing two nuclei of equal size (Fig. 5 A, c and d) as reported previously (Tatsumoto et al., 1999; Kimura et al., 2000). However, the more striking observation is the accumulation of cells showing aberrant nuclei with nuclear invaginations and micronuclei (Fig. 5 A, e and f), and also of cells arrested in prometaphase with scattered chromosomes (Fig. 5 A, g and h). Immunostaining revealed that Mad2 was present at some kinetochores of condensed chromosomes in prometaphase-arrested cells, suggesting the absence of MT attachment to these kinetochores (Fig. 5 B). Quantification of the numbers of the cells showing these phenotypes revealed statistically significant increases in the number of cells showing each phenotype upon Ect2-N1 expression compared with GFP expression (10.2 vs. 4.4% for binucleated cells, 13.2 vs. 3.3% for cells with abnormal nuclei, and 20 vs. 9.6% for cells in prometaphase; Fig. 5 C). Accumulation in prometaphase and aberrant nuclear shape were also noted in cells expressing CRIB-Pak, a protein motif capable of trapping the active form of Cdc42 (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200408085/DC1).
Figure 5.

Effects of overexpression of Ect2-N1 on mitosis of HeLa cells. (A) Mitotic phenotype. HeLa cells synchronized in S phase were transfected with pEGFP (a and b) or pEGFP-Ect2-N1 (c–h). The cells were fixed 120 min after nocodazole removal and subjected to fluorescence microscopy for GFP (a, c, and e, green), anti–β-tubulin staining (g, green), and DAPI (a–h, blue). The Ect2-N1-expressing cells show the binucleate phenotype (c and d), the multinucleate phenotype with macro- and micro-nuclei (e and f), or the prometaphase arrest (g and h). (B) Mad2 staining. The Ect2-N1–expressing cells were treated as in A and stained for CENP-A (red), Mad2 (green), and chromosomes (DAPI, blue). Positive Mad2 staining was detected at some kinetochores. (C) Quantitative analysis of the mitotic phenotypes of the cells expressing Ect2-N1. These experiments were repeated seven times, and the numbers of cells showing each mitotic phenotype were determined in more than 200 cells each experiment. Results shown are the mean ± SEM (n = 7). *, P < 0.05 versus each corresponding subpopulation of the pEGFP-transfected cells. Bars: (A) 10 μm; (B) 2 μm.
Next, we expressed MgcRacGAPR386A in mitotic cells and examined the effect of MgcRacGAP inactivation on mitosis essentially in the same manner as described for Ect2-N1. Expression of MgcRacGAPR386A not only induced binucleate cells containing two nuclei of equal size (Fig. 6 A, a and b), as reported previously (Hirose et al., 2001), but also caused the accumulation of cells with aberrant nuclei with nuclear invaginations and micronuclei (Fig. 6 A, c and d) and the accumulation of cells arrested in prometaphase with scattered chromosomes (Fig. 6 A, e and f). Again, immunostaining revealed that Mad2 was present at the kinetochores of some condensed chromosomes in prometaphase-arrested cells (Fig. 6 B). Quantification revealed that 9.4, 6.3, and 21.6% of the MgcRacGAPR386A-transfected cells were binucleate, multinucleate with aberrant shape, and arrested in prometaphase, respectively (Fig. 6 C). We found that expression of Cdc42G12V resulted in the production of comparable populations of cells exhibiting these three mitotic phenotypes (Fig. 6 C). These results, together with those observed in the pull-down assay (Fig. 1 C), have verified the participation of MgcRacGAP in the regulation of Cdc42 in mitosis.
Figure 6.

Effects of overexpression of MgcRacGAPR386A on mitosis of HeLa cells. (A) Mitotic phenotypes. HeLa cells synchronized in S phase were transfected with either pEGFP alone or pEGFP and pME18S-MgcRacGAPR386A (a–f). At 120 min after nocodazole removal, the cells were fixed and subjected to fluorescence microscopy for GFP (a and c, green), anti–β-tubulin staining e, green), and DAPI (a–f, blue). MgcRacGAPR386A-expressing cells show the binucleate phenotype (a and b), the multinucleated phenotype with macro- and micro-nuclei (c and d), or the prometaphase arrest (e and f). (B) Mad2 staining. The MgcRacGAPR386A-expressing cells were treated as in A and stained for CENP-A (red), Mad2 (green), and chromosomes (DAPI, blue). Positive Mad2 staining was detected at some kinetochores. (C) Quantitative analysis of the mitotic phenotypes of the cells expressing MgcRacGAPR386A. The mitotic phenotype of HeLa cells expressing GFP, MgcRacGAPR386A (MRG-R386A), or Cdc42G12V was examined in more than 200 cells for each experiment, and the percentages of cells showing prometaphase arrest, binucleate, or aberrant multinucleate phenotypes were determined. Results represent the mean ± SEM (n = 7). *, P < 0.05 versus each corresponding subpopulation of the pEGFP-transfected cells. Bars: (A) 10 μm; (B) 2 μm.
Cell cycle–associated changes in the spatial localization of Cdc42
The aforementioned results strongly suggest that Cdc42 works spatio-temporally in the mitosis of mammalian cells. To investigate the localization of endogenous Cdc42 during the cell cycle, we stained for Cdc42 in HeLa cells in different phases of mitosis. We tested the specificity of the antibody against Cdc42 by Western blot analysis of HeLa cell lysates. This antibody recognized a single band in HeLa cell lysates, the intensity of which could be increased in a sample enriched in Cdc42 using GST-CRIB-Pak beads (Fig. 7 A). The specificity of the antibody was further verified by Cdc42 RNAi, as the intensity of the band recognized by this antibody decreased with depletion of the endogenous protein (Fig. 2 A). This antibody was then used for immunofluorescence studies to determine the localization of endogenous Cdc42 in mitotic HeLa cells. The most prominent feature observed using this antibody was the appearance of a spindle shape signal in metaphase cells (Fig. 7 B). Costaining for β-tubulin revealed that this signal was localized along MTs of the mitotic spindle and that the signal was more concentrated at the minus-end region of these MTs. The addition of either the immunogenic peptide (Fig. 7 C) or recombinant GST-Cdc42 (not depicted) abolished this signal, suggesting that it represented endogenous Cdc42. Furthermore, staining for Rho under identical conditions did not reveal any specific signal (unpublished data). The Cdc42 signal localized in the spindle MTs of prometaphase cells, moved to the central spindle as cells progressed through anaphase to telophase, and finally concentrated in the intercellular bridge formed between two daughter cells at the end of cytokinesis (Fig. 7 B). When costaining for MgcRacGAP was conducted, the MgcRacGAP signal was also seen on spindle MTs from prometaphase to telophase and partially colocalized with the Cdc42 signal. However, the signals for the two molecules began to separate in late anaphase, and, at the end of cytokinesis, the MgcRacGAP signal no longer overlapped with that of Cdc42; the former concentrated in the Fleming body and the latter on the bridge on either side of it.
Figure 7.

Localization of Cdc42 in mitotic cells. (A) Specificity of the antibody to Cdc42. One fiftieth of HeLa cell extracts from one 10-cm dish (lane 1) or one fourth of the pull-down precipitates of lysates from four dishes of HeLa cells at metaphase using the GST-CRIB-Pak (lane 2) were probed with the monoclonal anti-Cdc42 antibody. (B) Cdc42 localization in mitotic HeLa S3 cells. Randomly growing HeLa S3 cells were preextracted for 1 min with 0.5% Triton X-100 in PHEM, fixed, and stained for Cdc42 (green), MgcRacGAP (red), and chromosomes (DAPI, blue). Immunofluorescence images of the cells in each phase of mitosis are shown. (C) Competition experiments. The cells were treated as in B and subjected to staining for Cdc42 (red) and chromosomes (DAPI, blue) in the presence of the immunogenic peptide. A single representative cell in metaphase is shown (a, merged image; b, staining for Cdc42 alone). (D) Effects of MgcRacGAPR386A or Ect2-N1 expression on distribution of Cdc42 on the metaphase spindle. The cells were cotransfected with MgcRacGAPR386A together with EGFP (a–d) to allow the identification of transfected cells, or with EGFP-Ect2-N1 (e–h) and treated as in B. Chromosomes were stained with DAPI (a, c, e, and g, blue). Cdc42 was stained with monoclonal anti-Cdc42 antibody and detected with goat anti–mouse IgG coupled to Alexa-647 and pseudo-colored to green (a–d) or with donkey anti–mouse IgG coupled to Alexa-594 shown in red (e–h). MgcRacGAP is shown in red (a–d). EGFP-Ect2-N1 is shown in green (e–h). Two different optical sections of a single cell are shown each in a and b and c and d. Bars, 10 μm.
To test whether or not the distribution of the Cdc42 signal on the spindle MTs was under regulation of Ect2 and MgcRacGAP, we expressed dominant-negative mutants of these molecules in mitotic HeLa cells and performed Cdc42 immunostaining (Fig. 7 D). In the cells transfected with MgcRacGAPR386A, the Cdc42 signal was observed all along the spindle MTs, reaching the vicinity of the kinetochores, where the MgcRacGAP signal was also observed (Fig. 7 D; 65% of 20 transfected cells). In contrast, when EGFP-Ect2-N1 transfection was performed, the Ect2-N1 signal was distributed from the vicinity of the kinetochores on chromosomes to the plus-end region of the mitotic spindle, and the Cdc42 signal became more limited to the minus-end region of the spindle in the vicinity of the centrosomes (61% of 23 transfected cells).
Discussion
We recently conducted a series of experiments using toxin B and various Rho GTPase mutants, and examined their role in mitosis (Yasuda et al., 2004). The results of these experiments indicate that Cdc42 acts on its downstream target, mDia3, and facilitates bi-orient attachment and stabilization of spindle MTs to kinetochores. Here, we have adopted several strategies to extend this study and, further, to clarify the pathway regulating Cdc42 in mitosis. First, we used the pull-down assay for GTP-Cdc42, and found accumulation of GTP-Cdc42 in metaphase of HeLa S3 cells, which is consistent with the proposed action of Cdc42 in metaphase. Second, we expressed dominant-negative forms of Ect2 and MgcRacGAP, a GEF and a GAP of Rho GTPases in mitosis, respectively, or depleted Ect2 by RNAi, and found that the aforementioned accumulation of Cdc42 in metaphase was abolished by these procedures. Third, we performed RNAi for Cdc42 and Ect2 or expressed the dominant-negative mutants of Ect2 and MgcRacGAP, and found that these procedures cause the prometaphase delay similar to that found in the previous study. Finally, using immunofluorescence, we found that a subpopulation of Cdc42 is associated with the spindle MTs and that its localization is regulated by the activities of Ect2 and MgcRacGAP. These results strongly argue that Ect2 and MgcRacGAP regulate the activation and function of Cdc42 in mitosis.
Regulation of Cdc42 in mitosis by Ect2 and MgcRacGAP is intriguing, given previous findings that the two molecules regulate Rho in mitosis (Tatsumoto et al., 1999; Kimura et al., 2000; Jantsch-Plunger et al., 2000; Hirose et al., 2001; Minoshima et al., 2003). These results, together with our current findings, indicate that the same GEF and GAP can regulate two different Rho GTPases, Cdc42 and Rho, in different phases of mitosis. Both Ect2 and MgcRacGAP are present in interphase nuclei, are released to the cytoplasm after nuclear envelope breakdown, associate with the spindle MTs, and change their localization during cell division (Tatsumoto et al., 1999; Hirose et al., 2001). These results indicate that they associate with different proteins in different phases of mitosis and/or are modified differently. It is tempting to think that such interactions and/or modifications determine not only their localization but also their specificity. For example, the NH2-terminal domain of Ect2 that we used in these experiments contains the tandem BRCT domains. It was reported recently that the tandem BRCT domains have affinity for phospho-proteins (Yu et al., 2003). Therefore, it is likely that the BRCT domain of Ect2 binds to specific phosphorylated proteins, which in turn provide signals that dictate the specificity of Ect2. Ect2 might bind to different phospho-proteins in metaphase and telophase, which then may provide the phase-specific specificity of this exchanger. Moreover, a subpopulation of Ect2 is phosphorylated early in mitosis (Tatsumoto et al., 1999; Kimura et al., 2000). Experimental evidence for, and an implication of such an interaction and modification, are clearer for MgcRacGAP. Recently, Ban et al. (2004) reported that MgcRacGAP binds PRC1, a mitotic CDK substrate, and that this binding inhibits MgcRacGAP activity toward Cdc42 in metaphase. This finding can explain how Cdc42 is activated in metaphase in spite of its colocalization with MgcRacGAP on the metaphase spindle (Fig. 7 B). It is also known that covalent modification of MgcRacGAP changes its substrate specificity. Minoshima et al. (2003) found that phosphorylation of MgcRacGAP by Aurora kinase B induces latent GAP activity toward RhoA, and this activity is important for down-regulation of Rho at the end of cytokinesis. Thus, it appears that both Ect2 and MgcRacGAP work on Cdc42 around metaphase, whereas they act on RhoA late in mitosis. It is intriguing that the expression of MgcRacGAPR386A elevated the level of GTP-Cdc42 in prometaphase but tended to suppress the Cdc42 activation in metaphase. We do not have an experimental explanation for the latter effect. It may be a secondary effect due to the prometaphase arrest induced by this MgcRacGAP mutant. In addition, Somers and Saint (2003) found association of a Drosophila MgcRacGAP orthologue, RacGAP50C, with a Drosophila Ect2 orthologue, Pebble, during mitosis. Therefore, it is possible that overexpressed MgcRacGAPR386A binds to endogenous Ect2 and affects its activity.
Here, we performed RNAi for Cdc42 and Ect2 and found that depletion of either protein induces prometaphase delay, which is essentially similar to that found in cells subjected to toxin B treatment, overexpression of Cdc42 mutants, and RNAi for mDia3 (Yasuda et al., 2004). Furthermore, we found that expression of MgcRacGAPR386A also interfered with the transition to metaphase in a similar way. This, although apparently paradoxical, is consistent with our finding that both dominant-active and dominant-negative Cdc42 mutant induced the delay in prometaphase, and can be explained by misorientation of the spindle MTs by ectopic expression of an excess amount of active Cdc42. Although dominant-active and dominant-negative Cdc42 mutants induce the opposing actin phenotypes, they cause the same phenotype with respect to MT targeting in interphase cells (Etienne-Manneville and Hall, 2001). These results not only corroborated the role of Cdc42 in the MT attachment to kinetochores but also that Ect2 and MgcRacGAP regulate this Cdc42 function. In contrast, previous studies have shown that interfering with Ect2 and MgcRaGAP causes cytokinesis failure, which led the authors to suggest that Ect2 and MgcRacGAP are linked mainly with RhoA (Tatsumoto et al., 1999; Jantsch-Plunger et al., 2000; Kimura et al., 2000; Hirose et al., 2001). However, though not explicitly described in the previous papers, the tetraploid nucleus in these cells was abnormal in shape. Furthermore, Van de Putte et al. (2001) reported that homozygous disruption of the gene for MgcRacGAP resulted in death of E3.5–4 embryos due to the failure of chromosomal segregation. In addition, expression of active forms of Pak, an effector of Cdc42, resulted in the production of cells with multiple spindles (Vadlamudi et al., 2000), and an endogenous Pak isoform, Pak 1, was shown to localize at the kinetochore in mitotic cells (Li et al., 2002). Recently, Tatsumoto et al. (2003) used Xenopus laevis mitotic extracts and examined in vitro the effect on mitosis of anti-Ect2 antibody as well as dominant-negative forms of Ect2 and Rho GTPases. They found that addition of either anti-Ect2 antibody or dominant-negative Ect2 or Cdc42 interfered with normal progression of metaphase by causing many misaligned chromosomes, a phenotype very similar to that found in HeLa cells in this study. These results add further support to the role of Ect2 and Cdc42 in mitosis.
Thus, our current findings indicate that Cdc42 is essential in mitosis. Curiously, however, Chen et al. (2000) established a Cdc42 null cell line from ES cells by disruption of the Cdc42 gene, and reported that the Cdc42-null ES cells can proliferate normally. These findings suggest a possibility that other Cdc42-related GTPases such as TC10 and Chp may compensate for the absence of Cdc42. Indeed, the mitotic phenotype in Cdc42 RNAi cells appear milder than that found in toxin B–treated cells. Recently, we found that RNAi for all of the five Cdc42-related GTPases induces a more dramatic phenotype than that of RNAi for Cdc42 alone (unpublished data).
An interesting question is where Cdc42 is localized and functions in mitosis. Our immunofluorescence study showed that Cdc42 localizes on the mitotic spindle of HeLa cells during prometaphase and metaphase and is concentrated in the central spindle in anaphase and in the bridge MTs in the midbody at the end of cell division. This Cdc42 localization is consistent with the reported localization of Ect2 during cell division (Tatsumoto et al., 1999). Association of these molecules with spindle MTs was also suggested by a recent proteome study of isolated midbody preparation (Skop et al., 2004). Interestingly, Ect2-N1 accumulates in the vicinity of the kinetochores of metaphase cells (Fig. 7 D). Ect2 may generate GTP-bound Cdc42 around the kinetochores of metaphase chromosomes. Together, our results provide evidence that Ect2 and MgcRacGAP regulate the activation and function of Cdc42 spatio-temporally in mitosis, the former catalyzing the guanine nucleotide exchange of Cdc42 to activate Cdc42 in metaphase, and the latter facilitating the GTP hydrolysis of Cdc42 in prometaphase.
Materials and methods
Materials
Rabbit polyclonal antibody, P-1 to Cdc42 and C-20 to Ect2, and mouse mAb, B-8 to Cdc42 and 26C4 to RhoA, were purchased from Santa Cruz Biotechnology, Inc. Mouse mAb 05–389 to Rac1 and Tub 2.1 to β-tubulin were purchased from Upstate Biotechnology and Sigma-Aldrich, respectively. CREST serum recognizing CENP-A was provided by T. Mimori (Kyoto University, Kyoto, Japan). Polyclonal antibody to MgcRacGAP and cDNA for MgcRacGAPR386A were reported previously (Hirose et al., 2001). All other materials used were of reagent grade.
Plasmids, siRNA preparation, and transfection
pGEX4T1-CRIB-Pak has been described previously (Matsuo et al., 2002). Ect2 cDNA was provided by T. Miki (National Institutes of Health, Bethesda, MD). pCEV32F-Ect2-N1 has been described previously (Kimura et al., 2000). pEGFP-Ect2-N1 was generated by digestion of pCEV32F-Ect2-N1 with BamHI and EcoRI and inserted into BglII–EcoRI–digested pEGFP-C1. pEGFP-EB1, pEGFP-histone H2Bk, pEGFP-Cdc42G12V, and pDsRed2-histone H2Bk have been described previously (Yasuda et al., 2004).
To generate siRNA against Ect2 and Cdc42, we used the BLOCK-iT RNAi-TOPO Transcription and the BLOCK-iT Dicer RNAi kits (Invitrogen). In brief, the NH2-terminal 1,000 nucleotides of Ect2, starting from the first methionine, were amplified by PCR using the primers ATGGCTGAAAATAGTGTATTAACATCCACT and ATTTCTTGAGCTCAGGAGTATTTGCCTTTT. The full coding sequence of Cdc42 (G25K, Homo sapiens) was amplified by using the primers ATGCAGACAATTAAGTGTGTTGTTGTGGGCGA and TCATAGCAGCACACACCTGCGGCTCTTCTT. The PCR fragments obtained were linked to the T7 promoter and amplified again by PCR. The T7-linked PCR products were used for transcription of single stranded RNA and subsequently annealed with complementary RNA to obtain double stranded RNA. As control RNAi, we used RNA duplex for the Escherichia coli LacZ gene as provided by the manufacturer. The RNA duplexes obtained were cleaved into 21–23 nucleotide fragments with Dicer enzyme.
Transfection was performed using Lipofectamine-Plus Reagent (Invitrogen). A total of 1 or 4 μg of plasmid DNA was used to transfect HeLa cells in a well of a 6-well plate or a 10-cm dish, respectively. To transfect siRNA into HeLa cells, we used Lipofectamine 2000 (Invitrogen). For immunofluorescence, we used 0.3 μg of siRNA together with 0.5 μg of pEGFP-histone H2B and 6 μl of Lipofectamine 2000 diluted in Opti-MEM. The mixture was added to one well of a 6-well plate and incubated for 2 h at 37°C with 5% CO2. For time-lapse imaging, the cells were transfected with pEGFP-EB1 and pDsRed2-histone H2Bk, together with 0.3 μg of siRNA for either Ect2 or Cdc42 or both. The medium was replaced by DME containing 10% FCS and allowed to incubate for 48 h before observation.
Cell culture and synchronization
HeLa S3 cells were maintained in DME supplemented with 10% FCS and antibiotics at 37°C and with 5% CO2. Cells were synchronized at early S phase by the double thymidine block as described previously (Bostock et al., 1971). In brief, cells were seeded at a density of 2 × 105 per dish onto 10-cm dishes. After overnight culture, the medium was replaced by DME containing 10 mM thymidine and 5% FCS, and the cells were incubated for 12 h. The thymidine-containing medium was removed and the cells were washed twice with PBS without divalent cations. The cells were further cultured in DME containing 10% FCS for 10 h, and then subjected to the second thymidine block for 12 h. The cells were washed twice with PBS and placed in fresh DME containing 10% FCS. Cells were collected in G2 phase, prometaphase, metaphase, telophase, and G1 phase as described previously (Kimura et al., 2000). The cells were either frozen immediately for pull-down assays or used for immunofluorescence. In overexpression experiments, we prolonged the time of the second thymidine block to 15 h, during which we transfected cells using Lipofectamine Plus, 8 to 11 h after the addition of thymidine. For cell cycle analysis, we cotransfected cells with siRNA and pEGFP-histone H2Bk, stained the cells with propidium iodide, and subsequently examined the progression of cells expressing EGFP-histone H2Bk by flow cytometry (Darzynkiewicz, 1994).
Pull-down assays
Recombinant GST-Pak-CRIB was prepared and conjugated with glutathione-Sepharose 4B (Amersham Biosciences) as described previously (Matsuo et al., 2002). Frozen HeLa S3 cells were suspended in the lysis buffer containing 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 2 mM MgCl2, 1% NP-40, and 10% glycerol supplemented with 10 mM NaF, 1 mM NaVO4, 100 μg/ml PMSF, 5 μg/ml leupeptin, and 5 μg/ml pepstatin. The cell suspension was incubated for 15 min at 4°C with continuous agitation and centrifuged at 18,000 g for 15 min at 4°C. Supernatant was saved and sonicated on ice for 1 s 10 times. Protein concentration was determined by the Lowry method, and 800 μg of the supernatant protein was incubated with 40 μg of GST-CRIB-Pak for 30 min at 4°C. The beads were spun down and washed three times with the lysis buffer. One fourth or one eighth of the precipitates were subjected to SDS-PAGE to determine the amounts of GTP-Rac and GTP-Cdc42, respectively, and one twenty fifth of the supernatant was used to determine the total amounts of Rac and Cdc42. Separated proteins were transferred onto nitrocellulose membranes (Schleicher & Schuell; BA 83). The membranes were blotted with antibodies to Cdc42 (P-1) and Rac1. After the density of each band was determined, the amount of GTP-Cdc42 was calculated by (the density of each GTP-Cdc42 band × 8)/(the density of the total Cdc42 band in the G2 phase of each blot × 25), and expressed as a percentage of the total amount. The amount of GTP-Rac was calculated similarly. The pull-down assay for RhoA was performed as described previously (Kimura et al., 2000).
Time-lapse live imaging
HeLa cells were seeded on 35-mm glass-bottomed dishes (MatTek) and after overnight culture the cells were transfected with pEGFP-EB1 and pDsRed2-histone H2Bk, together with siRNA of Ect2 or Cdc42 or both, as described in the section Plasmids, siRNA preparation, and transfection. After 48 h, the dish was placed on a temperature-controlled stage maintained at 37°C with 5% CO2. Live-imaging was performed on an inverted microscope (model DMIRE2; Leica) with 63× NA 1.3 lenses equipped with a high pressure lamp (Xenon). EGFP, DsRed, and brightfield images were taken with a camera (model CoolSNAP HQ) driven by AS MDW software (Leica). Sequential time-lapse images were acquired every 2.5 min for control RNAi or at 5-min intervals for Ect2 and Cdc42 RNAi. For the cells shown in Fig. 4 (A and B), one Z section was recorded at each time interval. For the cell shown in Fig. 4 C, a collection of 20 Z sections at 0.5-μm step intervals were acquired. The images obtained from Fig. 4 (B and C) were processed by deconvolution using a nonblind method and analyzed with AS MDW and Deblur software (Leica).
Immunofluorescence
Cells collected at various times of mitosis were washed with PBS before fixation with 3.7% PFA in PBS at 37°C for 15 min. The cells were washed three times with PBS and permeabilized with 0.5% Triton X-100 in PBS for 5 min, followed by three washes with PBS. For kinetochore and Mad2 staining, the cells were preextracted with 0.5% Triton X-100 in PHEM buffer (60 mM Pipes, 25 mM Hepes, 10 mM EGTA, and 1 mM Mg-acetate, pH 6.9) for 1 min on ice just before fixation with 3.7% PFA in PBS (Dujardin et al., 1998). After immersion in 100 mM glycine in PBS for 30 min, samples were incubated with 3% BSA in PBS for 30 min. The samples were incubated for 1 h at RT or 4°C overnight with indicated combinations of the following primary antibodies: anti–β-tubulin (1:200), anti-Mad2 (Covance; 1:100), and CREST serum (1:500). After three washes with PBS, the samples were incubated with appropriate secondary antibodies coupled to Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647 (Molecular Probes) and DAPI (WAKO). The samples were washed three times with PBS before mounting on glass slides. For detection of Cdc42, cells on the coverslips were rinsed with PHEM solution and extracted with 0.5% Triton X-100 in PHEM for 1 min before fixation with 3.7% PFA in PBS at RT. Antibodies to Cdc42 and MgcRacGAP were used at 1:200 and 1:1,000 dilution, respectively. Immunofluorescence for mDia3 has been described previously (Yasuda et al., 2004).
For high-resolution imaging, fluorescence images were obtained with a microscope (model IX70; Olympus) using oil immersion objective lenses (PlanApo 60, NA 1.4; and PlanApo 100, NA 1.4) and high-selectivity filters under the control of softWoRx software. Serial optical section data (15–30 focal planes at 0.5-μm intervals for 60× or 0.2-μm intervals for 100× objectives) were collected on a Peltier-cooled charge-coupled device (Photometrics) and computationally processed by a three-dimensional deconvolution method (Agard et al., 1989).
Online supplemental material
Fig. S1 shows the mitotic phenotype of HeLa cells expressing CRIB-Pak. Videos 1–3 show mitosis of HeLa cells transfected with control siRNA, Ect2 siRNA, and Cdc42 siRNA, respectively. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb. 200408085/DC1.
Acknowledgments
We thank J. Monypenny for critical reading of the manuscript; A. Fujita, N. Watanabe, T. Furuyashiki, and T. Ishizaki for discussion; S. Tsukita for use of a DeltaVision system; M. Yanagida for helpful advice; K. Nonomura for technical assistance; and T. Arai, H. Nose, and Y. Kitagawa for secretarial assistance.
F. Oceguera-Yanez was a recipient of the Monbukagakushou scholarship. This work was supported by grants from a Grant-in-Aid for Specially Promoted Research to S. Narumiya and a Grant-in-Aid for Scientific Research (B) to T. Haraguchi and Y. Hiraoka from the Ministry of Education, Culture, Sports, Science and Technology of Japan; a grant from the Ministry of Health, Labor and Welfare of Japan; and a Core Research for Evolutionary Science and Technology grant from the Japan Science and Technology Agency to T. Haraguchi and Y. Hiraoka.
K. Kimura's present address is Dept. of Ophthalmology, Yamaguchi University School of Medicine, Yamaguchi 755-8505, Japan.
Abbreviations used in this paper: CRIB, Cdc42-Rac–interacting binding domain; DH, Dbl homology; GAP, GTPase activating protein; GEF, guanine nucleotide exchange factor; MT, microtubule; PH, pleckstrin homology; RNAi, RNA interference.
References
- Agard, D.A., Y. Hiraoka, P. Shaw, and J.W. Sedat. 1989. Fluorescence microscopy in three dimensions. Methods Cell Biol. 30:353–377. [DOI] [PubMed] [Google Scholar]
- Agnel, M., L. Roder, C. Vola, and R. Griffin-Shea. 1992. A Drosophila rotund transcript expressed during spermatogenesis and imaginal disc morphogenesis encodes a protein which is similar to human Rac GTPase-activating (racGAP) proteins. Mol. Cell. Biol. 12:5111–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktories, K., G. Schmidt, and I. Just. 2000. Rho GTPases as targets of bacterial protein toxins. Biol. Chem. 381:421–426. [DOI] [PubMed] [Google Scholar]
- Ban, R., Y. Irino, K. Fukami, and H. Tanaka. 2004. Human mitotic spindle-associated protein PRC1 inhibits MgcRacGAP activity toward Cdc42 during the metaphase. J. Biol. Chem. 279:16394–16402. [DOI] [PubMed] [Google Scholar]
- Bostock, C.J., D.M. Prescott, and J.B. Kirkpatrick. 1971. An evaluation of the double thymidine block for synchronizing mammalian cells at the G1-S border. Exp. Cell Res. 68:163–168. [DOI] [PubMed] [Google Scholar]
- Chen, F., L. Ma, M.C. Parrini, X. Mao, M. Lopez, C. Wu, P.W. Marks, L. Davidson, D.J. Kwiatkowski, T. Kirchhausen, et al. 2000. Cdc42 is required for PIP(2)-induced actin polymerization and early development but not for cell viability. Curr. Biol. 10:758–765. [DOI] [PubMed] [Google Scholar]
- Cleveland, D.W., Y. Mao, and K.F. Sullivan. 2003. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 112:407–421. [DOI] [PubMed] [Google Scholar]
- Darzynkiewicz, Z. 1994. Cell cycle analysis by flow cytometry. Cell Biology: A Laboratory Handbook. Vol. 1. J.E. Celis, editor. Academic Press, New York. 261–271.
- Dujardin, D., U.I. Wacker, A. Moreau, T.A. Schroer, J.E. Rickard, and J.R. De Mey. 1998. Evidence for a role of CLIP-170 in the establishment of metaphase chromosome alignment. J. Cell Biol. 141:849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2001. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCζ. Cell. 106:489–498. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2002. Rho GTPases in cell biology. Nature. 420:629–635. [DOI] [PubMed] [Google Scholar]
- Hirose, K., T. Kawashima, I. Iwamoto, T. Nosaka, and T. Kitamura. 2001. MgcRacGAP is involved in cytokinesis through associating with mitotic spindle and midbody. J. Biol. Chem. 276:5821–5828. [DOI] [PubMed] [Google Scholar]
- Jantsch-Plunger, V., P. Gönczy, A. Romano, H. Schnabel, D. Hamill, R. Schnabel, A.A. Hyman, and M. Glotzer. 2000. CYK-4: A Rho family GTPase activating protein (GAP) required for central spindle formation and cytokinesis. J. Cell Biol. 149:1391–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima, T., K. Hirose, T. Satoh, A. Kaneko, Y. Ikeda, Y. Kaziro, T. Nosaka, and T. Kitamura. 2000. MgcRacGAP is involved in the control of growth and differentiation of hematopoietic cells. Blood. 96:2116–2124. [PubMed] [Google Scholar]
- Kimura, K., T. Tsuji, Y. Takada, T. Miki, and S. Narumiya. 2000. Accumulation of GTP-bound RhoA during cytokinesis and a critical role of ECT2 in this accumulation. J. Biol. Chem. 275:17233–17236. [DOI] [PubMed] [Google Scholar]
- Kishi, K., T. Sasaki, S. Kuroda, T. Itoh, and Y. Takai. 1993. Regulation of cytoplasmic division of Xenopus embryo by rho p21 and its inhibitory GDP/GTP exchange protein (rho GDI). J. Cell Biol. 120:1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F., L. Adam, R.K. Vadlamudi, H. Zhou, S. Sen, J. Chernoff, M. Mandal, and R. Kumar. 2002. p21-activated kinase 1 interacts with and phosphorylates histone H3 in breast cancer cells. EMBO Rep. 3:767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi, I., Y. Hamaguchi, H. Fujimoto, N. Morii, M. Mishima, and S. Narumiya. 1993. A rho-like protein is involved in the organisation of the contractile ring in dividing sand dollar eggs. Zygote. 1:325–331. [DOI] [PubMed] [Google Scholar]
- Matsuo, N., M. Hoshino, M. Yoshizawa, and Y. Nabeshima. 2002. Characterization of STEF, a guanine nucleotide exchange factor for Rac1, required for neurite growth. J. Biol. Chem. 277:2860–2868. [DOI] [PubMed] [Google Scholar]
- Miki, T., C.L. Smith, J.E. Long, A. Eva, and T.P. Fleming. 1993. Oncogene ect2 is related to regulators of small GTP-binding proteins. Nature. 362:462–465. [DOI] [PubMed] [Google Scholar]
- Minoshima, Y., T. Kawashima, K. Hirose, Y. Tonozuka, A. Kawajiri, Y.C. Bao, X. Deng, M. Tatsuka, S. Narumiya, W.S. May Jr., et al. 2003. Phosphorylation by Aurora B converts MgcRacGAP to a RhoGAP during cytokinesis. Dev. Cell. 4:549–560. [DOI] [PubMed] [Google Scholar]
- Moon, S.Y., and Y. Zheng. 2003. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 13:13–22. [DOI] [PubMed] [Google Scholar]
- Prokopenko, S.N., A. Brumby, L. O'Keefe, L. Prior, Y. He, R. Saint and H.J. Bellen. 1999. A putative exchange factor for Rho1 GTPase is required for initiation of cytokinesis in Drosophila. Genes Dev. 13:2301–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skop, A.R., H. Liu, J. Yates III, B.J. Meyer, and R. Heald. 2004. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science. 305:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers, W.G., and R. Saint. 2003. A RhoGEF and Rho family GTPase-activating protein complex links the contractile ring to cortical microtubules at the onset of cytokinesis. Dev. Cell. 4:29–39. [DOI] [PubMed] [Google Scholar]
- Tatsumoto, T., X. Xie, R. Blumenthal, I. Okamoto, and T. Miki. 1999. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J. Cell Biol. 147:921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumoto, T., H. Sakata, M. Dasso, and T. Miki. 2003. Potential roles of the nucleotide exchange factor ECT2 and Cdc42 GTPase in spindle assembly in Xenopus egg cell-free extracts. J. Cell. Biochem. 90:892–900. [DOI] [PubMed] [Google Scholar]
- Toure, A., O. Dorseuil, L. Morin, P. Timmons, B. Jegou, L. Reibel, and G. Gacon. 1998. MgcRacGAP, a new human GTPase-activating protein for Rac and Cdc42 similar to Drosophila rotundRacGAP gene product, is expressed in male germ cells. J. Biol. Chem. 273:6019–6023. [DOI] [PubMed] [Google Scholar]
- Vadlamudi, R.K., L. Adam, R.A. Wang, M. Mandal, D. Nguyen, A. Sahin, J. Chernoff, M.C. Hung, and R. Kumar. 2000. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J. Biol. Chem. 275:36238–36244. [DOI] [PubMed] [Google Scholar]
- Van de Putte, T., A. Zwijsen, O. Lonnoy, V. Rybin, M. Cozijnsen, A. Francis, V. Baekelandt, C.A. Kozak, M. Zerial, and D. Huylebroeck. 2001. Mice with a homozygous gene trap vector insertion in mgcRacGAP die during pre-implantation development. Mech. Dev. 102:33–44. [DOI] [PubMed] [Google Scholar]
- Wang, Y.L. 2001. The mechanism of cytokinesis: reconsideration and reconciliation. Cell Struct. Funct. 26:633–638. [DOI] [PubMed] [Google Scholar]
- Wittmann, T., A. Hyman, and A. Desai. 2001. The spindle: a dynamic assembly of microtubules and motors. Nat. Cell Biol. 3:E28–E34. [DOI] [PubMed] [Google Scholar]
- Yasuda, S., F. Oceguera-Yanez, T. Kato, M. Okamoto, S. Yonemura, Y. Terada, T. Ishizaki, and S. Narumiya. 2004. Cdc42 and mDia3 regulate microtubule attachment to kinetochores. Nature. 428:767–771. [DOI] [PubMed] [Google Scholar]
- Yu, X., C.C.S. Chini, M. He, G. Mer, and J. Chen. 2003. The BRCT domain is a phospho-protein binding domain. Science. 302:639–642. [DOI] [PubMed] [Google Scholar]
- Zheng, Y. 2001. Dbl family guanine nucleotide exchange factors. Trends Biochem. Sci. 26:724–732. [DOI] [PubMed] [Google Scholar]