

Abstract
Contributions of actin-related proteins (Arp) 2 and 3 nucleotide state to Arp2/3 complex function were tested using nucleotide-binding pocket (NBP) mutants in Saccharomyces cerevisiae. ATP binding by Arp2 and Arp3 was required for full Arp2/3 complex nucleation activity in vitro. Analysis of actin dynamics and endocytosis in mutants demonstrated that nucleotide-bound Arp3 is particularly important for Arp2/3 complex function in vivo. Severity of endocytic defects did not correlate with effects on in vitro nucleation activity, suggesting that a critical Arp2/3 complex function during endocytosis may be structural rather than catalytic. A separate class of Arp2 and Arp3 NBP mutants suppressed phenotypes of mutants defective for actin nucleation. An Arp2 suppressor mutant increased Arp2/3 nucleation activity. Electron microscopy of Arp2/3 complex containing this Arp2 suppressor identified a structural change that also occurs upon Arp2/3 activation by nucleation promoting factors. These data demonstrate the importance of Arp2 and Arp3 nucleotide binding for nucleating activity, and Arp3 nucleotide binding for maintenance of cortical actin cytoskeleton cytoarchitecture.
Introduction
Spatial and temporal regulation of actin polymerization are critical for cellular processes such as cell migration and endocytosis (Welch and Mullins, 2002; Engqvist-Goldstein and Drubin, 2003). Because nucleation is rate limiting for actin polymerization, regulation of this step controls when and where actin is assembled in cells. An important nucleator of actin filaments is the Arp2/3 complex, a seven subunit complex that includes two actin-related proteins (Arps) 2 and 3 (Welch and Mullins, 2002). The Arp2/3 complex nucleates actin filaments de novo, and from the sides of preexisting filaments, generating a branched filament array (Blanchoin et al., 2000) resembling those observed at the leading edge of certain motile cells (Svitkina and Borisy, 1999). Nucleation promoting factors (NPFs), such as members of the Wiskott-Aldrich syndrome protein (WASP) family of proteins, activate Arp2/3 complex nucleation activity, allowing regulated actin assembly that can generate forces in cells (Welch and Mullins, 2002). Elucidating the mechanisms that regulate Arp2/3 complex function is essential for understanding the dynamics of actin-based force generation.
Nucleotide binding plays a critical role in regulation of actin dynamics. ATP binding by actin promotes actin filament assembly by closing the nucleotide-binding pocket (NBP). ATP hydrolysis and Pi release results in a transition from the “closed” to the “open” state, promoting actin filament disassembly (Belmont et al., 1999a; Sablin et al., 2002). Similarly, Arp2 and Arp3 bind ATP, and the Arp2/3 complex requires ATP for nucleation (Dayel et al., 2001; Le Clainche et al., 2001). Arp2 ATP hydrolysis was linked to both the dissociation of actin filament branches (Le Clainche et al., 2003), and to nucleation (Dayel and Mullins, 2004). In the crystal structure of nucleotide-free Arp2/3 complex, the Arp2 and Arp3 NBPs resemble that of actin in the “open” conformation (Robinson et al., 2001). Thus, ATP binding may close the NBPs of Arp2 and Arp3 such that they can serve as a nucleus for actin polymerization.
In addition to regulating the association of the Arp2/3 complex with actin, nucleotide binding by Arp2/3 complex is linked to NPF binding. ATP-bound Arp2/3 exhibits higher affinity for NPFs than ADP-bound complex (Dayel et al., 2001). Furthermore, Arp2 ATP binding was reported to be enhanced by NPFs, and to be required for efficient Arp2/3 complex activation (Le Clainche et al., 2001). Similar to ATP, actin filaments also enhance Arp2/3 complex affinity for NPFs (Marchand et al., 2001). Because Arp2 and Arp3 are splayed apart in the crystal structure of the inactive Arp2/3 complex (Robinson et al., 2001), it is possible that the cooperative binding of ATP, NPFs, and actin filaments bring Arp2 and Arp3 together to form a template dimer for nucleation. Cryo-electron microscopy of the Arp2/3 complex in actin filament branches revealed that a large conformational change in the Arp2/3 complex represented in the crystal model is required for Arp2/3 activation (Volkmann et al., 2001). These conformational changes are likely to be influenced by the nucleotide state of Arp2 and Arp3.
Although ATP has been demonstrated to be important for Arp2/3 nucleation in vitro, the contributions of ATP-bound Arp2 or Arp3 to in vitro and in vivo function have not been determined individually. Here, we generated NPB mutations of S. cerevisiae Arp2 and Arp3 to explore the individual contributions of nucleotide to the function of each protein.
Results
Identification of Arp2 and Arp3 mutants that affect nucleotide binding
To test the function of ATP binding by Arp2 and Arp3, we generated 26 site-directed mutations that changed conserved residues in the NBPs of these two proteins (Fig. 1; Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200408177/DC1). We attempted to make mutations that would affect nucleotide affinity, hydrolysis, and exchange. Mutants were chosen based on previous mutations of actin (Wertman et al., 1992; Chen et al., 1995; Belmont and Drubin, 1998; Belmont et al., 1999b) and on predictions based on the actin crystal structure (Kabsch et al., 1990; Vorobiev et al., 2003). Residue numbers used to refer to mutants correspond to the amino acid position in the yeast actin sequence. Mutants were integrated into the endogenous ARP2 and ARP3 chromosomal loci and were assessed for growth on different media (Table S1). Immunoblotting revealed that mutants expressed wild-type levels of Arp2 and Arp3 (unpublished data). Although viable arp2Δ and arp3Δ mutant strains have been observed (Winter et al., 1999), deletion of either gene resulted in lethality under our growth conditions. Surprisingly, all but one of the integrated Arp2 and Arp3 mutants were viable, suggesting that they retain partial function, and allowing further characterization of their effects.
Figure 1.

Arp2 and Arp3 residues chosen for mutagenesis. Crystal structure of actin and NBPs of actin, Arp2, and Arp3 (Kabsch et al., 1990; Robinson et al., 2001). Subdomains of actin are indicated with Roman numerals. Because subdomains I and II of Arp2 are not present in the crystal structure, the backbone of actin was substituted. Altered residues, ATP (blue), and Ca2+ (orange) are identified by colors. Residue numbers correspond to the yeast actin peptide sequence.
Because ATP is required for Arp2/3 nucleation activity in vitro (Dayel et al., 2001; Le Clainche et al., 2001), we predicted that mutants affecting ATP binding would decrease Arp2/3 function in vivo. Two mutants predicted to lower nucleotide binding affinity, arp3-D11A and arp3-G302Y, showed growth defects at high temperature and high osmolarity (Table S1). The arp3-D11A mutant was predicted to lower nucleotide affinity by removing an aspartic acid that interacts with divalent cation, whereas the arp3-G302Y mutant was predicted to prevent nucleotide binding by introducing a tyrosine into the adenine-binding pocket. Interestingly, the analogous Arp2 mutations, arp2-D11A and arp2-G302Y, exhibited less severe growth phenotypes. To further analyze the effects of these mutants, we crossed arp3-G302Y and arp2-G302Y to several arp2 and arp3 alleles, respectively. In addition, we crossed these mutants to las17-11, a temperature-sensitive allele of the gene encoding a yeast WASP-like NPF (Duncan et al., 2001). arp2-G302Y and arp3-G302Y showed synthetic lethality with each other and with las17-11 (Table I). In addition, arp2-G302Y and arp3-G302Y exhibited synthetic lethality with arp3-D11A and arp2-D11A, respectively (Table I). Thus, the arp2-D11A, arp3-D11A, arp2-G302Y, and arp3-G302Y mutants impaired Arp2/3 function in vivo, possibly indicating a defect in nucleotide binding.
Table I. Genetic interactions of Arp2 and Arp3 mutants.
| arp2-G302Y[WT] x | Double mutant phenotype | arp3-G302Y[ts, ss, fs] x | Double mutant phenotype |
|---|---|---|---|
| arp3-D11A | SL | arp2-D11A | SL |
| arp3-T14A | SS (37°C) | arp2-T14A | SS (30°C) |
| arp3-Q137A | nd | arp2-Q137A | NE |
| arp3-Q137E | nd | arp2-Q137E | SS (25°C) |
| arp3-D157E | SS (NaCl) | arp2-D157E a | Sup (fs) |
| arp3-V159N | NE | arp2-V159N | NE |
| arp3-G302Y | SL | arp2-G302Y | SL |
| arp3-F306A | NE | arp2-Y306A a | Sup (ts,fs) |
| las17-11 | SL | las17-11 | SL |
| arp2-Y306A[WT] x | Double mutant phenotype | arp3-F306A[WT] x | Double mutant phenotype |
| arp3-D11A | Sup (ss, fs) | arp2-D11A | Sup (ss) |
| arp2-G13C b | Sup (ts, fs) | arp3-G15C b | Sup (ss, fs) |
| las17-11 | Sup (fs) | las17-11 | SS (30°C) |
Genetic interactions were examined by dissecting at least seven tetrads for each cross and identifying double mutants using linked auxotrophic markers. The phenotypes of starting strains are shown in brackets. SL, synthetic lethal at 25°C; SS, synthetic sick; NE, no effect; Sup, suppression; ts, temperature sensitive; ss, salt sensitive; fs, formamide sensitive; nd, not determined. Growth under the condition shown in parentheses changes in the double mutant.
Suppression is of arp3-G302Y phenotype.
Intragenic interaction.
Surprisingly, we identified two mutants, arp2-D157E and arp2-Y306A, that partially suppressed the arp3-G302Y phenotype, with arp2-Y306A being the stronger suppressor (Table I). Suppression was not allele specific because arp2-Y306A also partially suppressed arp3-D11A and las17-11 (Table I). In addition, arp2-Y306A exhibited intragenic suppression of arp2-G13C (Table I). The analogous Arp3 mutation, arp3-F306A, suppressed both arp2-D11A and arp3-G15C (Table I). Thus, arp2-Y306A and arp3-F306A represented a class of NBP mutant that appeared to increase the activity of the Arp2/3 complex.
ATP binding to Arp2 and Arp3 mutants
To determine if the D11A, G302Y, and Y(F)306A mutations affected nucleotide binding, we purified the Arp2/3 complex from these mutants (Fig. 2 A). Similar amounts of purified protein were obtained for each strain and most of these mutant complexes showed normal subunit compositions. However, we were unable to determine the stoichiometry of Arc40 due to its abnormal mobility on SDS-PAGE gels (Pan et al., 2004). Interestingly, Arp2/3 complex from arp2-D11A exhibited noticeable Arp3 degradation, indicating that Arp2 can contribute to the stability of Arp3 (Fig. 2 A).
Figure 2.

ATP binding of Arp2 and Arp3 mutants. (A) Coomassie staining of purified mutant Arp2/3 complexes. Asterisk indicates Arp3 degradation. (B) ATP cross-linking to purified Arp2/3 complex mutants. Arp2/3 complex subunits were separated by SDS-PAGE and visualized by autoradiography (top) and Coomassie staining (bottom). Asterisks indicate Arp3 degradation. See Table II for quantification. (C and D) Etheno-ATP binding to purified Arp2/3 complex mutants. Data points represent averages ± SEM from two experiments. See Table II for K d values.
The ability of mutant complexes to bind ATP was assessed using a photo-cross-linking assay previously used to examine ATP binding to the Arp2/3 complex (Dayel et al., 2001; Le Clainche et al., 2001). For wild-type yeast Arp2/3 complex, α-[32P]ATP was cross-linked to Arp2 and Arp3, and addition of the Arp2/3-activating WCA domain of Las17 did not substantially affect this cross-linking, similar to what was observed for Acanthamoeba Arp2/3 complex (Fig. 2 B; Dayel et al., 2001). The arp2-G302Y and arp3-G302Y mutations dramatically decreased ATP cross-linking to Arp2 and Arp3, respectively, which is consistent with the prediction that this change sterically inhibits nucleotide binding (Fig. 2 B). These mutations did not affect ATP cross-linking by the nonmutated Arp subunit, suggesting that ATP binding by Arp2 and Arp3 are independent, although we cannot rule out subtle effects on conformation. Surprisingly, the arp2-Y306A and arp3-F306A mutations also decreased ATP cross-linking to Arp2 and Arp3, respectively, despite their ability to suppress other mutants. However, Arp2-Y306A ATP cross-linking was reproducibly increased to ∼70% of wild type by Las17 WCA (Fig. 2 B). The arp2-D11A and arp3-D11A mutations had a lesser effect on nucleotide cross-linking, suggesting that these mutants have milder effects on ATP binding (Fig. 2 B).
We confirmed the ATP binding defects of our mutants using a fluorescent derivative of ATP, etheno-ATP, which increases in fluorescence upon binding to the Arp2/3 complex. Although this assay is more quantitative than the cross-linking assay, it cannot be used to distinguish between binding to Arp2 or Arp3 in the wild-type Arp2/3 complex. We found that wild-type yeast Arp2/3 complex binds to etheno-ATP with a K d of 2.3 μM (Fig. 2 C). Arp2/3 complex from arp3-G302Y and arp3-F306A was saturated with etheno-ATP at ∼25% the fluorescence of wild-type Arp2/3 complex, further suggesting that Arp3-G302Y and Arp3-F306A did not bind ATP under these conditions (Fig. 2 C). The residual fluorescence increase is likely due to Arp2 etheno-ATP binding, with a calculated K d of ∼20 μM. This value agrees with previous results showing a low affinity of Arp2 for ATP in the absence of an NPF (Le Clainche et al., 2001). arp3-D11A Arp2/3 complex showed a greater fluorescence increase, but exhibited a 15-fold higher K d, suggesting that Arp3-D11A bound etheno-ATP with lower affinity (Fig. 2 C). In contrast, arp2 mutant complexes exhibited similar affinity to wild-type Arp2/3 complex and only moderately decreased the fluorescence at saturation (Fig. 2 D). Thus, the fluorescence increase is mostly due to Arp3 etheno-ATP binding, with a calculated K d of ∼2 μM, similar to the previously determined value (Dayel et al., 2001). Overall, the cross-linking and etheno-ATP binding experiments demonstrated that the D11A, G302Y, and Y(F)306A mutants lower the affinity of Arp2 and Arp3 for ATP, with the G302Y mutant being most severely defective.
Arp3 nucleotide binding is required for normal actin organization and endocytosis
The Arp2/3 complex localizes to yeast cortical actin patches (Moreau et al., 1996; Winter et al., 1997), and Arp2/3 complex mutants show defects in actin patch polarity (Moreau et al., 1996; Madania et al., 1999; Winter et al., 1999) and in endocytosis (Moreau et al., 1996, 1997). To test the importance of Arp2 and Arp3 nucleotide binding in vivo, we first examined actin cytoskeleton organization in fixed cells using rhodamine phalloidin. At 25°C, actin patches in arp2-D11A, arp3-D11A, and arp3-G302Y mutants were more depolarized than in wild-type cells (Fig. 3 A, Table II). In addition, ∼10–20% of arp3-G302Y cells and a few percent of arp2-D11A and arp3-D11A cells exhibited cable-like actin aggregates, similar to those previously observed in arp3-2 mutant cells (Winter et al., 1997). Actin patches became almost completely depolarized when arp3-D11A and arp3-G302Y cells were shifted to the restrictive temperature of 37°C for 1 h (Fig. 3 A). Consistent with previous findings (Winter et al., 1999), actin cables were present in all mutants at all temperatures. In contrast to arp3-G302Y, actin patches in arp2-G302Y were of normal size and were polarized at both 25°C and 37°C (Fig. 3 A). The Arp2/3 complex localized to actin patches in both arp2 and arp3 mutants, although with slightly less intensity than in wild-type cells (Fig. 3 B). We were unable to examine arp3-G302Y localization because of synthetic sickness with HA-tagged ARC18. Interestingly, actin organization and Arp2/3 localization in arp2-Y306A and arp3-F306A mutants were indistinguishable from wild type (unpublished data), suggesting that these mutants did not decrease Arp2/3 activity.
Figure 3.

Actin cytoskeleton and endocytosis defects of Arp2 and Arp3 mutants. (A) Rhodamine phalloidin staining of yeast expressing indicated Arp2 and Arp3 mutants. Cells were grown at 25°C, or shifted to 37°C for 1 h. Arrows indicate depolarized cells. Arrowheads indicate abnormal actin aggregates. (B) Arp2/3 complex localization in Arp2 and Arp3 mutants. Actin and Arp2/3 were visualized by indirect immunofluorescence after growth at 25°C. HA-tagged Arc18 was used to immunolocalize the Arp2/3 complex. Arrowheads indicate examples of colocalization. (C) Analysis of endocytosis by fluorescence microscopy using LY uptake. Cells were incubated with LY for 1 h at 25°C. Quantification is in Table II.
Table II. Summary of Arp2 and Arp3 mutant biochemical and cellular phenotypes.
| Allele
|
Growth
|
ATP cross-linking
|
Etheno-ATP
|
Actin polarization
|
LY uptake
|
Abp1 motility (lifetime) |
Sla1 motility (lifetime) |
Nucleation activity
|
|---|---|---|---|---|---|---|---|---|
| % | Kd (μM) | % | % | % | % | |||
| WT | + | 100 | 2.3 ± 0.1 | 98 (90) | 97 | 94.5 ± 2.1 (15 ± 3 s) |
85.4 ± 5.1 (33 ± 10 s) |
1.0 |
| arp2-D11A | ss | 82.8 | 2.0 ± 0.1 | 72 (59) | 57 | 21.5 ± 5.4 | 9.2 ± 4.4 | 0.11 |
| arp2-G302Y | + | 16.9 | 1.8 ± 0.1 | 98 (90) | 91 | 75.9 ± 8.2 (24 ± 5 s) |
51.5 ± 1.3 (69 ± 24 s) |
0.28 |
| arp2-Y306A | + | 23.1 | 2.4 ± 0.2 | nd | 97 | 94.3 ± 3.4 | 89.1 ± 3.5 (33 ± 8 s) |
1.12 |
| arp3-D11A | ts, fs, ss | 68.1 | 34.5 ± 2.2 | 43 (7) | 14 | 14.5 ± 9.4 | 3.3 ± 2.9 | 0.28 |
| arp3-G302Y | ts, fs, ss | 17.1 | 22.0 ± 4.4 | 18 (4) | 15 | 6.2 ± 4.9 (29 ± 6 s) |
1.7 ± 2.9 (148 ± 52 s) |
0.24 |
| arp3-F306A | + | 29.5 | 16.7 ± 4.2 | nd | 88 | 94.3 ± 3.0 | 78.2 ± 1.7 | 0.96 |
| arp3-Y306A; arp3-G302Y | ss | nd | 1.5 × 106 | nd | 85 | 21.8 ± 1.7 (29 ± 6 s) |
11.0 ± 9.3 (98 ± 27 s) |
0.21 |
Characterization of Arp2 and Arp3 nucleotide-binding mutants. Growth: growth phenotypes on different media. ts, temperature sensitive (37°C); ss, salt sensitive; fs, formamide sensitive. ATP cross-linking: % of wild-type signal in the absence of WCA on Arp2 for arp2 mutants and Arp3 for arp3 mutants. Etheno-ATP K d: average K d ± SEM calculated from data in Fig. 2 (C and D) as described in Materials and methods. Actin polarization: calculated as % of small-budded cells containing <5 patches in the rear of the mother cell at 25°C and 37°C (parentheses). Actin was visualized with rhodamine phalloidin. LY (Lucifer yellow) uptake: measured visually as the % of the cells exhibiting bright vacuolar staining (Fig. 3 C, WT) versus faint or lack of staining (Fig. 3 C, arp3-G302Y). Abp1 motility and Sla1 motility: average % of motile patches ± SD as determined from at least 60 patches for each strain. Parentheses: average lifespan ± SD of patches in seconds as determined from at least 20 patches for each strain. Nucleation activity: fraction of barbed ends generated relative to wild type. nd, not determined.
Because the Arp2/3 complex and actin patches have been implicated in yeast endocytosis (Moreau et al., 1996, 1997; Kaksonen et al., 2003), we tested endocytic function by visualizing uptake of the fluid phase dye, Lucifer yellow (LY) at 25°C. The arp3-D11A and arp3-G302Y mutants showed a dramatic reduction in LY staining in the vacuole (Fig. 3 C). However, the arp2-Y306A and arp3-F306A mutants showed similar LY internalization to wild-type cells (Table II). The arp2-Y306A mutant suppressed the LY internalization defect of arp3-G302Y (Fig. 3 C). Importantly, LY uptake was only moderately impaired in arp2-G302Y and arp2-D11A mutant cells, which is consistent with the more mild effect on growth and actin organization (Fig. 3 C). Therefore, Arp3 nucleotide binding is more important compared with Arp2 nucleotide binding for endocytosis, the major function of cortical actin patches.
Arp3 nucleotide binding is required for Abp1 and Sla1 internalization
We next determined which steps in endocytic internalization are affected in Arp3 mutants. An endocytic pathway has been elucidated in which the behavior of yeast cortical patches is correlated with invariant changes in the protein composition of these patches over time (Kaksonen et al., 2003). Early components of this pathway, such as the endocytic adaptor Sla1, assemble as patches at the plasma membrane and remain stably associated with the membrane until they are internalized after ∼20–30 s. Late patch components, such as actin, Arp2/3 complex, and the actin filament binding protein, Abp1, are recruited to Sla1 containing patches later, and movement of actin and Abp1 off the plasma membrane coincides with Sla1 internalization. The Arp2/3 complex has previously been shown to be important for actin patch motility (Winter et al., 1997), but its importance in the context of the recently defined endocytic pathway had not yet been investigated.
Abp1-GFP, a marker for filamentous actin, and Sla1-GFP were crossed to the D11A, G302Y, and Y(F)306A mutants to visualize the dynamics of these proteins in live mutant cells. Similar results were obtained using either Sac6-GFP or Abp1-GFP as a marker for actin (unpublished data), demonstrating that Abp1-GFP is a suitable marker for filamentous actin in these cells. The arp2-D11A, arp3-D11A, and arp3-G302Y mutants significantly decreased internalization of both Abp1 and Sla1 patches at 25°C (Fig. 4, A and B; Table II; Videos 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200408177/DC1). This internalization defect resulted in significantly longer lifetimes for Sla1 cortical patches in these arp2 and arp3 mutants (Table II). In contrast, internalization of Abp1 and Sla1 patches was only moderately reduced in the arp2-G302Y mutant (Fig. 4, A and B, Videos 1 and 2). Thus, Abp1 and Sla1 internalization defects correlated with endocytosis defects observed by LY uptake, which is consistent with the conclusion that patch internalization represents endocytic internalization (Kaksonen et al., 2003). The arp2-Y306A mutant, while exhibiting wild-type Abp1 and Sla1 patch internalization, partially suppressed the internalization defects of arp3-G302Y, which is consistent with its suppression of the LY internalization defect (Fig. 4 A, Table II).
Figure 4.
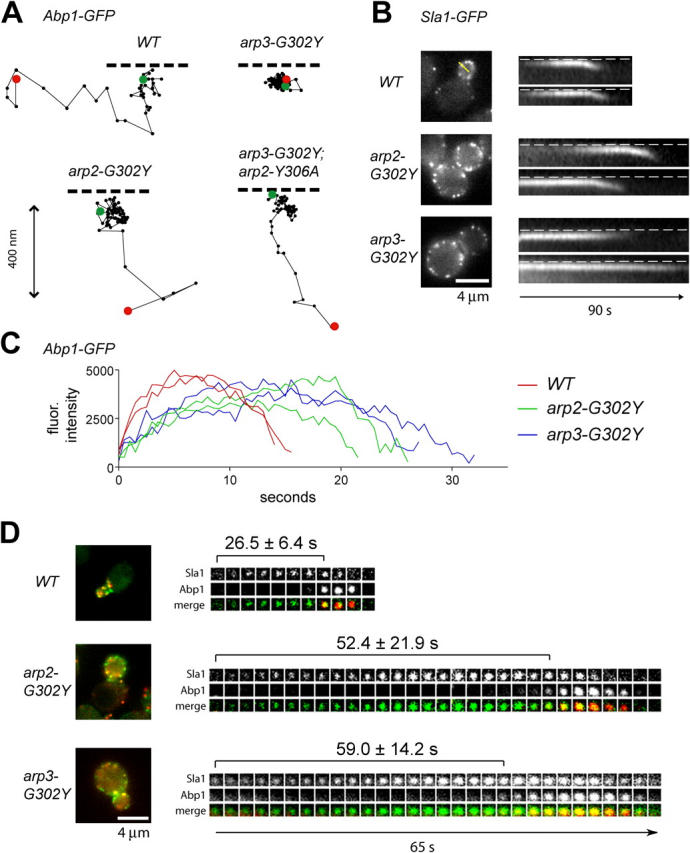
Arp2 and Arp3 mutants affect actin patch protein internalization. (A) Tracking of individual cortical Abp1 patches. Abp1-GFP was visualized every 0.25 s and patch movement traces were obtained for the entire life of patches. Green and red dots indicate first and last positions, respectively. Dotted line indicates plasma membrane. (B) Internalization of Sla1 patches. Single frames (left) and kymographs (right) of time-lapse images collected at 0.5-s intervals using a line drawn perpendicular to the plasma membrane (e.g., yellow). Dotted line: plasma membrane. (C) Fluorescence intensity of individual Abp1-GFP patches measured over time. (D) Single frames (left) and time series of single cortical patches (right) of cells expressing Abp1-GFP (red) and Sla1-CFP (green). Time-lapse between images was ∼4.55 s. Average times and SDs between Sla1 appearance and Abp1 recruitment were calculated from at least 20 patches.
The internalization defects of Arp3 mutants could have resulted from loss of spatial or temporal regulation of actin assembly at endocytic sites. Interestingly, the rate of Abp1 patch assembly was slightly decreased in both arp2-G302Y and arp3-G302Y (Fig. 4 C), resulting in longer Abp1 patch lifetimes in these mutants (Table II). In addition, the onset of actin assembly was significantly delayed in both arp2-G302Y and arp3-G302Y relative to the initial appearance of Sla1 patches (Fig. 4 D). Thus, nucleotide-bound Arp2 and Arp3 are required to initiate actin polymerization at endocytic sites with the proper timing. However, the internalization defects observed in arp3-G302Y could not be explained by actin assembly defects.
Arp2 and Arp3 mutants form defective actin comet tails in sla2Δ mutants
To further investigate the basis for the Arp3 mutant internalization defect, we analyzed the effects of the arp2-G302Y and arp3-G302Y mutants on actin polymerization at endocytic sites by taking advantage of the sla2Δ mutant phenotype. Deletion of the SLA2 gene blocks Sla1 internalization and results in the formation of actin comet tails that associate with Sla1-containing patches at the plasma membrane (Kaksonen et al., 2003). Clusters of actin comet tails can be observed in sla2Δ cells expressing GFP-tagged actin, and the treadmilling rate can be measured by the directional FRAP (Kaksonen et al., 2003).
Surprisingly, photo-bleaching of actin comet tails in sla2Δ arp2-G302Y or sla2Δ arp3-G302Y mutants revealed that the rate of actin treadmilling was only slightly reduced compared with the rate in wild-type cells (Fig. 5 A). This was consistent with the relatively normal assembly of actin at endocytic sites described above. Importantly, however, the continuity of actin tails in sla2Δ arp3-G302Y cells was severely disrupted. Whereas actin tails in sla2Δ mutants appeared smooth and continuous, all sla2Δ arp3-G302Y actin tails exhibited a broken structure (Fig. 5 B, Video 3, available at http://www.jcb.org/cgi/content/full/jcb.200408177/DC1). In these mutants, clumps of actin appeared to separate from the plasma membrane and to move into the cytoplasm at a continuous rate (Fig. 5 C). This phenotype was also observed for 40% of sla2Δ arp2-G302Y tails (Fig. 5 B and C), although the remaining tails appeared similar to wild-type. Thus, the arp3-G302Y mutant, which has an endocytic internalization defect, more severely affected the continuity of actin comet tails.
Figure 5.

Arp2 and Arp3 mutants affect the continuity of sla2 Δ actin comet tails. (A) Quantification of actin treadmilling rates using FRAP in sla2Δ cells expressing the indicated Arp mutant and Act1-GFP. Average treadmilling rates and SDs were calculated from 10 separate actin tail clusters. (B) Confocal images of whole cells (left) and magnified views of actin comet tail clusters (right). Arrowheads indicate actin clumps. (C) Kymograph of time-lapse images collected at 1-s intervals using a line drawn perpendicular to the plasma membrane (e.g., gray line, B). A section of the actin comet tail cluster (e.g., white box, B) was photo-bleached after 10 s. Arrowheads indicate where the actin network appears to separate from the plasma membrane.
In vitro nucleation activity of Arp2 and Arp3 mutants
To determine whether the more severe endocytic defects of Arp3 mutants versus Arp2 mutants reflected lower Arp2/3 nucleation activity, we used the pyrene-actin assembly assay. The in vitro nucleation activity of mutant Arp2/3 complexes activated by full-length Las17 (Rodal et al., 2003) was examined. Unless otherwise noted, we used 500 μM ATP in our assays to approximate physiological conditions. Surprisingly, the Arp2/3 complex from arp2-G302Y, which had only a subtle effect in cells, and the Arp2/3 complex from arp3-G302Y and arp3-D11A, which caused severe effects in cells, exhibited a similar decrease in activity (Fig. 6, A and B). The nucleation activity of these mutant Arp2/3 complexes was equivalent to that exhibited when the concentration of wild-type Arp2/3 complex was lowered 20-fold (Fig. 6 A). Arp2/3 complex from arp2-D11A showed the lowest nucleation activity, which is consistent with this mutant affecting Arp3 stability as well as Arp2 nucleotide binding (Fig. 6 B). The nucleation activities of arp2-G302Y and arp3-G302Y Arp2/3 complex were similar to each other with different concentrations of NPF, nucleotide, and actin, as well as with the NPFs Pan1 and Abp1 (Fig. 6, B and C, not depicted). In addition, arp2-G302Y and arp3-G302Y Arp2/3 complexes exhibited a similar reduction in the number of Y-branches formed compared with wild-type Arp2/3 complex (Fig. 6 D). Therefore, the endocytic defects of the arp3-G302Y mutant did not correlate with effects on Arp2/3 nucleation or branch formation activities.
Figure 6.
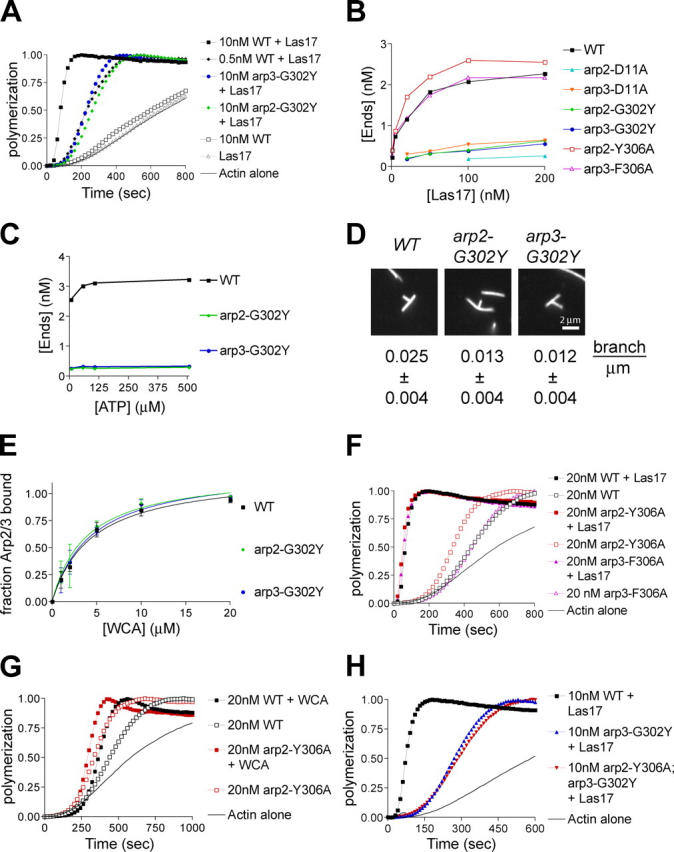
Effects of Arp2 and Arp3 mutants on Arp2/3 complex nucleation and branching. (A and F–H) Nucleation activity of Arp2 and Arp3 mutants. 2 μM actin (5% pyrene labeled) was polymerized with the indicated concentration of Arp2/3 complex and, when indicated, 50 nM Las17 or 400 nM WCA. 500 μM ATP was present in nucleation reactions unless otherwise stated. (B and C) Barbed end concentrations at 50% polymerization generated by Arp2/3 (20 nM) and different concentrations of Las17 (B) or with 50 nM Las17 and 7, 57, 107, or 507 μM ATP (C). (D) Branching activity of Arp2 and Arp3 mutants. Actin filaments polymerized with 20 nM mutant Arp2/3 complex (2 nM wild-type Arp2/3) and 200 nM Las17 were visualized with rhodamine phalloidin. Average branching efficiency ± SD was calculated as branch number per micrometer filament length, corrected for differences in nucleation activity. (E) WCA binding to Arp2/3 complex mutants. 40 nM Arp2/3 complex was incubated with various concentrations of Las17 WCA-coated beads and the fraction of bound Arp2/3 was calculated by quantifying Arp3 left in the supernatant by immunoblotting. Data points represent averages ± SEM from three experiments. K d values of 4.5 ± 0.9 μm, 3.9 ± 1.0 μm, and 4.5 ± 1.0 μm were calculated for wild-type, arp2-G302Y, and arp3-G302Y Arp2/3 complexes, respectively.
Interestingly, we found that whereas wild-type Arp2/3 nucleation activity was saturated at 200 nM Las17, arp2-G302Y and arp3-G302Y were not, suggesting that these mutants might decrease nucleation activity in part by lowering the affinity for NPF (Fig. 6 B). To test this hypothesis, we pulled down Arp2/3 complex with various amounts of WCA-coated beads. Wild-type Arp2/3 complex bound Las17 WCA with a K d of ∼4 μM, slightly higher than that reported for WCA from WASP and N-WASP (0.2–2 μM; Dayel et al., 2001; Le Clainche et al., 2001; Marchand et al., 2001; Panchal et al., 2003). Surprisingly, both arp2-G302Y and arp3-G302Y Arp2/3 complexes bound Las17 WCA with similar affinity to the wild-type Arp2/3 complex (Fig. 6 E). Furthermore, neither arp2-G302Y nor arp3-G302Y significantly affected Arp2/3 affinity for the sides of actin filaments (unpublished data). Thus, the low activity of these mutants was not due to reduced NPF affinity.
Because arp2-Y306A and arp3-F306A suppressed Arp2/3 mutant phenotypes in vivo, we purified Arp2/3 complex from arp2-Y306A and arp3-F306A to determine the biochemical basis for this suppression. Despite lowering affinity for ATP (Fig. 2), arp3-F306A Arp2/3 complex exhibited wild-type nucleation activity, whereas the arp2-Y306A Arp2/3 complex increased nucleation activity with the NPF Las17, exhibiting an activity equivalent to a fourfold higher concentration of wild-type Arp2/3 complex (Fig. 6, B and F). In addition, arp2-Y306A exhibited two- to fourfold higher activity in the absence of an NPF, similar to that of wild-type Arp2/3 complex activated with the less active Las17 WCA (Fig. 6 G). Despite this increase in activity, the arp2-Y306A mutant did not restore nucleation activity to Arp2/3 complex containing arp3-G302Y (Fig. 6 H), and it failed to rescue Arp3 nucleotide binding of arp3-G302Y (Fig. 2 C), suggesting that in vivo suppression by the arp2-Y306A mutant does not simply restore nucleotide binding to Arp3.
The arp2-Y306A suppressor results in a conformational change that mimics Arp2/3 complex activation
We next used electron microscopy and image analysis to identify conformational changes that occur upon activation of wild-type Arp2/3 complex. In addition, because the Arp2/3 complex containing the arp2-Y306A mutant exhibited constitutive activity similar to wild-type Arp2/3 complex activated by Las17 WCA (Fig. 6 G), we tested whether the arp2-Y306A mutation mimics the Arp2/3 complex activation step induced by NPF binding. Non-activated wild-type Arp2/3 complex, the arp2-Y306A Arp2/3 mutant, and wild-type Arp2/3 complex bound to Las17 WCA, were imaged by electron microscopy in negative stain (Fig. 7). Single-particle analysis resulted in three reconstructions at ∼2.5 nm resolution (Fig. 7 E), considerably higher than the 3.9 nm resolution previously reported for the yeast Arp2/3 complex (Volkmann et al., 2001). Fitting the atomic model of the nonactivated bovine Arp2/3 complex (Robinson et al., 2001) into the three-dimensional maps revealed that the overall morphology of all three reconstructions resembles that of the crystal structure (Fig. 7 A). However, differences in the density distribution between the activated and the nonactivated wild-type Arp2/3 reconstructions are clearly visible in the Arp2 and Arc15 regions (Fig. 7, B and D). This observation shows for the first time a distinct structural change upon Arp2/3 complex activation by WCA. Interestingly, no significant differences were observed when the nonactivated arp2-Y306A mutant reconstruction was compared with the activated wild-type Arp2/3 map. Thus, the structural changes induced by NPF binding in the wild-type Arp2/3 complex are similar to those observed in the mutant complex in the absence of NPF. These structural similarities explain the close resemblance of the nucleation activities of the mutant and the activated wild-type Arp2/3 complex (Fig. 6 G) and suggest that arp2-Y306A suppresses other mutants by causing the Arp2/3 complex to resemble the active conformation.
Figure 7.
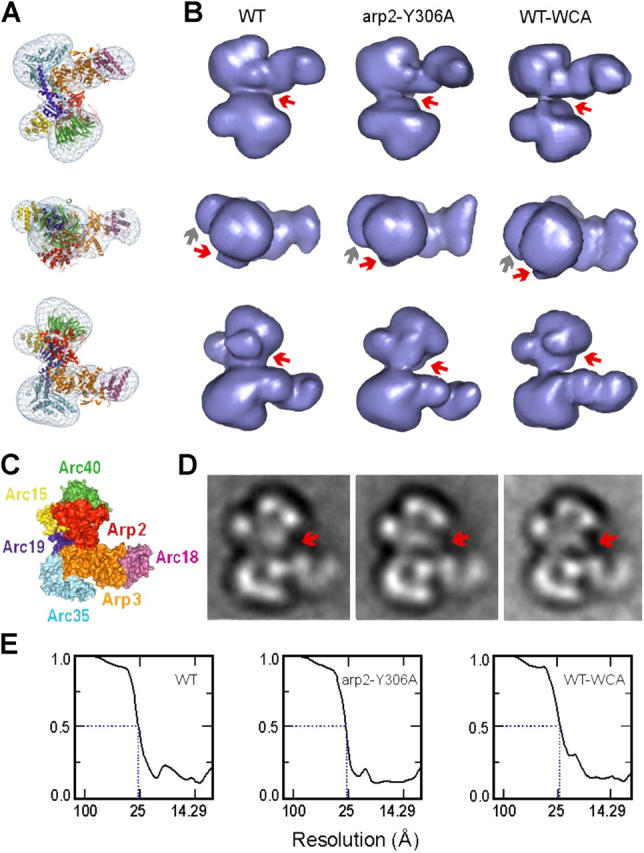
Single-particle reconstructions of Arp2/3 complexes. (A) Docking of the nonactivated bovine Arp2/3 crystal structure (ribbon diagram) into the reconstruction of nonactivated wild-type yeast Arp2/3 complex (chicken-wire representation). Color scheme follows C. The view in the second row is rotated by 90° horizontally from that in the first row; the view in the third row is rotated by 180° horizontally from that in the first row. (B) Three views of the reconstructions (solid blue surface representation) from nonactivated wild-type Arp2/3 complex (WT), nonactivated arp2-Y306A mutant (arp2-Y306A), and activated wild-type Arp2/3 complex (WT-WCA). The views match those in A. Red arrows indicate the region of Arp2 that shows differences between WT and WT-WCA/arp2-Y306A. Gray arrows indicate the differences in the Arc15 region. (C) Solvent exposed surface of the crystal structure; view matches third row in A. (D) Projection views of the three Arp2/3 reconstructions. Red arrows indicate the Arp2 region, highlighting differences seen in B. (E) Fourier shell correlation graphs for the three reconstructions calculated from two randomly selected half sets of each respective data set. The 0.5 criterion indicates ∼2.5 nm resolution for all reconstructions.
Discussion
We have generated mutants that affect either Arp2 or Arp3 nucleotide binding. Based on the effects of these mutants on endocytosis and in vitro nucleation activity, we present a model for the functions of nucleotide-bound Arp2 and Arp3 in Fig. 8. First, ATP-bound Arp2 and Arp3 are both required for full activation of the Arp2/3 complex by NPFs in vitro (Fig. 8 A). Moreover, a structural change in Arp2 accompanies activation of the Arp2/3 complex by an NPF, as observed by electron microscopy. The resulting nucleation activity is important for efficiently initiating actin polymerization at endocytic sites (Fig. 8 B, i). Once actin filaments are nucleated, actin assembly can proceed, albeit more slowly, with minimal Arp2/3 nucleation activity. Finally, Arp2/3 complex, particularly nucleotide binding by Arp3, plays a yet to be defined role, possibly anchoring actin filaments to each other and to the plasma membrane, to allow polymerization forces to be harnessed for endocytic internalization (Fig. 8, A and B, ii). The evidence supporting this model is discussed below.
Figure 8.
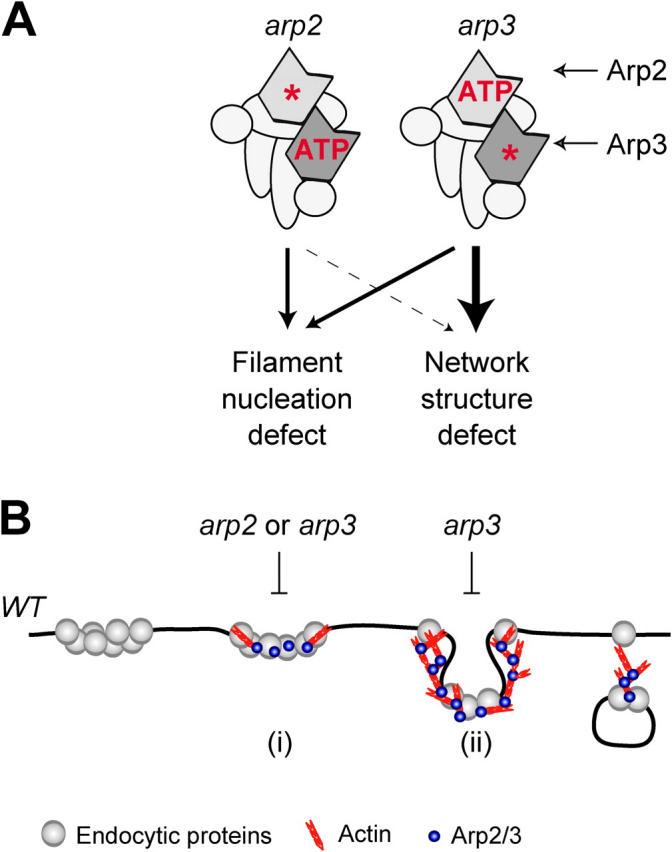
Model for nucleotide's role in Arp2/3 complex function. (A) Proposed relative contributions of Arp2 and Arp3 nucleotide binding to actin filament nucleation and a yet to be determined structural activity. (B) Proposed steps in an endocytic pathway (Merrifield et al., 2002; Kaksonen et al., 2003) affected by Arp2 and Arp3 nucleotide-binding mutants. Both Arp2 and Arp3 mutants affect the initiation and the rate of actin assembly at endocytic sites (i), but Arp3 mutants more dramatically inhibit internalization of endocytic proteins as a consequence of defects in actin network structure and integrity (ii).
Importance of Arp2 and Arp3 nucleotide binding for actin filament nucleation
Of the mutants we characterized, arp2-G302Y and arp3-G302Y most dramatically inhibited ATP binding to Arp2 and Arp3, respectively. Previous studies reported that ATP binding is required for Arp2/3 complex nucleation activity, but these studies were unable to tease apart the individual contributions of ATP binding to Arp2 versus Arp3 (Dayel et al., 2001; Le Clainche et al., 2001). Here, we demonstrated using the G302Y mutants that ATP-bound Arp2 and Arp3 both contribute to Arp2/3 nucleation activity in vitro. Importantly, raising the ATP concentration to physiological levels did not increase the in vitro nucleation activity of these mutant complexes, suggesting that their nucleation activities in vivo resemble those of nucleotide-free Arp2 and Arp3 complexes.
Previous studies suggested that nucleotide-bound Arp2 and Arp3 are important for WCA binding (Dayel et al., 2001). Interestingly, arp2-G302Y and arp3-G302Y Arp2/3 complexes bound WCA with similar affinity to the wild-type complex, demonstrating that the decreased activity of these mutants is not due to reduced NPF binding. A recent study probed conformational states of the human Arp2/3 complex using fluorescence resonance energy transfer (FRET) between fluorophores attached to different subunits of the complex (Goley et al., 2004). Using increase in FRET to monitor WCA binding, it was demonstrated that the arp2-D11A and arp3-D11A mutants reduce the ability of WCA to induce conformational changes in the Arp2/3 complex (Goley et al., 2004). Previous studies using mutations in WCA also suggested the existence of distinct WCA-binding and WCA-activation steps during Arp2/3 activation (Marchand et al., 2001; Panchal et al., 2003). These results support a model in which NPF-mediated Arp2/3 activation requires ATP bound to Arp2 and Arp3.
Interestingly, the arp3-D11A mutant, which was expected to bind ATP in our nucleation assays, had a similar nucleation defect to the arp3-G302Y mutant. This result suggested that the arp3-D11A mutation caused structural changes in the Arp2/3 complex even when nucleotide was bound to Arp3. Consistent with this interpretation, both the arp3-D11A and arp3-G302Y mutants prevented conformational changes in human Arp2/3 complex that occur upon Arp3 nucleotide binding as assessed by FRET (Goley et al., 2004). Thus, the arp3-D11A mutant appears to uncouple conformational changes in the Arp2/3 complex from Arp3 nucleotide binding, mimicking the nucleotide-free arp3-G302Y mutant. The similarity of the in vivo and in vitro phenotypes of the arp3-D11A and arp3-G302Y mutants suggests that defects in these mutants are specifically related to Arp3 nucleotide binding. In addition, the dramatic endocytic phenotypes seen in arp3-D11A and arp3-G302Y demonstrate the importance of this conformational change to Arp2/3 function in vivo.
Mechanism of Arp2/3 complex activation
Using genetic crosses of our Arp2 and Arp3 mutants, we found that arp2-Y306A and arp3-F306A are suppressors of Arp2/3 complex defects. Interestingly, Arp2/3 complex purified from arp2-Y306A, but not from arp3-F306A, increased the nucleation activity of the Arp2/3 complex with and without Las17. This difference in activity was also seen in vivo, as arp2-Y306A, but not arp3-F306A, was able to suppress las17-11, identifying a possible functional difference between Arp2 and Arp3. Because the NBP is critical for regulating actin structure, and because changes in the Arp2 region were visible in electron microscopy reconstructions of wild-type Arp2/3 complex upon activation, we speculated that the arp2-Y306A mutant causes a conformational change that increases activities of the Arp2/3 complex.
Because arp2-Y306A Arp2/3 complex exhibited similar nucleation activity to wild-type Arp2/3 complex activated by Las17 WCA, we examined the conformation of this mutant by comparing its structure to both inactive and WCA-bound wild-type Arp2/3 complex. Indeed, in three-dimensional reconstructions, arp2-Y306A Arp2/3 complex resembled wild-type Arp2/3 complex activated by Las17 WCA, showing a similar difference in the region corresponding to Arp2. Previous evidence demonstrating that NPFs enhance Arp2 nucleotide binding support a conformational change in Arp2 upon NPF binding (Le Clainche et al., 2001). Because it has been proposed that the Arp2/3 complex must undergo a large conformational change to bring Arp2 and Arp3 together to form a template dimer (Robinson et al., 2001; Volkmann et al., 2001), we suggest that a conformational change in Arp2 is a critical step in this process.
It was surprising that a mutant that lowers affinity for ATP results in Arp2/3 activation. However, this lower affinity might indicate greater conformational flexibility of the NBP, allowing Arp2 to adopt an active conformation in the absence of an NPF. Interestingly, changing F306 in yeast actin to tyrosine increased the specificity of yeast actin for ATP versus GTP (Wen et al., 2002), possibly indicating a more compact conformation. We also found that in addition to the arp2-Y306A mutant, the arp2-D157E mutant also suppressed arp3 mutants. In actin, this mutation has been shown to increase the nucleotide exchange rate (Wolven et al., 2000), suggesting a more open conformation for the NBP. We speculate that the conformational changes observed in the arp2-Y306A Arp2/3 complex result from greater conformational flexibility of the Arp2 NBP.
Function of Arp2 and Arp3 nucleotide binding during endocytosis: general biological implications
Analysis of actin organization and endocytosis in arp2-G302Y and arp3-G302Y demonstrated an important functional difference between nucleotide binding by Arp2 versus Arp3 in vivo. The arp3-G302Y mutant caused severe endocytic defects, whereas arp2-G302Y showed only slight defects. However, the in vitro nucleation activities of these two mutants were indistinguishable. Importantly, the arp3-G302Y mutant assembled actin filaments at endocytic sites as well as arp2-G302Y, indicating that nucleation of actin filaments alone is not sufficient to drive endocytic internalization. We speculate that the structure or integrity of the cortical actin network at endocytic sites is disrupted in Arp3 mutants.
How might Arp3 contribute to actin cortical patch integrity and function? Because Arp3 nucleotide binding influences the conformation of the Arp2/3 complex (Goley et al., 2004), Arp3 mutants are likely to affect multiple interactions of the Arp2/3 complex, possibly with one or several of the four known yeast NPFs, Las17, Myo3/5, Pan1, and Abp1. Interestingly, Myo5 appears in patches immediately preceding internalization, suggesting that its interaction with the Arp2/3 complex plays an important role in vesicle formation (Jonsdottir and Li, 2004). Although wild-type, arp2-G302Y, and arp3-G302Y Arp2/3 complexes bound Las17 WCA with similar affinity, conformational changes upon actin filament binding and Arp2 ATP hydrolysis might alter interactions with NPFs subsequent to branch formation (Marchand et al., 2001; Volkmann et al., 2001; Le Clainche et al., 2003; Dayel and Mullins, 2004). Connections between NPFs and the Arp2/3 complex in Y-branches might anchor branched actin filaments to the plasma membrane allowing forces to be applied for endocytic internalization. In addition, interactions with NPFs could be important for maintaining branched actin filament networks, such as those recently observed in actin structures isolated from yeast (Young et al., 2004). The phenotype of arp3-G302Y sla2Δ actin tails, in which clumps of actin appeared to separate from the plasma membrane, supports a defect in the integrity of plasma membrane attachment, actin network integrity, or both. Discontinuous actin comet tails have also been observed for NPF mutant “hopping” Listeria in vivo, and using a reconstituted motility system in vitro, although the molecular basis for this phenomenon is not understood (Lasa et al., 1997; Bernheim-Groswasser et al., 2002). Further analysis of the molecular interactions of Arp2 and Arp3 mutants is required to determine the mechanism by which Arp3 mutants affect the functionality of actin patches.
Because the arp2-G302Y and arp3-G302Y mutants affected in vitro nucleation activity indistinguishably, the similarities between these mutants provided insight into the function of Arp2/3 complex nucleation. Both mutants exhibited a considerable delay in the onset of actin polymerization at endocytic sites. This delay could have resulted from a lower nucleation rate or a delay in the recruitment of the Arp2/3 complex to endocytic sites. In addition, we cannot rule out the possibility of Arp2/3-independent actin polymerization at these sites. Interestingly, once initiated, the rate of actin assembly at endocytic sites in arp2-G302Y and arp3-G302Y mutants was only slightly lower in otherwise normal cells or in sla2Δ cells. This observation was similar to the moderate decrease in motility rates for Listeria ActA mutants that significantly decreased in vitro nucleation activity (Skoble et al., 2000). Therefore, Arp2/3 nucleation activity in these contexts appears more important for initiating actin polymerization than for sustaining it.
In summary, we found that analysis of the yeast endocytic pathway provided a sensitive assay for distinguishing functions of the Arp2/3 complex. The similarity of the effects of our NBP mutants of the yeast Arp2/3 complex to the analogous mutants made in the human Arp2 and Arp3 (Goley et al., 2004), and similarity in the recruitment of Arp2/3 complex to endocytic sites in yeast and mammalian cells (Kaksonen et al., 2003; Merrifield et al., 2004), suggest that our findings are directly applicable to the function of the Arp2/3 complex in multicellular eukaryotes.
Materials and methods
Strains, media, and genetic methods
Yeast strains and plasmids used in this study are listed in Tables S2 and S3, available at http://www.jcb.org/cgi/content/full/jcb.200408177/DC1.
Construction of mutant and tagged strains
An ARP2::URA3 integration construct obtained from a previous study (Madania et al., 1999), pIMW18-U, and an ARP3::LEU2 integration construct, pDD1698, were mutagenized using QuikChange XL site-directed mutagenesis kit (Stratagene). Mutants were integrated into DDY2918 and DDY2919 as described previously (Wertman et al., 1992). Integration of the correct allele was confirmed by change in restriction site or sequencing. ARC18 containing a COOH-terminal HA tag with a TEV protease site was generated using pBG265 (a gift from B. Goode, Brandeis University, Waltham, MA) and was integrated into the endogenous locus.
Protein purification
Arp2/3 complex was purified using an HA-tagged Arc18 subunit and was stored in HEKG5 (20 mM Hepes, pH 7.5, 1 mM EDTA, 50 mM KCl, 5% glycerol) buffer. Full-length Las17 was purified as described previously (Rodal et al., 2003). GST-tagged Las17WCA was purified from bacteria using pDD1122. Details of these purifications and actin purification procedures are provided in the supplemental materials.
ATP binding assays
ATP cross-linking was performed using 5 μM ATP supplemented with 100 μCi α-[32P]ATP. Purified Arp2/3 complex (0.43 μM) with or without WCA (10 μM) in KMEI (50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 10 mM imidazole, pH 7.0) was equilibrated with ATP and irradiated at a 1-cm distance with 254 nm UV light for 5 min using a hand-held Mineralight lamp. Samples were subjected to SDS-PAGE and visualized/quantified using film or a Typhoon 9400 Phosphorimager (Molecular Dynamics). Etheno-ATP binding was measured at 18°C using excitation and emission wavelengths of 350 and 410 nm, respectively. Increasing amounts of etheno-ATP were added to 0.6 μM Arp2/3 complex in KMEI + 0.2 M acrylamide, and the background fluorescence of unbound etheno-ATP was subtracted, as described previously (Dayel et al., 2001). K d values were calculated by fitting the data to a one-site binding equation using GraphPad Prism (GraphPad Software, Inc.).
Microscopy
Phalloidin staining, immunofluorescence, and LY uptake were performed as described previously (Belmont and Drubin, 1998). For live cell imaging, log phase cells grown at 25°C were adhered to the surface of a Con A–coated coverslip, inverted onto a glass slide, and sealed with vacuum grease (Dow Corning). All imaging was done at RT in synthetic media. Single fluorophore images (Abp1-GFP, Sla1-GFP, and rhodamine phalloidin) were obtained using a microscope (model TE300; Nikon), 100×/NA 1.4 objective, and an Orca-100 Camera (Hamamatsu) controlled by Image Pro Plus software (Phase 3 Imaging). Dual fluorophore images (Abp1-GFP/Sla1-CFP) were obtained using a microscope (model IX81; Olympus), 100×/NA 1.4 objective, and an Orca-ER Camera (Hamamatsu), controlled by Slidebook 4 software (Intelligent Imaging Innovations, Inc.). Dual fluorophore movies were obtained using a CFP-YFP filter set (JP4, Chroma) and motorized excitation and emission filter wheels (Sutter Instruments). Actin comet tails were observed using a confocal microscope (model 510; Carl Zeiss MicroImaging, Inc.) with a 63×/NA 1.4 objective. Photobleaching was performed by scanning a small region three times at full laser intensity at 488 nm, followed by continued image acquisition at low laser intensity. Images used to track individual Abp1 patches were smoothed by 3 × 3 pixel averaging and then thresholded with an adaptive thresholding algorithm, which takes into account the local intensity level. Patch positions were determined by calculating the center of fluorescence intensity for each patch. The patch positions of each frame were then connected to trace patch trajectories. Patch intensity represents the sum of the intensity of each pixel making up an individual Abp1 patch. Custom-made software was used for all patch tracking calculations. Processing of individual images and movies was performed using Image J software (http://rsb.info.nih.gov/ij/).
Antibodies
Arp2 and Arp3 were detected by immunoblotting using antibodies sc-11968 and sc-11973 (Santa Cruz Biotechnology, Inc.). Anti-HA 12CA5 (Roche) was used for immunofluorescence with Arc18-HA.
Nucleation, branching, and WCA binding assays
Actin assembly reactions were performed using 2 μM actin (5% pyrene labeled) as described previously (Duncan et al., 2001). Barbed end concentrations were calculated from the rate of actin assembly at 50% polymerization using a rate constant of 11.6 μM−1 s−1 (Higgs et al., 1999). Branched filaments were observed by stabilizing actin filaments after polymerization was complete with equimolar rhodamine phalloidin (Molecular Probes) and were processed for imaging as described previously (Amann and Pollard, 2001). Branching was quantified by first calculating total filament length using Fovea Pro 3.0 (Reindeer Graphics, Inc.) for Adobe Photoshop 7.0 (Adobe Systems, Inc.), and then counting branches manually. More than 8,000 or 13,000 μm of filaments were analyzed for wild-type or Arp2/3 mutants, respectively. WCA binding was measured using glutathione beads (Sigma-Aldrich) coated with GST-WCA. Binding was performed at RT for 30 min in G buffer (5 mM Tris, pH 7.5, 0.2 mM ATP, 0.2 mM CaCl2, 0.2 mM DTT) supplemented with 50 mM KCl, 2 mM MgCl2, and 0.5 mM ATP. Amounts of bound Arp3 were quantified by immunoblotting using a Typhoon 9400 Phosphorimager (Molecular Dynamics) and K d values were calculated using GraphPad Prism (GraphPad Software, Inc.).
Electron microscopy
50 nM Arp2/3 complex (wild-type or arp2-Y306A mutant) was diluted in F buffer (2 mM imidazole, pH 7.0, 50 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.2 mM DTT, 0.1 mM ATP, 0.02% azide), applied to a carbon coated EM grid and stained with 2% uranyl acetate. 500 nM Las17 WCA was mixed with the wild-type Arp2/3 complex for generating the activated Arp2/3 samples. Images were obtained under low-dose conditions using a Tecnai 12 microscope (FEI electron optics) equipped with a Lab6 filament at 120 kV at a nominal magnification of 67,000× and at ∼1-μm defocus. The micrographs were digitized on a SCAI scanner (Z/I Imaging) at 7-μm raster and compressed to a final pixel size of 0.6 nm.
Image processing
Interactive selection from 16, 17, and 30 micrographs yielded 2,774, 2,654, and 2,330 particles from nonactivated wild-type Arp2/3, nonactivated arp2-Y306A mutant, and wild-type yeast Arp2/3 with WCA, respectively. The particles were processed and analyzed using the EMAN (Ludtke et al., 1999) and CoAn (Volkmann and Hanein, 1999; Volkmann and Hanein, 2003) software packages. A correction for the contrast transfer function was applied for all images. Particles were first sorted into 89 classes according to their mutual similarity without the use of an external reference. Subsets of these views were used to generate initial references using a Fourier-based common-line approach. The initial models were iteratively refined against the data. Convergence was achieved after 7–10 iterations. In addition to these reference-free models, a density map calculated from the bovine Arp2/3 crystal structure was used for separate refinements, which converged to the same reconstructions as the data-based models. To ensure the model independence of the differences between activated and nonactivated complex, the final reconstruction of the activated complex was used as a starting model with the nonactivated data and vice versa. In both cases, the distinct features of the reconstructions were recovered after a few rounds of refinement. A similar calculation was performed with the nonactivated and the mutant complex, also with the result that the characteristic features were recovered after a few iterations. The crystal structure of the nonactivated bovine Arp2/3 complex was docked into each of the reconstructions using CoAn. The correlation between the docked structure and the reconstructions was between 84 and 85% at 2.5-nm resolution.
Online supplemental material
Supplemental videos showing Abp1, Sla1 dynamics (Videos 1 and 2), and actin treadmilling in sla2Δ cells (Video 3); tables showing Arp2/3 mutant growth phenotypes (Table S1), yeast strains used in this study (Table S2), and plasmids used in this study (Table S3); and supplemental materials and methods are available online at http://www.jcb.org/cgi/content/full/jcb.200408177/DC1.
Acknowledgments
We thank S. Almo and K. Shokat for advice on which mutations to make and for helpful discussions about the effects of these mutations. We also thank B. Goode, B. Winsor, and members of the Drubin lab (M. Duncan, C. Toret, and J. Toshima) for providing reagents. Finally, we thank C. Le Clainche and E. Goley for critically reading the manuscript and members of the Drubin, Welch, Hanein, and Volkmann labs for helpful discussions.
This work was supported by National Institutes of Health (NIH) grants GM50399 and GM42759 to D.G. Drubin and GM59609 to M. Welch. The electron microscopy and image analysis was supported by the NIH Cell Migration Consortium (U54 GM646346) and NIH grant P01 GM66311 for D. Hanein and N. Volkmann.
Abbreviations used in this paper: Arp, actin-related protein; FRET, fluorescence resonance energy transfer; LY, Lucifer yellow; NBP, nucleotide-binding pocket; NPF, nucleation promoting factor; WASP, Wiskott-Aldrich syndrome protein.
References
- Amann, K.J., and T.D. Pollard. 2001. The Arp2/3 complex nucleates actin filament branches from the sides of pre-existing filaments. Nat. Cell Biol. 3:306–310. [DOI] [PubMed] [Google Scholar]
- Belmont, L.D., and D.G. Drubin. 1998. The yeast V159N actin mutant reveals roles for actin dynamics in vivo. J. Cell Biol. 142:1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont, L.D., A. Orlova, D.G. Drubin, and E.H. Egelman. 1999. a. A change in actin conformation associated with filament instability after Pi release. Proc. Natl. Acad. Sci. USA. 96:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont, L.D., G.M. Patterson, and D.G. Drubin. 1999. b. New actin mutants allow further characterization of the nucleotide binding cleft and drug binding sites. J. Cell Sci. 112(Pt 9):1325–1336. [DOI] [PubMed] [Google Scholar]
- Bernheim-Groswasser, A., S. Wiesner, R.M. Golsteyn, M.F. Carlier, and C. Sykes. 2002. The dynamics of actin-based motility depend on surface parameters. Nature. 417:308–311. [DOI] [PubMed] [Google Scholar]
- Blanchoin, L., K.J. Amann, H.N. Higgs, J.B. Marchand, D.A. Kaiser, and T.D. Pollard. 2000. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature. 404:1007–1011. [DOI] [PubMed] [Google Scholar]
- Chen, X., J. Peng, M. Pedram, C.A. Swenson, and P.A. Rubenstein. 1995. The effect of the S14A mutation on the conformation and thermostability of Saccharomyces cerevisiae G-actin and its interaction with adenine nucleotides. J. Biol. Chem. 270:11415–11423. [DOI] [PubMed] [Google Scholar]
- Dayel, M.J., and R.D. Mullins. 2004. Activation of Arp2/3 complex: addition of the first subunit of the new filament by a WASP protein triggers rapid ATP hydrolysis on Arp2. PLoS Biol. 2:E91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayel, M.J., E.A. Holleran, and R.D. Mullins. 2001. Arp2/3 complex requires hydrolyzable ATP for nucleation of new actin filaments. Proc. Natl. Acad. Sci. USA. 98:14871–14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan, M.C., M.J. Cope, B.L. Goode, B. Wendland, and D.G. Drubin. 2001. Yeast Eps15-like endocytic protein, Pan1p, activates the Arp2/3 complex. Nat. Cell Biol. 3:687–690. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein, A.E., and D.G. Drubin. 2003. Actin assembly and endocytosis: from yeast to mammals. Annu. Rev. Cell Dev. Biol. 19:287–332. [DOI] [PubMed] [Google Scholar]
- Goley, E.D., S.E. Rodenbusch, A.C. Martin, and M.D. Welch. 2004. Critical conformational changes in the Arp2/3 complex are induced by nucleotide and nucleation promoting factor. Mol. Cell. 16:269–279. [DOI] [PubMed] [Google Scholar]
- Higgs, H.N., L. Blanchoin, and T.D. Pollard. 1999. Influence of the C terminus of Wiskott-Aldrich syndrome protein (WASp) and the Arp2/3 complex on actin polymerization. Biochemistry. 38:15212–15222. [DOI] [PubMed] [Google Scholar]
- Jonsdottir, G.A., and R. Li. 2004. Dynamics of yeast Myosin I: evidence for a possible role in scission of endocytic vesicles. Curr. Biol. 14:1604–1609. [DOI] [PubMed] [Google Scholar]
- Kabsch, W., H.G. Mannherz, D. Suck, E.F. Pai, and K.C. Holmes. 1990. Atomic structure of the actin:DNase I complex. Nature. 347:37–44. [DOI] [PubMed] [Google Scholar]
- Kaksonen, M., Y. Sun, and D.G. Drubin. 2003. A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell. 115:475–487. [DOI] [PubMed] [Google Scholar]
- Lasa, I., E. Gouin, M. Goethals, K. Vancompernolle, V. David, J. Vandekerckhove, and P. Cossart. 1997. Identification of two regions in the N-terminal domain of ActA involved in the actin comet tail formation by Listeria monocytogenes. EMBO J. 16:1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Clainche, C., D. Didry, M.F. Carlier, and D. Pantaloni. 2001. Activation of Arp2/3 complex by Wiskott-Aldrich syndrome protein is linked to enhanced binding of ATP to Arp2. J. Biol. Chem. 276:46689–46692. [DOI] [PubMed] [Google Scholar]
- Le Clainche, C., D. Pantaloni, and M.F. Carlier. 2003. ATP hydrolysis on actin-related protein 2/3 complex causes debranching of dendritic actin arrays. Proc. Natl. Acad. Sci. USA. 100:6337–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke, S.J., P.R. Baldwin, and W. Chiu. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128:82–97. [DOI] [PubMed] [Google Scholar]
- Madania, A., P. Dumoulin, S. Grava, H. Kitamoto, C. Scharer-Brodbeck, A. Soulard, V. Moreau, and B. Winsor. 1999. The Saccharomyces cerevisiae homologue of human Wiskott-Aldrich syndrome protein Las17p interacts with the Arp2/3 complex. Mol. Biol. Cell. 10:3521–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand, J.B., D.A. Kaiser, T.D. Pollard, and H.N. Higgs. 2001. Interaction of WASP/Scar proteins with actin and vertebrate Arp2/3 complex. Nat. Cell Biol. 3:76–82. [DOI] [PubMed] [Google Scholar]
- Merrifield, C.J., M.E. Feldman, L. Wan, and W. Almers. 2002. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 4:691–698. [DOI] [PubMed] [Google Scholar]
- Merrifield, C.J., B. Qualmann, M.M. Kessels, and W. Almers. 2004. Neural Wiskott-Aldrich syndrome protein (N-WASP) and the Arp2/3 complex are recruited to sites of clathrin-mediated endocytosis in cultured fibroblasts. Eur. J. Cell Biol. 83:13–18. [DOI] [PubMed] [Google Scholar]
- Moreau, V., A. Madania, R.P. Martin, and B. Winsor. 1996. The Saccharomyces cerevisiae actin-related protein Arp2 is involved in the actin cytoskeleton. J. Cell Biol. 134:117–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau, V., J.M. Galan, G. Devilliers, R. Haguenauer-Tsapis, and B. Winsor. 1997. The yeast actin-related protein Arp2p is required for the internalization step of endocytosis. Mol. Biol. Cell. 8:1361–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, F., C. Egile, T. Lipkin, and R. Li. 2004. ARPC1/Arc40 mediates the interaction of the Arp2/3 complex with WASP family activators. J. Biol. Chem. 279:54629–54636. [DOI] [PubMed] [Google Scholar]
- Panchal, S.C., D.A. Kaiser, E. Torres, T.D. Pollard, and M.K. Rosen. 2003. A conserved amphipathic helix in WASP/Scar proteins is essential for activation of Arp2/3 complex. Nat. Struct. Biol. 10:591–598. [DOI] [PubMed] [Google Scholar]
- Robinson, R.C., K. Turbedsky, D.A. Kaiser, J.B. Marchand, H.N. Higgs, S. Choe, and T.D. Pollard. 2001. Crystal structure of Arp2/3 complex. Science. 294:1679–1684. [DOI] [PubMed] [Google Scholar]
- Rodal, A.A., A.L. Manning, B.L. Goode, and D.G. Drubin. 2003. Negative regulation of yeast WASp by two SH3 domain-containing proteins. Curr. Biol. 13:1000–1008. [DOI] [PubMed] [Google Scholar]
- Sablin, E.P., J.F. Dawson, M.S. VanLoock, J.A. Spudich, E.H. Egelman, and R.J. Fletterick. 2002. How does ATP hydrolysis control actin's associations? Proc. Natl. Acad. Sci. USA. 99:10945–10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoble, J., D.A. Portnoy, and M.D. Welch. 2000. Three regions within ActA promote Arp2/3 complex-mediated actin nucleation and Listeria monocytogenes motility. J. Cell Biol. 150:527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1999. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann, N., and D. Hanein. 1999. Quantitative fitting of atomic models into observed densities derived by electron microscopy. J. Struct. Biol. 125:176–184. [DOI] [PubMed] [Google Scholar]
- Volkmann, N., and D. Hanein. 2003. Docking of atomic models into reconstructions from electron microscopy. Methods Enzymol. 374:204–225. [DOI] [PubMed] [Google Scholar]
- Volkmann, N., K.J. Amann, S. Stoilova-McPhie, C. Egile, D.C. Winter, L. Hazelwood, J.E. Heuser, R. Li, T.D. Pollard, and D. Hanein. 2001. Structure of Arp2/3 complex in its activated state and in actin filament branch junctions. Science. 293:2456–2459. [DOI] [PubMed] [Google Scholar]
- Vorobiev, S., B. Strokopytov, D.G. Drubin, C. Frieden, S. Ono, J. Condeelis, P.A. Rubenstein, and S.C. Almo. 2003. The structure of nonvertebrate actin: implications for the ATP hydrolytic mechanism. Proc. Natl. Acad. Sci. USA. 100:5760–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, M.D., and R.D. Mullins. 2002. Cellular control of actin nucleation. Annu. Rev. Cell Dev. Biol. 18:247–288. [DOI] [PubMed] [Google Scholar]
- Wen, K.K., X. Yao, and P.A. Rubenstein. 2002. GTP-yeast actin. J. Biol. Chem. 277:41101–41109. [DOI] [PubMed] [Google Scholar]
- Wertman, K.F., D.G. Drubin, and D. Botstein. 1992. Systematic mutational analysis of the yeast ACT1 gene. Genetics. 132:337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter, D., A.V. Podtelejnikov, M. Mann, and R. Li. 1997. The complex containing actin-related proteins Arp2 and Arp3 is required for the motility and integrity of yeast actin patches. Curr. Biol. 7:519–529. [DOI] [PubMed] [Google Scholar]
- Winter, D.C., E.Y. Choe, and R. Li. 1999. Genetic dissection of the budding yeast Arp2/3 complex: a comparison of the in vivo and structural roles of individual subunits. Proc. Natl. Acad. Sci. USA. 96:7288–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolven, A.K., L.D. Belmont, N.M. Mahoney, S.C. Almo, and D.G. Drubin. 2000. In vivo importance of actin nucleotide exchange catalyzed by profilin. J. Cell Biol. 150:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, M.E., J.A. Cooper, and P.C. Bridgman. 2004. Yeast actin patches are networks of branched actin filaments. J. Cell Biol. 166:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]