

Abstract
Ultradian rhythms are widespread phenomena found in various biological organisms. A typical example is the defecation behavior of the nematode Caenorhabditis elegans, which repeats at about 45-sec intervals. To elucidate the mechanism, we studied flr-1 mutants, which show very short defecation cycle periods. The mutations also affect some food-related functions, including growth rate, the expulsion step of defecation behavior, and the regulation of the dauer larva (a nonfeeding, special third-stage larva) formation in the unc-3 (Olf-1/EBF homolog) background. The flr-1 gene encodes a novel ion channel belonging to the DEG/ENaC (C. elegans degenerin and mammalian epithelial sodium channel) superfamily. A flr-1∷GFP (green fluorescent protein) fusion gene that can rescue the flr-1 mutant phenotypes is expressed only in the intestine from embryos to adults. These results suggest that FLR-1 may be a component of an intestinal regulatory system that controls the defecation rhythm as well as other functions.
Ultradian rhythms, i.e., rhythms having shorter periods than a day, are found in diverse aspects of many biological organisms (1–3). The biological importance of various ultradian rhythms has been discussed. For instance, Ca2+ oscillations play a role in fertilization, etc. (4), whereas metabolic oscillations might function as a synchronizing clock (5). Defects in the heartbeat rhythm result in a disease called heartbeat arrhythmia (6), and an irregular REM/non-REM sleep cycle can cause insomnia (7). The courtship song of Drosophila, which has a species-specific rhythm, stimulates mating behavior (8). The temporal perception of humans, in which patients with Parkinson’s disease have abnormalities, may be controlled by neural oscillations in the brain (9). Thus, it is worth elucidating the mechanisms of generation and regulation of ultradian rhythms. Because there are probably several different mechanisms, dissection of individual rhythms will become important to classify them and to find basic rules.
The defecation behavior of the nematode Caenorhabditis elegans provides a typical and interesting example of an ultradian rhythm, which repeats with a period of approximately 45 sec (10). The period stays approximately constant at temperatures from 19°C to 30°C, while it is modulated by food concentration (11). Each defecation behavior consists of three sequential motor steps: posterior body-wall muscle contraction (pBoc), anterior body-wall muscle contraction (aBoc), and expulsion (Exp). Cell-ablation experiments showed that AVL neuron and AVL and DVB neurons contribute to mediate the signals of aBoc and Exp, respectively (12).
Mutants in genes flr-1, flr-3, and flr-4 (class 1 flr mutants) show very short defecation cycle periods (2). They were isolated first by resistance to fluoride ion (13). However, the defecation cycle abnormality was noticed when some alleles of flr-1 and flr-4 were isolated in a screen for mutants showing this phenotype (2). Besides the two phenotypes, they have the following characteristics: (i) slow growth rate and small brood size, (ii) abnormality in dauer larva formation in the unc-3 background (ref. 13 and this study), and (iii) frequent loss of the Exp step and weak muscle contraction in the pBoc step (2). Molecular genetic studies on those mutants will help elucidate the mechanism of regulation of the ultradian rhythm.
In this paper we first show that flr-1 mutants have diverse abnormalities. Then we demonstrate that flr-1 gene encodes an ion channel belonging to the degenerin (DEG)/amiloride-sensitive epithelial sodium channel (ENaC) superfamily, and that FLR-1 acts in the intestine, based on the expression pattern of a flr-1∷GFP (green fluorescent protein) fusion gene that can rescue the mutant phenotypes. Finally, we discuss how FLR-1, acting in the intestine, can control the defecation rhythm as well as other diverse functions.
MATERIALS AND METHODS
Strains.
All the strains used in this study derive from C. elegans var. Bristol N2. The mutations used were flr-1(sa96, ut1, ut2, ut4, ut6, ut11), flr-2(ut5), flr-5(ut73), unc-3(e151), and unc-3(cn4146). Animals were grown at 20°C as described (14).
Assays of Phenotypes.
Defecation behaviors were observed at 23°C essentially as described (2). The defecation motor steps of class 1 flr mutants and class 2 class 1 double mutants sometimes became too weak to observe even the pBoc step, and the animals looked as if they ceased defecation behavior for a long time. Hence, we did not include defecation interval lengths longer than 100 sec in the statistics. Such cases are indicated by asterisks in Figs. 1 and 2. Extra-long defecation intervals of N2 animals on the plates containing 400 μg/ml of NaF also were omitted in the statistics, although pBoc did not look weak in this case. Dauer formation (15), growth rate, and fluoride sensitivity (13) were assayed as described.
Figure 1.

Defecation interval length and the percentage of defecation behaviors having the Exp step. The graphs show the relationship between the length of each defecation interval (abscissa) and that of the next interval (ordinate). The bars on the right of the graphs represent the percentage of defecation behaviors that show the Exp step (Exp/pBoc). We used plates containing 400 μg/ml of NaF in the experiments shown in the middle two graphs and plates without NaF in other experiments. The numbers above the graphs show the average defecation cycle period ± SD (number of defecation intervals measured) and Exp/pBoc (number of pBoc observed). See Materials and Methods for the meaning of the ∗. Data not shown are the following: flr-1 alleles other than ut11, 34.0 ± 15.4 sec (106)* and 31% (121) for flr-1(ut4); 26.6 ± 8.5 sec (116) and 6% (126) for flr-1(ut6); 24.7 ± 6.5 sec (107)* and 64% (120) for flr-1(sa96). Another class 2 flr mutation flr-2(ut5): 55.4 ± 7.7 sec (101) and 96% (111) for flr-2(ut5), and 37.6 ± 12.1 sec (111) and 60% (121) for flr-2(ut5); flr-1(ut11). Unlike L1 larvae, young adults survived on the plates containing 400 μg/ml of NaF for at least 3 days.
Figure 2.

Cloning of flr-1 gene and cDNA. (A) Genetic and physical map around flr-1 gene. A genomic DNA fragment flanking the transposon Tc1 that caused the flr-1(ut11) mutation, hybridized to the yeast artificial chromosome Y50B10 and the cosmid C15A2. An overlapping cosmid F02D10 and its subclone pMT21–3 rescued the flr-1 phenotypes by microinjection. The arrow shows the direction of transcription of the only complete ORF contained in pMT21–3. Sl, SalI site; Sc, SacI site. (B, Top) The structure of the 8.5-kb genomic clone (pMT21–3) that rescued the flr-1 phenotypes. The 17 exons are indicated by boxes. The trans-splice leader SL1 and the poly(A) tail also are shown. (Middle) The structure of pMTG25–1, which codes for a GFP(F64L S65T)-tagged FLR-1 protein. (Bottom) The structure of pMTG24–5, a flr-1∷GFP(S65C) fusion gene containing a nuclear localization signal. (C) Rescue of various phenotypes of flr-1(ut11) by pMTG25–1.
Cloning of flr-1 Gene and cDNA.
We cloned a 2.8-kb BamHI genomic DNA fragment (pTA1) containing the Tc1 transposon that caused the flr-1(ut11) mutation, by the transposon-tagging method. A DNA fragment (probe TA2) flanking the Tc1 hybridized to yeast artificial chromosome clone Y50B10 and the cosmid clone C15A2. Because C15A2 contained only part of the predicted flr-1 gene, we used an overlapping cosmid F02D10 and its 8.5-kb SacI–SalI fragment [pMT21–3; cloned in pBluescript KS+ (Stratagene)] for the rescue experiments. By screening about 106 plaques from R. Barstead’s mixed stage cDNA library (42) with the probe TA2, we obtained four partial-length cDNA clones, of which the longest one (λMK5–2) consisted of 920 bp. The missing, 5′ part of the flr-1 cDNA was obtained by reverse transcription–PCR.
Transgenic Lines.
Microinjection was carried out as described (16). For the rescue experiments, the rol-6(su1006) dominant marker plasmid pRF4 (17) and the DNA to be tested were coinjected into the gonad of flr-1(ut2) animals at concentrations of 99 ng/μl and 1 ng/μl, respectively. Multiple independent lines were established from each injection. The extrachromosomal array was transferred to the flr-1(ut11) unc-3(e151) double mutant by cross to test the rescue of the synthetic dauer-constitutive phenotype. To make strains that carry a flr-1∷GFP construct (pMTG25–1 or pMTG24–5), we injected a mixture of pRF4 and the construct at concentrations of 5 ng/μl and 95 ng/μl, respectively, to N2 animals. The extrachromosomal array containing pMTG25–1 was transferred to flr-1(ut11) and flr-1(ut11) unc-3(e151) animals by cross to test the rescue of the flr-1 phenotypes.
Reporter Fusion.
pMTG25–1 was made with the pPD95.77 vector, courtesy of A. Fire (Carnegie Institution of Washington, Baltimore). The GFP gene in pPD95.77 was modified by site-directed mutagenesis (F46L S65T). pMTG25–1 was designed to produce a protein, in which the C terminus of FLR-1 was connected to GFP through linker amino acid residues. For this purpose, we used a PCR primer f1-XG315 (GTCTAGAACCTCCAATTAATTGTGATTTGAATATGG), which contains a sequence complementary to the 3′ part of the flr-1 coding region, the linker sequence, and an XbaI site to connect to the GFP cDNA. pMTG24–5 was made by cloning the 3.2-kb PvuII–SalI fragment of pMT21–3 in the vector pPD95.70 cleaved by SmaI and SalI. GFP fluorescence was observed as described (18).
Determination of Mutation Sites by DNA Sequencing.
The flr-1 coding region was amplified with Expand High Fidelity PCR System (Boehringer Mannheim) from single animals of flr-1 mutants as described (19). The PCR products were cloned, and at least two independent clones for each PCR product were sequenced with a dye-terminator sequencing system (Perkin–Elmer).
RESULTS
Fluoride Ion Increases the Defecation Cycle Period of Wild-Type Animals But Not That of flr-1 Mutants.
Fig. 1 shows the defecation interval length and the percentage of defecation behaviors having the Exp step. The graphs represent the relationship between each defecation interval length and the next interval length, to demonstrate the characteristics of the fluctuation. Wild-type (N2) animals under normal conditions (Top Left) had an average defecation interval length of about 45 sec with a relatively small SD. Almost all of the defecation behaviors had the Exp step. In contrast, flr-1 mutants (Top Right and legend) usually had short defecation intervals of 20–30 sec, but occasionally showed longer intervals with large variation. The Exp step was observed in less than half of the defecation behaviors. Similar results have been reported for N2 (10) and flr-1 (2).
Because loss-of-function mutations in class 1 flr genes lead to abnormally short defecation cycle periods (2), we tested whether fluoride ion, whose lethal effect is antagonized by class 1 flr mutations, increases the defecation cycle period of N2. When we transferred N2 animals from plates without fluoride ion to those containing 400 μg/ml of NaF, they initially showed a normal defecation cycle period [47.5 ± 6.7 sec (n = 134)] and nearly complete presence of the Exp step [95% (n = 147)] for the first 30 min. However, 30–60 min after the transfer (Fig. 1, Middle Left), they showed about a 30% longer defecation cycle period and frequent skip of the Exp step. In contrast, the flr-1(ut11) mutant (Middle Right) was not affected by fluoride ion, within the experimental error.
Class 2 flr Mutations Do Not Suppress the Defecation Abnormalities of flr-1 Mutants.
Class 2 flr mutations, which map in genes flr-2 and flr-5, suppress the slow growth of flr-1 (13). Hence, we checked whether they also suppress the defecation abnormalities. Class 2 flr mutants, flr-2(ut5) and flr-5(ut73), had essentially the same characteristics as N2 (Fig. 1, Bottom Left and legend). In contrast, the double mutants flr-2;flr-1 and flr-5;flr-1 showed essentially the same phenotypes as flr-1 single mutants (Fig. 1, Bottom Right and legend). Namely, the defecation abnormalities of flr-1 are not suppressed by class 2 flr mutations.
flr-1 Mutants Show Abnormalities in Dauer Formation in the unc-3 Background.
In a previous paper (13) we mentioned briefly that class 1 flr unc-3 double mutants form dauer larvae more easily than unc-3 single mutants. The dauer larva is a special third-stage larva that can survive under harsh conditions (20). C. elegans regulates dauer larva formation by sensing the concentrations of food and pheromone with amphid sensory neurons (21). Because the abnormality in dauer regulation suggests disorder in neural functions, we investigated the phenomenon in detail. As shown in Table 1, flr-1 unc-3 double mutants produced many dauer larvae at 20°C under nondauer-forming conditions, whereas none of the single mutants did, except unc-3(cn4146), which produced some dauers. Class 2 flr mutants, flr-2(ut5) and flr-5(ut73), produced only a few dauer larvae in the unc-3 background. Class 2 class 1 unc-3 triple mutants resembled class 2 unc-3 double mutants rather than class 1 unc-3 double mutants in the dauer larva formation. We therefore conclude that class 2 flr mutations suppress the dauer-constitutive phenotype of flr-1 unc-3.
Table 1.
Synthetic dauer-constitutive phenotype of flr-1
| Genotype | Percentage dauer larvae (n)* |
|---|---|
| flr-1(ut11) unc-3(el51) | 63% (347) |
| flr-1(ut11) unc-3(cn4146) | 40% (446) |
| flr-1(ut1) unc-3(el51) | 46% (180) |
| flr-1(ut1) unc-3(cn4146) | 49% (284) |
| flr-2(ut5); unc-3(el51) | 5% (301) |
| flr-5(ut73); unc-3(el51) | 2% (808) |
| flr-2(ut5); flr-1(ut11) unc-3(el51) | 0% (236) |
| flr-5(ut73); flr-1(ut11) unc-3(el51) | 3% (256) |
Shown is the percentage of dauer larvae produced at 20°C under non-dauer-forming conditions, i.e., plenty of food and sparse population. All of the flr-1 unc-3 strains produced fewer dauer larvae (5–19%) at 25°C than 20°C. Wild-type animals, all the single-mutants except unc-3(cn4146), and the double mutants flr-2; flr-1 and flr-5; flr-1 produced less than 1% dauer larvae (n > 100), while unc-3(cn4146) produced 8% dauer larvae (n = 218).
Number of animals scored.
flr-1 Gene Encodes an Ion Channel of the DEG/ENaC Superfamily.
We cloned flr-1 gene and cDNA (see Materials and Methods). An 8.5-kb SacI–SalI fragment from the cosmid F02D10 (pMT21–3, Fig. 2A) rescued all the flr-1 phenotypes (data not shown). The flr-1 cDNA consists of 1,984 bases with the SL1 splice leader sequence (22) at the 5′ end (Fig. 2B). A homology search revealed that the FLR-1 protein has similarity to degenerins of C. elegans such as DEG-1, MEC-4, MEC-10, UNC-105, and UNC-8 (23–27), the α-, β-, and γ-subunits of the mammalian ENaC (28, 29), and the ligand-gated cation channels amiloride-sensitive FMRFamide-activated sodium channel (FaNaCh) and acid-sensing ionic channel (ASIC) (30, 31). The most closely related sequence was C27C12.5, a gene predicted by the C. elegans genome project. All of these proteins including FLR-1 have two potential membrane-spanning domains, MSDI and MSDII, and extracellular cysteine-rich domains, CRDII and CRDIII (Fig. 3). FLR-1, like ENaCs, lacked an additional CRD, CRDI, which is present in DEGs. FLR-1 was 27.1%, 25.7%, and 22.9% identical in a similarity region including MSDI, and 34.6%, 38.5%, and 34.6% identical in MSDII to MEC-4, the β-subunit of ENaC, and FaNaCh, respectively. Thirteen cysteine residues in the extracellular region are conserved among those channels, suggesting conservation of the tertiary structure. The serine-rich C-terminal intracellular domain, consisting of 187 unique amino acid residues, contains many potential phosphorylation sites (32) (Fig. 3). FLR-1 lacks the consensus of the Nedd4-binding site (33), which is present in ENaCs for the regulation of the amount in the apical membrane in a renal epithelium (34).
Figure 3.

Sequence alignment of FLR-1 and other ion channels of the DEG/ENaC superfamily. Shown is a comparison of the predicted amino acid sequences between FLR-1, the C. elegans mechanosensory channel MEC-4 (24), the human amiloride-sensitive epithelial sodium channel beta subunit (ENaCbe) (28), and the snail amiloride-sensitive FMRFamide peptide-gated channel (FaNaCh) (30). The residues similar in three or four proteins are boxed. The numbers in parentheses show those of amino acids omitted. Two putative membrane-spanning domains (MSDI and MSDII) and the extracellular Cys-rich domains (CRDII and CRDIII) are shown by lines with arrowheads. A broken line with arrowheads shows the similarity region including MSDI. N and + indicate five potential N-glycosylation sites in the extracellular region and potential phosphorylation sites in the C-terminal intracellular domain of FLR-1, respectively. The latter consist of the consensus sequences for cyclic AMP-dependent protein kinase (R R X S/T), cyclic GMP-dependent protein kinase (R/K R/K X S/T), and Ca2+/calmodulin-dependent protein kinase II (R X X S/T) (32). The mutation sites of (ut1, ut4, ut6), ut2, and sa96 are indicated by ∗ together with the allele names and the resultant amino acid substitution. The Tc1-insertion site in ut11 is indicated by an arrowhead.
flr-1 Mutation Sites Show That the Overall Design of the DEG/ENaC Superfamily Is Conserved.
We identified the mutational changes in six independently derived alleles (Fig. 3). Mutations in ut1, ut4, and ut6 (G30R) and that in ut2 (G438D) were located at Gly residues conserved in all of the DEG/ENaC proteins. MEC-4 and MEC-10 have loss-of-function mutations just at the position corresponding to ut2 and near the position corresponding to ut1, ut4, and ut6 (25, 35). The mutation G30R corresponds to the human mutation in pseudohypoaldosteronism type I that disrupts ENaC function (36). These results suggest that the functional design of the FLR-1 ion channel is similar to that of DEGs and ENaCs. The mutation sa96 was located at a semiconserved Gly residue in the predicted extracellular loop (G288R). The Tc1 insertion site of ut11 also was located in the extracellular loop.
flr-1∷GFP Fusion Genes Are Expressed Only in the Intestine.
To investigate the tissue and subcellular localization of FLR-1, we examined the expression of a flr-1∷GFP fusion gene pMTG25–1 (Fig. 2B) in wild-type animals. This construct fully rescued all of the flr-1 phenotypes (Fig. 2C), which indicates that the product was functional and present at a proper time in the cells that require the FLR-1 function for the wild-type phenotypes. Comma stage embryos began to express the GFP-tagged protein in the E lineage cells (Fig. 4H), and animals retained the intestinal expression until the adult stage (see Fig. 4 I and J for a 3-fold stage embryo and an L1 larva, respectively). The fusion protein accumulated at the cell membrane, especially the membrane facing the inner lumen and part of the lateral membrane intervening the intestinal cells (Fig. 4J). The localization is consistent with the FLR-1 function as an ion channel. Thus, the intestinal expression seems to be sufficient for the function of FLR-1, unless pMTG25–1 produces a nonfluorescent protein having the FLR-1 activity outside of the intestine. Another flr-1∷GFP fusion product (pMTG24–5, Fig. 2B) also was localized exclusively in the intestine (Fig. 4G) but mostly in the cell nuclei, because of the nuclear localization signal and the lack of the FLR-1 membrane spanning domains. This experiment, together with the control experiment (Fig. 4F) without a flr-1∷GFP fusion, shows that the autofluorescence of the gut granule is negligible.
Figure 4.
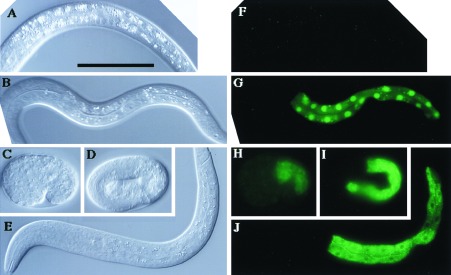
Expression of GFP-tagged FLR-1 protein. (A–E) Nomarski images and (F–J) epifluorescence images of the corresponding animals. (A and F) L1 larva of N2 (control). Autofluorescence of gut granules can be seen only faintly in F. (B and G) L1 larva of N2 carrying pMTG24–5. All of the intestinal nuclei show strong GFP fluorescence, whereas weak fluorescent dots in the cytoplasm in G may be gut granules. (C and H) Comma stage embryo of N2 carrying pMTG25–1. (D and I) Three-fold stage embryo of N2 carrying pMTG25–1. (E and J): L1 larva of N2 carrying pMTG25–1. (Scale bar represents 50 μm.)
DISCUSSION
FLR-1 Encodes a Novel Ion Channel of the DEG/ENaC Superfamily.
In this study we showed that flr-1 gene of C. elegans, which controls defecation cycle periods, encodes a protein belonging to the DEG/ENaC superfamily. The amino acid sequence, the mutation sites, and the intracellular localization strongly suggest that FLR-1 acts as an ion channel. FLR-1, together with some predicted gene products in C. elegans, forms a novel subfamily in the DEG/ENaC superfamily, as shown by alignment studies (ref. 37; M. Driscoll, personal communication), where FLR-1 corresponds to F02D10.5 in ref. 37.
Mechanism of the Action of Fluoride Ion in Relation to the Function of the FLR-1 Ion Channel.
Considering the fluoride-resistant phenotype of flr-1 mutants, fluoride ion can kill the wild-type animal, (a) by enhancing the activity of FLR-1, possibly acting on a phosphatase or a trimeric G-protein (13) in a hypothetical signal transduction system that controls the FLR-1 activity, (b) by elevating membrane potential by the inhibition of Na+,K+-ATPase as AlF4− (38), whereas loss of FLR-1, probably a cation channel, acts against this effect, or (c) by functioning in the uptake of fluoride ion, which is toxic only if it enters the intestinal cells. In this study we showed that fluoride ion increases the defecation interval length in a flr-1-dependent manner. This result is consistent with a, which also is supported by the presence of many putative phosphorylation sites in the C-terminal domain of FLR-1. Theory b has difficulty in explaining the insensitivity of the defecation cycle periods of flr-1 mutants to fluoride ion, while theory c has difficulty in explaining the relationship between the increase of defecation cycle periods by fluoride ion and the decrease by flr-1 mutations. However, more experiments are required to obtain a definitive conclusion.
Class 2 flr Mutations Suppress Only Part of the flr-1 Phenotypes.
Of the flr-1 phenotypes, those in growth and dauer regulation are suppressed by class 2 flr mutations, while those in fluoride sensitivity, defecation cycle, and Exp are not (ref. 13 and this study). The results can be interpreted in two ways. One interpretation is that FLR-1 achieves the former functions by suppressing the activity of class 2 flr gene products, whereas class 2 flr genes do not play an essential role in the latter functions. Another interpretation would be that the action of FLR-1 is always somehow antagonized by that of class 2 flr gene products, and that the threshold between wild-type and mutant phenotypes is different between the former and the latter functions.
flr-1 Mutations Affect Dauer Larva Formation.
flr-1 mutations cause abnormality in dauer larva formation in the unc-3 (this study), osm-1, and tax-2 (unpublished results) background. unc-3 codes for a putative transcription factor of the Olf-1/EBF family that is expressed in ASI sensory neurons and transiently in developing motor neurons (39). Mutants in osm-1 and tax-2 also have defects in ASI neurons as well as some other neurons (40, 41). Because the killing of both ASI and ADF neurons leads to the constitutive formation of dauer larvae (21), flr-1 mutations may suppress the dauer-inhibitory signal in ADF neurons. flr-1 mutations also may affect AVL and DVB motor neurons, because the partial loss of the Exp step in flr-1 mutants can be explained by the effect on those neurons or defecation muscles.
FLR-1 Probably Acts in the Intestine.
FLR-1 seems to act in the intestine to achieve its functions, because a flr-1∷GFP fusion gene that rescues the flr-1 phenotypes is expressed only in the intestine. Strictly speaking, it is possible that the flr-1∷GFP fusion gene also is expressed in another tissue but that the expression cannot be detected by fluorescence. Such possibilities include removal of the GFP-coding region by alternative splicing, selective degradation of the GFP part at the mRNA or protein level, and failure in the posttranslational reaction that produces the fluorescent group. However, such tissue specificity has not been reported in C. elegans. In accordance with the intestinal expression of flr-1, a flr-4∷GFP fusion gene also is expressed in the intestine, besides the isthmus of the pharynx, and a pair of head neurons (unpublished result).
FLR-1 May Control Food-Related Functions.
The reduction of the FLR-1 activity causes partial loss of the Exp step, frequent dauer larva formation, and slow growth. Hence, it can adapt the animal to food-deficient conditions. The short defecation cycle period of flr-1 mutants is not so contradictory, because the efficiency of defecation in flr-1 mutants is low, as seen from their constipated phenotype (unpublished result). Although the pleiotropic phenotype of flr-1 may arise from an abnormal state of the intestine, it is also possible that FLR-1 might regulate those functions by receiving a signal from the environment or from ingested food in the intestine.
Possible Mechanisms of the Control of the Defecation Rhythm by FLR-1 Ion Channel.
In this study we showed that the FLR-1 ion channel probably acts in the intestine and control defecation cycle periods. One possible mechanism is that it directly controls the rhythmic behavior in the intestine as an ion channel, by changing membrane potential like SCN5A Na+ channel, HERG K+ channel, and IsK+KvLQT1 K+ channel in the heartbeat rhythm (6) or by regulating the concentration of an ion in the cytoplasm like the inositol 1,4,5-trisphosphate receptor/Ca2+ release channel in Ca2+ oscillations (4). Alternatively, it is also possible that FLR-1 affects a gut function and has an indirect influence on some developmental or neural functions required for the normal rhythmic behavior. For instance, it might affect the secretion of substances like gastrointestinal hormones in mammals, which, in turn, control the defecation rhythm in another organ. Thus, it is important to know in which organ the defecation rhythm is generated. Furthermore, how FLR-1 changes the defecation interval length must be elucidated. This study provides a basic framework for such studies.
Acknowledgments
We thank J. H. Thomas for flr-1(sa96), R. Hosono for unc-3(cn4146), A. Fire for pPD95.77 and pPD95.70, A. Coulson for cosmid clones, R. Barstead for the C. elegans cDNA library, and the C. elegans Genome Sequence Consortium for C. elegans genome sequences. We are also grateful to M. Driscoll, K. Iwasaki, E. Jorgensen, and J. H. Thomas for personal communication, and M. Chalfie, R. Hosono, I. Mori, Y. Moriyama, Y. Ohshima, and members of our lab for useful suggestions and discussions. Some nematode strains were obtained from the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources. This research was supported by grants from the Ministry of Education, Science, and Sports of Japan (03680217, 04270219, 04454615, 05263227, 05269102, 08878117, and 09480190 to I.K. and 07278105 to T.I.) and by a Japan Society for the Promotion of Science Research Fellowship (2266) to M.T.
ABBREVIATIONS
- pBoc
posterior body-wall muscle contraction
- Exp
expulsion
- DEG
degenerin
- ENaC
amiloride-sensitive epithelial sodium channel
- MSD
membrane-spanning domain
- CRD
cysteine-rich domain
- GFP
green fluorescent protein
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AB012617).
References
- 1. Hall J. Trends Neurosci. 1995;18:230–240. doi: 10.1016/0166-2236(95)93908-g. [DOI] [PubMed] [Google Scholar]
- 2.Iwasaki K, Liu D W C, Thomas J H. Proc Natl Acad Sci USA. 1995;92:10317–10321. doi: 10.1073/pnas.92.22.10317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iwasaki K, Thomas J H. Trends Genet. 1997;13:111–115. doi: 10.1016/s0168-9525(97)01059-7. [DOI] [PubMed] [Google Scholar]
- 4.Miyazaki S, Shirakawa H, Nakada K, Honda Y. Dev Biol. 1993;158:62–78. doi: 10.1006/dbio.1993.1168. [DOI] [PubMed] [Google Scholar]
- 5.Hess B, Boiteux A. Annu Rev Biochem. 1971;40:237–258. doi: 10.1146/annurev.bi.40.070171.001321. [DOI] [PubMed] [Google Scholar]
- 6.Attali B. Nature (London) 1996;384:24–25. doi: 10.1038/384024a0. [DOI] [PubMed] [Google Scholar]
- 7.Weitzman E D. Annu Rev Neurosci. 1981;4:381–417. doi: 10.1146/annurev.ne.04.030181.002121. [DOI] [PubMed] [Google Scholar]
- 8.Hall J C, Rosbach M. Annu Rev Neurosci. 1988;11:373–393. doi: 10.1146/annurev.ne.11.030188.002105. [DOI] [PubMed] [Google Scholar]
- 9.Ivry R B. Curr Opin Neurobiol. 1996;6:851–857. doi: 10.1016/s0959-4388(96)80037-7. [DOI] [PubMed] [Google Scholar]
- 10.Thomas J H. Genetics. 1990;124:855–872. doi: 10.1093/genetics/124.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu D W, Thomas J H. J Neurosci. 1994;14:1953–1962. doi: 10.1523/JNEUROSCI.14-04-01953.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McIntire S, Jorgensen E, Kaplan J, Horvitz H R. Nature (London) 1993;364:337–341. doi: 10.1038/364337a0. [DOI] [PubMed] [Google Scholar]
- 13.Katsura I, Kondo K, Amano T, Ishihara T, Kawakami M. Genetics. 1994;136:145–154. doi: 10.1093/genetics/136.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas J H, Birnby D A, Vowels J J. Genetics. 1993;134:1105–1117. doi: 10.1093/genetics/134.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mello C C, Kramer J M, Stinchcomb D, Ambros V. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kramer J M, French R P, Park E-C, Johnson J J. Mol Cell Biol. 1990;10:2081–2089. doi: 10.1128/mcb.10.5.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chalfie M, Tu Y, Euskirchen G, Ward W W, Prasher D C. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 19.Williams B D, Schrank B, Huynh C, Shownkeen R, Waterston R H. Genetics. 1992;131:609–624. doi: 10.1093/genetics/131.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riddle D L, Albert P S. In: C. elegans II. Riddle D L, Blumenthal T, Meyer B J, Priess J R, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1997. pp. 739–768. [Google Scholar]
- 21.Bargmann C I, Horvitz H R. Science. 1991;251:1243–1246. doi: 10.1126/science.2006412. [DOI] [PubMed] [Google Scholar]
- 22.Krause M, Hirsh D. Cell. 1987;49:753–761. doi: 10.1016/0092-8674(87)90613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chalfie M, Wolinsky E. Nature (London) 1990;345:410–416. doi: 10.1038/345410a0. [DOI] [PubMed] [Google Scholar]
- 24.Driscoll M, Chalfie M. Nature (London) 1991;349:588–593. doi: 10.1038/349588a0. [DOI] [PubMed] [Google Scholar]
- 25.Huang M, Chalfie M. Nature (London) 1994;367:467–470. doi: 10.1038/367467a0. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Schrank B, Waterston R H. Science. 1996;273:361–364. doi: 10.1126/science.273.5273.361. [DOI] [PubMed] [Google Scholar]
- 27.Tavernarakis N, Shreffler W, Wang S, Driscoll M. Neuron. 1997;18:107–119. doi: 10.1016/s0896-6273(01)80050-7. [DOI] [PubMed] [Google Scholar]
- 28.Canessa C M, Schild L, Buell G, Thorens B, Gautschi I, Horisberger J-D, Rossier B C. Nature (London) 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 29.Voilley N, Lingueglia E, Champigny G, Mattei M-G, Waldmann R, Lazdunski M, Barbry P. Proc Natl Acad Sci USA. 1994;91:247–251. doi: 10.1073/pnas.91.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lingueglia E, Champigny G, Lazdunski M, Barbry P. Nature (London) 1995;378:730–733. doi: 10.1038/378730a0. [DOI] [PubMed] [Google Scholar]
- 31.Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. Nature (London) 1997;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- 32.Smart T G. Curr Opin Neurobiol. 1997;7:358–367. doi: 10.1016/s0959-4388(97)80063-3. [DOI] [PubMed] [Google Scholar]
- 33.Staub O, Dho S, Henry P, Correa J, Ishikawa J, McGlade J, Rotin D. EMBO J. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- 34.Snyder P M, Price M P, McDonald F J, Adams C M, Volk K A, Zeiher B G, Stokes J B, Welsh M J. Cell. 1995;83:969–978. doi: 10.1016/0092-8674(95)90212-0. [DOI] [PubMed] [Google Scholar]
- 35.Hong K, Driscoll M. Nature (London) 1994;367:470–473. doi: 10.1038/367470a0. [DOI] [PubMed] [Google Scholar]
- 36.Gründer S, Firsov D, Chang S S, Jaeger N F, Gautschi I, Schild L, Lifton R P, Rossier B C. EMBO J. 1997;16:899–907. doi: 10.1093/emboj/16.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corey D P, García-Añoveros J. Science. 1996;273:323–324. doi: 10.1126/science.273.5273.323. [DOI] [PubMed] [Google Scholar]
- 38.Missiaen L, Wuytack F, De Smedt H, Vrolix M, Casteels R. Biochem J. 1988;253:827–833. doi: 10.1042/bj2530827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prasad B C, Ye B, Zackhary R, Schrader K, Seydoux G, Reed R R. Development (Cambridge, UK) 1998;125:1561–1568. doi: 10.1242/dev.125.8.1561. [DOI] [PubMed] [Google Scholar]
- 40.Perkins L A, Hedgecock E M, Thomson J N, Culotti J G. Dev Biol. 1986;117:456–487. doi: 10.1016/0012-1606(86)90314-3. [DOI] [PubMed] [Google Scholar]
- 41.Coburn C M, Bargmann C I. Neuron. 1996;17:695–706. doi: 10.1016/s0896-6273(00)80201-9. [DOI] [PubMed] [Google Scholar]
- 42.Barstead R J, Waterston R H. J Biol Chem. 1989;264:10177–10185. [PubMed] [Google Scholar]