

Abstract
EphA4 signaling has recently been implicated in the regulation of synapse formation and plasticity. In this study, we show that ankyrin repeat-rich membrane spanning (ARMS; also known as a kinase D–interacting substrate of 220 kD), a substrate for ephrin and neurotrophin receptors, was expressed in developing muscle and was concentrated at the neuromuscular junction (NMJ). Using yeast two-hybrid screening, we identified a PDZ (PSD-95, Dlg, ZO-1) domain protein, α-syntrophin, as an ARMS-interacting protein in muscle. Overexpression of α-syntrophin induced ARMS clustering in a PDZ domain–dependent manner. Coexpression of ARMS enhanced EphA4 signaling, which was further augmented by the presence of α-syntrophin. Moreover, the ephrin-A1–induced tyrosine phosphorylation of EphA4 was reduced in C2C12 myotubes after the blockade of ARMS and α-syntrophin expression by RNA interference. Finally, α-syntrophin–null mice exhibited a disrupted localization of ARMS and EphA4 at the NMJ and a reduced expression of ARMS in muscle. Altogether, our findings suggest that ARMS may play an important role in regulating postsynaptic signal transduction through the syntrophin-mediated localization of receptor tyrosine kinases such as EphA4.
Introduction
Eph and tropomyosin-related kinase (Trk) receptors are two families of receptor tyrosine kinases (RTKs) that are involved in the crucial processes of neural development, including neuronal survival, axon guidance, synapse formation, and regulation of synaptic plasticity (for reviews see Flanagan and Vanderhaeghen, 1998; Kullander and Klein, 2002; Huang and Reichardt, 2003). Recently, accumulating evidence has begun to reveal the functions of these molecules at the neuromuscular junction (NMJ). TrkB protein is expressed in skeletal muscle and is concentrated at the NMJ, and an important requirement of TrkB signaling in NMJ stabilization has been suggested (Gonzalez et al., 1999). Similarly, the prominent expression and enrichment of two EphA receptors, EphA4 and EphA7, are also detected at postsynaptic NMJ (Lai et al., 2001). Like TrkB, EphA receptors have been implicated in NMJ formation and/or maintenance (Lai et al., 2001, 2004). The downstream signaling of these two families of RTKs in muscle has just begun to be elucidated.
Ankyrin repeat-rich membrane spanning (ARMS), also known as a kinase D–interacting substrate of 220 kD, was identified as a novel downstream substrate for protein kinase D, Trk, and Eph receptors (Iglesias et al., 2000; Kong et al., 2001). The expression pattern of ARMS overlaps with Trk and Eph receptors in postmitotic neurons, and it was proposed to play a role in axon guidance during neural network establishment (Kong et al., 2001). Recently, ARMS was shown to mediate sustained MAPK signaling elicited by neurotrophins, implicating ARMS as an important target for RTK signaling (Arévalo et al., 2004). ARMS is a multidomain protein, and analysis of the ARMS sequence revealed a class I PDZ (PSD-95, Dlg, ZO-1)-binding motif, “RESIL,” at its COOH terminus, raising the intriguing possibility that ARMS may interact with PDZ proteins. In a variety of cellular contexts, PDZ proteins function as scaffolds, orchestrating signal transduction complexes by clustering signaling components (such as ion channels, neurotransmitters, and cytokine receptors) into appropriate subcellular compartments (for review see Sheng and Sala, 2001). Consequently, PDZ proteins are thought to regulate crucial cellular processes via protein localization. The disruption of PDZ interactions perturbs protein localization and cell function (Simske et al., 1996; Kaech et al., 1998). At neuronal synapses, the PDZ domain protein PSD-95 interacts with the N-methyl-d-aspartate receptor, the Shaker type K+ channel, and neuronal nitric oxide synthase (nNOS), forming a multimolecular complex that has important implications in long-term potentiation (Kim et al., 1995; Kornau et al., 1995; Brenman et al., 1996; Migaud et al., 1998; Christopherson et al., 1999). Similarly, the induction of long-term depression in cerebellar Purkinje cells also involves regulated PDZ interactions between GluR2 and GRIP/PICK1 (Xia et al., 1999, 2000). Because ARMS functions as an important RTK downstream target and contains a PDZ-binding motif, PDZ proteins may be involved in ARMS-mediated RTK signaling.
In this study, we show that the transcript and protein levels of ARMS are regulated throughout muscle development and that ARMS is progressively clustered at the NMJ during the first week of postnatal development. As part of the effort to understand the function of ARMS in muscle, we performed a yeast two-hybrid screen by using the PDZ domain–binding tail of ARMS as bait. A single PDZ domain protein, α-syntrophin, was identified as its binding partner. The interaction between the two proteins was PDZ domain dependent, and α-syntrophin induced ARMS cluster formation in a PDZ and pleckstrin homology (PH) 1 domain–dependent manner. Furthermore, α-syntrophin enhanced EphA4-induced janus kinase (Jak) and signal transducer and activator of transcription (Stat) tyrosine phosphorylation in an ARMS-dependent manner. On the other hand, the ephrin-A1–induced tyrosine phosphorylation of EphA4 in differentiated C2C12 myotubes was impaired when the expression of ARMS and α-syntrophin was inhibited by small interference RNA (siRNA). Finally, analysis of α-syntrophin–null mice suggests that α-syntrophin modulates the localization and expression levels of ARMS and EphA4 at the NMJ.
Results
ARMS is expressed in developing muscle
To investigate the expression profile of ARMS in muscle during development, we first examined the ARMS transcript by Northern blot analysis using a probe directed to the 5′ end of ARMS mRNA. A single, prominent ARMS transcript of ∼7 kb was detected in rat embryonic muscle, and its abundance gradually decreased with the progression of development (Fig. 1 A). A similar developmental profile of ARMS transcript was observed with mouse muscle tissues (Fig. 1 B). Using an antibody that specifically recognized the COOH-terminal fragment of ARMS, we detected a prominent protein band of ∼220 kD in muscle throughout development (Fig. 1 C).
Figure 1.
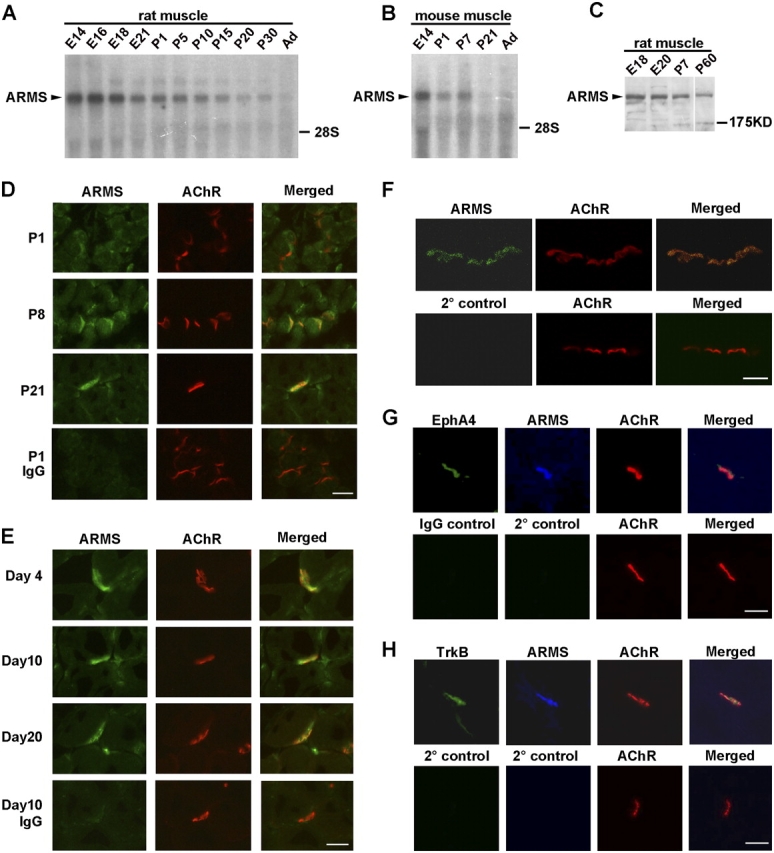
ARMS was expressed in developing muscle and was localized postsynaptically at the NMJ. ARMS transcript was analyzed in rat (A) and mouse (B) muscle at various developmental stages (embryonic day [E] 14 to postnatal day [P] 30 and adult [Ad]) by Northern blot analysis. (C) Western blot of ARMS protein expression in developing muscle. (D and E) Immunohistochemical analysis of ARMS and AChR in rat gastrocnemius muscle during development and at different time periods after denervation. Bars, 20 μm. (F) Longitudinal sections (2 μm) of adult rat sternomastoid muscle were stained with ARMS antibody and α-BTX to reveal the localization of ARMS and AChR, respectively. Bar, 5 μm. (G and H) Adult rat gastrocnemius muscle sections were stained with ARMS antibody and α-BTX, followed by the addition of fluorescein-labeled EphA4 or TrkB antibody. Bars, 20 μm.
We then investigated the localization of ARMS protein in developing muscle by immunohistochemistry. ARMS was present on the sarcolemma in postnatal day (P) 1 rat gastrocnemius muscle. By P8, the protein became more concentrated at the junctional sites, and by P21 it was mostly colocalized with acetylcholine receptor (AChR) clusters (Fig. 1 D). ARMS and AChR staining of longitudinal sections of adult rat sternomastoid muscle also revealed a colocalization of the two proteins at the NMJ (Fig. 1 F). To confirm the postsynaptic localization of ARMS, we denervated rat gastrocnemius muscle and examined the distribution of ARMS on muscle fibers up to 20 d after denervation, when the presynaptic axon terminals should have degenerated and retracted. Significant ARMS staining could still be observed at junctional sites 20 d after denervation, indicating that ARMS was localized postsynaptically (Fig. 1 E). In addition, we found that ARMS was colocalized with EphA4 and TrkB receptors, which are two RTKs that were previously reported to cluster postsynaptically at the NMJ at around P8 (Fig. 1, G and H; Gonzalez et al., 1999; Lai et al., 2001). These findings suggest that TrkB, EphA4, and ARMS are all clustered at postsynaptic regions and are similarly regulated throughout development.
Library screening identifies α-syntrophin as an ARMS-interacting protein
To identify which PDZ proteins might interact with ARMS at the NMJ, we performed a yeast two-hybrid screening using the COOH terminus of ARMS (last 100 amino acids) that contained the potential PDZ-binding motif as bait to screen a P12 mouse muscle cDNA library. 31 clones were identified as putative candidates, and 20 of them contained the full coding sequence of the PDZ protein α-syntrophin. We confirmed the authenticity of the clones by transforming them back into yeast with the ARMS COOH-terminal fragment.
In addition to α-syntrophin, two other isoforms, β1- and β2-syntrophin, are also expressed in muscle. These syntrophins share high homology in their protein domains with α-syntrophin (Adams et al., 1995; Ahn et al., 1996). We found that all three syntrophin isoforms bound to the ARMS COOH-terminal fragment in yeast, although to different extents (Fig. 2 A). The PDZ-binding motif RESIL at the ARMS cytoplasmic tail contains the consensus sequence S/T-X-V/I/L that preferentially interacts with the class I PDZ domain present in syntrophin (Gee et al., 1998, 2000; Schultz et al., 1998). The basic amino acid (R) at the −4 position and acidic residue (E) at the −3 position may further enhance the binding affinity between the two motifs (Wiedemann et al., 2004). We found that ARMS lacking the last three amino acids in the RESIL motif (ARMS COOH-terminal ΔSIL) failed to interact with α-syntrophin in yeast, demonstrating that the PDZ-binding motif is required for ARMS–syntrophin interaction (Fig. 2 A). Conversely, when we mutated the syntrophin PDZ domain by substituting two highly conserved residues in the PDZ GLGI loop with alanines (GLGI→GLAA; α-syntrophin PDZ mutant [mPDZ]), the ARMS–syntrophin binding was eliminated. Thus, the syntrophin PDZ domain was also required for ARMS–syntrophin interaction (Fig. 2 B). We confirmed that the wild-type and mutated proteins were expressed at similar levels in these experiments (unpublished data). In quantitative assays, β-galactosidase activity was detected only in the presence of both intact ARMS PDZ-binding motifs and wild-type syntrophin PDZ domains (Fig. 2 C). Together with the observation of cell growth on His−/Trp−/Leu− selective plates (Fig. 2 D), these results demonstrate that ARMS and syntrophin bind to each other through PDZ domain–mediated interactions in yeast and that α-syntrophin binds to ARMS more strongly than β-syntrophins.
Figure 2.
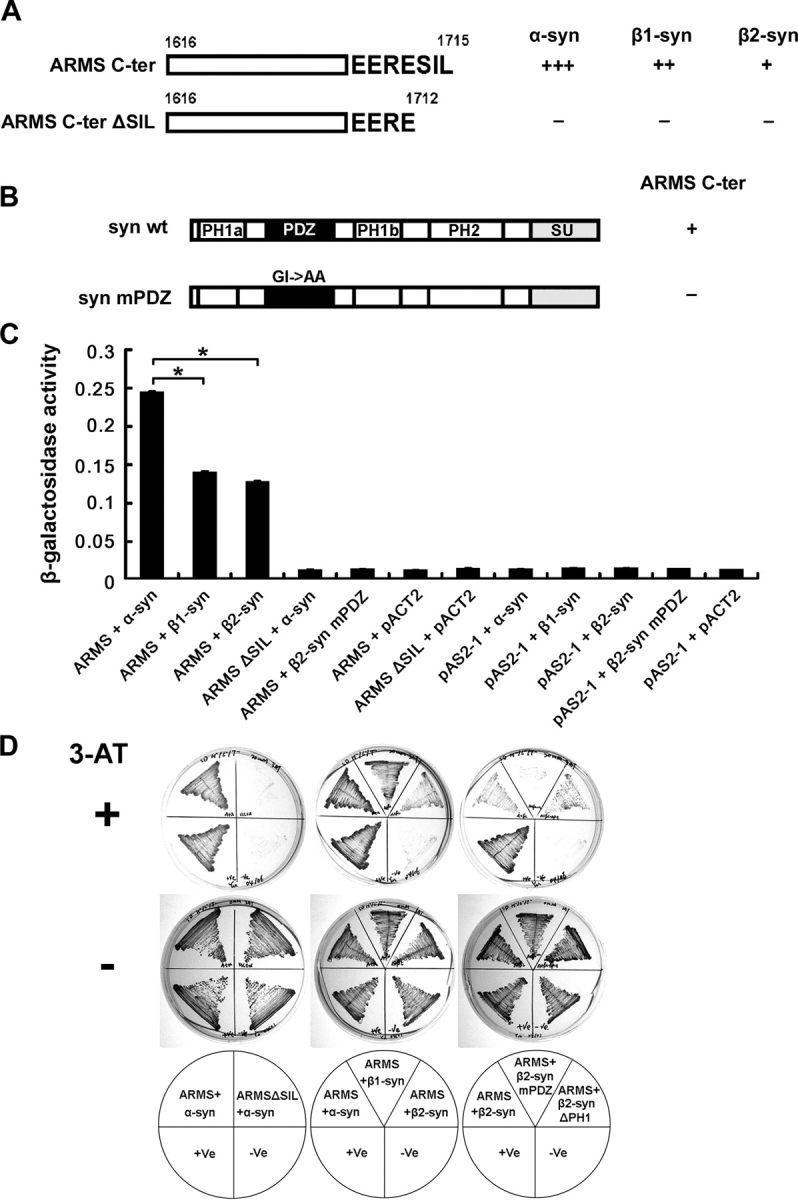
Yeast library screening identified α-syntrophin as an ARMS-interacting protein. (A) α-, β1-, and β2-syntrophins interacted with ARMS COOH terminus (ARMS C-ter, aa 1616–1715) in yeast. Deletion of SIL from the COOH terminus (ARMS C-ter ΔSIL, aa 1616–1712) abolished the protein interaction. +++, very strong; ++, strong; +, moderate; −, no interaction. (B) α-, β1-, and β2-syntrophins are composed of two pleckstrin homology domains (PH1, which is split into PH1a and PH1b by PDZ domain, and PH2), one PDZ domain, and one syntrophin unique domain (SU). Mutation of GLGI loop in syntrophin PDZ domain to GLAA (syn mPDZ) disrupted ARMS–syntrophin interaction. (C) Quantitative β-galactosidase activity assay indicated that α-syntrophins bind to ARMS more strongly than do β-syntrophins in yeast. Arbitrary units were used for the y axis to indicate the relative activity. Each data point represents the mean ± SEM; n = 3; *, P < 0.005. (D) Growth analysis of various yeast transformants on His−/Trp−/Leu− selective plates. In the presence of 20 mM 3-amino-1,2,4-triazole (3-AT), only yeast that expressed interacting proteins grew (top). As a control, all yeast transformants grew normally in the absence of the inhibitor (middle). Bottom panel shows the combinations of different constructs that were transformed into the yeast. +Ve (yeast transformed with pTD1-1 and pVA3-1 plasmids) served as a positive control for this yeast two-hybrid system. −Ve (yeast transformed with pTD1-1 and pAS2-1 plasmids) served as a negative control.
ARMS and α-syntrophin form complexes in mammalian cells and are colocalized at developing NMJs
Next, we tested whether ARMS and α-syntrophin interact in mammalian cells. HA-tagged α-syntrophin and ARMS full-length constructs were transiently transfected into COS7 cells. Total proteins were subjected to immunoprecipitation by anti-ARMS antibody, followed by immunoblotting with anti-HA antibody. HA–α-syntrophin was coimmunoprecipitated with ARMS from the cell lysates (Fig. 3 A), and, conversely, ARMS was coimmunoprecipitated with syntrophin from cell lysates using anti–α-syntrophin antibody (Fig. 3 B). As a specificity control, this antibody did not pull down ARMS protein when ARMS was expressed in COS7 cells alone (unpublished data). These results show that ARMS and α-syntrophin form a complex in transfected mammalian cells.
Figure 3.
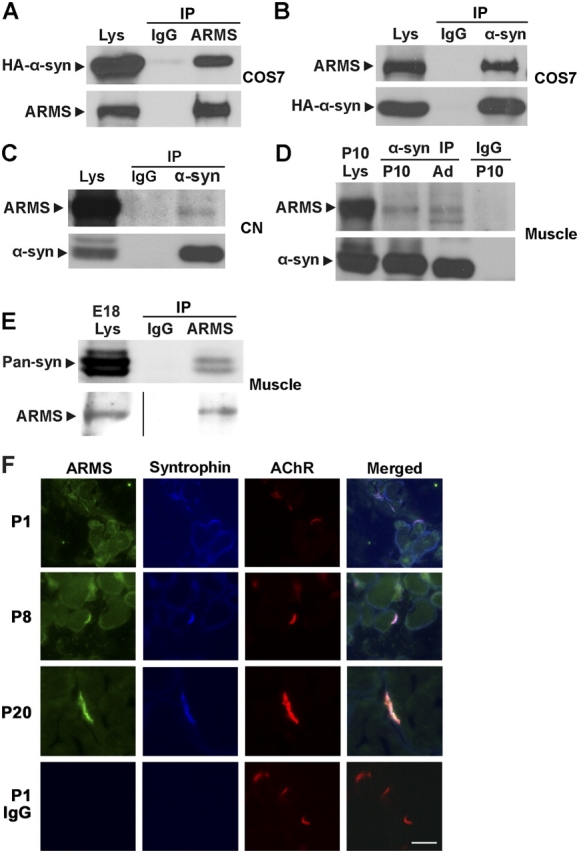
α-Syntrophin interacted and colocalized with ARMS. (A) ARMS and HA-tagged α-syntrophin were overexpressed in COS7 cells. Cell lysates were subjected to immunoprecipitation with ARMS 892 antiserum followed by Western blots using α-HA antibody. (B) Reciprocal coimmunoprecipitation using anti–α-syntrophin antibody. (C) Cell lysates from 14 DIV cultured cortical neurons (CN) were subjected to immunoprecipitation using anti–α-syntrophin antibody. (D and E) Membrane proteins from rat muscle were prepared and subjected to coimmunoprecipitation, demonstrating that ARMS and syntrophin interacted in vivo. (F) Immunohistochemical analysis of ARMS protein in rat gastrocnemius muscle at various developmental stages. P1 rat muscle stained with the FITC or AMCA-conjugated secondary antibodies served as a negative control. Bar, 20 μm.
To determine whether the interaction between ARMS and α-syntrophin occurred in vivo, lysate from cultured rat cortical neurons (14 days in vitro [DIV]) was subjected to immunoprecipitation using anti–α-syntrophin antibody. ARMS protein was detected in the immunoprecipitates (Fig. 3 C). Similar coimmunoprecipitation experiments using embryonic muscle lysates indicated that ARMS interacted with α-syntrophin in muscle (Fig. 3, D and E).
Like ARMS, α-syntrophin is concentrated at the NMJ in adult muscle, and its distribution is temporally regulated throughout muscle development (Kramarcy and Sealock, 2000). To compare the localization of ARMS and syntrophin in developing muscle, we colabeled rat gastrocnemius muscle sections of different developmental stages with ARMS antibody and pan-syntrophin antibody SYN1351. Syntrophin protein was colocalized with ARMS at junctional sites in P8 and P20 muscle, and its temporal expression pattern closely resembled that of ARMS (Fig. 3 F).
α-Syntrophin induces ARMS clustering in mammalian cells
We investigated whether α-syntrophin was able to regulate ARMS localization, which is a function attributed to many other PDZ proteins. COS7 cells were transfected with HA-tagged α-syntrophin and ARMS, and the distribution of ARMS and syntrophin was examined by immunocytochemistry. In cells expressing either syntrophin or ARMS alone, the proteins were found to be uniformly distributed. Strikingly, ARMS protein formed dense clusters when coexpressed with α-syntrophin, but colocalized clustering of α-syntrophin was not observed (Fig. 4 A). To eliminate the possibility that ARMS might block the access of syntrophin antibody to its epitope, we stained the cells with anti-HA antibody. Again, no significant syntrophin clusters were observed (unpublished data). In an attempt to determine the subcellular localization of ARMS clusters, COS7 cells that overexpressed both ARMS and α-syntrophin were costained with ARMS antibody and with specific markers for different intracellular organelles. We found that ARMS colocalized with the Golgi marker Rab8 and partially with the Golgi marker GM130 (unpublished data).
Figure 4.
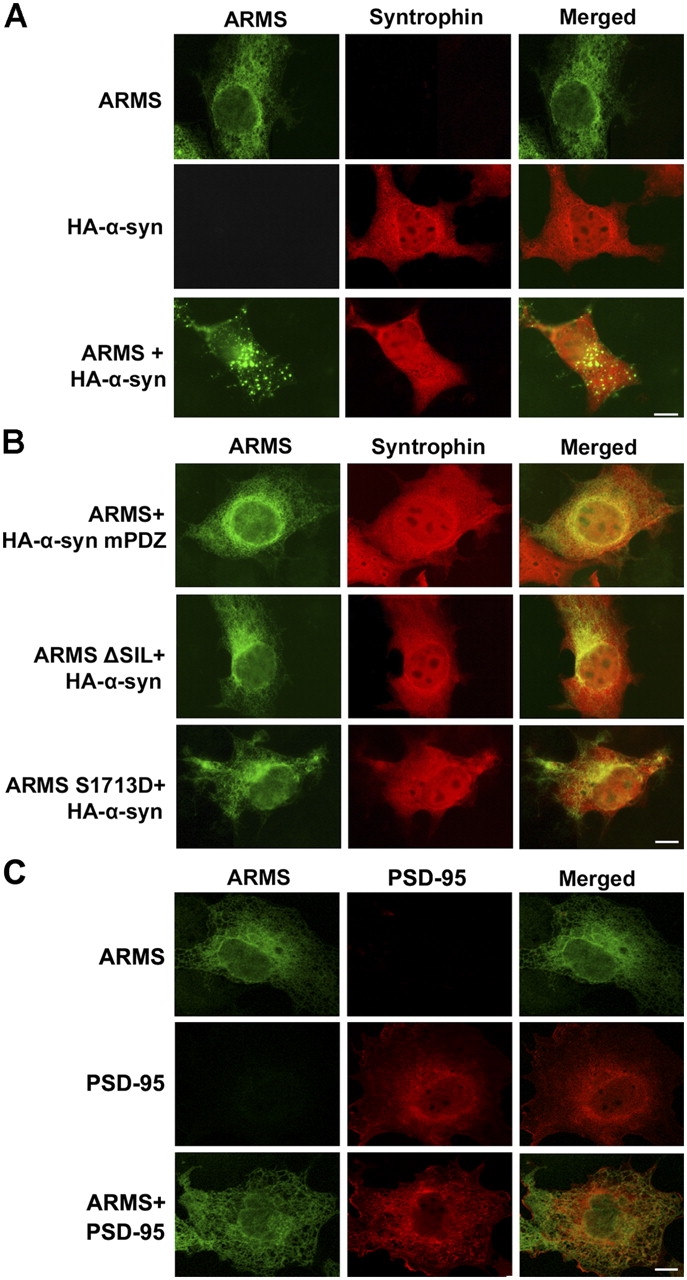
α-Syntrophin induced ARMS clustering in a PDZ domain–dependent manner. (A) Immunostaining of COS7 cells transfected with either ARMS, HA–α-syntrophin, or both ARMS and HA–α-syntrophin. α-Syntrophin (SYN1351 antibody staining, red) induced ARMS clustering (ARMS antibody staining, green) in cotransfected cells, whereas α-syntrophin itself remained diffused. (B) Mutations in either α-syntrophin PDZ domain (HA-α-syn mPDZ) or ARMS COOH terminus (ARMS ΔSIL and ARMS S1713D) abolished ARMS cluster formation. The immunostaining was performed as stated in A. (C) PSD-95 did not induce ARMS clustering when they were coexpressed in COS7 cells. Bars, 10 μm.
We expressed various ARMS and syntrophin mPDZ to see if ARMS clustering in these cells was dependent on PDZ domain interaction. The expression of α-syntrophin mPDZ or ARMS PDZ-binding motif mutant (ΔSIL) did not lead to ARMS clustering. We also tested the protein clustering of another ARMS mutant, ARMS S1713D, in which the serine residue within the ARMS PDZ-binding motif was mutated to aspartate. Mutation of the essential serine residue at position −2 of α-syntrophin PDZ ligands (equivalent to S1713) leads to the loss of their binding to the PDZ domain (Schultz et al., 1998; Wiedemann et al., 2004). As shown in Fig. 4 B, the mutation of S1713 to aspartate abolished the ability of ARMS to form clusters, suggesting that modifications by the potential phosphorylation of the PDZ-binding motif of ARMS may modulate its interaction with syntrophin. The results show that ARMS binding to the PDZ domain of α-syntrophin is required for ARMS clustering (Fig. 4 B). Furthermore, because an acidic residue at position −2 may mimic phosphorylation, the interaction may be regulated by posttranslational modification.
To show that the syntrophin-induced ARMS clustering is specific, we coexpressed ARMS with another PDZ protein, PSD-95, in COS7 cells. Although PSD-95 associated with ARMS in our coimmunoprecipitation experiment, it could not induce obvious ARMS clustering in COS7 cells (Fig. 4 C and not depicted).
β2-Syntrophin, but not β1-syntrophin, interacts with and clusters ARMS in mammalian cells
In yeast, both β1- and β2-syntrophins bind to the COOH terminus of ARMS, although the interactions were weaker than those with α-syntrophin. To test if β1- and β2-syntrophins also regulated ARMS localization, we coexpressed the proteins in COS7 cells and examined the localization of ARMS and syntrophin. We also performed coimmunoprecipitation to test if ARMS and β-syntrophins form complexes in these cells. Surprisingly, we observed robust ARMS clustering in cells that overexpressed β2-syntrophin, but not in cells that overexpressed β1-syntrophin (Fig. 5 C). Consistent with this result, β2-syntrophin, but not β1-syntrophin, was coimmunoprecipitated with ARMS from the cell lysate (Fig. 5, A and B). Although the syntrophin band that was detected in the immunoprecipitate was relatively weak, it was consistently detected in cells that were transfected with β2-syntrophin, but not with β1-syntrophin. Like α-syntrophin, the induction of ARMS clustering by β2-syntrophin is dependent on PDZ domain–mediated interactions. Mutations in either the β2-syntrophin PDZ domain or the ARMS PDZ-binding motif completely abolished protein interaction and ARMS cluster formation (unpublished data).
Figure 5.

β2-Syntrophin, but not the β1 isoform, interacted with and clustered ARMS, and the PH1 domain was required for syntrophin-induced ARMS clustering. (A and B) Immunoprecipitation of ARMS and HA–β-syntrophins from cotransfected COS7 cell homogenates. HA–β2-syntrophin (B), but not HA–β1-syntrophin (A), was coimmunoprecipitated with ARMS 892 antiserum. (C) Immunostaining of ARMS and HA–β-syntrophin–overexpressing cells. Only HA–β2-syntrophin, but not HA–β1-syntrophin, elicited ARMS clustering. Bar, 10 μm. (D) PH1-deleted HA–β2-syntrophin (HA-β2-syn ΔPH1) was coimmunoprecipitated with ARMS. (E) HA–β2-syn ΔPH1 did not induce ARMS clustering. Bar, 10 μm.
PH1 domain is required for syntrophin-induced ARMS cluster formation
We also examined the involvement of the syntrophin PH1 domain in ARMS clustering. Although β2-syntrophin with PH1 domain deletion (β2-syntrophin ΔPH1) was coimmunoprecipitated with ARMS, it did not induce ARMS cluster formation in these cells (Fig. 5, D and E). When β2-syntrophin ΔPH1 and ARMS COOH-terminal constructs were transformed into yeast, growth on selective plates and activation of β-galactosidase was observed, confirming an interaction between the two proteins (Fig. 2 D and not depicted). Coexpression of α-syntrophin ΔPH1 and ARMS in COS7 cells also failed to induce ARMS cluster formation (unpublished data). Therefore, the PH1 domain is required for ARMS clustering in syntrophin-expressing cells.
EphA4 is associated with ARMS and phosphorylates both ARMS and α-syntrophin
ARMS was previously shown to be tyrosine phosphorylated upon ephrin-B2 treatment (Kong et al., 2001). Notably, ARMS and EphA4 exhibit similar expression patterns at junctional sites in developing muscle (Lai et al., 2001; Fig. 1, D and G). We investigated whether EphA4 interacted with ARMS in muscle. Co-immunoprecipitation showed that the EphA4 receptor was associated with ARMS in vitro (Fig. 6, A and B) and in cortical neurons and rat muscle (Fig. 6, C and D). Although we also observed syntrophins in the same coimmunoprecipitation (Fig. 3 E), further studies still need to be conducted to confirm the formation of a ternary complex of these three proteins. The association between EphA4 and ARMS was independent of EphA4 kinase activity, as ARMS interacted equally well with both wild-type and kinase dead (KD) EphA4 (Fig. 6 E). Moreover, the overexpression of wild-type, but not KD, EphA4 induced the tyrosine phosphorylation of ARMS (Fig. 6 F). Similarly, the tyrosine phosphorylation of α-syntrophin was increased in the presence of wild-type EphA4 receptors, although α-syntrophin did not interact with EphA4 itself (Fig. 6 G). EphA4 did not induce significant phosphorylation in β1- and β2-syntrophins, indicating that the phosphorylation is isoform specific (unpublished data). Nevertheless, the association between ARMS and α-syntrophin was not affected by their phosphorylation status, as syntrophin was similarly immunoprecipitated with ARMS in the presence of either wild-type or KD EphA4 proteins (Fig. 6 E). Moreover, we did not observe the clustering of ARMS, α-syntrophin, or EphA4 when EphA4/ARMS or EphA4/α-syntrophin were coexpressed in COS7 cells, suggesting that ARMS or syntrophin cannot induce EphA4 clustering (unpublished data).
Figure 6.
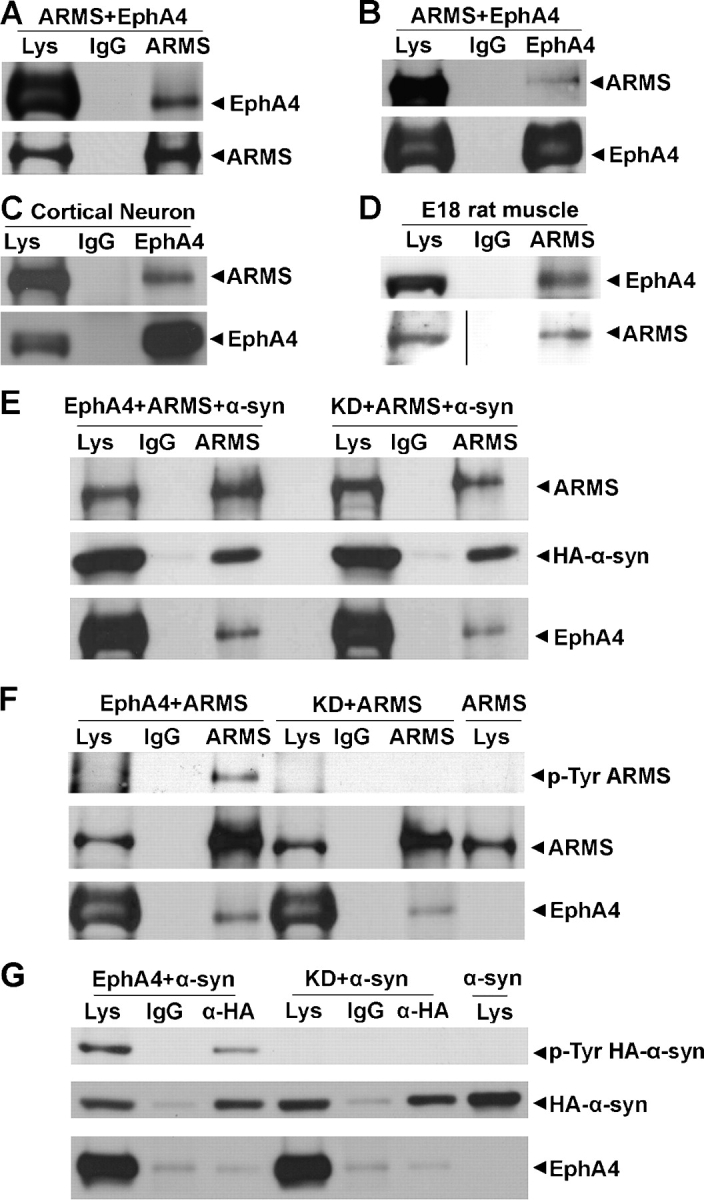
EphA4 interacted with ARMS, phosphorylated ARMS, and α-syntrophin. (A) Cell lysates were prepared from COS7 cells overexpressing ARMS and EphA4, were immunoprecipitated with ARMS 892 antiserum, and were immunoblotted with EphA4 antibody. (B) Reciprocal coimmunoprecipitation using EphA4 antibody. (C and D) In vivo coimmunoprecipitation was performed using 14 DIV cultured cortical neurons and membrane proteins from rat muscle, respectively. (E) ARMS and α-syntrophin were overexpressed in COS7 cells together with wild-type EphA4 or its KD mutant. Cell lysates were immunoprecipitated using ARMS antibody and were immunoblotted with ARMS, HA, or EphA4 antibody. (F and G) EphA4 tyrosine-phosphorylated ARMS and α-syntrophin in COS7 cells.
α-Syntrophin enhances the EphA4-induced Jak/Stat signaling in an ARMS-dependent manner
Our laboratory recently demonstrated that the activation of EphA4 receptors increases the tyrosine phosphorylation of Jak and Stat proteins (Lai et al., 2004). To assess the functional implications of the interaction between EphA4 and ARMS, we first examined whether ARMS was involved in EphA4-induced Jak and Stat activation. Consistent with our published results, the overexpression of EphA4 in COS7 cells enhanced the tyrosine phosphorylation of endogenous Jak2, tyrosine kinase (Tyk) 2, and Stat1 proteins, as revealed by immunoblots with antibodies that specifically recognize the phosphorylated Tyr1007/1008 of Jak2, Tyr1054/1055 of Tyk2, and Tyr701 of Stat1, respectively (Fig. 7 A). Interestingly, when ARMS was coexpressed with EphA4 in these cells, the tyrosine phosphorylation of Jak2 and Tyk2 kinases was greatly enhanced (250 and 170% increases, respectively), and a significant increase in Stat1 tyrosine phosphorylation (50% increase) was also observed (Fig. 7, A and B). When ARMS was coexpressed with EphA4 KD mutant, no increase in the tyrosine phosphorylation of Jak/Stat proteins was detected (Fig. 7, A and B).
Figure 7.

ARMS and α-syntrophin enhanced the EphA4-induced Jak/Stat activation. (A) COS7 cells were transfected with EphA4, EphA4 KD mutant, or ARMS, and cell lysate was immunoblotted with antibodies against phosphorylated Tyr1007/1008 of mouse Jak2, Tyr1054/1055 of human Tyk2, Tyr701 of Stat1, and Thr202/Tyr204 of ERK1/2. p-Tyr; phosphorylated Tyr. (B) Quantification of the Western blots. y axis represents the fold change of tyrosine phosphorylation. n = 3; *, P < 0.05; **, P < 0.005. (C) COS7 cells were transfected with EphA4, ARMS, α-syntrophin, or α-syntrophin mPDZ, and cell lysate was immunoblotted with antibodies against phosphorylated Tyr1007/1008 of mouse Jak2, Tyr1054/1055 of human Tyk2, Tyr701 of Stat1, and Thr202/Tyr204 of ERK1/2. (D) Quantification of the Western blots. n = 3; *, P < 0.05; **, P < 0.005. Error bars represent the SEM.
Because α-syntrophin is a binding partner of ARMS and potentially regulates ARMS localization, we asked whether α-syntrophin is also involved in EphA4 signaling. In the presence of EphA4, the overexpression of α-syntrophin further augmented the increase in the tyrosine phosphorylation of endogenous Jak2, Tyk2, and Stat1 proteins caused by ARMS (80, 50, and 30% increases, respectively; Fig. 7, C and D). This enhancement required an association between ARMS and α-syntrophin because the α-syntrophin mPDZ was ineffective in enhancing the EphA4-mediated signaling (Fig. 7, C and D). Moreover, α-syntrophin (or its mutant) alone was unable to enhance EphA4 signaling (Fig. 7, C and D). These results indicate that α-syntrophin cooperates with ARMS to enhance EphA4-induced Jak/Stat signaling.
To further verify that ARMS and α-syntrophin regulate EphA4 signaling in muscle, we transfected differentiated C2C12 myotubes with siRNA against ARMS or α-syntrophin and induced EphA4 receptor activation with preclustered ephrin-A1-Fc chimera. In cells transfected with the control oligos, ephrin-A1 activated EphA4 normally, whereas the tyrosine phosphorylation of EphA4 was greatly decreased when the expression of ARMS and α-syntrophin was reduced by siRNA transfection (Fig. 8, A and B). The impaired tyrosine phosphorylation of Eph receptors was also revealed by an antibody that recognized two phosphorylated tyrosine residues in the juxtamembrane region of EphA3 (Shamah et al., 2001; unpublished data), further supporting the notion that ARMS and α-syntrophin are important regulators in Eph receptor signaling.
Figure 8.

Tyrosine phosphorylation of EphA4 in differentiated C2C12 cells was impaired after the blockade of ARMS and α-syntrophin expression by siRNA. (A) Differentiated C2C12 muscle cells were transfected with siRNA against ARMS and were treated with 5 μg/ml ephrin-A1-Fc chimera for 10 min. Decreased tyrosine phosphorylation of EphA4 was observed in the cells transfected with ARMS siRNA compared with the control oligo. (B) α-Syntrophin siRNA was transfected at the same condition as the ARMS siRNA. Reduced tyrosine phosphorylation of EphA4 was also observed. Un, untransfected cells that were not treated with ephrin-A1-Fc. For quantification data, the y axis represents the relative level of EphA4 tyrosine phosphorylation. Tyrosine phosphorylation in cells transfected with control oligos was normalized to 100%. n = 3; *, P < 0.005. Error bars represent the SEM.
Aberrant localization of ARMS and EphA4 at the NMJ in syntrophin knockout mice
The absence of α-syntrophin in skeletal muscle leads to abnormal NMJ morphology and the decreased expression of several other proteins, such as AChR, acetylcholine esterase, nNOS, utrophin, and aquaporin-4 (Kameya et al., 1999; Adams et al., 2000, 2001; Neely et al., 2001). Because α- and β2-syntrophins interact with ARMS and induce ARMS clustering, we examined the impact of syntrophin loss in genetically altered mice lacking α- and β2-syntrophins (Adams et al., 2000, 2004). Longitudinal sections of sternomastoid muscle from adult syntrophin knockout mice were stained with ARMS antiserum (Fig. 9 A). In wild-type and β2-syntrophin knockout mice, ARMS and AChR were expressed at normal levels and were localized to the NMJ, indicating that either β2-syntrophin was not essential for the localization of ARMS (Fig. 9 A; Adams et al., 2004) or, alternatively, that α-syntrophin compensates for the loss of β2-syntrophin (Fig. 9 A). In contrast, the absence of α-syntrophin or of both α- and β2-syntrophins led to a dramatic decrease in ARMS staining at the NMJ (53 and 55% decreases, respectively; Fig. 9, A and D). In these knockout mice, synaptic AChR formed discontinuous clusters at the NMJ, and residual ARMS proteins had a similar distribution (Fig. 9 A). Western blot analysis also revealed a reduced ARMS expression in these knockout mice (Fig. 9 E). We then investigated the localization of EphA4 in syntrophin-null mice. The EphA4 staining was normal and well colocalized with AChR clusters in β2-syntrophin–null muscle (Fig. 9 B). In contrast, both EphA4 and AChR showed significantly lower staining intensities in α-syntrophin and α,β2-syntrophin–null tissues, and the staining boundary between synaptic and extrasynaptic regions was lost (for EphA4, 39 and 41% decreases, respectively; Fig. 9, B and D). Nevertheless, the total EphA4 protein level in the syntrophin−/− muscle was not significantly affected (Fig. 9 E), perhaps as a result of the expression of EphA4 in nonmuscle tissues (e.g., blood vessels) that are not affected by the absence of α- or β2-syntrophin.
Figure 9.
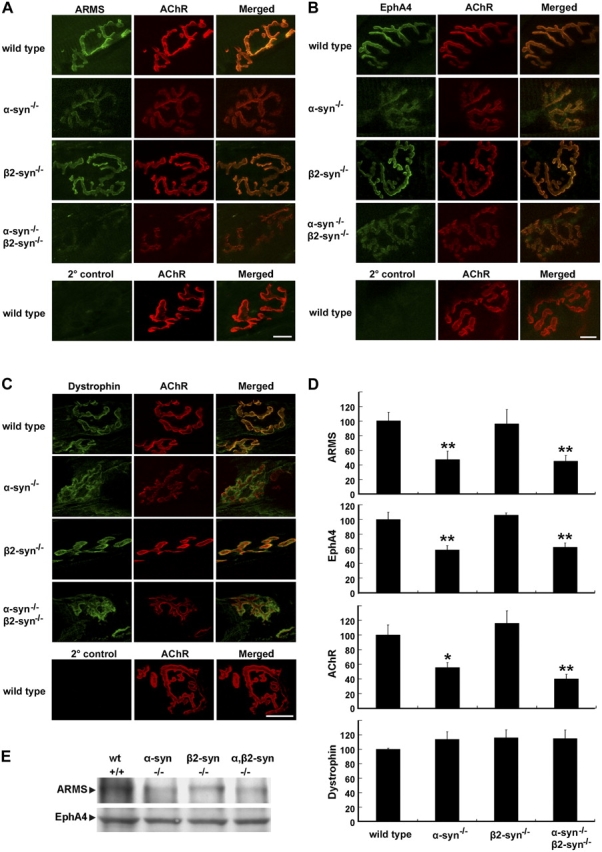
Abnormal synaptic localization of ARMS and EphA4 in syntrophin-null mice. (A) Longitudinal sections of sternomastoid muscle were double stained with α-BTX and ARMS antibody (A), EphA4 antibody (B), or dystrophin antibody (C). Wild-type sections were stained with FITC-conjugated secondary antibody and α-BTX to show the background levels. Bars (A and B), 10 μm; (C), 20 μm. (D) Quantitative analysis showed reduced junctional staining of ARMS and EphA4 in α- and α,β2-syntrophin–null mice, but not for dystrophin. y axis indicated the knockout NMJ compared with that in the wild-type NMJ, which was normalized to 100%. n = 5; *, P < 0.05; **, P < 0.01. Error bars represent the SEM. (E) Membrane proteins of gastronemius muscle from syntrophin-null mice and wild-type littermates were prepared and subjected to Western blot with ARMS and EphA4 antibodies.
Dystrophin was shown to be normally localized at the NMJ of α-, β2-, and α,β2-syntrophin–null mice in gastrocnemius muscle (Adams et al., 2000, 2004). We found that, in contrast to the decreased staining of ARMS and EphA4 at the NMJ of α- and α,β2-syntrophin–null mice, the level of staining intensity for dystrophin was not reduced in these mutant muscles (Fig. 9, C and D).
We also examined ARMS and syntrophin localizations in EphA4-null mice. Unlike the aberrant pattern observed in syntrophin−/− mice, both ARMS and syntrophins were normally expressed and localized at the NMJ in EphA4−/− muscle (unpublished data).
Discussion
The spatial and temporal patterns of ARMS expression in developing muscle closely resemble those of Eph and Trk receptors
ARMS was initially identified as a transmembrane protein that is phosphorylated on tyrosine residues in response to ephrin and neurotrophin stimulation (Kong et al., 2001). It was proposed to play key roles in neurotrophin- and ephrin-mediated neuronal outgrowth and in axon guidance during neural development and neuronal regeneration (Kong et al., 2001). Apart from their essential functions in neural development and patterning, neurotrophin and ephrin signals have also been implicated in NMJ development. One of the most convincing results comes from the study of TrkB receptors at the NMJ, in which TrkB signaling at postsynaptic muscle was shown to stabilize AChR clusters (Gonzalez et al., 1999). Like TrkB, the expression of Eph receptors in muscle was also characterized; EphA4 receptor interacts with and tyrosine phosphorylates cortactin, an actin-binding protein implicated in NMJ formation and maintenance (Lai et al., 2001). These observations suggest that Trk and Eph receptors play a critical role in NMJ development and/or maintenance (Lai and Ip, 2003a,b).
In this study, we demonstrated that ARMS was also expressed in skeletal muscle and was specifically localized at the NMJ. Analysis of ARMS protein revealed an interesting postsynaptic expression pattern that closely resembles that of the RTKs (TrkB, EphA4, and EphA7) that were previously described in developing muscle (Gonzalez et al., 1999; Lai et al., 2001). Both ARMS and the RTKs are diffusely distributed on the sarcolemma of muscle in newborn rats. With the progression of development, the proteins become more concentrated at the NMJ. Colocalization of ARMS and EphA4/TrkB clusters is evident at postsynaptic junctional sites in adult muscle. Together with the biochemical evidence showing the interaction between ARMS and RTKs (Kong et al., 2001; Arévalo et al., 2004; Fig. 6), these observations strongly suggest that the expression of ARMS is temporally and spatially coregulated with Eph and Trk receptors during muscle development. As an evolutionally conserved substrate for Trk and Eph receptors, ARMS may play an important role in modulating ephrin/neurotrophin signaling at the NMJ.
ARMS enhances Eph receptor signaling by increasing EphA4-induced Jak and Stat phosphorylation
What might be the functions of ARMS at the NMJ? We previously reported that the activation of EphA4 induces the tyrosine phosphorylation of Jak and Stat, which are two novel downstream effectors of the Eph receptor signal transduction pathway (Lai et al., 2004). In the presence of ARMS, we observed a significant increase in the EphA4-induced tyrosine phosphorylation of Jak kinases and Stat1 proteins. On the other hand, the tyrosine phosphorylation of EphA4 in differentiated C2C12 myotubes was reduced when the expression of ARMS and α-syntrophin was impaired (Figs. 7 and 8). ARMS–syntrophin may regulate the oligomerization of EphA4 in response to ephrin-A1, which is important for signal transduction downstream of the RTK. Alternatively, because ARMS contains multiple protein–protein interaction domains (Kong et al., 2001) and α-syntrophin is a well-known scaffold protein, these proteins may function as docking sites for the downstream effectors of EphA4 by recruiting them to the signaling complex during Eph-activated signal transduction. This hypothesis is consistent with the recent findings that ARMS interacts with and recruits CrkL to the Trk receptor during sustained MAPK activation (Arévalo et al., 2004). Because of its proximity to EphA4 and TrkB at the developing NMJ, ARMS, together with α-syntrophin, may coordinate the molecular events that are essential for synapse development. Because ARMS itself is also tyrosine phosphorylated after the activation of Eph and Trk receptors, it will be interesting to determine whether the phosphorylation of ARMS is required for its regulation of ephrin and neurotrophin signaling.
Syntrophins interact with ARMS and regulate ARMS localization
ARMS contains a consensus PDZ domain–binding motif on its COOH terminus that is predicted to have a high affinity for class I PDZ domains (Kong et al., 2001; for review see Sheng and Sala, 2001). Using yeast two-hybrid and biochemical studies, we identified α- and β2-syntrophins (two class I single PDZ domain–containing proteins) as interacting partners, whereas β1-syntrophin interacted only with ARMS COOH terminus in yeast but not with full-length ARMS in mammalian cells. In agreement with these observations, only α- and β2-syntrophins, but not β1-syntrophin, were able to cluster ARMS in heterologous systems. Interestingly, although the three syntrophin isoforms are all expressed in skeletal muscle, they have different expression profiles and localization during muscle development. Specifically, both α- and β2-syntrophins are initially diffusely distributed on the sarcolemma during early postnatal stages (Fig. 3 E; Kramarcy and Sealock, 2000). Later in development, the two syntrophins become gradually concentrated at the postsynaptic junctional sites, and at P12 they form visible clusters at the NMJ (Kramarcy and Sealock, 2000). In contrast, β1-syntrophin is more diffuse on the sarcolemma as well as in various nonmuscle tissues, and it is absent from most adult muscle fibers, except for the type IIb fibers in gastrocnemius (Peters et al., 1997; Kramarcy and Sealock, 2000). This differential expression pattern of syntrophin isoforms at the NMJ and their selective interactions with ARMS are consistent with the observed concentration of ARMS at the postsynaptic junctional sites. With the specific localization at the NMJ, α- and β2-syntrophins may help to anchor ARMS proteins to the synaptic dystrophin–glycoprotein complex, thus stabilizing ARMS protein clusters at the NMJ. On the other hand, the difference in syntrophin expression levels in developing muscle (i.e., higher expression of α-syntrophin compared with β-syntrophins) may explain why only α-syntrophin was retrieved from the yeast two-hybrid screening.
Because syntrophins can induce ARMS clustering in vitro, and their expression pattern in muscle closely resembles that of ARMS and RTKs such as EphA4 and TrkB, we investigated whether syntrophins regulate ARMS localization in vivo. We found that the absence of α-syntrophin causes severe ARMS defects at the NMJ. Moreover, although α- and β2-syntrophins have the comparable ability to cluster ARMS in transfected COS7 cells, ARMS localization at the NMJ is not dependent on β2-syntrophin in vivo. This finding suggests that only α-syntrophin is crucial in localizing ARMS to the synapse. In fact, a previous study has shown that α-syntrophin clusters at the mouse NMJ earlier than β2-syntrophin (Kramarcy and Sealock, 2000). At P8 (when ARMS clusters become detectable), α-syntrophin is already enriched at the NMJ (Fig. 3 F), whereas synaptic clusters of β2-syntrophin are not visible until P12 (Kramarcy and Sealock, 2000 ). Therefore, α-syntrophin may play central roles in recruiting ARMS to the NMJ, whereas the action of β2-syntrophin on ARMS clustering is redundant. Alternatively, ARMS may be recruited to the NMJ by other synaptic proteins, whereas α-syntrophin stabilizes ARMS clusters at later developmental stages via interaction with the dystrophin–glycoprotein complex. This, in turn, maintains the stability of multiple protein complexes at the NMJ. The aberrant localization of junctional EphA4 clusters in α-syntrophin–null mice also suggests that the normal EphA4 localization at junctional sites is dependent on the presence of α-syntrophin. However, because no interaction between EphA4 and syntrophin was detected (Fig. 6 G and unpublished data), the current results do not distinguish between a change in the EphA4 staining pattern either directly caused by the syntrophin loss and subsequent mislocalization of the Eph–ARMS–syntrophin complex or indirectly caused by the mislocalization of other scaffolding proteins as a result of syntrophin deficiency.
As an important scaffold protein, α-syntrophin has many interacting partners at the NMJ, such as nNOS (Brenman et al., 1996; Hashida-Okumura et al., 1999; Hillier et al., 1999), sodium and water channels (Gee et al., 1998; Schultz et al., 1998; Adams et al., 2001; Neely et al., 2001), and Ser/Thr kinases (Hasegawa et al., 1999; Lumeng et al., 1999). It is possible that ARMS competes with these proteins to bind to α-syntrophin because there is limited α-syntrophin at the NMJ during a specific developmental stage. For example, α-syntrophin plays an important role in recruiting nNOS to the sarcolemma, but nNOS is absent from the crests of the folds despite the presence of α-syntrophin. Kramarcy and Sealock (2000) suggested that a protein with high affinity for α-syntrophin competes with nNOS at this site. If ARMS localizes to the crest, it is very likely to be such a candidate, as ARMS has a PDZ-binding motif that binds preferentially and with high affinity to the PDZ domain in α-syntrophin (Gee et al., 1998, 2000; Schultz et al., 1998). Further study of the precise localization of ARMS at the NMJ will be informative.
In summary, our biochemical and genetic evidence strongly suggests that α-syntrophin plays an important role in the regulation of ARMS protein localization during NMJ differentiation. It is likely that α-syntrophin mediates the enhancement of Eph signaling by serving as a scaffold protein in recruiting protein complexes to a specific subcellular localization, such as the subsynaptic region in muscle. Together with the potential involvement of ARMS in RTK signaling, these results suggest that α-syntrophin coordinates essential RTK-signaling events at the NMJs via interaction with ARMS, which may represent an important step in NMJ development. Confirmation of the in vivo functional role of ARMS in neurotrophin and ephrin signaling at the NMJ awaits studies using ARMS mutant mice. These studies will undoubtedly provide important insights into the functional role of ARMS as well as the significance of its interaction with syntrophins in regulating the development and maintenance of the NMJ.
Materials and methods
Surgical procedure and Northern blot
The denervation surgery and sham operation on rats were performed as previously described (Ip et al., 1996). Total RNA was extracted from rat and mouse hindlimb muscle, followed by formaldehyde RNA gel analysis as previously described (Ip et al., 1996). A 32P-labeled cDNA fragment corresponding to the NH2-terminal region (from start codon to nt +1713) of ARMS was used to hybridize the RNA blots.
Yeast two-hybrid screening
100 ARMS COOH-terminal amino acids were fused to the pAS2-1 GAL4-binding domain to generate the bait construct. The P12 mouse muscle cDNA library that was constructed in λ phage was provided by J.S. Chamberlain (University of Washington, Seattle, WA). To screen for ARMS-interacting proteins, 200 μg of library plasmids were introduced into a Y190 strain that was pretransformed with the bait construct. A total of 4.4 × 105 transformants were screened. The positive plasmids were selected for ABI 310 automatic sequencing. O-nitrophenyl-β-d-galactopyranoside–based β-galactosidase activity assay was performed according to the manufacturer's protocol (Yeast Protocols Handbook; CLONTECH Laboratories, Inc.). 3-amino-1,2,4-triazole was used to inhibit the leaky expression of HIS3 genes in the Y190 strain.
DNA constructs
All ARMS and syntrophin mutants were generated using PCR-directed mutagenesis. For cloning purposes, EcoRI and NotI sites were incorporated into 5′- and 3′-ARMS primers, and EcoRI and XhoI sites were incorporated into 5′- and 3′-syntrophin primers. After amplification, the PCR products were digested and cloned into pCDNA3 or pCDNA3-3 HA vectors.
Antibodies
Rabbit polyclonal antibodies against ARMS, α-syntrophin, dystrophin, and mouse mAb SYN1351 were previously described (Froehner et al., 1987; Kramarcy et al., 1994; Peters et al., 1997; Kong et al., 2001). Chicken anti-TrkB antibody was provided by L.F. Reichardt (University of California, San Francisco, San Francisco, CA). Rhodamine- and FITC-conjugated anti–mouse or rabbit secondary antibodies were purchased from Molecular Probes, Inc. AMCA AffiniPure donkey anti–mouse IgG was purchased from Jackson ImmunoResearch Laboratories. Polyclonal anti-HA antibodies, EphA4 antibody (sc-921), and Jak2 antibody (sc-278) were purchased from Santa Cruz Biotechnology, Inc., and the mAb that recognizes the α-isoform of Stat1 was obtained from Zymed Laboratories. Antibodies against phosphotyrosine residues, phosphorylated Tyr701 Stat1, Tyr1004/1005 Tyk2, and threonine/tyrosine-phosphorylated extracellular signal–regulated kinase (ERK) 1/2 were purchased from Cell Signaling, Inc., and antibody against phosphorylated Tyr1007/1008 Jak2 was purchased from Upstate Biotechnology.
Transient transfection, immunoprecipitation, and siRNA transfection
The COS7 cells were maintained in DME (GIBCO BRL) and were supplemented with 10% heat-inactivated FBS. Transient transfection was performed using a lipofectamine transfection protocol (Invitrogen). Subsequent preparation of cell lysates and immunoprecipitation were performed as previously described (Fu et al., 2001).
Day 2 C2C12 myotubes were transfected with siRNA according to the manufacturer's protocol (Invitrogen). Oligos were designed by Invitrogen. The sequences are as follows: GGG CAA UGU GGA AAU AGU GAA AGA A against ARMS; GGG CGA AUU GGU AAA GAA GUA AGA A as its control oligo; AAG GUA UGA AUG UGC CGA UCU GCG C against α-syntrophin; and AAG GUG GUA UGU AAC CGU CUA GCG C as its control oligo. Ephrin-A1-Fc (R&D Systems) was aggregated at 0.1 mg/ml 45 min before use with 0.1 mg/ml of goat anti–human Fcγ antibody (Jackson ImmunoResearch Laboratories). C2C12 myotubes were treated with 5 μg/ml ephrin-A1-Fc for 10 min before the cell lysates were collected.
Immunocytochemistry and immunohistochemistry
For COS7 immunostaining, transfected cells were fixed with 4% PFA for 15 min, incubated with 10% FBS, and permeabilized by 0.1% Triton X-100. The dishes were incubated with primary antibody at 4°C overnight and were then incubated with secondary antibodies at RT for 1 h. Dishes were analyzed under a fluorescence microscope (model DMRA; Leica) using a 40×/1.00 oil PH3 objective lens (model PL FLUOTAR; Leica) and a digital camera (model 2.3.1; SPOT).
Hindlimb muscle from rats of different developmental stages and gastrocnemius muscle of denervated rats were frozen in isopentane/liquid nitrogen. Muscle sections were mounted onto gelatin/poly-l-lysine–coated slides. Longitudinal sections of sternomastoid muscle were prepared as described previously (Adams et al., 2000). They were fixed with 2% PFA plus 5% sucrose for 15 min at RT. After being blocked with 10% FBS, the sections were permeabilized with 0.1% Triton X-100 (for longitudinal sections, 0.5% Triton X-100 was used), followed by incubation with primary antibody at 4°C for 2 d. After conjugation of secondary antibody and rhodamine-conjugated α-bungarotoxin (Molecular Probes, Inc.), sections were mounted with mowiol (Calbiochem) and were analyzed under a fluorescence or confocal microscope (model Fluoview BX-61; Olympus). Zenon Rabbit IgG Labeling Kits were purchased from Molecular Probes, Inc., and immunohistochemistry was performed according to the manufacturer's protocol. For 40-μm longitudinal sections of sternomastoid muscle, images were taken under a confocal microscope using a 100×/1.30 oil objective lens (UPlanFl), and final images were obtained by merging 10 focal planes.
All images were taken at RT by MetaMorph Version 5.0r1 or Olympus Fluoview Version 4.0 software.
EphA4 knockout mice were provided by P. Bartlett (The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia).
Quantitative and statistical analysis
Immunoblots were scanned, and the band intensity was measured using ImageJ software (National Institutes of Health). Quantitative analysis for Fig. 9 D was performed as previously described (Adams et al., 2004). All statistical analysis was performed using an unpaired t test.
Acknowledgments
We are grateful to P. Bartlett, J. Chamberlain, and L. Reichardt for the EphA4 knockout mice, P12 mouse muscle cDNA library, and chicken anti-TrkB antibody, respectively. We also thank R. Au and J. Cheung for excellent technical assistance, and M. Zhang, F. Ip, W.-Y. Fu, and A. Fu for helpful discussions.
This study was supported by the Research Grants Council of Hong Kong Special Administrative Region (HKUST 6103/00M, 6131/02M, 2/99C, and 3/03C); the Area of Excellence Scheme of the University Grants Committee (AoE-B-01); the Hong Kong Jockey Club; National Institutes of Health grants to S.C. Froehner and M.V. Chao (NS21072 and HD23315); and a Laurie Deierlein/American Brain Tumor Association postdoctoral fellowship to J.C. Arévalo, N.Y. Ip, and K.O. Lai are Croucher Foundation Senior Research Fellow and Research Fellow, respectively.
S. Luo and Y. Chen contributed equally to this paper.
Abbreviations used in this paper: AChR, acetylcholine receptor; ARMS, ankyrin repeat-rich membrane spanning; DIV, day in vitro; ERK, extracellular signal–regulated kinase; Jak, janus kinase; KD, kinase dead; mPDZ, PDZ mutant; NMJ, neuromuscular junction; nNOS, neuronal nitric oxide synthase; P, postnatal day; PDZ, PSD-95, Dlg, ZO-1; PH, pleckstrin homology; RTK, receptor tyrosine kinase; siRNA, small interference RNA; Stat, signal transducer and activator of transcription; Trk, tropomyosin-related kinase.
References
- Adams, M.E., T.M. Dwyer, L.L. Dowler, R.A. White, and S.C. Froehner. 1995. Mouse alpha 1- and beta 2-syntrophin gene structure, chromosome localization, and homology with a discs large domain. J. Biol. Chem. 270:25859–25865. [DOI] [PubMed] [Google Scholar]
- Adams, M.E., N. Kramarcy, S.P. Krall, S.G. Rossi, R.L. Rotundo, R. Sealock, and S.C. Froehner. 2000. Absence of α-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol. 150:1385–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, M.E., H.A. Mueller, and S.C. Froehner. 2001. In vivo requirement of the α-syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J. Cell Biol. 155:113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, M.E., N. Kramarcy, T. Fukuda, A.G. Engel, R. Sealock, and S.C. Froehner. 2004. Structural abnormalities at neuromuscular synapses lacking multiple syntrophin isoforms. J. Neurosci. 24:10302–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, A.H., C.A. Freener, E. Gussoni, M. Yoshida, E. Ozawa, and L.M. Kunkel. 1996. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J. Biol. Chem. 271:2724–2730. [DOI] [PubMed] [Google Scholar]
- Arévalo, J.C., H. Yano, K.K. Teng, and M.V. Chao. 2004. A unique pathway for sustained neurotrophin signaling through an ankyrin-rich membrane-spanning protein. EMBO J. 23:2358–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman, J.E., D.S. Chao, S.H. Gee, A.W. McGee, S.E. Craven, D.R. Santillano, Z. Wu, F. Huang, H. Xia, M.F. Peters, et al. 1996. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 84:757–767. [DOI] [PubMed] [Google Scholar]
- Christopherson, K.S., B.J. Hillier, W.A. Lim, and D.S. Bredt. 1999. PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 274:27467–27473. [DOI] [PubMed] [Google Scholar]
- Flanagan, J.G., and P. Vanderhaeghen. 1998. The ephrins and Eph receptors in neural development. Annu. Rev. Neurosci. 21:309–345. [DOI] [PubMed] [Google Scholar]
- Froehner, S.C., A.A. Murnane, M. Tobler, H.B. Peng, and R. Sealock. 1987. A postsynaptic M r 58,000 (58K) protein concentrated at acetylcholine receptor-rich sites in Torpedo electroplaques and skeletal muscle. J. Cell Biol. 104:1633–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, A.K.Y., W.Y. Fu, J. Cheung, K.W.K. Tsim, F.C.F. Ip, J.H. Wang, and N.Y. Ip. 2001. Cdk5 is involved in neuregulin-induced acetylcholine receptor expression at the neuromuscular junction. Nat. Neurosci. 4:374–381. [DOI] [PubMed] [Google Scholar]
- Gee, S.H., R. Madhavan, S.R. Levinson, J.H. Caldwell, R. Sealock, and S.C. Froehner. 1998. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J. Neurosci. 18:128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee, S.H., S. Quenneville, C.R. Lombardo, and J. Chabot. 2000. Single-amino acid substitutions alter the specificity and affinity of PDZ domains for their ligands. Biochemistry. 39:14638–14646. [DOI] [PubMed] [Google Scholar]
- Gonzalez, M., F.P. Ruggiero, Q. Chang, Y.J. Shi, M.M. Rich, S. Kraner, and R.J. Balice-Gordon. 1999. Disruption of TrkB-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron. 24:567–583. [DOI] [PubMed] [Google Scholar]
- Hasegawa, M., A. Cuenda, M.G. Spillantini, G.M. Thomas, V. Buee-Scherrer, P. Cohen, and M. Goedert. 1999. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J. Biol. Chem. 274:12626–12631. [DOI] [PubMed] [Google Scholar]
- Hashida-Okumura, A., N. Okumura, A. Iwamatsu, R.M. Buijs, H.J. Romijn, and K. Nagai. 1999. Interaction of neuronal nitric-oxide synthase with alpha1-syntrophin in rat brain. J. Biol. Chem. 274:11736–11741. [DOI] [PubMed] [Google Scholar]
- Hillier, B.J., K.S. Christopherson, K.E. Prehoda, D.S. Bredt, and W.A. Lim. 1999. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science. 284:812–815. [PubMed] [Google Scholar]
- Huang, E.J., and L.F. Reichardt. 2003. Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72:609–642. [DOI] [PubMed] [Google Scholar]
- Iglesias, T., N. Cabrera-Poch, M.P. Mitchell, T.J. Naven, E. Rozengurt, and G. Schiavo. 2000. Identification and cloning of Kidins220, a novel neuronal substrate of protein kinase D. J. Biol. Chem. 275:40048–40056. [DOI] [PubMed] [Google Scholar]
- Ip, F.C., A.K. Fu, K.W. Tsim, and N.Y. Ip. 1996. Differential expression of ciliary neurotrophic factor receptor in skeletal muscle of chick and rat after nerve injury. J. Neurochem. 67:1607–1612. [DOI] [PubMed] [Google Scholar]
- Kaech, S.M., C.W. Whitfield, and S.K. Kim. 1998. The LIN-2/LIN-7/LIN-10 complex mediates basolateral membrane localization of the C. elegans EGF receptor LET-23 in vulval epithelial cells. Cell. 94:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameya, S., Y. Miyagoe, I. Nonaka, T. Ikemoto, M. Endo, K. Hanaoka, Y. Nabeshima, and S. Takeda. 1999. alpha1-syntrophin gene disruption results in the absence of neuronal-type nitric-oxide synthase at the sarcolemma but does not induce muscle degeneration. J. Biol. Chem. 274:2193–2200. [DOI] [PubMed] [Google Scholar]
- Kim, E., M. Niethammer, A. Rothschild, Y.N. Jan, and M. Sheng. 1995. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature. 378:85–88. [DOI] [PubMed] [Google Scholar]
- Kong, H.Y., J. Boulter, J.L. Weber, C. Lai, and M.V. Chao. 2001. An evolutionarily conserved transmembrane protein that is a novel downstream target of neurotrophin and ephrin receptors. J. Neurosci. 21:176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau, H.C., L.T. Schenker, M.B. Kennedy, and P.H. Seeburg. 1995. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 269:1737–1740. [DOI] [PubMed] [Google Scholar]
- Kramarcy, N.R., and R. Sealock. 2000. Syntrophin isoforms at the neuromuscular junction: developmental time course and differential localization. Mol. Cell. Neurosci. 15:262–274. [DOI] [PubMed] [Google Scholar]
- Kramarcy, N.R., A. Vidal, S.C. Froehner, and R. Sealock. 1994. Association of utrophin and multiple dystrophin short forms with the mammalian M(r) 58,000 dystrophin-associated protein (syntrophin). J. Biol. Chem. 269:2870–2876. [PubMed] [Google Scholar]
- Kullander, K., and R. Klein. 2002. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell Biol. 3:475–486. [DOI] [PubMed] [Google Scholar]
- Lai, K.O., and N.Y. Ip. 2003. a. Central synapse and neuromuscular junction: same players, different roles. Trends Genet. 19:395–402. [DOI] [PubMed] [Google Scholar]
- Lai, K.O., and N.Y. Ip. 2003. b. Postsynaptic signaling of new players at the neuromuscular junction. J. Neurocytol. 32:727–741. [DOI] [PubMed] [Google Scholar]
- Lai, K.O., F.C. Ip, J. Cheung, A.K. Fu, and N.Y. Ip. 2001. Expression of Eph receptors in skeletal muscle and their localization at the neuromuscular junction. Mol. Cell. Neurosci. 17:1034–1047. [DOI] [PubMed] [Google Scholar]
- Lai, K.O., Y. Chen, H.M. Po, K.C. Lok, K. Gong, and N.Y. Ip. 2004. Identification of the Jak/Stat proteins as novel downstream targets of EphA4 signaling in muscle: implications in the regulation of acetylcholinesterase expression. J. Biol. Chem. 279:13383–13392. [DOI] [PubMed] [Google Scholar]
- Lumeng, C., S. Phelps, G.E. Crawford, P.D. Walden, K. Barald, and J.S. Chamberlain. 1999. Interactions between beta 2-syntrophin and a family of microtubule-associated serine/threonine kinases. Nat. Neurosci. 2:611–617. [DOI] [PubMed] [Google Scholar]
- Migaud, M., P. Charlesworth, M. Dempster, L.C. Webster, A.M. Watabe, M. Makhinson, Y. He, M.F. Ramsay, R.G. Morris, J.H. Morrison, T.J. O'Dell, and S.G. Grant. 1998. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 396: 433–439. [DOI] [PubMed] [Google Scholar]
- Neely, J.D., M. Amiry-Moghaddam, O.P. Ottersen, S.C. Froehner, P. Agre, and M.E. Adams. 2001. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. USA. 98:14108–14113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, M.F., M.E. Adams, and S.C. Froehner. 1997. Differential association of syntrophin pairs with the dystrophin complex. J. Cell Biol. 138:81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, J., U. Hoffmuüller, G. Krause, J. Ashurst, M.J. Macias, P. Schmieder, J. Schneider-Mergener, and H. Oschkinat. 1998. Specific interactions between the syntrophin PDZ domain and voltage-gated sodium channels. Nat. Struct. Biol. 5:19–24. [DOI] [PubMed] [Google Scholar]
- Shamah, S.M., M.Z. Lin, J.L. Goldberg, S. Estrach, M. Sahin, L. Hu, M. Bazalakova, R.L. Neve, G. Corfas, A. Debant, and M.E. Greenberg. 2001. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 105:233–244. [DOI] [PubMed] [Google Scholar]
- Sheng, M., and C. Sala. 2001. PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 24:1–29. [DOI] [PubMed] [Google Scholar]
- Simske, J.S., S.M. Kaech, S.A. Harp, and S.A. Kim. 1996. LET-23 receptor localization by the cell junction protein LIN-7 during C. elegans vulval induction. Cell. 85:195–204. [DOI] [PubMed] [Google Scholar]
- Wiedemann, U., P. Boisquerin, R. Leben, D. Leitner, G. Krause, K. Moelling, R. Volkmer-Engert, and H. Oschkinat. 2004. Quantification of PDZ domain specificity, prediction of ligand affinity and rational design of super-binding peptides. J. Mol. Biol. 343:703–718. [DOI] [PubMed] [Google Scholar]
- Xia, J., X. Zhang, J. Staudinger, and R.L. Huganior. 1999. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 22:179–187. [DOI] [PubMed] [Google Scholar]
- Xia, J., H.K. Chung, C. Wihler, R.L. Huganir, and D.J. Linden. 2000. Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron. 28:499–510. [DOI] [PubMed] [Google Scholar]