

Abstract
Stress granules (SGs) are cytoplasmic aggregates of stalled translational preinitiation complexes that accumulate during stress. GW bodies/processing bodies (PBs) are distinct cytoplasmic sites of mRNA degradation. In this study, we show that SGs and PBs are spatially, compositionally, and functionally linked. SGs and PBs are induced by stress, but SG assembly requires eIF2α phosphorylation, whereas PB assembly does not. They are also dispersed by inhibitors of translational elongation and share several protein components, including Fas-activated serine/threonine phosphoprotein, XRN1, eIF4E, and tristetraprolin (TTP). In contrast, eIF3, G3BP, eIF4G, and PABP-1 are restricted to SGs, whereas DCP1a and 2 are confined to PBs. SGs and PBs also can harbor the same species of mRNA and physically associate with one another in vivo, an interaction that is promoted by the related mRNA decay factors TTP and BRF1. We propose that mRNA released from disassembled polysomes is sorted and remodeled at SGs, from which selected transcripts are delivered to PBs for degradation.
Introduction
In response to environmental stress, eukaryotic cells reprogram their translational machinery to allow the selective expression of proteins required for viability in the face of changing conditions. During stress, mRNAs encoding constitutively expressed “housekeeping” proteins are redirected from polysomes to discrete cytoplasmic foci known as stress granules (SGs), a process that is synchronous with stress-induced translational arrest (Anderson and Kedersha, 2002; Kedersha and Anderson, 2002). Both SG assembly (Kedersha et al., 1999) and translational arrest (Krishnamoorthy et al., 2001) are initiated by the phosphorylation of translation initiation factor eIF2α, which reduces the availability of the eIF2–GTP–tRNAMet ternary complex that is needed to initiate protein translation. Drugs that stabilize polysomes (e.g., emetine) cause SG disassembly, whereas drugs that dismantle polysomes (e.g., puromycin) promote the assembly of SGs, indicating that mRNA moves between polysomes and SGs (Kedersha et al., 2000). These results suggest that SGs are sites of mRNA triage at which mRNP complexes are monitored for integrity and composition and are then routed to sites of reinitiation, degradation, or storage (Anderson and Kedersha, 2002; Kedersha and Anderson, 2002). During stress, mRNA continues to be directed to sites of reinitiation, but in the absence of eIF2–GTP–tRNAMet, it shuttles back to SGs, where it accumulates (Kedersha et al., 2000). mRNAs within SGs are not degraded, making them available for rapid reinitiation in cells that recover from stress. The observation that labile mRNAs are stabilized during stress (Laroia et al., 1999; Bolling et al., 2002) suggests that some aspect of the mRNA degradative process is disabled during the stress response. Thus, the accumulation of mRNA at SGs may be a consequence of both stress-induced translational arrest and stress-induced mRNA stabilization.
Although the process of stress-induced mRNA stabilization is poorly understood, it likely involves the inactivation of one or more mRNA decay pathways. Two major mechanisms of mRNA degradation are active in eukaryotic cells (Decker and Parker, 2002). In the first pathway, deadenylated transcripts are degraded by a complex of 3′–5′ exonucleases known as the exosome. In vitro studies using cell extracts reveal that some mRNAs bearing adenine/uridine-rich destabilizing elements (AREs) in their 3′ untranslated regions are degraded by this 3′–5′ exosome-dependent pathway (Jacobs et al., 1998; Chen et al., 2001; Mukherjee et al., 2002). The second pathway entails the removal of the seven-methyl guanosine cap from the 5′ end of the transcript by the DCP1–DCP2 complex (Long and McNally, 2003; Jacobson, 2004), allowing 5′–3′ exonucleolytic degradation by XRN1 (Stevens, 2001). In yeast, components of this 5′–3′ decay pathway are concentrated at discrete cytoplasmic foci known as processing bodies (PBs; Sheth and Parker, 2003). Yeast genetic studies reveal that mRNA decay intermediates accumulate at PBs when normal decay is blocked, suggesting that PBs are sites of decapping and 5′–3′ degradation (Sheth and Parker, 2003). Studies in mammalian cells have revealed similar structures that contain DCP1/2, XRN1, GW182, and Lsm1–7 heptamer (Eystathioy et al., 2002, 2003; Ingelfinger et al., 2002; Cougot et al., 2004a,b; Yang et al., 2004). In mammalian cells, the targeted knockdown of XRN1 results in the accumulation of poly(A)+-containing mRNA at these sites, suggesting that this mRNA decay pathway is conserved in both lower and higher eukaryotes. Although the composition of GW bodies/PBs is somewhat different in lower and higher eukaryotes, because they share the ability to process mRNA, we will provisionally refer to these foci as PBs. Interestingly, metabolic inhibitors that promote (e.g., puromycin) or inhibit (e.g., emetine) the assembly of SGs in mammalian cells have similar effects on the assembly of both yeast and mammalian PBs. These results indicate that both SGs and PBs are sites at which mRNA accumulates after polysome disassembly.
In this study, we catalog the protein composition of SGs and PBs and report several links between these cytoplasmic subdomains. DCP1a/2 and GW182 are components of PBs but not of SGs, whereas most initiation factors (e.g., eIF3, eIF4G, and PABP-1) are components of SGs but not of PBs. In contrast, eIF4E, XRN1, Fas-activated serine/threonine phosphoprotein (FAST), and tristetraprolin (TTP) are found in PBs in unstressed cells but partially or completely relocalize to SGs in stressed cells. A single class of reporter mRNA is found in both SGs and PBs, suggesting that individual transcripts at different stages of processing may localize in each structure. Photobleaching studies reveal kinetically distinct classes of proteins within SGs and PBs: TTP, T cell intracellular antigen (TIA), and G3BP rapidly shuttle in and out of these structures, whereas putative scaffold proteins DCP1a, GW182, and FAST are relatively stable constituents of these structures. We propose a model wherein mRNA released from polysomes during stress is routed to SGs for triage, sorting, and mRNP remodeling, after which certain transcripts are selectively exported to associated PBs for degradation.
Results
SGs and PBs are induced by different stimuli
Previous studies have shown that the composition of SGs varies with the stimulus used to elicit their assembly; e.g., heat shock–induced SGs contain HSP27, whereas arsenite-induced SGs do not (Kedersha et al., 1999), and SGs containing G3BP (Ras-GSP SH3 domain–binding protein) have been described as lacking TIA-1 (Tourriere et al., 2003). Therefore, we used a number of SG-inducing stimuli to survey SG and PB composition. U2OS cells and HeLa cells were treated with arsenite (oxidative stress), clotrimazole (mitochondrial stress), or heat shock, and were stained for SG markers eIF4E (Fig. 1) and eIF3 and PB marker DCP1a. As shown in Fig. 1 (A and E), some unstressed cells contain DCP1a-positive PBs (yellow arrows), whereas others do not. Remarkably, eIF4E appears present in PBs together with DCP1a. Arsenite treatment (Fig. 1, B and F) induces both SGs (Fig. 1, white arrowheads) and PBs in all cells, and the great majority of the PBs appear clustered around SGs in both U2OS (Fig. 1 B) and HeLa (Fig. 1 F) cells. In contrast, cells treated with the mitochondrial poison clotrimazole (Fig. 1, C and G) or heat shock (Fig. 1, D and H) display SGs but do not show an increase in PBs, nor do PBs appear associated with SGs. We conclude that SGs and PBs are coordinately induced by arsenite, but that other stress stimuli induce SGs in cells lacking PBs.
Figure 1.
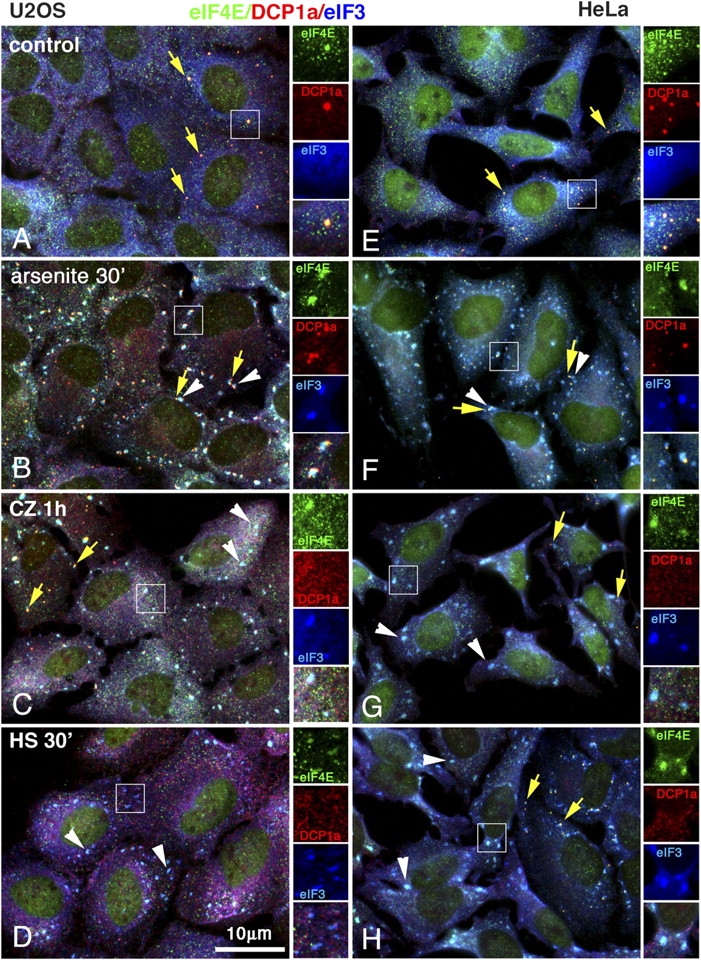
SGs and PBs in U2OS and HeLa cells. U2OS osteosarcoma (A–D) or HeLa (E–H) cells were untreated (A and E); exposed to 500 μM arsenite for 30 min (B and F); exposed to 20 μM clotrimazole (Sigma Aldrich) for 1 h (C and G); or exposed to heat (44°C) for 30 min (D and H). Cells were immediately fixed and stained for eIF4E, DCP1a, and eIF3. Yellow arrows indicate PBs; white arrowheads indicate SGs. In both cell lines, note that SGs are induced in cells lacking PBs upon clotrimazole (C and G) or heat shock treatment (D and H), whereas arsenite treatment induces both SGs and PBs that are juxtaposed (B and E). In each panel, the indicated inset is reproduced at the right as replicate views of the same field showing eIF4E, DCP1a, eIF3, and the merged view.
Shared versus unique protein components of SGs and PBs
The presence of eIF4E in PBs was unexpected. Therefore, we sought to confirm this result and determine whether other previously described SG components might also be present in PBs. We used DU145 cells, which had been previously used to analyze SG components (Kedersha et al., 2002), and induced SGs by the transient transfection of GFP-G3BP, an SG component whose expression induces the assembly of very large SGs readily amenable to microscopic analysis (Tourriere et al., 2003). GFP-G3BP (Fig. 2) induces the formation of large SGs (1–5 μm in diameter; Fig. 2, white arrowheads) that are typically irregular in shape and are frequently juxtaposed with PBs (Fig. 2, yellow arrows). GFP-G3BP transfectants were counterstained for the PB marker DCP1a and the SG marker TIA-1 (Fig. 2 A). DCP1a is found in PBs but is largely excluded from the SG, as shown by TIA-1 staining. This indicates that GFP-G3BP and TIA-1 are present in SGs but are excluded from PBs, whereas DCP1a is present in PBs but not in SGs. Similar analysis indicates that another PB component, XRN1 (Fig. 2 B), is present in both PBs and G3BP-induced SGs. Consistent with the data shown in Fig. 1, eIF4E (Fig. 2 C) is found in both SGs and PBs, whereas eIF4G is found in SGs but not in eIF4-positive PBs. Two approaches confirm that the eIF4E signal in PBs is not caused by antibody cross-reactivity with some PB protein: (1) a different eIF4E antibody gives identical results (unpublished data); and (2) transfected FLAG-tagged eIF4E reveals the same PB–SG distribution when detected using anti-FLAG (Fig. 2 D, blue). We conclude that eIF4E is present in both PBs and SGs. In contrast, eIF3b (Fig. 2 D) and PABP-1 (Fig. 2 E) are restricted to SGs. The TIA-1–interacting protein FAST (Fig. 2 F) exhibits a pattern similar to XRN1; i.e., it is predominantly associated with PBs and is weakly associated with SGs.
Figure 2.
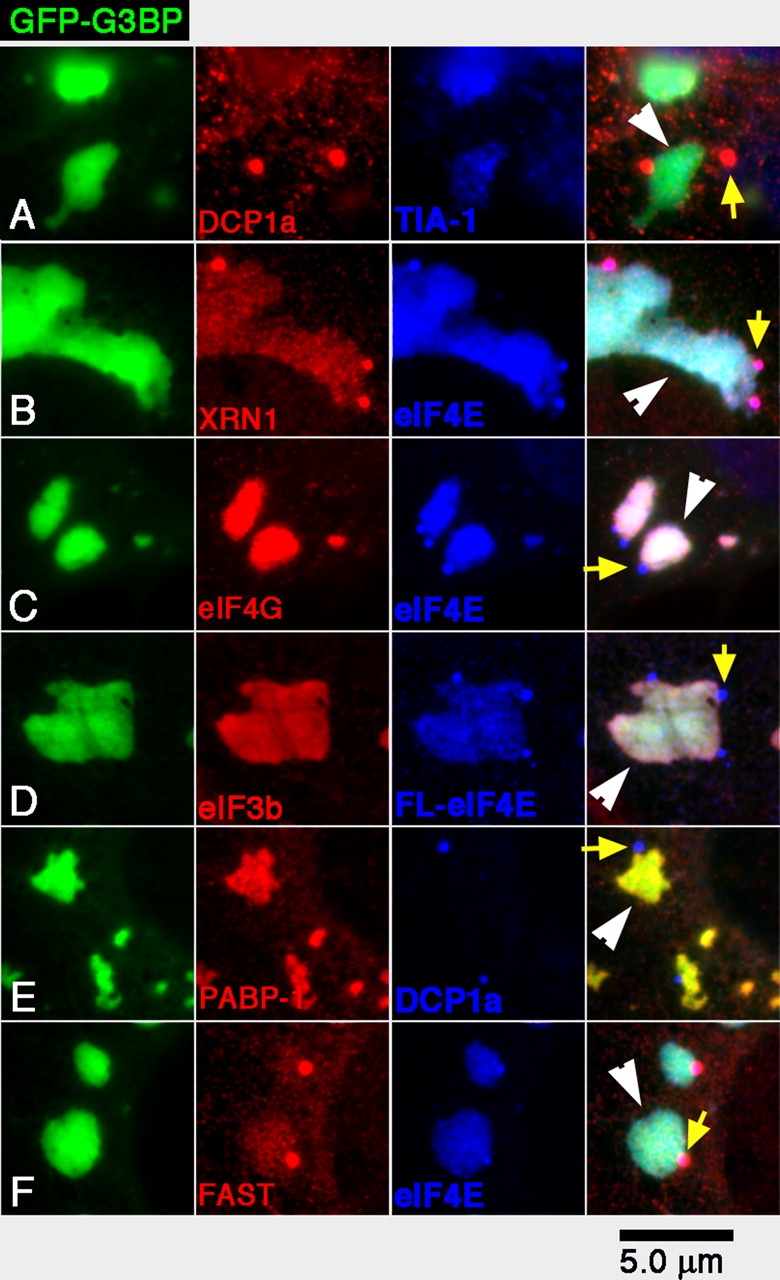
Distribution of proteins between G3BP-induced SGs and PBs. SGs were induced in DU145 cells by the transfection of GFP-G3BP and cells stained as indicated. In D, cells were cotransfected with FLAG-eIF4E and stained with anti-FLAG; (A) DCP1a and TIA-1; (B) XRN1 and eIF4E; (C) eIF4G and eIF4E; (D) eIF3b and FLAG-eIF4E; (E) PABP-1 and DCP1a; and (F) FAST and eIF4E. Yellow arrows indicate representative PBs; white arrowheads indicate SGs in the merged views.
To confirm that the SGs induced by G3BP overexpression are compositionally similar to SGs induced by stress, we exposed DU145 cells to oxidative stress using arsenite and stained for endogenous SG and PB markers (Fig. 3). Although arsenite-induced SGs are smaller than those induced by GFP-G3BP overexpression, the results are generally comparable. As shown in Fig. 3 A, DCP1a is confined to PBs (yellow arrow), eIF3b is confined to SGs (white arrowhead), and eIF4E is present in both structures. PABP-1 and TIA-1 are restricted to SGs, whereas XRN1 (Fig. 3 B) predominates in PBs, but a minor amount is detectable in SGs. eIF4G (Fig. 3 C), phospho-eIF2α (Fig. 3 D), and endogenous G3BP (Fig. 3 E) are only in SGs, whereas GW182 (Fig. 3 F) and FAST (Fig. 2 F) predominate in PBs. We conclude that G3BP, eIF4G, eIF3, phospho-eIF2α, and PABP-1 are restricted to SGs, whereas DCP1a and 2 (unpublished data) are confined to PBs. GW182 autoantibody staining suggests that it is present in both PBs and SGs (Fig. 3 F, green); however, anti-GW182 is not monospecific by Western blot analysis, and a GFP-tagged construct encoding most of GW182 (aa 313–1709) is only found in PBs (Yang et al., 2004). Thus, GW182 localizes to PBs, whereas its association with SGs remains inconclusive. Of considerable interest is the finding that XRN1, FAST, and eIF4E are present in both PBs and SGs. The dual SG–PB localization of each of these proteins was confirmed by using tagged constructs in transient transfection assays (Fig. 2 D and see Fig. 8, B–D). FAST interacts with TIA-1 and antagonizes the translational silencing of TIA-1 (Li et al., 2004b). In unstressed COS7 cells, most FAST is nuclear and is associated with mitochondria (Li et al., 2004a). Its presence in PBs and its relocalization to SGs may reflect its function as a translational regulator of TIA proteins.
Figure 3.
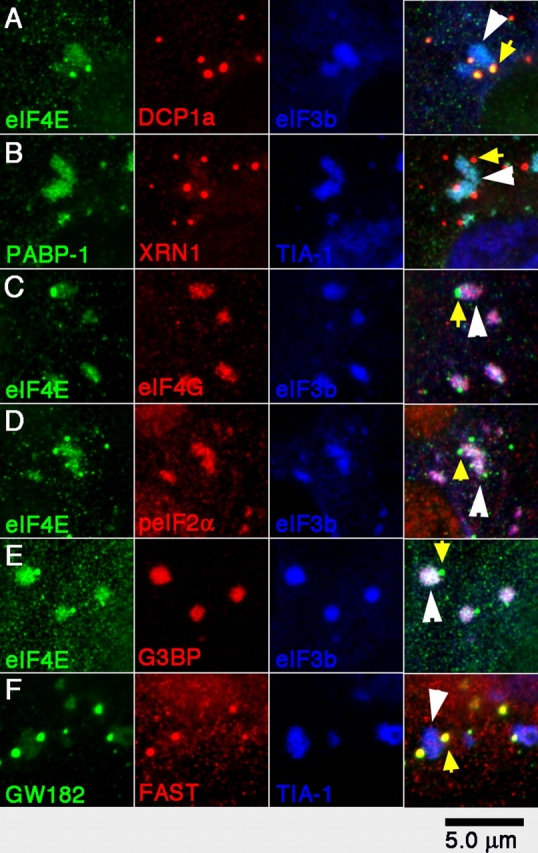
Distribution of proteins between arsenite-induced SGs and PBs. SGs were induced in DU145 cells by arsenite treatment, and cells were triple stained for the indicated proteins: (A) eIF4E, DCP1a, and eIF3b; (B) PABP-1, XRN1, and TIA-1; (C) eIF4E, eIF4G, and eIF3b; (D) eIF4E, phospho-eIF2α, and eIF3b; (E) eIF4E, G3BP, and eIF3b; and (F) GW182, FAST, and TIA-1. Yellow arrows indicate representative PBs; white arrowheads indicate SGs in the merged views.
Figure 8.

TTP and BRF1 promote fusion of SGs with PBs. COS7 cells triply transfected with GFP-G3BP as an SG marker, RFP-DCP1a as a PB marker, and one of the following: (A) vector; (B) FLAG-tagged FAST; (C) FLAG-XRN1; (D) FLAG-eIF4E; (E) TTP-myc; or (F) FLAG-BRF1. Yellow arrows indicate positions of representative PBs; white arrowheads indicate position of SGs.
PBs are present in AA cells that cannot phosphorylate eIF2α or assemble SGs
Little is known about the signaling pathways and specific molecular events that govern PB assembly, although their size and number increase when 5′–3′ mRNA decay is blocked (Sheth and Parker, 2003) and vary throughout the cell cycle (Yang et al., 2004). SG assembly requires the phosphorylation of eIF2α (Kedersha et al., 1999) and is mediated by the aggregation of one of several RNA-binding proteins, including TIA proteins (Gilks et al., 2004), Fragile X Mental Retardation protein (Mazroui et al., 2002), G3BP (Tourriere et al., 2003), and the survival of motor neurons protein (Hua and Zhou, 2004). We therefore asked whether PBs are present in mutant AA cells, in which the normal eIF2α allele has been replaced with a nonphosphorylatable mutant (S51A eIF2α) allele by homozygous replacement (Scheuner et al., 2001). As shown in Fig. 4 A, treatment of wild-type SS cells with arsenite results in robust SG assembly (white arrowheads), as assessed using three independent SG markers (eIF3b; G3BP; and TIA related [TIAR]). In contrast, no SG assembly is seen with any of these SG markers in arsenite-treated AA mouse cells (Fig. 4 A, right). Likewise, SGs are not induced in AA cells by any other treatments, including heat shock, puromycin treatment, or transfection with G3BP (unpublished data). Only the enforced expression of the phosphomimetic form of eIF2α generates SGs in AA cells (supplemental Fig. 1 in McEwen et al., 2005), demonstrating their competence to assemble SGs given this essential trigger.
Figure 4.

Role of eIF2α phosphorylation and Lsm4 expression in SG and PB formation. (A) Arsenite-treated wild-type (SS) and eIF2α S51A mutant (AA) MEFs stained for SG markers eIF3b, G3BP, and TIAR. (B) Arsenite-treated SS and AA MEFs stained for PB markers GW182 and DCP1a and the SG marker protein TIA-1. Yellow arrows indicate representative PBs; white arrowheads indicate SGs in the merged views. (C–E) DU145 or HT1080 cells were transfected with control siRNA or siRNA targeting Lsm4, processed for immunofluorescence, and examined for PBs and SGs. (C) Semiquantitative RT-PCR showing reduced expression of Lsm4 mRNA in Lsm4-siRNA–transfected HT1080 cells. (D) Percentage of cells containing visible PBs before (dark gray bars) or after (light gray bars) arsenite treatment. (E) Confocal micrographs of HT1080 cells stained for PB markers GW182 and DCP1a and SG marker TIA-1.
Staining arsenite-stressed control SS cells and mutant AA cells for PB marker proteins GW182 and DCP1a (Fig. 4) reveals that both cell lines display numerous PBs (Fig. 4 B, yellow arrows). In contrast, SGs (Fig. 4, white arrowheads) are induced in SS cells, as shown by TIA-1 staining, but are absent in AA cells treated similarly. To verify that these apparent PBs in both SS and AA cells behave normally, we confirmed that they were abolished upon treatment of the cells with emetine or cycloheximide (unpublished data). We conclude that PBs, unlike SGs, do not require the phosphorylation of eIF2α for their assembly.
PBs are induced by arsenite
As arsenite induces both PBs and SGs (Fig. 1), we asked whether the knockdown of PBs would affect SG assembly in response to arsenite. Several small interference RNAs (siRNAs) were used to knockdown different PB components (unpublished data), but only siRNA against Lsm4 was minimally effective in preventing PB assembly in response to arsenite. DU145 and HT1080 cells were transfected with control or Lsm4 siRNA, untreated or treated with arsenite, fixed and stained for PBs, scored microscopically, and counted. As shown in Fig. 4 C, RT-PCR reveals that efficient knockdown Lsm4 mRNA is obtained, which reduces PBs (Fig. 4, D [dark gray bars] and E). However, upon arsenite treatment, the percentage of cells with PBs increases markedly despite knockdown for Lsm4. In HT1080 cells, Lsm4 knockdown is able to reduce the percentage of PB-positive cells to <5% of control levels in the absence of stress (Fig. 4, D [right bars] and E). Arsenite treatment induces PBs in ∼75% of these cells, whereas >95% display SGs. As heat shock and clotrimazole also induce SGs in cells lacking PBs, the data indicate that assembly of SGs and PBs is regulated by distinct signaling pathways.
Physical juxtaposition and transient interactions between SGs and PBs
We were struck by the observation that arsenite-induced SGs appear juxtaposed with PBs and contain eIF4E but no other initiation factors (e.g., Figs. 1 and 3). Therefore, we investigated the kinetics of SG–PB assembly by using combinations of stress-inducing conditions. Fig. 5 shows HeLa cells subjected to different stresses and triple-stained for eIF3b (SG-specific marker), FAST (PBs), and eIF4E (found in both SGs and PBs). Untreated cells (Fig. 5 A) display few PBs (Fig. 5, yellow arrows), which appear as yellow dots because of the merge of green (eIF4E) and red (FAST) signals. The treatment of cells with arsenite for 30 min (Fig. 5 B) resulted in a dramatic increase in the number of PBs coordinate with robust SG assembly (Fig. 5, white arrowheads); remarkably, virtually all PBs were found adjacent to SGs, as shown in Fig. 1. SG and PB formation appear synchronously in response to shorter arsenite treatments.
Figure 5.
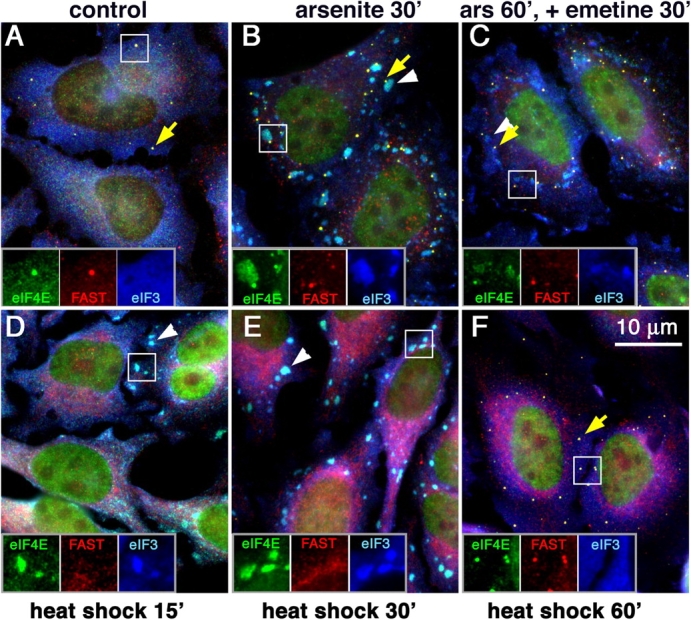
SG and PB assembly induced by different stresses. HeLa cells were subjected to different stresses and were stained for eIF4E, FAST, and eIF3. (A) Unstressed cells, some of which contain PBs (yellow arrow) but no SGs. (B) Arsenite (500 μM for 30 min) induces both SGs (white arrowhead) and PBs (yellow arrow). (C) Cells were treated with arsenite for 60 min, and 20 μg/ml emetine was added during the last 30 min. (D–F) Cells were subjected to heat shock (44°C) for 15 min (C), 30 min (D), or 60 min (E). Yellow arrows indicate representative PBs; white arrowheads indicate SGs in the merged views. In each panel, the indicated inset is reproduced at the bottom as replicate views of the same field showing eIF4E, FAST, and eIF3.
Disassembly of both SGs and PBs is enforced by emetine and cycloheximide, which are drugs that inhibit translational elongation and block the disassembly of polysomes, thereby preventing the translocation of mRNA into SGs and PBs (Kedersha et al., 2000; Sheth and Parker, 2003; Cougot et al., 2004a). As the size of both PBs and SGs should be proportional to the amount of mRNA within each, we determined whether adding emetine to arsenite-treated cells would cause the disassembly of SGs before PBs, or vice versa. Emetine addition to arsenite-treated cells followed by an additional 30-min incubation (Fig. 5 C) resulted in partial SG disassembly (Fig. 5, white arrowheads) without affecting PBs. A longer emetine treatment (1–2 h) completely dispersed SGs without affecting PBs (unpublished data), but the treatment of cells for 1 h with emetine in the absence of arsenite disassembled all the PBs (unpublished data). This indicates that emetine treatment disassembles SGs before disassembling PBs and that eIF4E is still present in PBs upon emetine-enforced SG disassembly. Cells that were exposed to heat shock (44°C) for 15 and 30 min (Fig. 5, D and E) displayed SGs in cells lacking PBs. Continued heat shock treatment for 1 h resulted in the disappearance of SGs and the appearance of PBs (Fig. 5 F). Thus, heat shock appears to trigger a coordinate sequence of events: an early and transient induction of SGs followed by a late induction of PBs. Remarkably, eIF4E distribution appears to shift between the two compartments under these conditions, suggesting that some eIF4E-bound mRNA may move from SGs to PBs during heat shock.
A single class of mRNA transcripts is present in both SGs and PBs
SGs are thought to be sites of mRNA sorting rather than decay (Kedersha and Anderson, 2002). As PBs are putative sites of 5′–3′ mRNA degradation (Sheth and Parker, 2003; Cougot et al., 2004a), the juxtaposition of the two structures during arsenite treatment (Fig. 1) and their sequential assembly/disassembly during heat shock (Fig. 5) suggests that mRNAs destined for decay are sorted in SGs and are subsequently transported into PBs. If so, a single class of mRNA transcripts should be detected in both SGs and PBs at different stages of its processing. To test this prediction, we expressed a β-globin mRNA containing the MS2-binding site in its 3′ untranslated region (pEF-7B-MS2bs) together with a fusion protein comprised of GFP, MS2 coat protein, and a nuclear localization signal (Fig. 6 A, GFP-MS2-NLS). Transfection of GFP-MS2 alone or with a globin reporter lacking the MS2-binding site resulted in a signal exclusively localized to the nucleus (Rook et al., 2000; unpublished data). When GFP-MS2 is cotransfected with the globin reporter containing the MS2-binding site, nuclear export of the tethered GFP signal is observed in 2–10% of transfected cells; only in cells expressing high amounts of globin-MS2 is the tethered GFP exported from the nucleus. Cytoplasmic GFP signal is found in SGs and PBs, as shown in Fig. 6 (B and C). The RNA-tethered signal is equally distributed between PBs (Fig. 6, yellow arrows) and SGs (Fig. 6, white arrowheads), as shown by colocalization with DCP1a and TIA-1 in Fig. 6 B and with XRN1 and eIF4E in Fig. 6 C. We conclude that a single class of mRNA localizes to both SGs and PBs.
Figure 6.

A single species of reporter mRNA is present in both SGs and PBs. (A) Schematic of the GFP-MS2–tethered mRNA reporter constructs used to visualize the subcellular localization of the globin-MS2 mRNA. (B and C) COS7 cells transiently transfected with both plasmids shown in A and counterstained for different SG and PB markers. (B) GFP-globin mRNA, PB marker DCP1a, and SG marker TIA-1. (C) GFP-globin mRNA, PB marker XRN1, and SG–PB marker eIF4E. Insets show enlargement of boxed areas with colors separated. Yellow arrows indicate representative PBs; white arrowheads indicate SGs in the merged views.
SG–PB interactions in real time using time-lapse microscopy
To investigate the physical interaction of PBs with SGs over time, we obtained a series of red (RFP) or green (GFP or YFP)-tagged proteins and transiently expressed various combinations in COS7 cells, which were subsequently viewed live using a heated stage and inverted confocal microscope. The extremely motile nature of PBs (Yang et al., 2004) required that each frame be made from a volume-rendered image derived from ∼10 z-axis sections (see Materials and methods) so as to visualize all of the PBs within each cell. Cells that were cotransfected with RFP-DCP1a (PB marker) and GFP–TIA-1 (SG marker) display spontaneous SGs in 30–70% of the transfectants, and these SGs frequently associate with one or more PBs. When followed over time (Fig. 7 A and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200502088/DC1), some “attached” PBs (Fig. 7 A, white arrowheads) remain stably bound to SGs. Other PBs (Fig. 7 A, yellow arrows) appear intermittently attached to SGs or move freely in the cytoplasm without interacting with SGs. “Free” or unbound PBs exhibit greater motility than SG-associated PBs, even when both types of PBs are present in the same cell. SGs exhibit fission, fusion, and occasional dispersal, which are properties consistent with ongoing sorting and export of their contents. Similar results are obtained when GFP-G3BP is used to induce and detect SG–PB interactions. The expression of FAST-YFP (Fig. 7, green) with RFP-DCP1a (Fig. 7, red) resulted in the incorporation of FAST into SGs in some cells but into PBs in other cells (Video 2, available at http://www.jcb.org/cgi/content/full/jcb.200502088/DC1). These data are consistent with the distribution of endogenous FAST, as shown in Figs. 1 and 2, and suggest that FAST (like eIF4E) may be present in both structures. Unfortunately, YFP-eIF4E constructs fail to recapitulate the localization of endogenous or FLAG-tagged eIF4E, so we are unable to examine its distribution between SGs and PBs in real time.
Figure 7.

Dynamics of SGs and PBs in vivo. COS7 cells cotransfected with RFP-DCP1a and either (A) GFP–TIA-1, (B) YFP-TTP, (C) GFP-G3BP plus empty myc-vector, or (D) GFP-G3BP and TTP-myc. Cells were observed at 37°C in real time by using confocal microscopy. Images from 10-min intervals are shown; Videos 1–5 depicts animation of these series (available at http://www.jcb.org/cgi/content/full/jcb.200502088.DC1). Each image is volume rendered from 10 Z-sections. Yellow arrows indicate PBs; white arrowheads indicate SGs.
We reasoned that SG–PB interaction may be influenced by the amount of mRNA being transported from the SG into the PB and hypothesized that the expression of TTP, an SG-associated protein that promotes mRNA decay (Stoecklin et al., 2004), might increase SG–PB interactions by increasing the amount of mRNA routed from SGs into PBs. As shown in Fig. 7 B and Video 3 (available at http://www.jcb.org/cgi/content/full/jcb.200502088/DC1), the expression of YFP-TTP with RFP-DCP1a results in the quantitative and stable association of PBs with SGs. Remarkably, the YFP-TTP SGs appear to encapsulate single or multiple PBs. Although fusion events between these conglomerate SG–PB structures were observed, fission events were rare. The data indicate that both the number and duration of SG–PB interactions is stabilized by the expression of TTP. As YFP-TTP is also diffusely present in the cytoplasm, making the borders of the SG difficult to determine, we sought to verify the ability of TTP to induce SG–PB fusion by using GFP-G3BP to induce SGs and test whether the coexpression of nonfluorescent TTP would alter the interaction of SGs with PBs. As shown in Video 4 (available at http://www.jcb.org/cgi/content/full/jcb.200502088/DC1) and Fig. 7 C, the coordinate expression of GFP-G3BP, RFP-DCP1a, and myc-tagged vector does not alter the relationship between GFP-G3BP SGs and PBs; both free and interacting structures are observed. However, cells expressing myc-tagged TTP with GFP-G3BP and RFP-DCP1a (Fig. 7 D and Video 5) display a nearly complete recruitment of PBs to SGs. Similar results were seen using GFP–TIA-1 as the SG inducer/marker, as the coexpression of GFP–TIA-1 with TTP promotes interactions between SGs and PBs (unpublished data).
We then asked whether FAST, XRN1, eIF4E, or the TTP-related protein BRF1 would promote interactions between GFP-G3BP SGs and PBs. Fig. 8 depicts GFP-G3BP and RFP-DCP1a cotransfected with one of the following: empty vector (Fig. 8 A), FAST-myc (Fig. 8 B), FLAG-XRN1 (Fig. 8 C), FLAG-eIF4E (Fig. 8 D), TTP-myc (Fig. 8 E), or FLAG-BRF1 (Fig. 8 F). Only TTP (Fig. 8 E) and its close homologue BRF1 (Fig. 8 F) are found to induce SG–PB fusion. Remarkably, BRF1 promotes the complete engulfment of large PBs by SGs, whereas in TTP transfectants, smaller, more numerous PBs are embedded in a single SG. Although not affecting the SG–PB relationship, eIF4E overexpression appears to reduce the size of PBs but increases their number (Fig. 8 D, red), whereas XRN1 expression results in fewer, larger PBs (Fig. 8 C, red). Altogether, the data indicate that the expression of different SG–PB components affects their size and interaction. Most important, the expression of TTP and BRF1, which functionally accelerate mRNA decay in these cells, also promote the interaction of SGs with PBs.
Dynamics of different SG–PB components in real time using FRAP
The TTP- and BRF1-induced fusion of SGs and PBs could either be direct (i.e., a physical linkage between SG–PB structural components) or indirect (i.e., by shunting more substrate mRNA destined for decay through SGs into PBs). To analyze the dynamic nature of SG and PB components within these structures, we used FRAP using GFP and/or YFP-tagged versions of different SG–PB-associated proteins. Previous studies (Kedersha et al., 2000) demonstrated that GFP–TIA-1 rapidly moves in and out of SGs. In these experiments, >90% recovery of the bleached signal occurred within 10 s. We used this system to analyze the FRAP kinetics of representative members of the “SG-only proteins” GFP–PABP-1 and GFP-G3BP, the “SG–PB shared proteins” YFP-TTP and FAST-YFP, and the “PB-only proteins” GFP-GW182 and YFP-DCP1a. As shown in Fig. 9 A, GFP–TIA-1 forms large, distinct SGs in response to arsenite treatment. A linear scan of the region containing a selected SG (Fig. 9 A, arrow) was obtained before bleaching (Fig. 9, pink tracing) and was subsequently bleached at the position indicated by the vertical yellow line. The dark blue line indicates the scan intensity taken immediately after bleaching. A scan taken 30 s later (Fig. 9, aqua tracing) reveals the complete recovery of GFP–TIA-1 fluorescence. The FRAP behavior of GFP–PABP-1, shown in Fig. 9 B, also recapitulates previous findings (Kedersha et al., 2000). GFP–PABP-1 exhibits slower and less complete recovery than does GFP–TIA-1 because only ∼60% of SG-associated GFP–PABP-1 fluorescence recovers after 30 s. This suggests that TIA-1 and PABP-1 are not quantitatively present in the same mRNP complexes.
Figure 9.

FRAP analysis of SG and PB proteins. COS7 cells were transfected with GFP–TIA-1 (A), GFP-PABP1 (B), YFP-TTP (C), GFP-G3BP (D), FAST-YFP (E), YFP-DCP1a (F), and GFP-GW182 (G). A two-dimensional scan was taken of each field before photobleaching, and a target SG or PB was selected (red arrows). Fluorescence intensity was obtained by using a linear scan centered around the target region (vertical yellow line); the prebleach scans (pink tracing) represent the mean of three separate scans; the dark blue tracing represents the scan taken immediately after the 1-s bleach; and the aqua tracing represents the scan taken 30 s later. Images are shown pseudocolored as indicated by the key shown in the bottom right panel.
TTP overexpression generates spontaneous SGs, and arsenite treatment induces TTP to leave SGs (Stoecklin et al., 2004) but not PBs (unpublished data), which is an effect dependent on TTP phosphorylation that mediates its binding to 14-3-3 (Stoecklin et al., 2004). YFP-TTP SG bleaching (Fig. 9 C) is followed by rapid and complete recovery; this result was consistently obtained in 10 cells and was the same when either large (probable SGs) or small foci (probable PBs) were bleached. Cells coexpressing RFP-DCP1a were used to verify the rapid kinetics of YFP-TTP that was unambiguously localized to PBs (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200502088.DC1). Thus, YFP-TTP rapidly moves in and out of both PBs and SGs, suggesting that TTP constitutes a transient tether between mRNA and the decay machinery.
SG-associated GFP-G3BP also displays rapid, complete recovery (Fig. 9 D) that is unaltered by arsenite treatment (unpublished data). FAST-YFP induces spontaneous SGs in 30–70% of transfectants and is localized to PBs in most of the remaining transfectants. As shown in Fig. 9 E, its FRAP kinetics are very slow, and recovery is minimal in all cells that were tested (n > 20) and unaltered by arsenite (unpublished data). The slow kinetics of FAST and its presence in both SGs and PBs suggests that it may play a scaffolding role in organizing SGs and PBs.
The overexpression of YFP-DCP1a induces very large PBs in many cells and more normal-sized PBs in others. PBs normally exhibit size variation depending on metabolic state (Sheth and Parker, 2003) and cell cycle (Yang et al., 2004). Fig. 9 F shows photobleaching of a medium-sized YFP-DCP1a PB, which exhibits kinetics similar to those of GFP–PABP-1. However, YFP-DCP1a FRAP kinetics is variable: the exchange rate is rapid in small PBs but is slower in larger PBs. GFP-GW182 exhibits very slow FRAP recovery (Fig. 9 G), similar to that of FAST. The transit time of PABP-1 and DCP1a is intermediate between that of TIA-TTP-G3BP and GW182-FAST, suggesting that PABP-1 and DCP1a either shuttle in and out of SGs and PBs independently of the RNA substrates or are removed from the transcripts during mRNP remodeling that occurs in tandem with their movement.
Discussion
Stress-induced phosphorylation of eIF2α results in stalled translational initiation such that actively translating ribosomes “run off” their transcripts, resulting in polysome disassembly concurrent with SG assembly (Anderson and Kedersha, 2002; Kedersha and Anderson, 2002). SG assembly is regulated by one or more RNA-binding proteins, including TIA-1 (Gilks et al., 2004), G3BP (Tourriere et al., 2003), Fragile X Mental Retardation protein (Mazroui et al., 2002), survival of motor neurons protein (Hua and Zhou, 2004), and/or TTP (Stoecklin et al., 2004). Another cytoplasmic mRNP domain termed the GW body was first visualized by using a patient-derived autoantisera reactive with GW182, a 182-kD RNA-binding protein (Eystathioy et al., 2002). GW bodies contain RNA, but unlike SGs, GW bodies are prominent in actively growing unstressed cells (Eystathioy et al., 2002). Convergent studies from several laboratories have shown that GW bodies contain proteins involved in mRNA degradation, including the decapping enzymes DCP1a and 2, a heptamer of Lsm proteins required for mRNA decapping, and the exonuclease XRN1 (Eystathioy et al., 2002, 2003; Ingelfinger et al., 2002; Cougot et al., 2004a,b; Yang et al., 2004). In Saccharomyces cerevisiae, the accumulation of nondegradable mRNAs at compositionally similar cytoplasmic foci (PBs) implicated these phylogenetically conserved foci in the process of mRNA degradation (Sheth and Parker, 2003). In mammalian cells, the interference RNA–mediated knockdown of XRN1 enhances the accumulation of poly(A)+ RNA at PBs, supporting the contention that these foci are sites of mRNA degradation (Cougot et al., 2004a).
Recently, dual immunofluorescence using antibodies against the SG marker TIA-1 and the PB marker DCP1a clearly showed that SGs and GW bodies/PBs are distinct and independent cytoplasmic structures (Cougot et al., 2004a), but the relationship between them has not been addressed. Our results confirm that SGs and PBs are compositionally and morphologically distinct entities, each of which can be assembled in the absence of the other and are compositionally distinct. However, there are strong spatial and functional links between SGs and PBs. First, oxidative stress induces the assembly of both SGs and PBs and promotes interactions between them. Second, time-lapse microscopy reveals that a subset of PBs is stably tethered to SGs, whereas another subset is independent and highly mobile within the cytoplasm. Third, several proteins (i.e., FAST, XRN1, eIF4E, and TTP) and mRNAs (i.e., globin-MS2 reporter) are found in both SGs and PBs. Fourth, SGs and PBs are induced to fuse by the overexpression of TTP or BRF1, which are RNA-binding proteins that promote mRNA decay and are components of both SGs and PBs. Finally, pharmacologic inhibitors of translational elongation promote the disassembly of both structures, suggesting that both PBs and SGs are assembled from translationally competent mRNA.
The SG–PB fusion induced by TTP and BRF1 suggests that these proteins regulate the dynamic interactions between SGs and PBs. Both TTP and BRF1 promote the degradation of mRNAs bearing ARE in their 3′ untranslated regions. TTP has been proposed to direct these transcripts to exosomes, which are degradative machines that promote 3′–5′ exonucleolytic degradation of deadenylated transcripts (Chen et al., 2001). The ability of TTP to promote interactions between PBs and SGs suggests that this class of destabilizing factor might also promote 5′–3′ mRNA degradation at the SG, which is consistent with recent data suggesting that TTP and BRF1 comprise molecular links between ARE-containing mRNAs and mRNA decay enzymes present in PBs (Lykke-Andersen and Wagner, 2005). Our data indicate that this molecular link has morphological as well as functional consequences. It is important to note that the SGs observed in the real-time experiments are induced by the overexpression of either TTP or G3BP. As such, they may not have the same composition and function as arsenite or heat-induced SGs. Nevertheless, the TTP- and BRF-1–induced stabilization of PB–SG interactions reveals a unique mechanism whereby this class of protein might regulate mRNA metabolism.
The presence of eIF4E in PBs is somewhat surprising, as it binds to the seven-methyl guanosine cap and is thought to protect the integrity of the cap (Ramirez et al., 2002; Liu et al., 2004). In S. cerevisiae, eIF4G and eIF4E are removed from mRNA before the recruitment of DCP1 and decapping (Tharun and Parker, 2001). Our data show that eIF4G, PABP, and eIF3 are present in SGs but not in PBs, suggesting that eIF4G is removed from the cap before its transit into PBs, whereas eIF4E remains bound to the cap. Because the rate at which eIF4E dissociates from capped mRNA is accelerated in the absence of eIF4G (Haghighat and Sonenberg, 1997), capped mRNA may be liberated within the PB, allowing DCP1a/2-mediated decapping.
We have proposed that SGs are sites of mRNA triage in which individual transcripts are sorted for storage, reinitiation, or degradation (Anderson and Kedersha, 2002; Kedersha and Anderson, 2002). This model predicts that those mRNAs targeted for decay will be exported from the SG to sites of mRNA decay such as PBs. The aforementioned interactions between SGs and PBs may allow mRNA to move from the SG to the PB. Two lines of evidence suggest the direction of this process. First, arsenite induces the formation of juxtaposed SGs and PBs, and subsequent emetine treatment forces the disassembly of SGs before the disassembly of PBs. Second, heat shock induces SG formation before PB formation. Initially, eIF4E is concentrated at SGs in cells lacking PBs, but in the continued presence of heat, SGs are disassembled, and PBs containing eIF4E are concomitantly assembled. These results imply (but do not mandate) that eIF4E is first incorporated into SGs and later translocates into PBs. As eIF3, eIF4G, PABP-1, small ribosomal subunits, and G3BP are found in SGs but not in PBs, these proteins must be removed from mRNA before its export from the SG. Because eIF4G and PABP-1 are directly involved in mRNA circularization, it is probable that mRNAs exported from SGs into PBs are decircularized before translocation, which is concurrent with their deadenylation (the activation step for mRNA decay by both mRNA decay pathways). Finally, as eIF4E and TTP are components of both SGs and PBs, these RNA-binding proteins may remain with mRNA as it moves from the SG to the PB.
In the model shown in Fig. 10, we posit that SGs contain transcripts routed from disassembling polysomes in accord with the absolute requirement for eIF2α phosphorylation in SG assembly. This idea is in agreement with the studies of Thomas et al. (2005), who demonstrated that newly synthesized mRNAs are not present in SGs. Error-containing transcripts selected for nonsense-mediated decay during the pioneer round of translation (before polysome assembly) may contribute to free PBs, as nonsense-mediated decay occurs via decapping and 5′–3′ decay (Maquat, 2002; Neu-Yilik et al., 2004) and is inhibited by cycloheximide. SGs induced by stress are likely to contain a mixture of transcripts, but SGs induced by the overexpression of different RNA-binding proteins (e.g., TIA, G3BP, and TTP) are likely to differ in their mRNA composition. For example, TIA-induced SGs are likely enriched for TIA-bound transcripts that are targeted for translational silencing, whereas TTP-induced SGs may be enriched for TTP-bound transcripts that are targeted for decay. Thus, TTP-induced SG–PB fusion occurs because TTP-induced SGs are assembled from mRNAs selected by TTP binding for rapid decay. Given the very rapid flux of TTP within SGs and PBs assessed by photobleaching, it is unlikely that TTP itself constitutes a stable component of either compartment. It is more likely that TTP serves to deliver its mRNA cargo to PBs by interacting with stable components of these particles (Lykke-Andersen and Wagner, 2005). However, FAST has the properties of a putative scaffold protein that might stabilize SG–PB interactions; it displays a very slow exchange rate, as measured by photobleaching, lacks known RNA binding motifs, nucleates both SGs and PBs upon overexpression, and interacts with TIA-1. Possibly, TTP or TTP-associated proteins promote SG–PB fusion by interacting either directly or indirectly with FAST to remodel the SG–PB scaffold.
Figure 10.

Hypothetical model of the relationship between SGs and PBs. Proteins found exclusively in SGs are shown in yellow; proteins found in both SGs and PBs are depicted in green; and proteins restricted to PBs are shown in blue type.
The data presented in this study establish that SGs and PBs are discrete cytoplasmic structures that share some protein and mRNA components as well as some functional properties. Both structures are induced by stress but are regulated by distinct signaling events, and each can exist without the other. PBs and SGs exhibit a high degree of motility when independent but appear less motile when they are tethered together, and their interaction is promoted by the mRNA-destabilizing protein TTP. The Janus-like juxtapositioning of SGs and PBs is reminiscent of the relationship between the nuclear gemini of coiled bodies and Cajal bodies (Dundr et al., 2004), a case in which the morphology of linked compartments arises from ordered, compartmentalized stages in nuclear small nuclear RNP biogenesis. The dynamic relation between SGs and PBs reiterates the importance of compartmentalization in regulating the fate of cytoplasmic mRNA.
Materials and methods
Cell lines
COS7, HeLa, and DU145 cells were obtained from the American Type Culture Collection, and U2OS cells were obtained from J. Blenis (Harvard Medical School, Boston, MA). HT1080 cells were obtained from C. Moroni (University of Basel, Basel, Switzerland). Cells were maintained in DME containing 10% FBS at 7.0% CO2.
Antibodies
Antibodies against eIF4E (monoclonal and rabbit polyclonal), eIF4G, eIF3b, myc, TIA-1, FXR1, and TIAR were obtained from Santa Cruz Biotechnology, Inc. Phospho-specific anti-eIF2α was obtained from StressGen Biotechnologies. Human autoantisera against GW182 was an index serum from a 48-yr-old female with mixed motor and sensory neuropathy, which was obtained from Advanced Diagnostics Laboratory. Antibodies against DCP1a and XRN-1 were previously described (Lykke-Andersen and Wagner, 2005). Antisera against FAST (anti–FAST-N) were described previously (Li et al., 2004a). Monoclonal anti-myc was a gift from L. Klickstein (Brigham and Woman's Hospital, Boston, MA). Anti-HA was obtained from Covance Research Products. Anti–PABP-1 was a gift from G. Dreyfuss (University of Pennsylvania, Philadelphia, PA). Anti-G3BP was a gift from I. Gallouzi (McGill University, Montreal, Canada). Anti-Dcp2was a gift of B. Seraphin (Centre de Genetique Moleculaire, Gif-sur-Yvette, France) and M. Kildejian (Rutgers University, Piscataway, NJ).
Plasmids
Plasmids encoding FLAG-DCP1a, FLAG-XRN1, and FLAG-DCP2 were previously described (Lykke-Andersen and Wagner, 2005). To make pEYFP-DCP1a, the human DCP1a cDNA was amplified from plasmid pcDNA3-Flag-DCP1a by using primers 5′-GTGCTCGAGCTGAGGCGCTGAGT-3′ and 5′-GTGGAATTCTCATAGGTTGTGGTTG-3′ and was ligated as an XhoI–EcoRI fragment into the Xho–EcoRI sites of pEYFP-C1 (CLONTECH Laboratories). To make mRFP-DCP1a, monomeric RFP (provided by R.Y. Tsien, Howard Hughes Medical Institute, University of California, San Diego, La Jolla, CA; Campbell et al., 2002) was amplified using primers 5′-ATTCATACCGGTCCACCATGGCCTCCTCCG-3′ and 5′-TAAATTCTCGAGAGGCGCCGGTGGAG-3′ and was ligated as an AgeI–XhoI fragment into the AgeI–XhoI sites of pEYFP-DCP1a, thereby replacing YFP. GFP-MS2-NLS was a gift from K. Kosik (University of California, Santa Barbara, Santa Barbara, CA) and was previously described (Rook et al., 2000). For pEF-7B-MS2bs, the T7-tagged rabbit β-globin gene containing a sixfold repeat of the MS2bs was excised from plasmid pcDNA3-7B-MS2bs as a HindIII/blunt–XbaI fragment and ligated into the NcoI/blunt–XbaI sites of pEF/myc/cyto (Invitrogen). The plasmid pcDNA3-TTP-mycHis was made as described previously (Stoecklin et al., 2004). For YFP-TTP, murine TTP cDNA was amplified from plasmid pcDNA3-TTP-mycHis by using primers 5′-TATCAAGCTTATGAATTCCGTTCC-3′ and 5′-TCAGATCCTCTTCTGAGATG-3′ digested with HindIII and XbaI and inserted into the HindIII–XbaI sites of pEYFP-C1 (CLONTECH Laboratories).
For pcDNA3-Flag-BRF1, the human BRF1 cDNA was excised as a BamHI/blunt–XbaI fragment from bsdHis-BRF1 (Stoecklin et al., 2002) and inserted into the BamHI/blunt–XbaI sites of pcDNA3-Flag-Bak (a gift from T. Chittenden, ImmunoGen, Inc., Cambridge, MA). For pcDNA3-Flag-eIF4E, eIF4E was amplified from plasmid pcDNA3-eIF4E (a gift from D. Dixon, University of South Carolina, Columbia, SC) using primers 5′-TTTGAATTCGCGACTGTCGAACCG-3′ and 5′-TGTTCTAGATTAAACAACAAACCTATTTTTAG-3′, digested with EcoRI and XbaI, and ligated into the EcoR–XbaI sites of pCDNA3-Flag. GFP-G3BP was a gift from J. Tazi (Centre National de la Recherche Scientifique, Montpellier, France). pGFP-GWaa313-1709 (Eystathioy et al., 2002) and FLAG-FAST (Li et al., 2004b) were described previously. For pEF-FAST-myc, FAST was amplified using primers 5′-CCACCATGGAATAGCCACCATGAGGAGGCCGCGGGGGGAA-3′ and 5′-ATAAGAATGCGGCCGCGCCCCCTTCAGGCCCCCAGCG-3′, digested with NcoI and NotI, and ligated into the NcoI–NotI sites of pEF-myc. To make FAST-YFP, the coding region of FAST was amplified using primers 5′-TGTGAGATCTAGTAGGAGGCCGCGGGGG-3′ and 5′-CCGAAGCTTGCCCCCTTCAGGCCC-3′, digested with BglII and HindIII, and ligated into pEF-YFP-N1 vector (CLONTECH Laboratories) that was digested with the same enzymes.
siRNA transfection
Du145 and HT1080 cells were transfected with 1.25 μl/ml of Lipofectamine 2000 and 100 nM siRNA duplexes for 48 h. Subsequently, cells were reseeded and, after 8 h, were transfected again with siRNA for another 40–44 h. siRNAs were designed using published recommendations (Reynolds et al., 2004) and were purchased from Ambion. The following target sequences (sense strand) were chosen: control siRNA (D0), 5′-GCAUUCACUUGGAUAGUAA-3′; and Lsm4 siRNA (L4), 5′-ACAACUGGAUGAACAUUAA-3′.
RT-PCR
HT1080 cells were transfected with siRNA D0 or L4. Total cytoplasmic RNA was extracted, and 5 μg RNA was used for reverse transcription using oligo-dT and MMLV-RT (Promega). cDNA was purified with the Qiaquick PCR purification kit (QIAGEN), and one tenth was used per PCR reaction using Taq polymerase (2.5 U/50 μl) and solution Q (QIAGEN). Annealing was performed at 56°C using primers 5′-CCTTGTCACTGCTGAAGACG-3 and 5′-GAGACTGTGGAGCGGAATC-3′ for the amplification of Lsm4 and 5′-GGTGGTCGGAAAGCTATC-3′ and 5′-GAGCTTCTTATAGACACCAG-3′ for the amplification of ribosomal protein S7 as a control. Parallel reactions were performed using 15, 20, 25, 30, and 35 PCR cycles, and the products were resolved by 1.5% agarose gel electrophoresis and were stained with ethidium bromide.
Fluorescence microscopy
Cells were stained and processed for fluorescence microscopy as previously described (Gilks et al., 2004). Conventional fluorescence microscopy was performed using a microscope (model Eclipse E800; Nikon) with epifluorescence optics and a digital camera (model CCD-SPOT RT; Diagnostic Instruments). The images were compiled using Adobe Photoshop software (v7.0).
Confocal microscopy
Cells transfected with combinations of GFP- and RFP-tagged vectors were viewed live at 37°C using an inverted microscope (model TE2000-U; Nikon) equipped with a 60× oil objective Cfi planapo lens (NA 1.40; Nikon) and a confocal system (model C-1; Nikon). Each image was volume rendered from 10 Z-stacks of 0.85-μm thickness using EZ-C1 software (Nikon). Timed series were acquired at a rate of 1 min per frame; each frame represents a volume-rendered image. Videos are shown in the supplemental material; frames taken 10 min apart are shown in Fig. 7. Videos were made using Adobe Image Ready software (v7.0) to animate the volume-rendered TIF images exported from the EZ-C1 software (Nikon).
FRAP photobleaching analysis
Fluorescently tagged constructs of SG and PB proteins were tested to determine whether they exhibited localization that was compatible with their endogenous or (in the case of TTP) FLAG-tagged counterparts; those failing to meet this criterion were not used. COS7 cells were transiently transfected with the indicated constructs using SuperFect (QIAGEN), were replated onto glass coverslips after 10 h of transfection, and were analyzed 38–46 h posttransfection. Transfectants were viewed using a 60× oil objective (NA 1.40) on an interactive laser cytometer (model Ultima; Meridian Instruments). Appropriate cells were located, and images were taken using a two-dimensional scanning mode before bleaching. Selected SGs or PBs (Fig. 9, arrows) were photobleached for 1 s at ∼0.5 mW of power using a beam radius of 0.7 μm and an excitation wavelength of 488 nm. Fluorescence emission was detected at 530 ± 15 nm. The results shown were representative of three independent transfections in which a total of >10 different cells were analyzed. In some cases (see Fig. S1), two-color scans were obtained by simultaneously exciting both fluorophores at 488 nm and separating the two emissions using a 575-nm dichroic filter and the appropriate emission filters (green emission 530 ± 15 nm; red emission >630 nm).
Online supplemental material
Fig. S1 shows photobleaching of PB-localized YFP-TTP. Video 1 shows the dynamics of GFP–TIA-1 SGs and PBs; Video 2 shows the dynamics of FAST-YFP and RFP-DCP1a PBs; Video 3 shows YFP-TTP and RFP-DCP1 PBs; and Video 4 shows GFP-G3BP SGs and RFP-DCP1 PBs, all in real time. Video 5 shows that TTP coexpression promotes fusion between GFP-G3BP SGs and RFP-DCP1 PBs. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200502088.DC1.
Acknowledgments
We thank R. Tsien for the mRFP construct, K. Kosik for GFP-MS2-NLS, J. Tazi for GFP-G3BP, M. Kiledjian and B. Serephin for antibodies, and H. Gilbert and M. Gurish for help with the confocal microscopy. We thank W. Li (Brigham and Women's Hospital, Boston, MA) for the FAST-myc construct and N. Gilks and members of the Anderson lab for lively discussions.
This work was supported by the National Institutes of Health grants AI50167, AR051472, and AI33600 (to P. Anderson); DK42394 and HL52173 (to R.J. Kaufman); GM 066811 (to J. Lykke-Andersen); HL32854 and HL070819 (to D.E. Golan) and the Canadian Institutes for Health Research Grant MOP-38034 (to M.J. Fitzler). M.J. Fitzler holds the Arthritis Society Chair at the University of Calgary. J. Lykke-Andersen is a Pew Scholar.
Abbreviations used in this paper: ARE, adenine/uridine-rich destabilizing elements; FAST, Fas-activated serine/threonine phosphoprotein; PB, processing body; SG, stress granule; siRNA, small interference RNA; TIA, T cell intracellular antigen; TIAR, TIA related; TTP, tristetraprolin.
References
- Anderson, P., and N. Kedersha. 2002. Stressful initiations. J. Cell Sci. 115:3227–3234. [DOI] [PubMed] [Google Scholar]
- Bolling, F., R. Winzen, M. Kracht, B. Bhebremedhin, B. Ritter, A. Wilhelm, K. Resch, and H. Holtmann. 2002. Evidence for general stabilization of mRNAs in response to UV light. Eur. J. Biochem. 269:5830–5839. [DOI] [PubMed] [Google Scholar]
- Campbell, R.E., O. Tour, A.E. Palmer, P.A. Steinbach, G.S. Baird, D.A. Zacharias, and R.Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 99:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.Y., R. Gherzi, S.E. Ong, E.L. Chan, R. Raijmakers, G.J. Pruijn, G. Stoecklin, C. Moroni, M. Mann, and M. Karin. 2001. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell. 107:451–464. [DOI] [PubMed] [Google Scholar]
- Cougot, N., S. Babajko, and B. Seraphin. 2004. a. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 165:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougot, N., E. van Dijk, S. Babajko, and B. Seraphin. 2004. b. ‘Cap-tabolism’. Trends Biochem. Sci. 29:436–444. [DOI] [PubMed] [Google Scholar]
- Decker, C., and R. Parker. 2002. mRNA decay enzymes: decappers conserved between yeast and mammals. Proc. Natl. Acad. Sci. USA. 99:12512–12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr, M., M.D. Hebert, T.S. Karpova, D. Stanek, H. Xu, K.B. Shpargel, U.T. Meier, K.M. Neugebauer, A.G. Matera, and T. Misteli. 2004. In vivo kinetics of Cajal body components. J. Cell Biol. 164:831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eystathioy, T., E.K. Chan, S.A. Tenenbaum, J.D. Keene, K. Griffith, and M.J. Fritzler. 2002. A phosphorylated cytoplasmic autoantigen, GW182, associates with a unique population of human mRNAs within novel cytoplasmic speckles. Mol. Biol. Cell. 13:1338–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eystathioy, T., A. Jakymiw, E.K. Chan, B. Seraphin, N. Cougot, and M.J. Fritzler. 2003. The GW182 protein colocalizes with mRNA degradation associated proteins hDcp1 and hLSm4 in cytoplasmic GW bodies. RNA. 9:1171–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks, N., N. Kedersha, M. Ayodele, L. Shen, G. Stoecklin, L.M. Dember, and P. Anderson. 2004. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell. 15:5383–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighat, A., and N. Sonenberg. 1997. eIF4G dramatically enhances the binding of eIF4E to the mRNA 5′-cap structure. J. Biol. Chem. 272:21677–21680. [DOI] [PubMed] [Google Scholar]
- Hua, Y., and J. Zhou. 2004. Survival motor neuron protein facilitates assembly of stress granules. FEBS Lett. 572:69–74. [DOI] [PubMed] [Google Scholar]
- Ingelfinger, D., D.J. Arndt-Jovin, R. Luhrmann, and T. Achsel. 2002. The human LSm1-7 proteins colocalize with the mRNA-degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA. 8:1489–1501. [PMC free article] [PubMed] [Google Scholar]
- Jacobs, J.S., A.R. Anderson, and R.P. Parker. 1998. The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J. 17:1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, A. 2004. Regulation of mRNA decay: decapping goes solo. Mol. Cell. 15:1–2. [DOI] [PubMed] [Google Scholar]
- Kedersha, N., and P. Anderson. 2002. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 30:963–969. [DOI] [PubMed] [Google Scholar]
- Kedersha, N.L., M. Gupta, W. Li, I. Miller, and P. Anderson. 1999. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2α to the assembly of mammalian stress granules. J. Cell Biol. 147:1431–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha, N., M.R. Cho, W. Li, P.W. Yacono, S. Chen, N. Gilks, D.E. Golan, and P. Anderson. 2000. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 151:1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha, N.L., S. Chen, N. Gilks, W. Li, I.J. Miller, J. Stahl, and P. Anderson. 2002. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol. Biol. Cell. 13:195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy, T., G.D. Pavitt, F. Zhang, T.E. Dever, and A.G. Hinnebusch. 2001. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol. 21:5018–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroia, G., R. Cuesta, G. Brewer, and R.J. Schneider. 1999. Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science. 284:499–502. [DOI] [PubMed] [Google Scholar]
- Li, W., N. Kedersha, S. Chen, N. Gilks, G. Lee, and P. Anderson. 2004. a. FAST is a BCL-X(L)-associated mitochondrial protein. Biochem. Biophys. Res. Commun. 318:95–102. [DOI] [PubMed] [Google Scholar]
- Li, W., M. Simarro, N. Kedersha, and P. Anderson. 2004. b. FAST is a survival protein that senses mitochondrial stress and modulates TIA-1-regulated changes in protein expression. Mol. Cell. Biol. 24:10718–10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S.W., X. Jiao, H. Liu, M. Gu, C.D. Lima, and M. Kiledjian. 2004. Functional analysis of mRNA scavenger decapping enzymes. RNA. 10:1412–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, R.M., and M.T. McNally. 2003. mRNA decay: x (XRN1) marks the spot. Mol. Cell. 11:1126–1128. [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen, J., and E. Wagner. 2005. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 19:351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat, L.E. 2002. Nonsense-mediated mRNA decay. Curr. Biol. 12:R196–R197. [DOI] [PubMed] [Google Scholar]
- Mazroui, R., M.E. Huot, S. Tremblay, C. Filion, Y. Labelle, and E.W. Khandjian. 2002. Trapping of messenger RNA by Fragile X Mental Retardation protein into cytoplasmic granules induces translation repression. Hum. Mol. Genet. 11:3007–3017. [DOI] [PubMed] [Google Scholar]
- McEwen, E., N. Kedersha, B. Song, D. Scheuner, N. Gilks, A. Han, J.J. Chen. P. Anderson, and R.J. Kaufman. 2005. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 10.1074/jbc.M412882200. [DOI] [PubMed]
- Mukherjee, D., M. Gao, J.P. O'Connor, R. Raijmakers, G. Pruijn, C.S. Lutz, and J. Wilusz. 2002. The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J. 21:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu-Yilik, G., N.H. Gehring, M.W. Hentze, and A.E. Kulozik. 2004. Nonsense-mediated mRNA decay: from vacuum cleaner to Swiss army knife. Genome Biol. 5:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, C.V., C. Vilela, K. Berthelot, and J.E. McCarthy. 2002. Modulation of eukaryotic mRNA stability via the cap-binding translation complex eIF4F. J. Mol. Biol. 318:951–962. [DOI] [PubMed] [Google Scholar]
- Reynolds, A., D. Leake, Q. Boese, S. Scaringe, W.S. Marshall, and A. Khvorova. 2004. Rational siRNA design for RNA interference. Nat. Biotechnol. 22:326–330. [DOI] [PubMed] [Google Scholar]
- Rook, M.S., M. Lu, and K.S. Kosik. 2000. CaMKIIalpha 3′ untranslated region-directed mRNA translocation in living neurons: visualization by GFP linkage. J. Neurosci. 20:6385–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner, D., B. Song, E. McEwen, C. Liu, R. Laybutt, P. Gillespie, T. Saunders, S. Bonner-Weir, and R.J. Kaufman. 2001. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell. 7:1165–1176. [DOI] [PubMed] [Google Scholar]
- Sheth, U., and R. Parker. 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 300:805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens, A. 2001. 5′-exoribonuclease 1: XRN1. Methods Enzymol. 342:251–259. [DOI] [PubMed] [Google Scholar]
- Stoecklin, G., M. Colombi, I. Raineri, S. Leuenberger, M. Mallaun, M. Schmidlin, B. Gross, M. Lu, T. Kitamura, and C. Moroni. 2002. Functional cloning of BRF1, a regulator of ARE-dependent mRNA turnover. EMBO J. 21:4709–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin, G., T. Stubbs, N. Kedersha, T.K. Blackwell, and P. Anderson. 2004. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 23:1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharun, S., and R. Parker. 2001. Targeting an mRNA for decapping: displacement of translation factors and association of the Lsm1p-7p complex on deadenylated yeast mRNAs. Mol. Cell. 8:1075–1083. [DOI] [PubMed] [Google Scholar]
- Thomas, M.G., L.J. Tosar, M. Loschi, J.M. Pasquini, J. Correale, S. Kindler, and G.L. Boccaccio. 2005. Staufen recruitment into stress granules does not affect early mRNA transport in oligodendrocytes. Mol. Biol. Cell. 16:405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourriere, H., K. Chebli, L. Zekri, B. Courselaud, J.M. Blanchard, E. Bertrand, and J. Tazi. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yang, Z., A. Jakymiw, M.R. Wood, T. Eystathioy, R.L. Rubin, M.J. Fritzler, and E.K. Chan. 2004. GW182 is critical for the stability of GW bodies expressed during the cell cycle and cell proliferation. J. Cell Sci. 117:5567–5578. [DOI] [PubMed] [Google Scholar]