

Abstract
Extracellular stimuli that activate cell surface receptors modulate glucocorticoid actions via as yet unclear mechanisms. Here, we report that the guanine nucleotide-binding protein (G protein)–coupled receptor-activated WD-repeat Gβ interacts with the glucocorticoid receptor (GR), comigrates with it into the nucleus and suppresses GR-induced transactivation of the glucocorticoid-responsive genes. Association of Gγ with Gβ is necessary for this action of Gβ. Both endogenous and enhanced green fluorescent protein (EGFP)–fused Gβ2 and Gγ2 proteins were detected in the nucleus at baseline, whereas a fraction of EGFP-Gβ2 and DsRed2-GR comigrated to the nucleus or the plasma membrane, depending on the exposure of cells to dexamethasone or somatostatin, respectively. Gβ2 was associated with GR/glucocorticoid response elements (GREs) in vivo and suppressed activation function-2–directed transcriptional activity of the GR. We conclude that the Gβγ complex interacts with the GR and suppresses its transcriptional activity by associating with the transcriptional complex formed on GR-responsive promoters.
Introduction
Glucocorticoids play a crucial role in the regulation of basal and stress-related homeostasis. They are necessary for maintenance of many important biological activities, such as the homeostasis of the central nervous and cardiovascular systems, the intermediary metabolism and the immune/inflammatory reaction (Chrousos, 2004). They also act as potent immunosuppressive and anti-inflammatory agents at “pharmacologic” doses, properties that make them irreplaceable therapeutic means for many inflammatory, autoimmune, allergic, and lymphoproliferative diseases (Kino et al., 2003a).
The actions of glucocorticoids are mediated by a ubiquitous intracellular receptor protein, the glucocorticoid receptor (GR), which functions as a hormone-activated transcription factor of glucocorticoid target genes (Kino et al., 2003a). The GR consists of three domains: the NH2-terminal or “immunogenic” domain, the central, DNA-binding domain (DBD), and the COOH-terminal, ligand-binding domain. The functions of the latter two domains have been studied extensively, whereas those of the immunogenic domain are less well known (Kino et al., 2003a). In the unliganded state, GR is located primarily in the cytoplasm, as part of hetero-oligomeric complexes containing heat shock proteins 90, 70, and 50, and, possibly, other proteins. After binding to its agonist ligand, the GR undergoes conformational changes, dissociates from the heat shock proteins, homodimerizes, and translocates into the nucleus through the nuclear pore via an active process (Kino et al., 2003a). There, the ligand-activated GR directly interacts with DNA sequences, the glucocorticoid response elements (GREs), in the promoter regions of target genes, or with other transcription factors via protein–protein interactions, indirectly influencing the activity of the latter on their target genes (Kino and Chrousos, 2002; Kino et al., 2003a). The GRE-bound GR stimulates the transcription rate of responsive genes by facilitating the formation of a transcription initiation complex, including the RNA polymerase II and its ancillary components via its activation function (AF)-1 and AF-2 domains (Kino et al., 2003a). The former is localized in the immunogenic domain, whereas the latter spans the entire ligand-binding domain.
Because glucocorticoids have a broad array of life-sustaining functions and play an important role in therapeutic interventions, changes of tissue sensitivity to glucocorticoids may be associated with and influence the course and therapy of many pathological states (Kino and Chrousos, 2002; Kino et al., 2003a). Such changes may present on either side of an optimal range, respectively, as glucocorticoid resistance or hypersensitivity, and may be generalized and/or tissue specific. Several autoimmune/inflammatory/allergic states, such as rheumatoid arthritis, osteoarthritis, Crohn's disease, ulcerative colitis and asthma, are often associated with resistance of the inflamed tissues to glucocorticoids (Chrousos, 1995; Kino and Chrousos, 2002). On the other hand, glucocorticoid hypersensitivity has been suggested in visceral obesity–related insulin resistance associated with components of the dysmetabolic syndrome, and in the acquired immunodeficiency syndrome caused by human immunodeficiency virus type 1 infection (Chrousos, 2000; Kino et al., 2003b). These changes in tissues' sensitivity to glucocorticoids associated with such pathological conditions may be possible by local modifications of GR functions, where altered concentrations/production of hormones, neurotransmitters, cytokines, growth factors and autacoids may play important roles (Kino and Chrousos, 2002; Kino et al., 2003a). The biological activities of such extracellular molecules are transduced into the intracellular compartment via their specific cell surface receptors (Cotecchia et al., 2004; Radeff-Huang et al., 2004). Binding of the compounds to their receptors activates signal-transducing molecules located on the cytoplasmic side of the plasma membrane, which subsequently communicate with downstream effector molecules, finally exerting a variety of biological effects on their target cells.
The heterotrimeric guanine nucleotide-binding proteins (G proteins) are downstream signal transducers for the G protein–coupled receptors (GPCRs), which form a large family consisting of >1,000 members (Cabrera-Vera et al., 2003). There are three subunits, Gα, Gβ, and Gγ, each of which is also composed of many isoforms (Hamm, 1998; Cabrera-Vera et al., 2003). Among them, Gβ contains a portion characterized as a seven times–repeated blade-like structure, called a WD repeat (Clapham and Neer, 1997; Smith et al., 1999). Crystallographic analyses revealed that all WD-repeats of Gβ are made up of four twisted β strands and are arranged in a ring, thus, forming a propeller-like structure (Clapham and Neer, 1997). Gβ is associated with Gγ through its NH2-terminal α helical portion and the fifth blade of the WD repeats.
The heterotrimeric complex consisting of Gα, Gβ, and Gγ is attached to the cytoplasmic surface of the plasma membrane through the prenylated Gγ (Clapham and Neer, 1997). This heterotrimeric complex is inactive in the GDP-bound state. Once ligands bind to their GPCRs, GDP on Gα is catalyzed to GTP, leading to dissociation of the GTP-bound Gα from the Gβ/Gγ heterodimer (Hamm, 1998). Liberated GTP-Gα and the Gβ/Gγ complex then interact with their downstream effector molecules and exert their biological actions (Clapham and Neer, 1997). The Gβ/Gγ complex binds to and modulates the activity of diverse molecules, such as several forms of potassium and calcium ion channels, the enzymes phospholipase A2 and Cβ, several adenylyl cyclases and the plasma membrane Ca2+ ATPase pump (Clapham and Neer, 1997).
The receptor for activated C-kinase 1 (Rack1), also a WD-repeat protein that harbors 42% amino acid similarity to Gβ with many conserved amino acids, plays regulatory roles in cell development and growth, as well as in the immune response and brain function (McCahill et al., 2002). Rack1 exerts its biological activities by binding to numerous partner molecules, including PKCb, phosphodiesterase 4-D5, Src family kinases, the Interleukin-3 and Interleukin-5, and granulocyte macrophage-colony stimulating factor receptors, and by regulating their activities as a scaffold molecule providing steric hindrance (McCahill et al., 2002). For example, Rack1 acts as a negative scaffold on the Src family kinases by binding to the phosphotyrosine-binding pocket of their Src homology-2 domains and by inhibiting their further association with downstream molecules (Chang et al., 2001; McCahill et al., 2002).
To look for partner molecules that may interact with GR and explain the local modification of GR activity in several physiological or pathological conditions, we performed a yeast two-hybrid screening assay using fragments of the GR immunogenic domain. We found that the WD repeat proteins Gβ2 and Rack1 are specific interactors of GR, negatively regulating its transcriptional activity. Because Gβ proteins play key roles in many signal transduction cascades, GR-associated Gβ might play a role in the development of target tissue resistance or hypersensitivity to glucocorticoids in states in which the G protein system is affected and the sensitivity to glucocorticoids is altered.
Results
Gβ1, Gβ2, and Rack1, all interact with GR(263-419)
The immunogenic domain of the human GR consists of 420 aa accounts for over a half of the entire molecule. Although it contains the AF-1 domain at amino acid positions 77–261, through which the GR communicates with components of the transcriptional machinery, the functions of the rest of the immunogenic domain are yet unknown. We performed a yeast two-hybrid screening assay using as bait a GR fragment spanning amino acids 263–419, located between the AF-1 domain and the DBD in a Jurkat cDNA library. Among over 85 independent interactors, we found two independent clones, which contained the COOH-terminal half of the human Gβ2 and Rack1 coding sequences, respectively (unpublished data). We confirmed the binding of GR(263-419) to full-length Gβ2, as well as its close isoform Gβ1, and Rack1, in a reconstituted yeast two-hybrid assay (Fig. 1). As expected, full-length Gβ1, Gβ2, and Rack1 all interacted with GR(263-419) in this system. Rack1(139-317), which spans blades 4–7 of Rack1, was also associated with this fragment of the GR (Fig. 1 A). To map the portion of Gβ2, which was responsible for binding to GR, we constructed a series of plasmids expressing the B42-activation domain fusions of the Gβ2 fragments, and examined their interactions to GR(263-419) fused to the LexA-DBD in the same yeast two-hybrid assay (Fig. 1 B). We found that amino acids 143–270 of Gβ2, which correspond to blades 3–5 of Gβ2, supported the interaction between Gβ2 and GR(263-419). A summary of the yeast two-hybrid assays examining the interactions of Gβ2 and Rack1 with GR(263-419) is shown in Fig. 1 C.
Figure 1.

Gβ1, Gβ2 and Rack1 interact with GR(263-419) in yeast two-hybrid assays. (A) Full-length Gβ1, Gβ2, and Rack1, as well as Rack1(139-317) interact with GR(263-420) in a yeast two-hybrid assay. EGY48 yeast cells were transformed with p8OP-LacZ, pLexA-GRα(263-419) and the indicated full-length Gβ1-, Gβ2-, Rack1-, or Rack1(139-317)-expressing pB42AD-derived plasmids. Bars represent mean ± SEM values of fold activation compared with the baseline. (B) Gβ2(143-270) (blades 3–5) interacts with GR(263-420) in a yeast two-hybrid assay. EGY48 yeast cells were transformed with p8OP-LacZ, pLexA-GRα(263-419) and the indicated Gβ2 fragment-expressing pB42AD plasmids. Bars represent mean ± SEM values of fold activation compared with the baseline. (C) Summary of yeast two-hybrid assays, which demonstrates domains of Gβ2 and Rack1 that are necessary for the interaction with GR(263-419).
Gβs suppress the transcriptional activity of GR
We next examined the effect of Gβ1, 2, and Rack1 on GR-induced transcriptional activity of the glucocorticoid-responsive mouse mammary tumor virus (MMTV) promoter in HCT116 cells (Fig. 2 A). Overexpression of these molecules dose-dependently suppressed the transcriptional activity of GR. Because Gβ2 showed the strongest suppressive effect, we focused on Gβ2 in further functional analyses. Gβ2 strongly suppressed the transcriptional activity of the wild-type GR, whereas it lost its suppressive effect on GR(Δ262-404), which is devoid of an interaction domain for Gβ2 (Fig. 2 B). Indeed, this GR deletion mutant demonstrated stronger transcriptional activity on the MMTV promoter than the wild-type GR, suggesting that endogenously expressed Gβ, Rack1, and/or other related molecules might down-regulate the transcriptional activity of the wild-type GR by physical association through this portion of GR. We also tested Gβ2 on the dexamethasone titration curve of the luciferase activity from the MMTV promoter-driven luciferase gene in HCT116 cells (Fig. 2 C). Overexpression of Gβ2 dramatically shifted the titration curve downward, indicating that Gβ2 suppressed GR-induced transactivation by acting at a step subsequent to ligand binding of the GR.
Figure 2.

Gβ1, Gβ2, and Rack1 suppress the transcriptional activity of GR on the MMTV promoter, and endogenous Gβ and Gγ are associated with GR in vivo. (A) Gβ1, Gβ2, and Rack1 dose dependently suppress the transcriptional activity of GR on the MMTV promoter in HCT116 cells. HCT116 cells were transfected with indicated amounts of Gβ1, Gβ2, or Rack1-expressing plasmids together with pRShGRα, pMMTV-Luc, and pSV40-β-Gal. Bars represent mean ± SEM values of the luciferase activity normalized for β-galactosidase activity in the absence or presence of 10−6 M of dexamethasone. (B) GR(Δ262-404) has stronger transcriptional activity than the wild-type GR and Gβ2 loses its suppressive effect on GR(Δ262-404)-induced transactivation in HCT116 cells. HCT116 cells were transfected with Gβ2-expressing plasmids and pRShGRα or pRShGRα(Δ262-404), together with pMMTV-Luc and pSV40-β-Gal. Bars represent mean ± SEM values of the luciferase activity normalized for β-galactosidase activity in the absence or presence of 10−6 M of dexamethasone. (*), P < 0.01; n.s., not significant, compared with the baseline. (C) Expression of Gβ2 shifts the dexamethasone titration curve of the luciferase activity from the pMMTV-Luc in HCT116 cells. HCT116 cells were transfected with pRShGRα, pMMTV-Luc, and pSV40-β-Gal in the absence or presence of Gβ2-expressing plasmid. Cells were then stimulated with increasing concentrations of dexamethasone. Open and closed circles represent mean ± SEM values of the luciferase activity normalized for β-galactosidase activity in the absence (open circle) or presence (closed circle) of the Gβ2 expression. (D–F) Abrogation of Gβ1 and Gβ2 enhanced dexamethasone-activated TAT activity (D) and suppressed mRNA (E) and protein (F) levels of Gβ1 and Gβ2 in HTC cells. HTC cells were transfected with control or Gβ1 and Gβ2 siRNAs and the cells were treated with 10−6 M of dexamethasone for 24 h. Cell lysate and total RNA were harvested and the TAT activity (D), Gβ1 and Gβ2 mRNA abundance (E) and protein levels of Gβ1 and Gβ2 in Western blots using their specific antibodies in the absence of dexamethasone (F), were determined. Bars represent mean ± SEM values of TAT activity (D) or fold induction of Gβ1 or Gβ2 mRNAs (E) in the absence or presence of 10−6 M of dexamethasone. (*), P < 0.01; n.s., not significant, compared with the baseline. (G) Endogenous Gβ and Gγ, but not Gαi, are associated with GR in vivo. HeLa cells were stimulated with 10−6 M of dexamethasone and coimmunoprecipitation was performed with control or anti-GRα antibody. After blotting the precipitated proteins on nitrocellulose membranes, the associated Gβ, Gγ, or Gαi was detected with their specific antibodies. Expression of Gβ, Gγ, Gαi, and GR was also examined in 10% whole homogenates in Western blots.
We also tested Gβ activity on a well-known endogenous glucocorticoid-responsive gene, the rat tyrosine aminotransferase (TAT). Glucocorticoids increase TAT enzymatic activity by stimulating the transcriptional rate of its gene via tandem GREs located in the promoter region (Thompson et al., 1966; Jantzen et al., 1987). In rat HTC cells, 10−6 M of dexamethasone increased TAT activity by fivefold, whereas transfection of short interfering RNAs (siRNAs) for Gβ1 and Gβ2 significantly enhanced dexamethasone-stimulated TAT activity (Fig. 2 D). These siRNAs reduced mRNA abundance of Gβ1 and Gβ2 (Fig. 2 E) and their protein levels (Fig. 2 F). These results indicate that endogenous Gβ1 and Gβ2 act as negative regulators of GR transactivation on an endogenous glucocorticoid-responsive gene.
The Gβ/Gγ heterodimer, but not Gαi, is associated with GR in vivo
To test if Gβ interacts with GR in vivo, we performed a regular coimmunoprecipitation assay using specific antibodies to Gβ, Gγ, Gαi, and GR in HeLa cells, which express all these molecules endogenously. Both Gβ and Gγ, but not Gαi, were coprecipitated with GR in the absence of dexamethasone, and addition of dexamethasone increased the precipitation of these molecules with GR (Fig. 2 G, top three gels). This effect of dexamethasone was not through induction of any of these three proteins, because their expression levels did not change throughout the experiment (Fig. 2 G, bottom four gels). These results indicate that the Gβ/Gγ heterodimer, but not Gαi, is a component of the complex formed with GR both in the absence and presence of glucocorticoids. Gβ2 mutants, G10K and Gβ2(34–340), both of which are defective in the association with Gγ (Garritsen et al., 1993), lost the wild-type Gβ suppressive effect on GR-induced transactivation in HCT116 cells (unpublished data), further confirming that Gβ2 acts as a heterodimer with Gγ to suppress the transcriptional activity of the GR. They also suggest that the overexpressed Gβ1 and Gβ2 acted on GR by forming complexes with endogenous Gγ.
Somatostatin suppresses GR-induced transcriptional activity by activating Gβ/Gγ in rat GH3 cells
Somatostatin binds its receptor and activates the Gβ/Gγ dimer by causing dissociation of the complex from GTP-bound Gαi (Law et al., 1991; Brown and Schonbrunn, 1993). Thus, we tested the effect of somatostatin on GR-induced transactivation in rat pituitary GH3 cells, which express endogenous somatostatin receptor and GR (Yatani et al., 1987; Levitan et al., 1991). These cells also express the Kv1.5 potassium channel, whose expression levels are positively regulated by glucocorticoids possibly though GREs located in its promoter region (Levitan et al., 1991). 24-h incubation of cells with 10−6 M of dexamethasone stimulated mRNA expression of the Kv1.5 potassium channel by eightfold and somatostatin suppressed this dexamethasone effect in a dose-dependent fashion (Fig. 3 A). Transfection of Gβ1 and Gβ2 siRNAs, which strongly suppressed the mRNA expression of these subunits (Fig. 3, B and C), enhanced dexamethasone-induced induction of Kv1.5 potassium channel mRNA and attenuated the suppressive effect of somatostatin (Fig. 3 A). These results indicate that somatostatin suppressed GR-induced transcriptional activity of the endogenous Kv1.5 potassium channel gene by activating the Gβ/Gγ complex.
Figure 3.
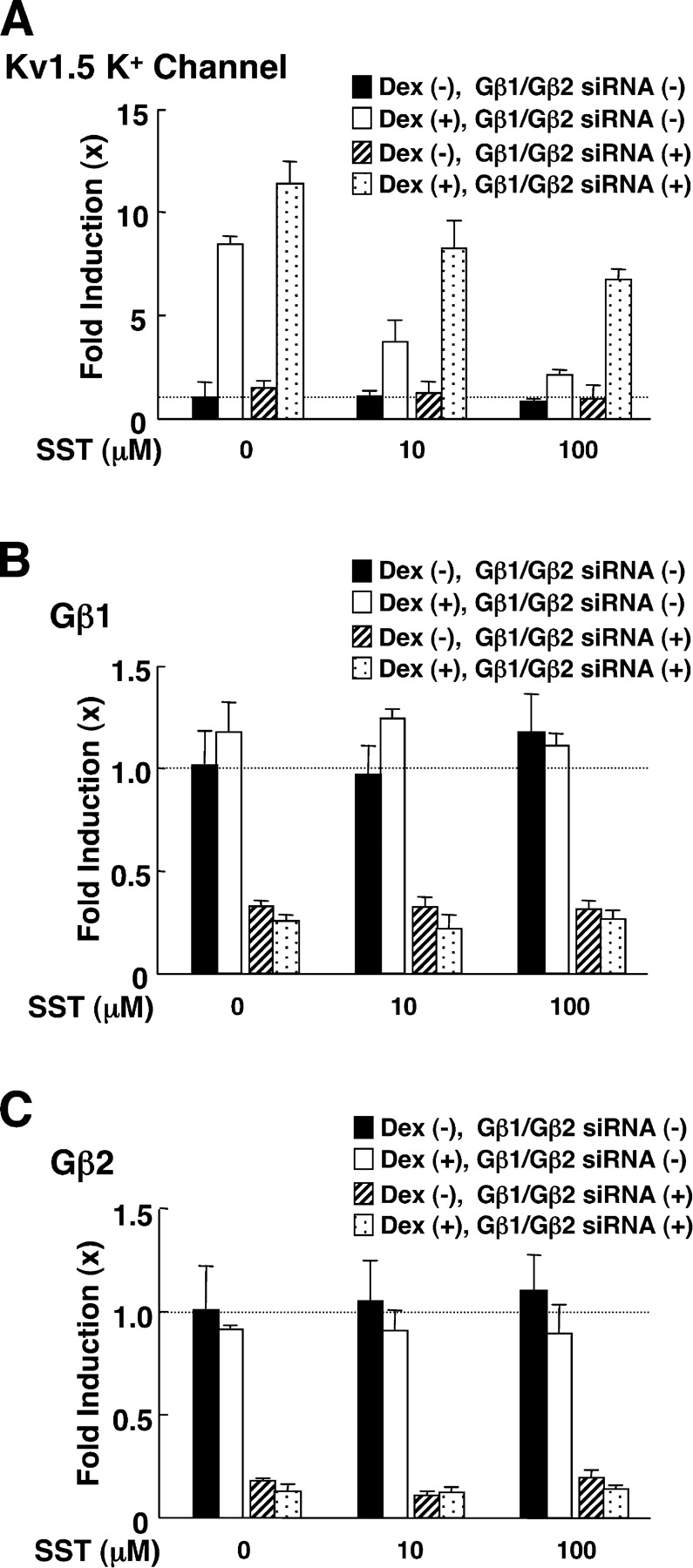
Somatostatin suppressed dexamethasone-stimulated transcriptional activity of the Kv1.5 potassium channel gene, whereas abrogation of Gβ1 and Gβ2 attenuated the somatostatin effect in GH3 cells. GH3 cells were transfected with control or Gβ1 and Gβ2 siRNAs, and were treated with 10−6 M of dexamethasone and/or the indicated amounts of somatostatin for 24 h. Total RNA was then purified from the cells and the amounts of Kv1.5 potassium channel (A), Gβ1 (B), Gβ2 (C), or RPLP0 mRNAs were determined by RT-PCR. Bars show mean ± SEM of their fold induction over baseline.
Gβ2 and Gγ2 are localized in the nucleus and Gβ2 translocates into the nucleus with GR
The trimeric G protein complexes are presumably exclusively located in the cytoplasmic surface of the plasma membrane in order to serve as intermediate communicators between membrane-integrated GPCRs and their downstream effector molecules (Clapham and Neer, 1997). Gγ plays a central role in anchoring the G protein complexes on the plasma membrane through a prenylated cysteine residue located amino acid 68 (in a CAAX motif; Clapham and Neer, 1997). Small amounts of G protein are also detected in caveolae, growth cones and cytoskeleton (Clapham and Neer, 1997). In contrast, GR is located in the cytoplasm in the absence of ligands and translocates into the nucleus in response to ligand binding (Kino et al., 2003a). The discrepancy in the subcellular localization of Gβ/Gγ and GR prompted us to examine the subcellular distribution of Gβ and Gγ (Fig. 4).
Figure 4.

Subcellular localization of Gβ2 and Gγ2 in HCT116 cells. (A) Endogenous Gβ and Gγ are visualized in the nucleus as well as in the cytoplasm/plasma membrane in HCT116 cells. Endogenous Gβ (left, top two panels) and Gγ (right, top two panels) were visualized by treatment with anti-Gβ or -Gγ2 antibodies, and FITC-labeled secondary antibody, and their confocal images were obtained. Nuclei were also stained with DAPI. Co-treatment of the samples with blocking peptides for anti-Gβ (left, bottom) or anti-Gγ2 (right, bottom) antibodies abolished their specific staining. Cells, expressing Gβ or Gγ exclusively in the cytoplasm, are indicated as “ℵ” and “a”, respectively, whereas cells retaining these molecules weakly or strongly in the nucleus are indicated as “ℑ” and “b”, and “ℜ” and “c”, respectively. (B) Endogenous Gβ and Gγ are detected in the nuclear fraction as well as in the cytoplasm and membrane fractions in HCT116 cells. HCT116 cells were lysed and their subcellular fractions were separated by centrifugation. 0.1 μg of protein of indicated subcellular fractions was run on SDS-PAGE gels, blotted to the nitrocellulose membranes, and Gβ and Gγ were visualized with their specific antibodies by reprobing the same membrane. Intracellular adhesion molecule 1 (ICAM1), α-tubulin, and Oct1, detected also by reprobing the same membrane with their specific antibodies, were respectively shown as positive controls for the membrane, cytoplasmic and nuclear fractions to indicate that the subcellular fractionation did not produce cross-contamination. (C and D) EGFP-Gβ2 was localized in the nucleus in addition to the cytoplasm, whereas EGFP-Gγ2 was detected in the nucleus and the cytoplasm, and at the plasma membrane in HCT116 cells. HCT116 cells were transfected with pEGFP-C-1-Gβ2 or -Gγ2, and the cells were fixed and their confocal images were obtained. Nuclei were also stained with DAPI. Representative images of EGFP-Gβ2 and -Gγ2 are respectively shown in C, whereas mean ± SEM values of their signal intensities in the nucleus and the cytoplasm obtained from over 20 cells are shown in D. (E and F) EGFP-Gβ2 translocated into the nucleus with DsRed2-GR in response to 10−6 M of dexamethasone in HCT116 cells. HCT116 cells were transfected with pEGFP-C1-Gβ2 and pDsRed2-GRα. Confocal images of EGFP-Gβ2 and DsRed2-GR were obtained before and 30 min after the treatment with 10−6 M of dexamethasone. Representative images are shown in D, whereas mean ± SEM values of signal intensities in the nucleus (black bars) and the cytoplasm (white bars) obtained from over 20 cells is shown in E. (G) EGFP-Gβ2 and DsRed2-GR are colocalized at the plasma membrane in response to somatostatin in HCT116 cells. HCT116 cells were transfected with pEGFP-C1-Gβ2, pDSRed2-GRα, and Gγ2- and SSTR2-expressing plasmids. Confocal images of EGFP-Gβ2 and DsRed2-GR were obtained before and 30 min after the treatment with 100 nM of somatostatin. Blue and orange arrows indicate signals of EGFP-Gβ2, DsRed2-GR, which are localized at the plasma membrane, whereas yellow arrows indicate their colocalization.
We first examined the localization of endogenous Gβ and Gγ in HCT116 cells by using specific antibodies in indirect immunofluorescent staining studies. The majority of endogenous Gβ and Gγ molecules were localized in the cytoplasm, however small fractions were also detected in the plasma membrane and the nucleus (Fig. 4 A, top). In some cells, both Gβ and Gγ were localized exclusively in the cytoplasm (cell “ℵ” and “a”), whereas other cells retained these molecules weakly (cell “ℑ” and “b”) or strongly (cell “ℜ” and “c”) in the nucleus (Fig. 4 A, middle). Treatment of the cells with blocking peptides in addition to their antibodies, completely abolished these specific staining patterns (Fig. 4 A, bottom). In Western blots, Gβ and Gγ were also detected in the cytoplasmic and nuclear fractions, as well as in the membrane fraction (Fig. 4 B).
To further examine the subcellular localization of Gβ and Gγ, we constructed plasmids expressing EGFP-fused Gβ2 or Gγ2 and examined their localization in the HCT116 cells (Fig. 4, C and D). Consistent with our results obtained with indirect immunofluorescence staining and Western blots, EGFP-Gβ2 was mainly distributed in the cytoplasm, with a relatively small fraction in the nucleus and a very small fraction at the plasma membrane (Fig. 4 C, left and Fig. 4 D). EGFP-Gγ2 was also detected in the cytoplasm, as well as at the plasma membrane, whereas a small fraction was detected in the nucleus (Fig. 4 C, right, and Fig. 4 D). The limited localization of EGFP-Gβ2 at the plasma membrane was not caused by a small number of binding sites at the plasma membrane, as coexpression of Gγ2 and the human somatostatin receptor type 2 (SSTR2) that provided binding sites for EGFP-Gβ2 at the plasma membrane, did not change the characteristic subcellular localization of EGFP-Gβ2 (unpublished data).
Gβ2 and GR comigrate either to the nucleus or to the plasma membrane in response to dexamethasone or somatostatin, respectively
Because the above results suggest that Gβ2 and GR might communicate in the cytoplasm as well as in the nucleus, we examined colocalization of these two molecules by overexpressing EGFP-Gβ2 and DsRed2-GR in HCT116 cells (Fig. 4, E and F). EGFP-Gβ2 was localized mainly in the cytoplasm in addition to the nucleus, whereas DsRed2-GR was exclusively distributed in the former subcellular fraction in the absence of glucocorticoids. 30-min incubation of the transfected cells with 10−6 M of dexamethasone induced nuclear translocation of both DsRed2-GR and EGFP-Gβ2 (Fig. 4, E and F).
Because very little Gβ2 was found at the plasma membrane and EGFP-Gβ2 comigrated from the cytoplasm into the nucleus with the GR in response to dexamethasone, we stimulated a GPCR to test the hypothesis that this treatment induces the plasma membrane localization of EGFP-Gβ in HCT116 cells. We coexpressed EGFP-Gβ2 and DsRed2-GR with Gγ2 and SSTR2, added 100 nM of somatostatin in the medium and examined the localization of EGFP-Gβ2 under the microscope. 30 min after addition of somatostatin, EGFP-Gβ2 accumulated at the plasma membrane (Fig. 4 G). EGFP-Gβ2 was not seen at the plasma membrane after 3 h, possibly because of internalization of the receptor complex into the cell (unpublished data). As expected, a small amount of DsRed2-GR also accumulated at the plasma membrane with EGFP-Gβ2. These results indicate that EGFP-Gβ2 migrated from the cytoplasm to the plasma membrane in response to stimulation of a G protein–coupled, cell surface receptor. They also indicate that some fractions of Gβ and GR are associated with each other in the cytoplasm and can translocate to either the nucleus or the plasma membrane following the activation of the GR or the cell surface GPCR.
Forced cytoplasmic localization of Gβ2 attenuates its suppressive effect on GR-induced transcriptional activity
To further examine the mechanism of Gβ2-induced suppression of GR transactivation, we constructed plasmids expressing a nuclear export signal (NES)- or NLS-fused Gβ2 and EGFP-Gβ2. The NES sequence used was that of the human protein kinase inhibitor α (Henderson and Eleftheriou, 2000), whereas the NLS sequence was from the SV40 large T antigen (Rihs et al., 1991). NES-fused EGFP-Gβ2 was localized exclusively in the cytoplasm, whereas NLS-fused EGFP-Gβ2 was located entirely in the nucleus (Fig. 5, A and B). As expected, NES-Gβ2 lost its suppressive effect on GR-induced transactivation, whereas NLS-Gβ2 demonstrated a stronger suppressive effect than the wild-type Gβ2 (Fig. 5 C). Wild-type, and NES- and NLS-fused Gβ2 were similarly expressed in HCT116 cells (Fig. 5 D). These results indicate that increased nuclear localization of Gβ2 correlates with its suppressive effect on GR-induced transcriptional activity.
Figure 5.

Forced cytoplasmic localization of Gβ2 attenuates the suppressive effect of the wild-type Gβ on GR-induced transactivation, whereas forced nuclear localization enhances it. (A and B) EGFP-fused NES-Gβ2 and NLS-Gβ2 are exclusively localized in the cytoplasm and the nucleus, respectively, in HCT116 cells. HCT116 cells were transfected with pEGFP-C1-NES-Gβ2- or pEGFP-C1-NLS-Gβ2-expressing plasmid. The cells were fixed and their confocal images were obtained. Representative images are shown in A, whereas mean ± SEM values of signal intensities in the nucleus (black bars) and the cytoplasm (white bars) obtained from over 20 cells are shown in B. (C) NES-Gβ2 loses the suppressive effect on GR transactivation, whereas NLS-Gβ2 has a stronger inhibitory effect than the wild-type Gβ2 on GR transactivation in HCT116 cells. HCT116 cells were transfected with pCDNA4His/MaxB-NES-Gβ2- or -NLS-Gβ2 together with pRShGRα, pMMTV-Luc, and pSV40-β-Gal. Bars represent mean ± SEM values of the luciferase activity normalized for β-galactosidase activity in the absence or presence of 10−6 M of dexamethasone. (*), P < 0.01; n.s., not significant, compared with the baseline. (D) Gβ2 wild-type and its fusions with NES or NLS are similarly expressed in HCT116 cells. HCT116 cells were transfected with pCDNA4His/MaxB-Gβ2, -NES-Gβ2-, or -NLS-Gβ2. The cells were lysed and the expression of wild-type, NES-, and NLS-fused Gβ2 was examined in a Western blots using anti-His antibody.
Gβ accumulates on GREs through GR in vivo
We, therefore, performed a chromatin immunoprecipitation (ChIP) assay in COS7 cells that had a stably integrated MMTV promoter in their genomic DNA. We expressed Gβ2 and GR in these cells and detected the coprecipitated MMTV GREs by the PCR with a specific primer pair (Fig. 6 A). Gβ2 was attracted to GREs when the wild-type GR was coexpressed with Gβ2, whereas Gβ2 was not precipitated with GREs in the presence of GR(Δ262-404), indicating that Gβ2 was attracted to GREs through physical interaction with the GR. In these cells, overexpression of Gβ2 suppressed the transcriptional activity of the chromatin-integrated MMTV promoter in a dexamethasone-dependent fashion (Fig. 6 B).
Figure 6.

Gβ2 is attracted to GREs and directly suppresses GR-induced transactivation by inhibiting its AF-2 function. (A) Gβ2 was attracted to the chromatin-integrated MMTV GREs via interaction with GR in COS7 cells. COS7 cells, which have genomically integrated MMTV-Luc, were transfected with Gβ2-expressing plasmid and pRShGRα or pRShGRα(Δ262-404). 24 h after addition of 10−6 M of dexamethasone, the cells were fixed and the ChIP reaction was performed with anti-Gβ or control antibodies. The portion of the MMTV promoter that contains two GREs was amplified by PCR with a specific primer pair. Two images obtained from separate gels were combined to produce the Input gel image. (B) Gβ2 suppresses the transcriptional activity of GR on the chromatin-integrated MMTV promoter in COS7 cells. COS7 cells with genomically integrated MMTV-Luc, were transfected with the indicated amounts of the Gβ2-expressing plasmid, together with pRShGRα and pSV40-β-Gal. Bars represent mean ± SEM values of the luciferase activity normalized for β-galactosidase activity in the absence or presence of 10−6 M of dexamethasone. (*), P < 0.01, compared with the baseline. (C) Gβ2 does not have intrinsic transcriptional activity. HCT116 cells were transfected with increasing amounts of GAL4 DBD-fused Gβ2-, SMRT-, or VP16-expressing plasmid together with pGAL4-E1B-Luc and pSV40-β-Gal. Bars represent mean ± SEM values of the luciferase activity normalized for β-galactosidase activity. (D) Gβ2 suppresses the AF2-directed transcriptional activity of GR, but not the AF-1–dependent transactivation in HCT116 cells. HCT116 cells were transfected with Gβ2-expressing plasmid, pMMTV-Luc, and pSV40-β-Gal together with pRShGRα, pRShGRα(Δ77-261), or pRShGRα(1-515). Bars represent mean ± SEM values of the luciferase activity normalized for β-galactosidase activity in the absence or presence of 10−6 M of dexamethasone. (*), P < 0.01; n.s., not significant, compared with the baseline.
Gβ2 does not have intrinsic transcriptional activity but suppresses the AF-2–directed transcriptional activity of the GR
To explore the mechanism of the transcriptional regulation of GR by Gβ2, we constructed a plasmid expressing the GAL4-fused Gβ2 and examined its transcriptional activity on the GAL4-responsive promoter-driven luciferase construct in HCT116 cells (Fig. 6 C). As positive controls, we used GAL4-fused VP16- and SMRT-expressing plasmids, which respectively demonstrate transactivating or transrepressing activity on this promoter (Nagy et al., 1997; Pazin et al., 1998). GAL4-Gβ2 did not change the basal transcriptional activity of the promoter at all, whereas GAL4-VP16 and -SMRT respectively activated or suppressed it, indicating that Gβ2 does not have intrinsic transcriptional activity.
We further examined the effect of Gβ2 on the transcriptional activity of the two GR mutants GR(Δ77-261) and (1-515) in HCT116 cells, which are respectively devoid of the AF-1 and AF-2 domains (Fig. 6 D). Gβ2 suppressed the transcriptional activity of GR(Δ77-261) similarly to its effect on the wild-type GR, whereas it did not affect the transcriptional activity of GR(1-515), indicating that Gβ2 acts on GR by suppressing AF-2 but not AF-1.
Discussion
The WD-repeat proteins Gβ1, Gβ2, and Rack1 interacted with the GR at the NH2-terminal region enclosed between amino acids 263 and 419; Gβ2 used blades 3–5 for this interaction. All three proteins suppressed the transcriptional activity of the GR on the glucocorticoid-responsive GRE-containing MMTV promoter and endogenous Gβ1 and Gβ2 functioned as negative regulators of glucocorticoid-induced stimulation of TAT enzymatic activity and Kv1.5 potassium channel mRNA expression. Gβ2 acted as a heterodimer with Gγ, and the Gβ/Gγ complex, but not Gαi, was associated with the GR in vivo both in the absence and presence of dexamethasone. Somatostatin suppressed GR-induced expression of the Kv1.5 potassium channel mRNA via activation of the Gβ/Gγ heterodimer. Gβ and Gγ were localized in the nucleus as well as in the cytoplasm/plasma membrane, and a fraction of Gβ2 and GR comigrated to the nucleus or the plasma membrane, respectively, in response to dexamethasone or somatostatin. Forced sequestration of Gβ2 away from the nucleus attenuated its suppressive effect on GR-induced transactivation. Furthermore, Gβ2 was attracted to GREs through the GR and suppressed GR-induced transcriptional activity by inhibiting the AF-2 domain.
Components of the G protein system are presumably strictly located at the inner surface of the plasma membrane attached to it through the prenylated Gγ (Clapham and Neer, 1997). However, there are previously reported exceptions (Kageyama et al., 1999; Jin et al., 2000; Zhang et al., 2001; Chen et al., 2004); for instance, one of the Gβs, Gβ5 was reported in the nucleus as well as in the cytoplasm and the membrane fraction of the cell, and this protein translocated into the nucleus in response to surface stimuli in neuronal cells (Zhang et al., 2001). Similarly, Gβ3 was found in the cytoplasmic pool and moved to the plasma membrane in response to stimulation of the β1 adrenergic receptor in rat cardiac cells (Kageyama et al., 1999). Furthermore, endogenous Gβ replaced with the GFP-fusion form in yeast cells was localized in the cytoplasm, in addition to the plasma membrane, and changed localization in response to a chemo-attractant (Jin et al., 2000). A distribution of Gβ1/Gγ2 similar to the one we found in our study was also reported recently (Chen et al., 2004). In addition to these previous reports, our results indicate that the localization of Gβ is more diffuse than previously thought, being distributed in the cytoplasm and the nucleus, as well as at the cytoplasmic surface of the plasma membrane. EGFP-Gβ2 comigrated with DsRed2-GR into the nucleus in response to dexamethasone, whereas it comigrated to the plasma membrane with the GR after addition of somatostatin. Together, these results indicate that Gβ can migrate between subcellular compartments, such as between the cytoplasm and the nucleus or the plasma membrane, depending on the type of stimuli and the responsive signaling molecules activating the cell. Thus, it appears that cytoplasmic Gβ may function as a reservoir pool, which supplies G proteins into various subcellular compartments according to the needs of the cell (Fig. 7).
Figure 7.

Interactions between the glucocorticoid and GPCR signaling systems at the level of the GR and the Gβ/Gγ subunits. In response to activation of a GPCR and the GR by their respective ligands, the Gβ/Gγ complex interacts with the GR and translocates with it into the cell nucleus where it suppresses glucocorticoid-induced transactivation. The inactivated Gβ/Gγ complex, normally located under the plasma membrane, may serve as an anchor to the nonligand-activated GR.
The Gβ/Gγ complex down-regulated the transcriptional activity of the GR in the nucleus. On the other hand, GR comigrated with Gβ to the plasma membrane, suggesting that membrane-located GR may regulate the activity of the Gβ/Gγ heterodimer on its downstream effector molecules at the plasma membrane. There are many reports indicating nongenomic or cell membrane actions of glucocorticoids; indeed, glucocorticoids increase the Na+-dependent uptake of glutamate, rapidly inhibit release of arginine vasopressin in rat hypothalamus, and transiently stimulate the activity of the Ca2+ channel (Sze and Iqbal, 1994; Liu et al., 1995; Zhu et al., 1998; Falkenstein et al., 2000). One of the steroid hormone receptors, the estrogen receptor was recently localized at the plasma membrane, and shown to regulate cAMP production through interaction with Gαi in neuronal cells (Navarro et al., 2003). Thus, it is possible that Gβ-associated GR might explain some of the nongenomic effects of glucocorticoids at the plasma membrane.
In our experiments, 24-h incubation of cells with somatostatin suppressed GR-induced transcriptional activity by stimulating the Gβ/Gγ heterodimer, possibly by increasing the fraction that can comigrate into the nucleus with the GR in response to dexamethasone. The underlying mechanism of this action is not understood, but it is possible that free Gβ/Gγ heterodimers produced after stimulation with somatostatin might have a greater chance to be associated with the GR, comigrate with it into the nucleus and finally suppress GR-induced transactivation. Somatostatin has anti-inflammatory activity, and glucocorticoids increase local production of somatostatin in inflamed tissues, a phenomenon that may explain part of their anti-inflammatory actions (Karalis et al., 1994, 1995). Our results of the suppressive effect of somatostatin on GR-induced transactivation complete a closed regulatory circuit between glucocorticoids and somatostatin, with the ligand-activated GR stimulating somatostatin and the GR-associated somatostatin-stimulated Gβ/Gγ complex suppressing the latter's effect.
Gβ physically interacted with the GR as a heterodimer with Gγ in the absence of dexamethasone, and addition of dexamethasone further increased the association of the Gβ/Gγ complex with the GR. The ligand-free GR forms a multi-protein complex with several heat shock proteins. In this complex, GR is also associated with other molecules, such as protein 14-3-3, Raf-1, and p53, which modulate GR-induced transcriptional activity (Kino and Chrousos, 2002). The Gβ/Gγ complex and Rack1 should be added to these GR-associated molecules, which suppress the transcriptional activity of the GR. Because Rack1 plays an important role in the regulation of signaling events induced by PKC, cAMP, and several growth factors and cytokines (McCahill et al., 2002), it is possible that these signaling cascades also regulate GR function through Rack1, as in the case of Gβ, where the upstream stimulus somatostatin suppresses GR-induced transcriptional activity.
Gβ together with Gγ was attracted to GREs through the GR, inhibiting AF-2. Gβ did not have any intrinsic transrepressive activity, and, hence, it is unlikely that it functions as an active repressor by attracting inhibitory molecules/complexes, such as, for instance, corepressors with histone deacetylase activity (Jones and Shi, 2003). Rather, Gβ/Gγ may act as a negative scaffold, similarly to Rack1, by binding to a critical portion of the GR and/or other transcriptional intermediate molecules that are attracted to the AF-2, blocking full activation of AF-2 and hence transcription stimulation by the ligand-bound GR.
Materials and methods
Plasmids
pLexA-GRα(263–419) was constructed by inserting the corresponding cDNA fragment of GRα(263–419) into pLexA (CLONTECH Laboratories, Inc.). pB42AD-Gβ1(1–340), -Gβ2(1–340), -Gβ2(1–143), -Gβ2(270–340), -Gβ2(143–226), -Gβ2(55–226), -Gβ2(183–270), and -Gβ2(226–310) were constructed by subcloning the corresponding cDNA fragments of Gβ1 or Gβ2 into pB42AD (CLONTECH Laboratories, Inc.). pB42AD-Rack1(1-317) and -Rack1(139-317) were gifts from D. Adam (University of Kiel, Kiel, Germany). pOP8-LacZ was purchased from CLONTECH Laboratories, Inc. pCDNA4His/MaxB-Gβ1 and -Gβ2 were constructed by inserting coding sequences of Gβ1 and Gβ2 into pCDNA4His/MaxB (Invitrogen). pEGFP-C1-Gβ2 and -Gγ2 were also produced by subcloning the coding sequences of Gβ2 and Gγ2 into pEGFP-C1 (CLONTECH Laboratories, Inc.). pCDNA4His/MaxB-NLS-Gβ2, -NES-Gβ2, or pEGFP-C1-NLS-Gβ2 and -NES-Gβ2 were constructed by inserting synthesized oligonucleotides corresponding to the NLS (AELIPEPPKKKRKVELGTA) of the SV40 large T antigen and the NES (NSNELALKLAGLDINKTE) of the human protein kinase inhibitor α into pCDNA4His/MaxB-Gβ2 and pEGFP-C1-Gβ2, respectively, at the portion in front of their Gβ2 coding sequence (Rihs et al., 1991; Henderson and Eleftheriou, 2000). pCDNA3-Rack1 was a gift from D. Adams. pDsRed2-GRα was constructed by inserting a coding sequence of the human GRα into pDsRed2 (CLONTECH Laboratories, Inc.). pRShGRα, pRShGRα (Δ262–404), (Δ77–261) and (1–515), and GAL-SMRT were all gifts from R.M. Evans (Salk Institute, La Jolla, CA). pCMV6-SSTR2, which expresses the human SSTR2, was a gift from S. Seino (University Kobe, Hyogo, Japan). pMMTV-Luc, which has the luciferase gene under the control of the MMTV promoter (which has four functional GREs) was donated from G.L. Hager (National Cancer Institute, Bethesda, MD). pGAL4-E1B-Luc, which has the luciferase gene under the control of 4 GAL4 response elements, was a gift from P.H. Driggers (Uniformed Services University of the Health Sciences, Bethesda, MD). GAL4-VP16 was a gift from Y. Shi (Harvard Medical School, Boston, MA). pSV40-β-Gal was purchased from Promega.
Yeast two-hybrid screening and assay
The yeast two-hybrid screening was performed using GR(263-419) as a bait in a human Jurkat cell cDNA library with the LexA system (CLONTECH Laboratories, Inc.). For a yeast two-hybrid assay, yeast strain EGY48 (CLONTECH Laboratories, Inc.) was transformed with pOP8-LacZ, pLexA-GRα (263–419), and indicated pB42AD-Gβ1, -Gβ2, and -Rack1 plasmids and β-galactosidase activity was measured in the cell suspension as previously described (Kino and Chrousos, 2003). The β-galactosidase activity was normalized for OD value at 600 nm. Fold induction was calculated by the ratio of adjusted β-galactosidase values of cells transformed with pLexA-derived bait plasmids versus pLexA in the presence of the same prey plasmid.
Cell cultures, transient transfections, and reporter assays
Human colon carcinoma HCT116, African green monkey kidney COS7, uterine cervical carcinoma HeLa, and rat pituitary GH3 cells were purchased from the American Type Culture Collection and maintained in MacCoy's 5A or DME media supplemented with 10% FBS, 50 U of penicillin, and 50 μg/ml of streptomycin. Rat hepatoma HTC cells were provided by J.I. Webster (National Institute of Mental Health, Bethesda, MD) and were cultured in DME with the same supplements. HCT116 and COS7 cells do not contain functional GR, whereas HeLa, HTC, and GH3 cells express the fully active GR (Levitan et al., 1991; Thompson et al., 1966; Kino et al., 2003c; Kino and Chrousos, 2003; Webster et al., 2003; De Martino et al., 2004).
HCT116 cells were transfected as previously described (Kino et al., 2003c). For the experiments using pMMTV-Luc or pGAL4-E1B-Luc as reporter plasmids, different amounts of Gβ1-, Gβ2-, Gγ2-, Rack1-, or GAL4-fused molecule-expressing plasmids were cotransfected with 0.5 μg/well of pRShGRα (for pMMTV-Luc), 1.5 μg/well of pMMTV-Luc or pGAL4-E1B-Luc and 0.5 μg/well of pSV40-β-Gal. Empty vectors were used to maintain the same amounts of transfected DNA. 10−6 M of dexamethasone was added to 24 h after transfection. The cells were harvested after an additional 24 h and luciferase and β-galactosidase assays were performed as previously described (Kino et al., 1999).
Introduction of Gβ1 and Gβ2 siRNAs into HTC and GH3 cells, the TAT assay, and the RT-PCR
The rat Gβ1 and Gβ2 siRNAs (5′-AAGCUCUGGGAUGUCCGAGAAdTdT-3′ and 5′-AACAUCUGCUCCAUCUAUAGUdTdT-3′, respectively), which respectively target nucleotides 625–645 and 355–375 of their coding regions, were produced by QIAGEN. The negative control siRNA (5′-UUCUCCGAACGUGUCACGUdTdT-3′) was also purchased from QIAGEN.
HTC and GH3 cells were transfected with siRNAs by using the Nucleofector System (Amaxa GmbH). In brief, 106 of HTC or GH3 cells were resuspended in solution R and T (Amaxa GmbH), respectively, and were mixed with 5 μg of indicated siRNAs. The electricity was applied with the Nucleofector Device (Amaxa GmbH) by using the protocol T-27 and T-20 for HTC and GH3 cells, respectively. We achieved nearly 80% transfection efficiencies in both cell lines with these combinations of solutions and protocols. 24 h after plating these cells in the 24-well plates, the cells were stimulated with 10−6 M of dexamethasone and/or different concentrations (0–100 nM) of somatostatin. After additional 24 h of incubation, cell lysates for the TAT assay and Western blots, and total RNA for the RT-PCR were harvested. TAT assays were performed as previously reported (Thompson et al., 1966; Webster et al., 2003).
The reverse transcription reaction was performed as previously described (De Martino et al., 2004). To detect mRNA levels of rat Gβ1, Gβ2, Kv1.5 potassium channel and control rat acidic ribosomal phosphoprotein P0 (RPLP0), primer pairs (Gβ1: forward: 5′-CAGCAGACAACCACGTTTAC-3′, reverse: 5′-CAGCCTGCATGTAGCATC-3′; Gβ2: forward: 5′-CTCATCATTTGGGACAGCTAC-3′, reverse: 5′-GTGATGATT-TGGTTGTCGTC-3′; Kv1.5 potassium channel: forward: 5′-CAACCTAGAAGGCTATCT-3, reverse: 5′-GTCGAAGAAGTATTCATTTC-3; RPLP0: forward: 5′-GACATGCTGCTGGCCAATAAG-3′; reverse: 5′-CAACATGTTCAGCAGTGTG-3′) were used. The RT-PCR reaction, consisting of heat activation of the Taq polymerase (10 min at 95°C) and the subsequent 60 PCR cycles (denaturing: 15 s at 95°C; annealing/extension: 1 min at 60°C) was performed in quadruplicate using the SYBR green PCR Master Mix (Applied Biosystems) in an ABI PRIZM 7900 SDS lightcycler (Applied Biosystems). Obtained CT (threshold cycle) values of Gβ1, Gβ2 and the Kv1.5 potassium channel were normalized for those of RPLP0 and their relative mRNA expressions were demonstrated as fold induction of the baseline. The dissociation curves of used primer pairs showed a single peak and samples after PCR reactions had a single expected DNA band in an agarose gel analysis (unpublished data).
Confocal microscopy analyses
HCT116 were cultured on poly-l-lysine–coated cover slides (for fixed cells) or in Delta-T Culture Dishes (Bioptechs Inc.; for live cells) and transfected and/or treated with the indicated plasmids/compounds. They were fixed with 4% PFA, and were subsequently mounted on glass slides with the Vectashield with DAPI (Vector Laboratories, Inc.), or were directly examined under the microscope. Emitted signals were recorded with the LSM510 meta/axiovert 200M microscope (Carl Zeiss MicroImaging, Inc.) stand at 19 ± 1.0°C for fixed samples and at 37 ± 0.5°C for samples cultured in Delta-T Culture Dishes at the NICHD Microscopy and Imaging Core (National Institute of Child Health and Human Development) with the assistance of V. Schram. The plan-apochromat 63× oil (1.4 NA, DIC, working distance = 0.17 mm; Carl Zeiss MicroImaging, Inc.) objective lens (Carl Zeiss MicroImaging, Inc.) was used with the lens immersion medium (Immersol 518FF, n = 1.518: Carl Zeiss MicroImaging, Inc.) for image acquisition. Confocal images were built point by point by collecting the intensities from the photo-multiplier tube using LSM 5 software version 3.2 (Carl Zeiss MicroImaging Inc.). Endogenous Gβ and Gγ were visualized with anti-Gβ and -Gγ2 antibodies (Santa Cruz Biotechnology, Inc.) followed by the FITC-labeled anti–rabbit IgG antibody (Santa Cruz Biotechnology, Inc.). To demonstrate the specificity of anti-Gβ and -Gγ2 antibodies, blocking peptides (Santa Cruz Biotechnology, Inc.) were co-administered with them.
Regular coimmunoprecipitation assay, subcellular fractionation, and Western blots
HeLa cells were treated with 10−6 M of dexamethasone or vehicle for 3 h. Cell lysis and coimmunoprecipitation were carried as previously described (Kino et al., 1999). Proteins were precipitated by anti-hGRα antibody or control rabbit IgG (Santa Cruz Biotechnology, Inc.) and the protein–antibody complexes were collected with protein Agarose A/G PLUS (Santa Cruz Biotechnology, Inc.). After blotting on nitrocellulose membranes, Gαi, Gβ, and Gγ were detected by anti-Gαi, -Gβ, or -Gγ2 antibodies, respectively (Santa Cruz Biotechnology, Inc.). To evaluate endogenously expressed GR, Gαi, Gβ, and Gγ, 10% of cell lysates used in the coimmunoprecipitation reaction were run on SDS-PAGE gels.
To compare endogenous levels of Gβ and Gγ residing in cytoplasmic, nuclear or membrane fractions, HCT116 cells were harvested and homogenated in buffer containing 50 mM Tris-HCl, pH 7.4, 25 mM KCl, 5 mM MgCl2, 0.5 mM dithiothreitol, 0.25 M sucrose and 1 Tab/50 ml Complete Tablet, and were centrifuged at 500 g for 5 min to obtain the whole homogenate. All the procedures were performed at 4°C. The whole homogenates were then centrifuged at 2,000 g for 15 min to harvest the nuclear fraction (pellet) and the supernatant was further centrifuged at 105,000 g for 1 h to separate the cytoplasmic (supernatant) and membrane (pellet) fractions. The nuclear and membrane fractions were washed once with the same buffer. The samples (0.1 μg of protein) were run on SDS-PAGE gels, transferred to the nitrocellulose membranes, and Gβ and Gγ were detected by reprobing the same membrane with anti-Gβ or -Gγ2 antibodies (Santa Cruz Biotechnology, Inc.), respectively. The intracellular adhesion molecule 1, α-tubulin and Oct1 were also respectively detected as positive controls for the membrane, cytoplasmic and nuclear fractions by reprobing the membrane with their specific antibodies (Santa Cruz Biotechnology, Inc.). To evaluate the expression levels of wild-type and NES-/NLS-fused Gβ2, plasmids expressing these molecules were transfected into HCT116 cells, and whole homogenates were prepared as described above. Expressed Gβ-related proteins were then separated on a SDS-PAGE gel, blotted, and visualized with anti-His antibody (Santa Cruz Biotechnology, Inc.).
ChIP assay
ChIP assay was performed in COS7/MMTV cells, which have the genomically integrated MMTV-luciferase gene, (COS7/MMTV), using a ChIP kit (Upstate Biotechnology) with minor modifications. COS7/MMTV cells were transfected with control plasmid, pRShGRα, or pRShGRα(Δ262-404) in the presence of pCDNA4His/MaxB-Gβ2, and were exposed to either 10−6 M of dexamethasone or vehicle for 5 h. The cells were then fixed, DNA and bound proteins were cross-linked, and ChIP assays were performed by coprecipitating the DNA–protein complexes with anti-GRα and -Gβ antibodies or rabbit control IgG (Santa Cruz Biotechnology, Inc.), as previously reported (De Martino et al., 2004). The promoter region −219 to −47 of the MMTV long terminal repeat was amplified from the prepared DNA samples using a primer pair: 5′-AACCTTGCGGTTCCCAG-3′ and 5′-GCATTTACATAAGATTTGG-3′. Amplified products were then run on a 3% agarose gel and visualized DNA bands were photographed.
Acknowledgments
We thank Drs. D. Adam, P.H. Driggers, R.M. Evans, G.L. Hager, S. Seino, and Y. Shi for providing their plasmids; and Mr. K. Zachman for his superb technical assistance.
Abbreviations used in this paper: AF, activation function; ChIP, chromatin immunoprecipitation; DBD, DNA-binding domain; G protein: guanine nucleotide-binding protein; GPCR, G protein–coupled receptor; GR, glucocorticoid receptor; GRE, glucocorticoid response element; MMTV, mouse mammary tumor virus; NES, nuclear export signal; RACK1, receptor for activated C-kinase 1; RPLP0, ribosomal phosphoprotein P0; siRNA, short interfering RNA; SSTR2, somatostatin receptor type 2; TAT, tyrosine aminotransferase.
References
- Brown, P.J., and A. Schonbrunn. 1993. Affinity purification of a somatostatin receptor-G-protein complex demonstrates specificity in receptor-G-protein coupling. J. Biol. Chem. 268:6668–6676. [PubMed] [Google Scholar]
- Cabrera-Vera, T.M., J. Vanhauwe, T.O. Thomas, M. Medkova, A. Preininger, M.R. Mazzoni, and H.E. Hamm. 2003. Insights into G protein structure, function, and regulation. Endocr. Rev. 24:765–781. [DOI] [PubMed] [Google Scholar]
- Chang, B.Y., M. Chiang, and C.A. Cartwright. 2001. The interaction of Src and RACK1 is enhanced by activation of protein kinase C and tyrosine phosphorylation of RACK1. J. Biol. Chem. 276:20346–20356. [DOI] [PubMed] [Google Scholar]
- Chen, S., E.J. Dell, F. Lin, J. Sai, and H.E. Hamm. 2004. RACK1 regulates specific functions of Gbetagamma. J. Biol. Chem. 279:17861–17868. [DOI] [PubMed] [Google Scholar]
- Chrousos, G.P. 1995. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 332:1351–1362. [DOI] [PubMed] [Google Scholar]
- Chrousos, G.P. 2000. The role of stress and the hypothalamic-pituitary-adrenal axis in the pathogenesis of the metabolic syndrome: neuro-endocrine and target tissue-related causes. Int. J. Obes. Relat. Metab. Disord. 24:S50–S55. [DOI] [PubMed] [Google Scholar]
- Chrousos, G.P. 2004. The glucocorticoid receptor gene, longevity, and the complex disorders of Western societies. Am. J. Med. 117:204–207. [DOI] [PubMed] [Google Scholar]
- Clapham, D.E., and E.J. Neer. 1997. G protein beta gamma subunits. Annu. Rev. Pharmacol. Toxicol. 37:167–203. [DOI] [PubMed] [Google Scholar]
- Cotecchia, S., L. Stanasila, D. Diviani, K. Bjorklof, O. Rossier, and F. Fanelli. 2004. Structural determinants involved in the activation and regulation of G protein-coupled receptors: lessons from the alpha1-adrenegic receptor subtypes. Biol. Cell. 96:327–333. [DOI] [PubMed] [Google Scholar]
- De Martino, M.U., N. Bhattachryya, S. Alesci, T. Ichijo, G.P. Chrousos, and T. Kino. 2004. The glucocorticoid receptor and the orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II interact with and mutually affect each other's transcriptional activities: implications for intermediary metabolism. Mol. Endocrinol. 18:820–833. [DOI] [PubMed] [Google Scholar]
- Falkenstein, E., H.C. Tillmann, M. Christ, M. Feuring, and M. Wehling. 2000. Multiple actions of steroid hormones -a focus on rapid, nongenomic effects. Pharmacol. Rev. 52:513–556. [PubMed] [Google Scholar]
- Garritsen, A., P.J. van Galen, and W.F. Simonds. 1993. The N-terminal coiled-coil domain of beta is essential for gamma association: a model for G-protein beta gamma subunit interaction. Proc. Natl. Acad. Sci. USA. 90:7706–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm, H.E. 1998. The many faces of G protein signaling. J. Biol. Chem. 273:669–672. [DOI] [PubMed] [Google Scholar]
- Henderson, B.R., and A. Eleftheriou. 2000. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp. Cell Res. 256:213–224. [DOI] [PubMed] [Google Scholar]
- Jantzen, H.M., U. Strahle, B. Gloss, F. Stewart, W. Schmid, M. Boshart, R. Miksicek, and G. Schutz. 1987. Cooperativity of glucocorticoid response elements located far upstream of the tyrosine aminotransferase gene. Cell. 49:29–38. [DOI] [PubMed] [Google Scholar]
- Jin, T., N. Zhang, Y. Long, C.A. Parent, and P.N. Devreotes. 2000. Localization of the G protein betagamma complex in living cells during chemotaxis. Science. 287:1034–1036. [DOI] [PubMed] [Google Scholar]
- Jones, P.L., and Y.B. Shi. 2003. N-CoR-HDAC corepressor complexes: roles in transcriptional regulation by nuclear hormone receptors. Curr. Top. Microbiol. Immunol. 274:237–268. [DOI] [PubMed] [Google Scholar]
- Kageyama, K., T. Murakami, K. Iizuka, K. Sato, K. Ichihara, Y. Tokumitsu, A. Kitabatake, and H. Kawaguchi. 1999. Translocation of G-protein beta3 subunit from the cytosol pool to the membrane pool by beta1-adrenergic receptor stimulation in perfused rat hearts. Biochem. Pharmacol. 58:1497–1500. [DOI] [PubMed] [Google Scholar]
- Karalis, K., G. Mastorakos, G.P. Chrousos, and G. Tolis. 1994. Somatostatin analogues suppress the inflammatory reaction in vivo. J. Clin. Invest. 93:2000–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karalis, K., G. Mastorakos, H. Sano, R.L. Wilder, and G.P. Chrousos. 1995. Somatostatin may participate in the antiinflammatory actions of glucocorticoids. Endocrinology. 136:4133–4138. [DOI] [PubMed] [Google Scholar]
- Kino, T., and G.P. Chrousos. 2002. Tissue-specific glucocorticoid resistance-hypersensitivity syndromes: multifactorial states of clinical importance. J. Allergy Clin. Immunol. 109:609–613. [DOI] [PubMed] [Google Scholar]
- Kino, T., and G.P. Chrousos. 2003. Tumor necrosis factor alpha receptor- and Fas-associated FLASH inhibit transcriptional activity of the glucocorticoid receptor by binding to and interfering with its interaction with p160 type nuclear receptor coactivators. J. Biol. Chem. 278:3023–3029. [DOI] [PubMed] [Google Scholar]
- Kino, T., A. Gragerov, J.B. Kopp, R.H. Stauber, G.N. Pavlakis, and G.P. Chrousos. 1999. The HIV-1 virion-associated protein vpr is a coactivator of the human glucocorticoid receptor. J. Exp. Med. 189:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino, T., M.U. De Martino, E. Charmandari, M. Mirani, and G.P. Chrousos. 2003. a. Tissue glucocorticoid resistance/hypersensitivity syndromes. J. Steroid Biochem. Mol. Biol. 85:457–467. [DOI] [PubMed] [Google Scholar]
- Kino, T., M. Mirani, S. Alesci, and G.P. Chrousos. 2003. b. AIDS-related lipodystrophy/insulin resistance syndrome. Horm. Metab. Res. 35:129–136. [DOI] [PubMed] [Google Scholar]
- Kino, T., E. Souvatzoglou, M.U. De Martino, M. Tsopanomihalu, Y. Wan, and G.P. Chrousos. 2003. c. Protein 14-3-3sigma interacts with and favors cytoplasmic subcellular localization of the glucocorticoid receptor, acting as a negative regulator of the glucocorticoid signaling pathway. J. Biol. Chem. 278:25651–25656. [DOI] [PubMed] [Google Scholar]
- Law, S.F., D. Manning, and T. Reisine. 1991. Identification of the subunits of GTP-binding proteins coupled to somatostatin receptors. J. Biol. Chem. 266:17885–17897. [PubMed] [Google Scholar]
- Levitan, E.S., L.M. Hemmick, N.C. Birnberg, and L.K. Kaczmarek. 1991. Dexamethasone increases potassium channel messenger RNA and activity in clonal pituitary cells. Mol. Endocrinol. 5:1903–1908. [DOI] [PubMed] [Google Scholar]
- Liu, X., C.A. Wang, and Y.Z. Chen. 1995. Nongenomic effect of glucocorticoid on the release of arginine vasopressin from hypothalamic slices in rats. Neuroendocrinology. 62:628–633. [DOI] [PubMed] [Google Scholar]
- McCahill, A., J. Warwicker, G.B. Bolger, M.D. Houslay, and S.J. Yarwood. 2002. The RACK1 scaffold protein: a dynamic cog in cell response mechanisms. Mol. Pharmacol. 62:1261–1273. [DOI] [PubMed] [Google Scholar]
- Nagy, L., H.Y. Kao, D. Chakravarti, R.J. Lin, C.A. Hassig, D.E. Ayer, S.L. Schreiber, and R.M. Evans. 1997. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 89:373–380. [DOI] [PubMed] [Google Scholar]
- Navarro, C.E., S.A. Saeed, C. Murdock, A.J. Martinez-Fuentes, K.K. Arora, L.Z. Krsmanovic, and K.J. Catt. 2003. Regulation of cyclic adenosine 3′,5′-monophosphate signaling and pulsatile neurosecretion by Gi-coupled plasma membrane estrogen receptors in immortalized gonadotrophin-releasing hormone neurons. Mol. Endocrinol. 17:1792–1804. [DOI] [PubMed] [Google Scholar]
- Pazin, M.J., J.W. Hermann, and J.T. Kadonaga. 1998. Promoter structure and transcriptional activation with chromatin templates assembled in vitro. A single Gal4-VP16 dimer binds to chromatin or to DNA with comparable affinity. J. Biol. Chem. 273:34653–34660. [DOI] [PubMed] [Google Scholar]
- Radeff-Huang, J., T.M. Seasholtz, R.G. Matteo, J.H. Brown, S. Cotecchia, L. Stanasila, D. Diviani, K. Bjorklof, O. Rossier, and F. Fanelli. 2004. G protein mediated signaling pathways in lysophospholipid induced cell proliferation and survival. Structural determinants involved in the activation and regulation of G protein-coupled receptors: lessons from the alpha1-adrenegic receptor subtypes. J. Cell. Biochem. 92:949–966. [DOI] [PubMed] [Google Scholar]
- Rihs, H.P., D.A. Jans, H. Fan, and R. Peters. 1991. The rate of nuclear cytoplasmic protein transport is determined by the casein kinase II site flanking the nuclear localization sequence of the SV40 T-antigen. EMBO J. 10:633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T.F., C. Gaitatzes, K. Saxena, and E.J. Neer. 1999. The WD repeat: a common architecture for diverse functions. Trends Biochem. Sci. 24:181–185. [DOI] [PubMed] [Google Scholar]
- Sze, P.Y., and Z. Iqbal. 1994. Regulation of calmodulin content in synaptic plasma membranes by glucocorticoids. Neurochem. Res. 19:1455–1461. [DOI] [PubMed] [Google Scholar]
- Thompson, E.B., G.M. Tomkins, and J.F. Curran. 1966. Induction of tyrosine alpha-ketoglutarate transaminase by steroid hormones in a newly established tissue culture cell line. Proc. Natl. Acad. Sci. USA. 56:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, J.I., L.H. Tonelli, M. Moayeri, S.S. Simons Jr., S.H. Leppla, and E.M. Sternberg. 2003. Anthrax lethal factor represses glucocorticoid and progesterone receptor activity. Proc. Natl. Acad. Sci. USA. 100:5706–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatani, A., J. Codina, R.D. Sekura, L. Birnbaumer, and A.M. Brown. 1987. Reconstitution of somatostatin and muscarinic receptor mediated stimulation of K+ channels by isolated GK protein in clonal rat anterior pituitary cell membranes. Mol. Endocrinol. 1:283–289. [DOI] [PubMed] [Google Scholar]
- Zhang, J.H., V.A. Barr, Y. Mo, A.M. Rojkova, S. Liu, and W.F. Simonds. 2001. Nuclear localization of G protein beta 5 and regulator of G protein signaling 7 in neurons and brain. J. Biol. Chem. 276:10284–10289. [DOI] [PubMed] [Google Scholar]
- Zhu, B.G., D.H. Zhu, and Y.Z. Chen. 1998. Rapid enhancement of high affinity glutamate uptake by glucocorticoids in rat cerebral cortex synaptosomes and human neuroblastoma clone SK-N-SH: possible involvement of G-protein. Biochem. Biophys. Res. Commun. 247:261–265. [DOI] [PubMed] [Google Scholar]