

Abstract
Proteins that in cells specifically bind to growing microtubule plus ends (+TIPs) are thought to play important roles in polarization of the cytoskeleton. However, most +TIPs do not show a bias of their microtubule-binding behavior toward different subcellular regions. Here, we examine the dynamics of the +TIP CLASP in migrating PtK1 epithelial cells. We find that, although CLASPs track microtubule plus ends in the cell body, they dynamically decorate the entire microtubule lattice in the leading edge lamella and lamellipodium. Microtubule lattice binding is mediated by the COOH-terminal region of the CLASP microtubule-binding domain and is regulated downstream of Rac1. Phosphorylation of sites in the NH2-terminal part of the microtubule-binding domain by glycogen synthase kinase 3β likely regulates the affinity of CLASPs for microtubule lattices. These results demonstrate the striking difference of the microtubule cytoskeleton in the lamella as compared with the cell body and provide the first direct observation of subcellular regulation of a microtubule-associated protein in migrating cells.
Introduction
The polarization of the microtubule (MT) cytoskeleton is essential for the directed migration of many cell types (Wittmann and Waterman-Storer, 2001; Andersen, 2005). This is reflected in the orientation of the MT-organizing center toward the direction of migration, as well as the bias of MT dynamic instability toward net growth in the leading edge lamella and lamellipodium. MT organization and assembly/disassembly dynamics in migrating cells are regulated downstream of Rho GTPases (Wittmann et al., 2003; Palazzo et al., 2004), which are central regulators of cell polarization and the actin cytoskeleton (Etienne-Manneville and Hall, 2002).
Recently, a diverse group of proteins called +TIPs, which in cells specifically bind near growing MT plus ends, have received much attention as potential regulators of MTs in cell polarization during migration. Different +TIPs have been shown to bind to each other in biochemical assays and are thus thought to form a complex at the end of growing MTs in cells (Galjart and Perez, 2003; Mimori-Kiyosue and Tsukita, 2003). +TIPs may regulate MT dynamic instability and possibly connect MTs to Rho GTPase signaling pathways (Fukata et al., 2002; Komarova et al., 2002a; Rogers et al., 2002). However, the molecular mechanisms by which +TIPs participate in polarizing the MT cytoskeleton are still poorly understood because most +TIPs, such as EB1 and CLIP-170, do not preferentially track specific subpopulations of MT plus ends in specialized cell regions.
Exceptions are the adenomatous polyposis coli protein (APC), which accumulates in clusters on a small subset of MT ends in protruding cell edges (Bienz, 2002) and CLASPs, homologues of Drosophila melanogaster orbit/mast, that were originally identified in mammalian cells through their interaction with CLIP-170 (Akhmanova et al., 2001). Recently, CLASPs have been shown to also bind EB1 and stabilize MTs in HeLa cells (Rogers et al., 2004; Mimori-Kiyosue et al., 2005). CLASPs have been reported to bind specifically to MT plus ends in fibroblast protrusions at monolayer wound edges and in the periphery of neuronal growth cones, suggesting that they may be important for regulating cytoskeletal polarization (Akhmanova et al., 2001; Lee et al., 2004). Here, we studied the in vivo dynamics of CLASP2 by time-lapse fluorescence microscopy in migrating PtK1 epithelial cells. At noncontacted edges of epithelial cell islands, PtK1 cells undergo a wound healing response and become highly polarized with larger and more persistent lamella/lamellipodia protrusions than fibroblasts (Wittmann et al., 2003; Gupton et al., 2005). We find that the affinity of CLASPs for MTs is spatially regulated, resulting in plus end tracking in the cell body and MT lattice binding in the lamella. This regulation occurs downstream of Rac1 and glycogen synthase kinase 3β (GSK3β) and is likely due to direct regulation of CLASP affinity for the MT lattice. Our results provide the first direct evidence of polarized regulation of a MT-associated protein (MAP) in migrating cells and show that a regulatory cascade can promote switching between +TIP and MAP behavior.
Results
CLASP-MT binding is spatially regulated in epithelial cells
To investigate the in vivo dynamics of CLASPs on MTs in migrating cells, we used PtK1 cells, a marsupial kidney epithelial cell line, that we used previously to characterize leading edge MT dynamic instability regulation (Wittmann et al., 2003). First, we examined the localization of endogenous CLASPs in PtK1 cells by immunofluorescence using an affinity-purified antibody raised against the CLASP homologue from Xenopus laevis (Fig. 1). This antibody specifically recognized a single protein band of ∼170 kD on immunoblots of crude PtK1 cell lysate (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1). Although cytoplasmic background in the cell body was relatively high and we observed the brightest staining on a perinuclear organelle likely to be the Golgi complex, it was often possible to discern labeling of individual MT plus ends in the cell body (Fig. 1 C). However, near the leading cell edge, MTs extending into the lamella and lamellipodium were labeled prominently along their lattices for 10 μm or more, which is inconsistent with classical +TIP behavior. The CLASP staining pattern in PtK1 cells at the edge of epithelial cell islands was similar to serum-stimulated fibroblasts at wound edges, where increased CLASP localization to leading edge MT plus ends has been observed (Akhmanova et al., 2001; Mimori-Kiyosue et al., 2005). However, extensive CLASP labeling along lamella MT lattices was specific to PtK1 cells, probably because these cells have much larger and more persistent lamellae than fibroblasts.
Figure 1.
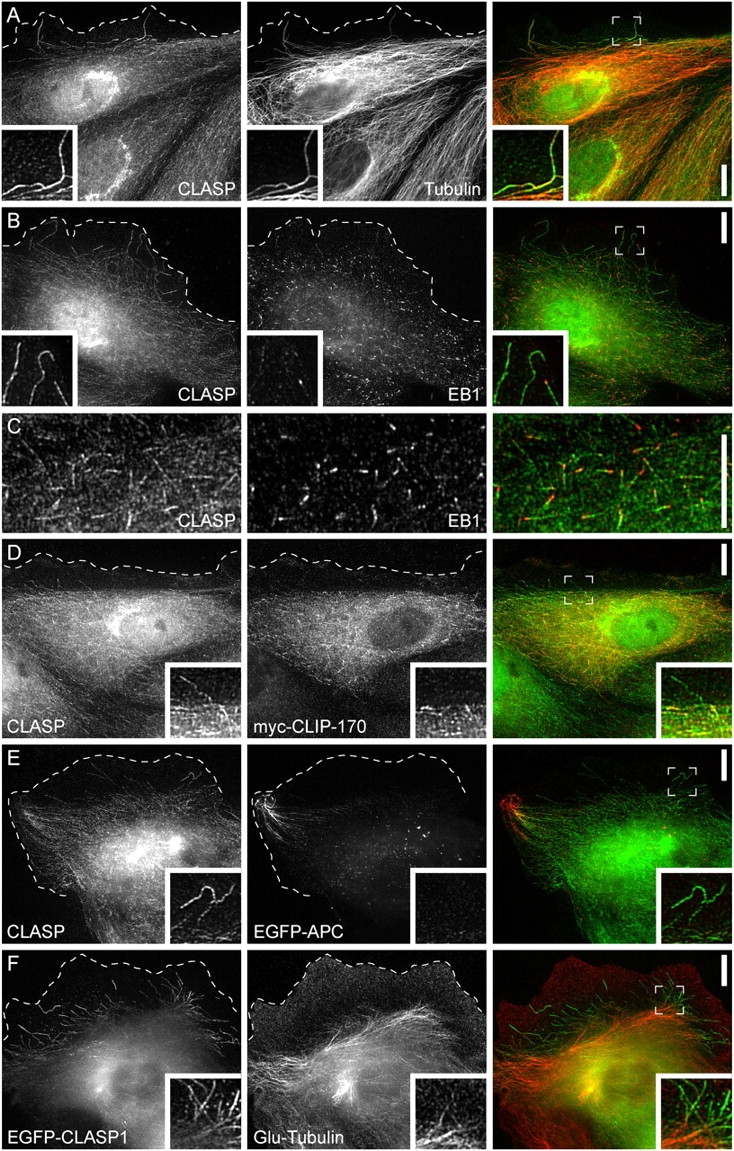
CLASPs are localized along MT lattices in the lamella and to MT plus ends in the cell body. (A) Immunofluorescence of CLASPs and MTs in PtK1 epithelial cells. Cells were stained with antibodies against the Xenopus CLASP homologue and MTs. (B) Immunofluorescence of CLASPs and EB1. (C) Higher magnification of CLASP- and EB1-MT plus end association in the cell body. EB1 is most concentrated at the very tip of the MT, whereas CLASPs are localized slightly behind EB1. (D) Immunofluorescence of CLASPs and expressed myc-tagged CLIP-170. (E) Immunofluorescence of CLASPs and expressed EGFP-tagged APC. None of the other +TIPs show specific accumulation on the lattice of lamella MTs. (F) Distribution of detyrosinated tubulin in a cell expressing EGFP-tagged CLASP1. Insets show lamella cell regions at higher magnification and the dashed line indicates the protruding cell edge. Bars, 10 μm.
We compared CLASP staining to the distribution of EB1, a “bona fide” +TIP that binds equally to all growing MT plus ends (Piehl and Cassimeris, 2003). EB1 staining mainly highlighted MT plus ends in the cell body, and, unlike CLASPs, very little EB1 was present on lamella MTs (Fig. 1 B). When we compared the localization of CLASPs and EB1 on MT plus ends in the cell body, we found that these two proteins did not colocalize (Fig. 1 C). EB1 staining was always brightest at the very tip of the MT, whereas CLASP staining occurred in a ∼1–2-μm stretch behind EB1 and was often excluded from the region of highest EB1 intensity. This implies that these two proteins may track MT plus ends by different mechanisms.
We also wanted to compare CLASP localization to that of CLIP-170 and APC. Because antibodies against these proteins are not functional in PtK1 cells, we expressed tagged constructs. At low expression levels, myc-tagged CLIP-170 localized with endogenous CLASP near MT plus ends in the cell body, but unlike CLASP, CLIP-170 was never specifically enriched on the lattice of lamella MTs (Fig. 1 D). EGFP-tagged APC associated with MTs only in a subset of cells, in which it formed clusters along MT plus ends in cell corners, similar to what has been reported in other cell types (Mimori-Kiyosue et al., 2000; Bienz, 2002). Only a subset of the CLASP-decorated lamella MTs also exhibited APC clusters (Fig. 1 E). Together, these results suggest that CLASP binding to MTs is very different from other +TIPs and is regionally regulated in cells. CLASP binding along MT lattices is favored in the lamella and lamellipodium, whereas CLASP binding near plus ends is favored in the cell body.
In fibroblasts, MTs oriented in the direction of migration often are posttranslationally modified such that they can be stained by antibodies against detyrosinated tubulin. These MTs have been shown to be stabilized against depolymerization by cold or nocodazole (Gundersen and Bulinski, 1988). Because CLASP localization on MT lattices clearly identified MTs in cell protrusions, we compared EGFP-CLASP1 distribution to the distribution of detyrosinated MTs (Fig. 1 F). In PtK1 cells, detyrosinated MTs were not particularly oriented and rarely entered the lamella, and CLASP-decorated MTs were almost never detyrosinated.
CLASPs track MT plus ends in the cell body and dynamically bind MT lattices in the lamella
Next, we examined the in vivo dynamics of CLASPs. In cells expressing low levels of either EGFP-tagged CLASP1 or CLASP2, the distribution of EGFP-tagged protein was very similar to what we observed by immunofluorescence (Fig. 1 F). Because, at higher expression levels, EGFP-tagged CLASPs bundled and decorated MTs in the cell body (unpublished data), we used a CLASP2 construct tagged with three EGFP moieties to achieve a better signal to noise ratio at low expression levels (Bulinski et al., 2001). In PtK1 cells, 3xEGFP-CLASP2 tracked growing MT plus ends in the cell body, but decorated the entire MT lattice in the lamella (Fig. 2, A and C; and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1). CLASP plus end comets tended to become longer as they approached the border between the cell body and the lamella. Remarkably, when individual CLASP comets crossed from the cell body into the lamella, the MT lattice became fully decorated immediately (Video 1). In cells coinjected with fluorescently labeled tubulin, it was evident that lattice binding occurred on all lamella MTs (Fig. 2 C) and 3xEGFP-CLASP2 remained associated with lamella MTs during all phases of dynamic instability (Video 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1).
Figure 2.
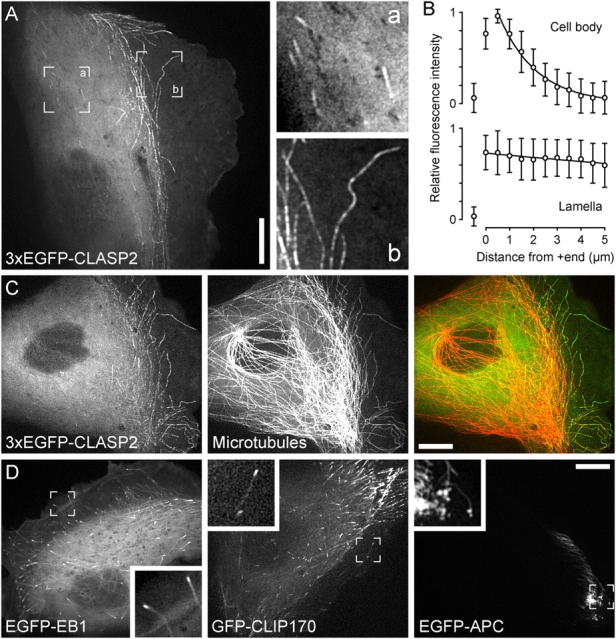
The dynamic association of CLASPs with MTs is spatially regulated in living cells. (A) PtK1 cell expressing 3xEGFP-CLASP2 showing plus end association in the cell body (a) and MT lattice binding in the lamella (b; see Video 1). Bar, 10 μm. (B) Quantification of 3xEGFP-CLASP2 fluorescence intensity along MT plus ends in the cell body or in the lamella. n = 42 MTs each. Mean (circles) and SD (error bars) are shown for individual data points. (C) Coinjection of fluorescent tubulin reveals that all lamella MTs are decorated with 3xEGFP-CLASP2 (Video 2). Bar, 10 μm. (D) The dynamics of other +TIPs in PtK1 cells are entirely different from CLASPs (Video 3). EGFP-EB1 and GFP-CLIP-170 are not specifically enriched on lamella MTs. EGFP-APC forms tight clusters on a small subset of MTs in cell corners. Insets show the indicated peripheral cell regions at higher magnification. Bar, 10 μm.
To confirm that the dynamic behavior of CLASPs in cells is unique, we observed GFP-tagged constructs of other +TIPs in live PtK1 cells (Fig. 2 D and Video 3, available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1). Both EGFP-EB1 and GFP-CLIP-170 tracked MT plus ends throughout the cell and were not specifically enriched on lamella MT lattices. In contrast, EGFP-APC formed dynamic clusters at the ends of few MTs in very small subcellular regions, and its overall dynamics were very different from EGFP-CLASP.
The binding behavior of CLASPs to MTs also correlated with distinct MT dynamic instability in these subcellular regions. MT plus end tracking by EGFP-tagged CLASP2 in the cell body identified a population of MTs that we previously did not observe in PtK1 cells due to the high MT density in the cell body (Wittmann et al., 2003). As described for other cells (Komarova et al., 2002b), these MTs grew very fast and persistently (16.9 ± 4.4 μm/min; see Fig. 4 H; Fig. S1 B). MT plus ends at the junction between the cell body and lamella, which we previously called “central MTs,” switched frequently between growing and shortening (Wittmann et al., 2003). When MTs entered the lamella and became “pioneer MTs,” they grew more persistently, but at a slower average rate than in the cell body (7.8 ± 3.8 μm/min; see Fig. 4 H; Fig. S1 B; Waterman-Storer and Salmon, 1997; Wittmann et al., 2003). Thus, CLASPs appeared to bind preferentially to the lattice of pioneer MTs and may contribute to the differential assembly/disassembly dynamics of this distinct MT population.
Figure 4.
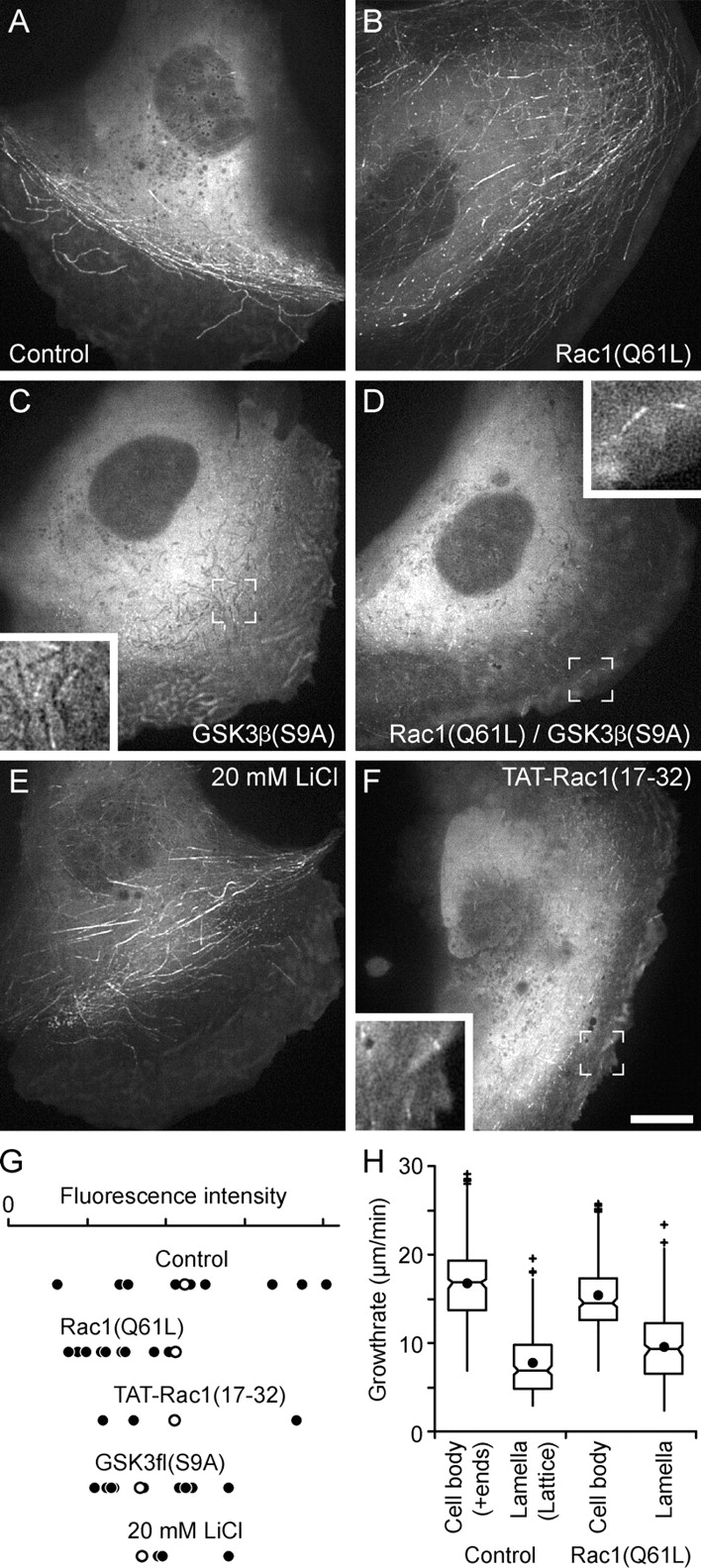
Regulation of CLASP-MT binding by Rac1 and GSK3β. (A) 3xEGFP-CLASP2 in a control PtK1 cell; (B) in a cell expressing constitutively active mRFP-Rac1(Q61L) (Video 5); (C) in a cell expressing constitutively active mRFP-GSK3β(S9A) (Video 7); and (D) in a cell expressing both mRFP-Rac1(Q61L) and mRFP-GSK3β(S9A). Rac1 activation induces CLASP binding to MT lattices in the cell body, whereas GSK3β inhibits lamella lattice binding but still allows some plus end tracking. (E) 3xEGFP-CLASP2 in a cell treated with 20 mM lithium chloride, which inhibits GSK3β and induces lattice binding in the cell body (Video 8). (F) 3xEGFP-CLASP2 in a cell treated with a Rac1 inhibitory peptide, TAT-Rac1(17–32), which largely inhibits lattice binding but still allows plus end tracking (Video 6). Insets show the indicated cell regions at higher magnification. Bar, 10 μm. (G) Quantification of the total EGFP fluorescence intensity of individual PtK1 cells. This demonstrates that the 3xEGFP-CLASP2 expression level was comparable under all conditions. Open circles identify the cells shown in A–F. (H) Distribution of growth rates in the cell body of control PtK1 cells, where CLASPs track MT plus ends, compared with the lamella, where CLASPs dynamically bind to the MT lattice, compared with MTs in similar cell regions in Rac1(Q61L)-expressing cells. Rac1 activation does not homogenize growth rates. n = 300 growth events for each condition. Data are presented as box-and-whisker plots indicating the 25th percentile (bottom boundary), median (middle line), 75th percentile (top boundary), nearest observations within 1.5 times the interquartile range (whiskers), 95% confidence interval of the median (notches), and outliers (+). Closed circles indicate the means.
To distinguish whether plus end tracking and MT lattice binding occurred through different mechanisms, we compared the CLASP distribution on these two populations of MTs. We measured the 3xEGFP-CLASP2 fluorescence intensity as a function of the distance from the MT plus end (Fig. 2 B). Consistent with our immunofluorescence results, in the cell body, the maximal association of 3xEGFP-CLASP2 with MT plus ends occurred proximal to the very tip and, on average, decayed exponentially with increasing distance and a half-length of ∼1 μm. This result is consistent with a first order dissociation reaction, and is qualitatively similar to EB1 dissociation kinetics (Tirnauer et al., 2002). In contrast, on lamella MTs, 3xEGFP-CLASP2 fluorescence intensity did not on average decrease significantly over several micrometers. Due to the low expression level, 3xEGFP-CLASP2 along lamella MTs appeared speckled, and the speckle pattern fluctuated continuously over time, indicating steady-state dissociation and association of CLASP2 on the MT lattice (Bulinski et al., 2001; Video 1).
FRAP reveals rapid CLASP binding kinetics on lamella MT lattices
MT lattice decoration could be explained by a higher affinity of CLASPs for MT lattices in the lamella, but might also be due to a failure of CLASP dissociation from the MT in the wake of tight binding at the MT plus end or to minus-end–directed lateral transport away from the plus end. To distinguish between these possibilities, we performed FRAP experiments (Fig. 3 and Video 4, available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1). The fluorescence recovery of EGFP-CLASP2 on lamella MTs was nearly complete and surprisingly fast with half-times on the order of 600 ms (Table I). 3xEGFP-CLASP2 recovered slightly more slowly, likely due to decreased cytoplasmic diffusion. Recovery rate constants were not significantly different in the plus end– or minus end–facing side of the bleach zone, indicating that neither directed transport nor lateral diffusion contribute to CLASP-MT lattice binding (Fig. 3 D). Thus, the difference in plus end tracking in the cell body versus lattice binding in the lamella is determined either by local differences in the MT lattice or by local regulation of the affinity of CLASPs for MT lattices.
Figure 3.

Photobleaching of EGFP-CLASP2 on MT lattices and growing plus ends. (A) FRAP of EGFP-CLASP2 on the MT lattice in the lamella (Video 4). Fluorescence recovers evenly in the entire bleached region. Bar, 2 μm. (B) FRAP of EGFP-CLASP2 on a growing plus end in the cell body. The arrow indicates the position of the terminus of CLASP fluorescence near the MT plus end at the time of bleaching. Note that recovery occurs transiently proximal to this site before the plus end comet treadmills away from its initial position. Bar, 2 μm. (C) Graphs of relative fluorescence recovery on MT lattices showing all data points, the average of exponential fits from individual experiments (n = 20; solid line) and the direct exponential fit of all data (dashed line). (D) Comparison of the recovery rate constant of EGFP-CLASP2 on MT lattices determined either in the center or toward either side of the bleached region. No significant difference could be detected by ANOVA. Data are presented as box-and-whisker plots indicating the 25th percentile (bottom boundary), median (middle line), 75th percentile (top boundary), nearest observations within 1.5 times the interquartile range (whiskers), and outliers (+).
Table I. FRAP of EGFP-CLASP2 on lamella MT lattices.
| k | Recovery | t 1/2 | n | |
|---|---|---|---|---|
| % | s | |||
| EGFP-CLASP2 | 1.21 ± 0.37 | 92.7 ± 5.1 | 0.63 ± 0.21 | 20 |
| towards −end of bleach zone | 1.25 ± 0.47 | 93.9 ± 4.0 | 0.62 ± 0.21 | 10 |
| center of bleach zone | 1.42 ± 0.45 | 91.5 ± 4.0 | 0.55 ± 0.23 | 10 |
| towards +end of bleach zone | 1.31 ± 0.38 | 92.7 ± 5.6 | 0.57 ± 0.17 | 10 |
| 3xEGFP-CLASP2 | 0.82 ± 0.30a | 86.5 ± 5.6 | 0.94 ± 0.29 | 20 |
| EGFP-CLASP2(340–1084) | 1.14 ± 0.31 | 95.7 ± 3.3 | 0.65 ± 0.18 | 20 |
| lithium chloride–treated | 0.74 ± 0.11a | 90.3 ± 2.5 | 0.96 ± 0.15 | 20 |
Values are mean ± SD. A subset of the EGFP-CLASP2 data was also analyzed at different positions along the MT in the bleached region to test for lateral transport. k, recovery rate constant; n, number of bleach events analyzed.
The recovery rate constants of 3xEGFP-CLASP2 compared to EGFP-CLASP2 and control compared to lithium chloride–treated EGFP-CLASP2(340–1084) were significantly different (P < 0.0001; t test).
Due to fast MT growth, it was technically difficult to photobleach EGFP-CLASP2 plus end comets in the cell body. However, we did achieve this a few times and in such cases observed recovery not only via new plus end growth, as we would have expected, but also at a short distance behind the MT end (Fig. 3 B and Video 4). This suggests that the underlying mechanism of CLASP plus end tracking involves binding to the MT lattice close to the plus end, which is thought to be structurally different from the rest of the MT lattice (Arnal et al., 2000), as opposed to copolymerization with tubulin dimer that was proposed for CLIP-170 (Diamantopoulos et al., 1999).
Regulation of CLASP2 through Rac1 and GSK3β
Next, we wanted to investigate how CLASP-MT binding is regionally regulated. The small GTPase Rac1 induces actin polymerization to drive leading edge protrusion. Because we previously found that Rac1 promotes MT net growth in the lamella (Wittmann et al., 2003), we sought to determine if Rac1 activity played a role in spatial regulation of CLASP-MT binding. In PtK1 cells expressing constitutively active Rac1(Q61L), 3xEGFP-CLASP2 binding to MTs became homogeneous throughout the cell (Fig. 4 B). In addition to plus end tracking, lattice binding now also occurred in the cell body, whereas growing MTs in the lamella displayed both lattice binding and more substantial plus end tracking than in control cells (Video 5, available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1). To inhibit endogenous Rac1, we used a cell permeable inhibitory peptide, TAT-Rac1(17–32) (van Hennik et al., 2003), which, as expected, induced the collapse of the lamella and lamellipodium. In these cells, MT lattice binding was greatly inhibited, but plus end tracking still occurred (Fig. 4 F and Video 6, available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1). These experiments demonstrate that the affinity of CLASPs for MT lattices is correlated with lamella formation and is promoted by Rac1 activation.
Because it has been reported that CLASP-MT binding is affected by GSK3β, which is specifically inactivated in cell protrusions (Akhmanova et al., 2001; Etienne-Manneville and Hall, 2003), we next examined whether GSK3β is involved in the regional regulation of CLASP-MT association. CLASPs contain several potential (S/T)XXX(S/T) GSK3β phosphorylation motifs (Cohen and Frame, 2001; Fig. 5 A) that are highly conserved in different vertebrate sequences. Indeed, GSK3β phosphorylated CLASP2 protein in vitro, and GSK3β inhibition in PtK1 cells resulted in a noticeable downshift of CLASPs on immunoblots of cell extracts (Fig. S1, C and D). In cells expressing constitutively active GSK3β(S9A), MT lattice binding was completely abolished (Fig. 4 C and Video 7, available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1). Plus end tracking, although reduced, still occurred in many cells. In contrast, lithium chloride, which inhibits GSK3β, induced ectopic MT lattice binding in the cell body (Fig. 4 E and Video 8, available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1). We also observed such ectopic lattice binding in EGFP-CLASP1–expressing PtK1 cells with a different GSK3β inhibitor, SB216763 (Fig. S1 E). In addition, the inhibition of protein phosphatases by 1 μM okadaic acid abolished MT lattice binding in the lamella but did not affect plus end tracking in the cell body (unpublished data). The total 3xEGFP-CLASP2 fluorescence intensity was comparable under different conditions, indicating that the observed differences in MT-binding behavior were not due to variations of the 3xEGFP-CLASP2 expression level (Fig. 4 G).
Figure 5.
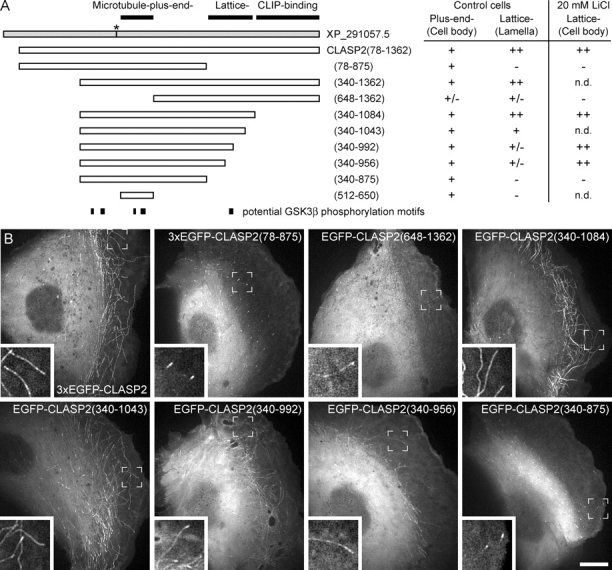
The COOH-terminal part of the CLASP-MT–binding domain mediates MT lattice binding in the lamella. (A) Diagram of the constructs used in this study and summary of their MT-binding behavior. Amino acid positions refer to the predicted human CLASP2 full-length sequence (GenBank/EMBL/DDBJ accession no. XP_291057.5; the asterisk indicates an 8-aa deletion in this sequence). (B) Localization of EGFP-tagged CLASP2 constructs to MTs in living PtK1 cells. Amino acids 340–875 are sufficient for plus end association, but residues 340–1084 are required for both plus end tracking and robust lattice binding to occur. Bar, 10 μm. Insets are higher magnifications of peripheral cell regions.
To determine the relationship of Rac1 and GSK3β in a tentative signaling pathway regulating CLASP-MT association, we expressed constitutively active Rac1(Q61L) together with constitutively active GSK3β(S9A). In this case, 3xEGFP-CLASP2 only tracked MT plus ends as in cells expressing GSK3β(S9A) alone (Fig. 4 D), indicating that GSK3β acts downstream of Rac1.
Because in control PtK1 cells the distribution of MT growth rates correlated strongly with the different MT-binding behavior of CLASPs in the cell body versus the lamella, we determined growth rates in the cell body and peripheral regions in Rac(Q61L)-expressing cells in which CLASP-MT binding was homogenous. In Rac(Q61L)-expressing cells, MTs still grew faster in the cell body (15.3 ± 3.9 μm/min) as compared with peripheral regions (9.5 ± 4.0 μm/min), very similar to what we observed for cell body versus lamella MTs in control cells (Fig. 4 H and Fig. S1 B). This finding indicates that MT growth rates are not regulated downstream of Rac1 and are not affected by CLASP binding to MT lattices, consistent with our previous results that Rac1 modulates lamella MT net growth primarily through regulation of the catastrophe frequency (Wittmann et al., 2003). Together, these results suggest a pathway in which inactivation of GSK3β downstream of Rac1 activation promotes the association of CLASPs with MT lattices.
The COOH-terminal region of the CLASP-MT–binding domain is required for high affinity MT lattice association
To determine whether MT lattice binding and plus end tracking resided in different parts of the CLASP2 molecule, we analyzed the localization of EGFP-tagged CLASP2 deletion mutants (Fig. 5). The shortest fragment that behaved indistinguishably from the full-length protein and displayed robust lamella MT lattice binding was EGFP-CLASP2(340–1084), which is roughly equivalent to the previously defined MT-binding region of CLASP1 (Maiato et al., 2003). This demonstrates that the COOH-terminal CLIP-170–binding domain is not required for either plus end tracking or lattice binding. Shorter constructs truncated at amino acid 875 lost all lattice-binding activity and only tracked growing plus ends throughout the cell (Fig. 5 and Video 9, available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1), and constructs terminating between residue 875 and 1084 showed gradually less lamella MT lattice association (Fig. 5). This demonstrates that the region between amino acid 875 and 1084 is required for CLASP-MT lattice binding, but not for plus end tracking. The recently reported minimal plus end tracking domain, EGFP-CLASP2(512–650) (Mimori-Kiyosue et al., 2005), was sufficient to track MT plus ends in PtK1 cells, and a construct lacking this domain, EGFP-CLASP2(648–1362), showed severely reduced plus end tracking and MT lattice binding, demonstrating that this domain is critical for all CLASP-MT association in cells (Fig. 5 B).
In FRAP experiments, the recovery rate of EGFP-CLASP2(340–1084) on lamella MT lattices was indistinguishable from EGFP-CLASP2 (Table I). Fluorescence of ectopic, lithium chloride–induced EGFP-CLASP2(340–1084) on MT lattices in the cell body still recovered rapidly, but significantly more slowly than on lamella MT lattices in control cells. Together with the observation that constitutively active GSK3β (S9A) also reduced plus end tracking in the cell body (Fig. 4), this suggests that GSK3β is gradually regulated in cells, resulting in a graded affinity of CLASPs for the MT lattice.
To establish whether the region between amino acids 875 and 1084 was directly involved in MT binding, we performed in vitro MT sedimentation assays with bacterially expressed, partially purified, histidine-tagged CLASP2 proteins. His6-CLASP2(340–1084) bound to MTs with high affinity (apparent KD = 0.1–0.25 μM; Fig. 6 A), which is similar to classic MAPs (Butner and Kirschner, 1991; Roger et al., 2004). Because we cannot exclude saturation at low MT concentrations, this value reflects an upper estimate of the true KD. In contrast, we could not detect any in vitro MT binding of His6-CLASP2(340–875) even at concentrations at which His6-CLASP2(340–1084) began to saturate available MTs (Fig. 6 B). This demonstrates that amino acids 875–1084 are required for high affinity CLASP2 binding to MT lattices.
Figure 6.

The COOH-terminal part of the CLASP-MT–binding domain is required for high affinity MT binding in vitro. (A) Immunoblot of a MT sedimentation assay of bacterially expressed His6-CLASP2(340–1084) at constant concentration incubated with increasing concentrations of MTs. Note the slightly smaller degradation product (∼70 kD), which does not bind to MTs. (B) Comparison of MT binding of His6-CLASP2(340–1084) and His6-CLASP2(340–875). 2.5 μM MTs were incubated with increasing amounts of expressed CLASP2 proteins. 1× corresponds to the concentration used in A. Gels were loaded with equivalent amounts of protein in each lane. Even at concentrations at which His6-CLASP2(340–1084) began to saturate MTs (8×), binding of His6-CLASP2(340–875) to MTs could not be detected. S, supernatant; P, pellet. Molecular masses are indicated in kilodaltons on the right.
Finally, we wanted to determine whether the most COOH-terminal GSK3β phosphorylation motif between amino acids 975 and 991 was critical to regulate CLASP-MT association. Because in cells EGFP-CLASP2(340–875) only tracked MT plus ends even in the presence of lithium chloride (Fig. 7 A), we anticipated this to be an important regulatory site (Fig. 7 B). However, lithium chloride still induced ectopic lattice binding of EGFP-CLASP2(340–956), which lacked the COOH-terminal GSK3β site (Fig. 7 A). In addition, a construct that lacked the NH2-terminal GSK3β motifs, EGFP-CLASP2(648–1362), but retained some residual MT-binding activity, did not respond to lithium chloride treatment (Fig. 7 A). To definitively test whether the regulation of lattice binding required the COOH-terminal GSK3β motif, we mutated the residues that constituted this site and were either conserved in all vertebrate CLASP sequences (S975 and S979) or only in CLASP2 homologues and zebrafish (S975, S979, T983, T987, and S991; Fig. 7 B). Both constructs tracked MT plus ends in the cell body and retained lamella MT lattice binding, demonstrating that another element between amino acids 875 and 1084 of the CLASP2 molecule is critical for the regulation of its affinity for MT lattices (Fig. 7 C). Together, these results show that the NH2-terminal part of the CLASP2 MT-binding domain (aa 512–650) is necessary for both plus end tracking in the cell body and MT lattice binding in the lamella and likely contains regulatory GSK3β phosphorylation sites, whereas the COOH-terminal part (aa 875–1084) is specifically required for high affinity binding to MT lattices in the lamella.
Figure 7.
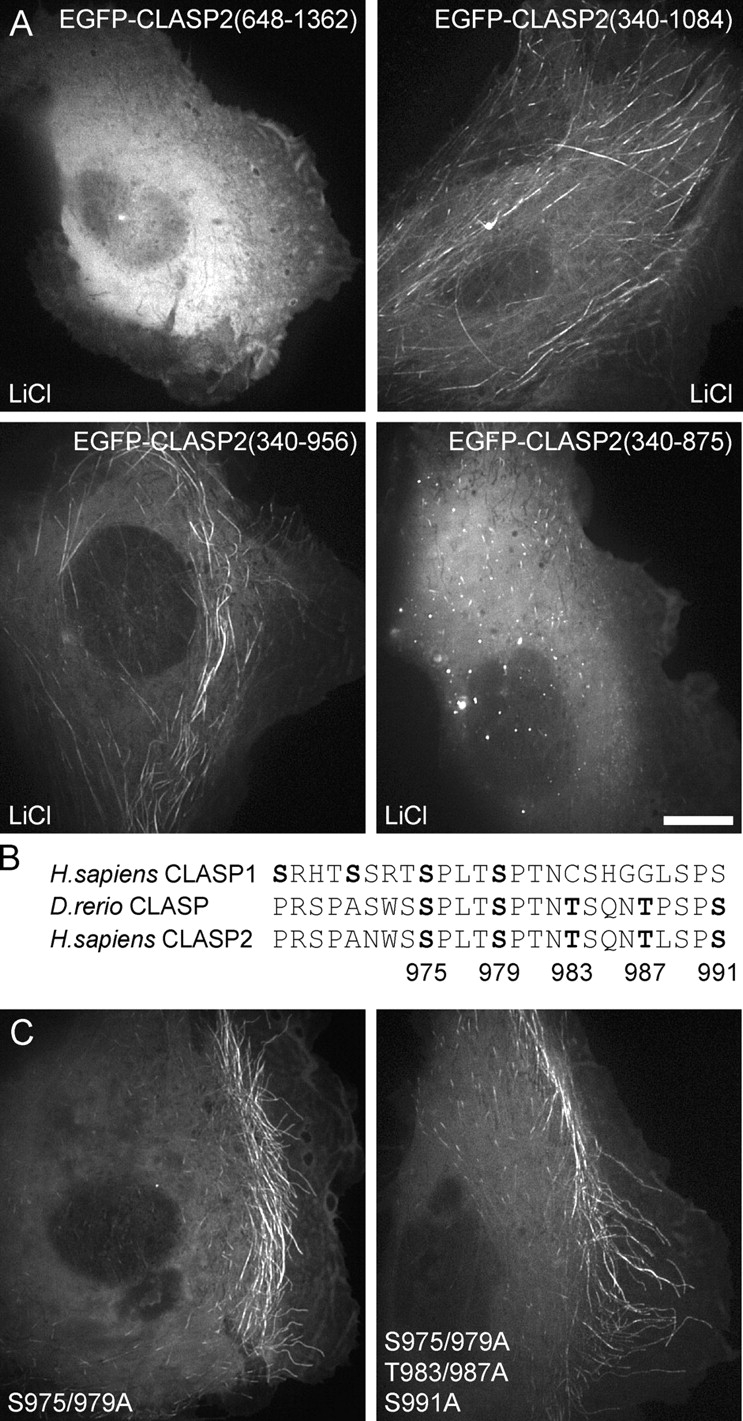
The regulation of CLASP affinity for MTs does not require the COOH-terminal GSK3β motif. (A) Localization of selected EGFP-tagged CLASP2 constructs in the presence of 20 mM lithium chloride to inhibit GSK3β. Lithium chloride still induces ectopic lattice binding of EGFP-CLASP2(340–956), which lacks the COOH-terminal GSK3β motif. (B) Protein sequence of the COOH-terminal GSK3β motif. (C) EGFP-CLASP2(340–1084) constructs, which have either the two most conserved or all five potentially GSK3β-phosphorylated residues mutated to alanine, still track MT plus ends in the cell body and bind MT lattices in the lamella. Bar, 10 μm.
Discussion
Subcellular regulation of CLASPs
In this study, we analyze the association of CLASPs with MTs in migrating PtK1 epithelial cells. At the edge of cell islands, these cells become highly polarized and migratory, extending a persistently protruding lamella and lamellipodium into their surroundings (Wittmann et al., 2003; Gupton et al., 2005). We find that in PtK1 cells the affinity of CLASPs for the MT lattice is locally regulated: in the cell body, CLASPs behave like other +TIPs and only bind near the plus ends of growing MTs. In the lamella, however, CLASPs behave like classic MAPs. They bind and dissociate along the MT lattice regardless of the polymerization state of the MT end. Other +TIPs characterized so far track growing plus ends throughout the cell. Polarized CLASP distribution has also been observed in wound edge fibroblasts (Akhmanova et al., 2001) and neuronal growth cones (Lee et al., 2004). In these cells, however, this was interpreted as increased plus end tracking of leading edge MTs. Extensive binding to the MT lattice has not been reported. Thus, stable CLASP-MT lattice binding may be specific for persistently polarized cells with stable cell–cell junctions such as our migrating epithelial cell model, and we have also observed this in human keratinocytes at the edge of cell islands (unpublished data).
To understand the mechanism of this polarization, we analyzed the regulation of CLASP-MT association downstream of Rac1 because we previously showed that Rac1 regulates lamella MT dynamic instability through p21-activated kinase and Op18/stathmin (Daub et al., 2001; Wittmann et al., 2003, 2004a). Here, we find that CLASP binding to the MT lattice is regulated through Rac1 as well. Inhibition of endogenous Rac1 reduced CLASP-MT lattice association, whereas expression of constitutively active Rac1(Q61L) enhanced binding to MT lattices throughout the cell. Thus, Rac1 is a regulator of multiple aspects of the MT cytoskeleton. Abelson tyrosine kinase might be an upstream regulator of these pathways, as it interacts genetically with the Drosophila CLASP homologue and can activate Rac1 (Lee et al., 2004; Sini et al., 2004).
We found that GSK3β, an emerging major regulator of cell polarity, also regulated CLASP-MT association. CLASPs are likely GSK3β substrates in vivo, and modulation of GSK3β activity affects CLASP plus end tracking in fibroblasts (Akhmanova et al., 2001). In the present study, we show that GSK3β inhibition induced CLASP-MT lattice association, which is consistent with low GSK3β activity in the lamella. In contrast, constitutively active GSK3β(S9A) completely abolished lattice binding, which is consistent with high GSK3β activity in the cell body. In the leading edge of astrocytes, GSK3β is phosphorylated and inactivated downstream of Cdc42 and Par6-PKCζ (Etienne-Manneville and Hall, 2003), and inactive, phospho-GSK3β is enriched in neuronal growth cones (Shi et al., 2004; Zhou et al., 2004). Unfortunately, we did not succeed in staining phosphorylated GSK3β in PtK1 cells and, thus, could not test this hypothesis directly. Our FRAP experiments indicate that GSK3β inhibition increased the association of CLASP with MT lattices compared with lamella MT lattice-bound CLASP in control cells, and constitutively active GSK3β(S9A) reduced CLASP plus end tracking below the control cell level. In addition, CLASP comets on cell body MT plus ends often grew longer as they approached the border between cell body and lamella. Together, this suggests a finely tuned regulatory gradient of GSK3β activity between these cell regions that results in a graded affinity of CLASPs for plus ends versus lattices. Although CLASPs contain multiple potential GSK3β phosphorylation sites, it is possible that CLASP regulation is not direct and instead requires an intermediate protein that is phosphorylated by GSK3β.
We do not know the molecular link between Rac1 and GSK3β, but our data suggest that CLASP-MT lattice binding through GSK3β inactivation occurs downstream of Rac1 activation. Although Cdc42-mediated GSK3β phosphorylation through direct interactions of GSK3β with Par6-PKCζ independent of Rac1 has been observed (Etienne-Manneville and Hall, 2003), the molecular mechanism of GSK3β inactivation is not completely understood. In PtK1 cells, PKC inhibition by GF109203X, which would lead to increased GSK3β activity by such a pathway, resulted in increased CLASP-MT lattice association (unpublished data), in opposition to what one would expect. In addition, a recent study shows that Rac1 activation downstream of Cdc42 is mediated by direct interaction of the Rac1-GEF Tiam with Par3 (Nishimura et al., 2005). Thus, both Rac1 and GSK3β are regulated downstream of the Par3-Par6-PKCζ polarity complex, and Rac1 and PKCζ likely act in parallel to control GSK3β activity and CLASP localization.
GSK3β also phosphorylates neuronal MAPs (Trivedi et al., 2005) and APC (Zumbrunn et al., 2001; Etienne-Manneville and Hall, 2003; Zhou et al., 2004), and there are interesting parallels between APC and CLASPs. Similar to CLASPs, GSK3β activation decreases and GSK3β inhibition increases APC-MT association. In cells, however, the localization and dynamics of APC and CLASPs are quite different. CLASPs associate with the lattice of all lamella MTs, whereas APC accumulates in dynamic clusters at the ends of only a small subset of MTs. This implies differences in the regulation of APC and CLASPs through GSK3β. APC and GSK3β are components of the WNT-signaling complex (Bienz, 2002) and APC is transported along MTs by a kinesin motor (Mimori-Kiyosue et al., 2000; Jimbo et al., 2002), which could explain these differences. It is not known whether CLASPs are part of a larger signaling complex, and we did not observe CLASP transport along MTs.
A molecular switch to regulate CLASP-MT affinity
To elucidate whether plus end tracking and MT lattice binding were distinct activities, we analyzed truncated CLASP constructs in PtK1 cells. Neither of these activities required the COOH-terminal CLIP-170–binding domain (Akhmanova et al., 2001). The NH2-terminal part of the CLASP-MT–binding domain (aa 512–650) was sufficient for plus end tracking in PtK1 cells, similar to what has been observed in HeLa cells (Mimori-Kiyosue et al., 2005). In addition, we identified a region at the COOH terminus of the MT-binding domain (aa 875–1084) that was indispensable for MT lattice binding in the lamella. Thus, the CLASP-MT–binding domain consists of subdomains with distinct activities. However, the functional relationship of these subdomains is complicated, as the NH2-terminal plus end tracking domain was also necessary for lamella MT lattice binding in cells, indicating that it is involved in direct CLASP-MT interactions. Indeed, some MT binding has been observed with the minimal plus end tracking construct in vitro (aa 512–650; Mimori-Kiyosue et al., 2005). In our experiments, however, the presence of the COOH-terminal subdomain (aa 875–1084) was necessary to bind taxol-stabilized MTs with high affinity in vitro. We did not observe any in vitro MT binding of a construct terminating at residue 875. Consequently, the NH2-terminal plus end tracking domain alone has a very low affinity for MT lattices. Because taxol-stabilized MTs do not have dynamic ends, the affinity of this domain for structurally distinct growing plus ends may be considerably higher. Together, these data indicate that the subcellular regulation of CLASP-MT association is likely a consequence of direct regulation of the affinity of CLASPs for MT lattices.
To switch between plus end tracking and MT lattice binding the COOH-terminal subdomain may contain a second MT-binding site that is required for high affinity lattice binding. Alternatively, it may instead be involved in regulating MT affinity of the NH2-terminal plus end tracking domain. Intramolecular interactions may regulate CLASP-MT binding, as it has been observed that the yeast CLASP homologue Stu1 can dimerize (Yin et al., 2002). We also find that the most COOH-terminal GSK3β phosphorylation motif was not involved in the regulation of CLASP-MT association in cells. Therefore, regulatory phosphorylation through GSK3β likely occurs on NH2-terminal phosphorylation sites, which overlap with the plus end tracking and EB1-binding domain (Mimori-Kiyosue et al., 2005). Thus, interactions with other +TIPs may also regulate CLASP association with MTs. However, because CLASP and EB1-staining showed little overlap on the plus ends of cell body MTs, such a +TIP complex has to be spatiotemporally regulated.
Role of CLASP-MT association in migrating cells
What is the functional significance of CLASP association with lamella MT lattices? In mitotic cells, CLASPs regulate the dynamics of kinetochore MTs (Maiato et al., 2003, 2005). Thus, CLASPs likely regulate MT dynamic instability also in polarized, migrating interphase cells. In HeLa cells, knockdown of CLASP1 and CLASP2 showed that CLASPs are important to promote rescues and to keep dynamic MT ends in close proximity to the cell edge (Mimori-Kiyosue et al., 2005). CLASP knockdown does not affect MT growth rates, consistent with our findings that growth rates were not regulated through CLASP-MT lattice association or Rac1 activation (Wittmann et al., 2003). However, in PtK1 cells, the persistent growth of pioneer MTs into the lamella is mainly regulated through a decreased catastrophe frequency downstream of Rac1 activation (Wittmann et al., 2003). Unlike PtK1 cells, HeLa cells do not possess an extensive lamella and effects of the cell boundary on MT dynamics may be more pronounced, possibly explaining these differences. In any case, it is easy to envisage how CLASP association with the MT lattice could affect transition frequencies and CLASP-MT lattice binding could stabilize MTs by increasing their growth persistence time in the lamella. Although CLASPs may mainly regulate the rescue frequency, regulation of the catastrophe frequency involves inactivation of Op18/stathmin through p21-activated kinases. Ultimately, the regulation of lamella MT dynamics downstream of Rac1 likely involves multiple pathways.
Materials and methods
Constructs and antibodies
pEGFP-CLASP1 and -CLASP2 were obtained from N. Galjart (Erasmus University, Rotterdam, Netherlands; Akhmanova et al., 2001). The CLASP2 sequence in this plasmid corresponds to aa 78–1362 of the predicted full-length human CLASP2 sequence (GenBank/EMBL/DDBJ accession no. XP_291057.5). CLASP2 tagged with multiple EGFPs was made by cloning an AgeI–MluI fragment of pEGFP-CLASP2 into XmaI–MluI-cut pEGFP-C1 (CLONTECH Laboratories, Inc.). CLASP2 deletion constructs were made by either subcloning fragments from internal EcoRI, SalI, XhoI, or BamHI sites or by PCR with Vent polymerase (New England Biolabs, Inc.). For bacterial expression, PCR products were cloned into pHAT2 (Peränen et al., 1996), expressed, and purified on TALON resin (CLONTECH Laboratories, Inc.) using standard protocols. Point mutations were generated with Quickchange II (Stratagene). Rac1(Q61L) and GSK3β (S9A) were subcloned into a pTriEx-4-mRFP-fusion vector (obtained from O. Pertz, The Scripps Research Institute, La Jolla, CA; mRFP was obtained from R. Tsien, University of California, San Diego, San Diego, CA; Campbell et al., 2002) to be able to monitor expression in living cells. pCB6GFP-CLIP-170 was obtained from H. Goodson (University of Notre Dame, Notre Dame, IN; Perez et al., 1999), pEGFP-APC from M. Bienz (University of Cambridge, Cambridge, UK; Rosin-Arbesfeld et al., 2001), and pEGFP-EB1 from L. Cassimeris (Lehigh University, Bethlehem, PA; Piehl and Cassimeris, 2003). The TAT-Rac1(17–32) peptide was obtained from P. Hordijk (University of Amsterdam, Amsterdam, Netherlands; van Hennik et al., 2003).
Affinity-purified rabbit polyclonal antibody against the Xenopus CLASP homologue Xorbit was obtained from E. Hannak and R. Heald (University of California, Berkeley, Berkeley, CA). Monoclonal EB1 antibody was purchased from BD Biosciences. For immunofluorescence with these antibodies, cells were fixed with −20°C methanol. Rabbit antiserum against detyrosinated tubulin was obtained from C. Bulinski (Columbia University, New York, NY).
Microtubule sedimentation assay
Taxol-stabilized microtubules were assembled as described previously (Wittmann et al., 1998), mixed with purified CLASP2 proteins in 80 mM K-Pipes, 1 mM EGTA, and 1 mM MgCl2. After 15-min incubation at RT, the microtubule-bound fraction was separated by centrifugation at 60,000 g. Equivalent amounts of supernatant and pellet were analyzed by immunoblot.
Microscopy and image analysis
PtK1 cells were cultured for microscopy and microinjected as described previously (Wittmann et al., 2003, 2004b). Time-lapse image series were acquired with a 100×/1.4 NA Plan Apo objective lens on a microscope (model TE-300; Nikon) equipped with a spinning disk confocal scan head (Ultra View; Yokogawa) and a cooled charged coupled device camera (model Orca II; Hamamatsu) as described previously (Adams et al., 2003). FRAP experiments were performed on a microscope (model DeltaVision RT; Applied Precision) equipped with a 488-nm laser module for photobleaching. Fluorescence recovery rate constants were obtained by least square fitting of the data to an exponential recovery model (Bulinski et al., 2001). All image processing was done with MetaMorph (Universal Imaging Corp.). For figures and supplemental videos, images were convolved with a 3 × 3 Gaussian low pass kernel to reduce camera noise and an unsharp mask (7 × 7 kernel size, scaling factor 0.5) to enhance the contrast of dim image features.
Online supplemental material
Fig. S1 shows the specificity of the CLASP antibody, analysis of MT growth rates in control and Rac1(Q61L)-expressing cells, in vitro CLASP phosphorylation by GSK3β, that GSK3β inhibition reduces the CLASP phosphorylation state in cells, and that GSK3β inhibition induces ectopic MT lattice binding in EGFP-CLASP1–expressing cells. Video 1 shows that 3xEGFP-CLASP2 in a PtK1 cell reveals MT plus end tracking in the cell body and dynamic lattice binding in the lamella. Video 2 shows 3xEGFP-CLASP2 and X-rhodamine-labeled tubulin. Video 3 shows a comparison of CLASP2, EB1, CLIP-170, and APC dynamics in PtK1 cells. Video 4 shows FRAP of EGFP-CLASP2 on lamella MT lattices and growing plus ends in the cell body. Video 5 shows 3xEGFP-CLASP2 in a PtK1 cell expressing constitutively active mRFP-Rac1(Q61L). Video 6 shows 3xEGFP-CLASP2 in a PtK1 cell treated with the Rac1 inhibitory peptide TAT-Rac1(17–32). Video 7 shows 3xEGFP-CLASP2 in a PtK1 cell expressing constitutively active mRFP-GSK3β(S9A). Video 8 shows 3xEGFP-CLASP2 in the presence of 20 mM lithium chloride. Video 9 shows that 3xEGFP-CLASP2(78–875) only tracks growing plus ends. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200412114/DC1.
Acknowledgments
We thank Niels Galjart, Peter Hordijk, Chloë Bulinski, Holly Goodson, Mariann Bienz, Lynne Cassimeris, Roger Tsien, Olivier Pertz, Eva Hannak, and Rebecca Heald for kind gifts of reagents and Applied Precision for letting us play with their new DeltaVision RT microscope.
This work was supported by National Institutes of Health grant GM61804 to C.M. Waterman-Storer.
Abbreviations used in this paper: APC, adenomatous polyposis coli protein; GSK3β, glycogen synthase kinase 3β; MAP; microtubule-associated protein; MT, microtubule.
References
- Adams, M.C., W.C. Salmon, S.L. Gupton, C.S. Cohan, T. Wittmann, N. Prigozhina, and C.M. Waterman-Storer. 2003. A high-speed multispectral spinning-disk confocal microscope system for fluorescent speckle microscopy of living cells. Methods. 29:29–41. [DOI] [PubMed] [Google Scholar]
- Akhmanova, A., C.C. Hoogenraad, K. Drabek, T. Stepanova, B. Dortland, T. Verkerk, W. Vermeulen, B.M. Burgering, C.I. De Zeeuw, F. Grosveld, and N. Galjart. 2001. Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell. 104:923–935. [DOI] [PubMed] [Google Scholar]
- Andersen, S.S. 2005. The search and prime hypothesis for growth cone turning. Bioessays. 27:86–90. [DOI] [PubMed] [Google Scholar]
- Arnal, I., E. Karsenti, and A.A. Hyman. 2000. Structural transitions at microtubule ends correlate with their dynamic properties in Xenopus egg extracts. J. Cell Biol. 149:767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz, M. 2002. The subcellular destinations of APC proteins. Nat. Rev. Mol. Cell Biol. 3:328–338. [DOI] [PubMed] [Google Scholar]
- Bulinski, J.C., D.J. Odde, B.J. Howell, E.D. Salmon, and C.M. Waterman-Storer. 2001. Rapid dynamics of the microtubule binding of ensconsin in vivo. J. Cell Sci. 114:3885–3897. [DOI] [PubMed] [Google Scholar]
- Butner, K.A., and M.W. Kirschner. 1991. Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 115:717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, R.E., O. Tour, A.E. Palmer, P.A. Steinbach, G.S. Baird, D.A. Zacharias, and R.Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 99:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, P., and S. Frame. 2001. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2:769–776. [DOI] [PubMed] [Google Scholar]
- Daub, H., K. Gevaert, J. Vandekerckhove, A. Sobel, and A. Hall. 2001. Rac/Cdc42 and p65PAK regulate the microtubule-destabilizing protein stathmin through phosphorylation at serine 16. J. Biol. Chem. 276:1677–1680. [DOI] [PubMed] [Google Scholar]
- Diamantopoulos, G.S., F. Perez, H.V. Goodson, G. Batelier, R. Melki, T.E. Kreis, and J.E. Rickard. 1999. Dynamic localization of CLIP-170 to microtubule plus ends is coupled to microtubule assembly. J. Cell Biol. 144:99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2002. Rho GTPases in cell biology. Nature. 420:629–635. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2003. Cdc42 regulates GSK-3β and adenomatous polyposis coli to control cell polarity. Nature. 421:753–756. [DOI] [PubMed] [Google Scholar]
- Fukata, M., T. Watanabe, J. Noritake, M. Nakagawa, M. Yamaga, S. Kuroda, Y. Matsuura, A. Iwamatsu, F. Perez, and K. Kaibuchi. 2002. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell. 109:873–885. [DOI] [PubMed] [Google Scholar]
- Galjart, N., and F. Perez. 2003. A plus-end raft to control microtubule dynamics and function. Curr. Opin. Cell Biol. 15:48–53. [DOI] [PubMed] [Google Scholar]
- Gundersen, G.G., and J.C. Bulinski. 1988. Selective stabilization of microtubules oriented toward the direction of cell migration. Proc. Natl. Acad. Sci. USA. 85:5946–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton, S.L., K.L. Anderson, T.P. Kole, R.S. Fischer, A. Ponti, S.E. Hitchcock-Degregori, G. Danuser, V.M. Fowler, D. Wirtz, D. Hanein, and C.M. Waterman-Storer. 2005. Cell migration without a lamellipodium: translation of actin dynamics into cell movement mediated by tropomyosin. J. Cell Biol. 168:619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimbo, T., Y. Kawasaki, R. Koyama, R. Sato, S. Takada, K. Haraguchi, and T. Akiyama. 2002. Identification of a link between the tumour suppressor APC and the kinesin superfamily. Nat. Cell Biol. 4:323–327. [DOI] [PubMed] [Google Scholar]
- Komarova, Y.A., A.S. Akhmanova, S. Kojima, N. Galjart, and G.G. Borisy. 2002. a. Cytoplasmic linker proteins promote microtubule rescue in vivo. J. Cell Biol. 159:589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova, Y.A., I.A. Vorobjev, and G.G. Borisy. 2002. b. Life cycle of MTs: persistent growth in the cell interior, asymmetric transition frequencies and effects of the cell boundary. J. Cell Sci. 115:3527–3539. [DOI] [PubMed] [Google Scholar]
- Lee, H., U. Engel, J. Rusch, S. Scherrer, K. Sheard, and D. Van Vactor. 2004. The microtubule plus end tracking protein orbit/MAST/CLASP acts downstream of the tyrosine kinase Abl in mediating axon guidance. Neuron. 42:913–926. [DOI] [PubMed] [Google Scholar]
- Maiato, H., E.A. Fairley, C.L. Rieder, J.R. Swedlow, C.E. Sunkel, and W.C. Earnshaw. 2003. Human CLASP1 is an outer kinetochore component that regulates spindle microtubule dynamics. Cell. 113:891–904. [DOI] [PubMed] [Google Scholar]
- Maiato, H., A. Khodjakov, and C.L. Rieder. 2005. Drosophila CLASP is required for the incorporation of microtubule subunits into fluxing kinetochore fibres. Nat. Cell Biol. 7:42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori-Kiyosue, Y., and S. Tsukita. 2003. “Search-and-capture” of microtubules through plus-end-binding proteins (+TIPs). J. Biochem. (Tokyo). 134:321–326. [DOI] [PubMed] [Google Scholar]
- Mimori-Kiyosue, Y., N. Shiina, and S. Tsukita. 2000. Adenomatous polyposis coli (APC) protein moves along microtubules and concentrates at their growing ends in epithelial cells. J. Cell Biol. 148:505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori-Kiyosue, Y., I. Grigoriev, G. Lansbergen, H. Sasaki, C. Matsui, F. Severin, N. Galjart, F. Grosveld, I. Vorobjev, S. Tsukita, and A. Akhmanova. 2005. CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J. Cell Biol. 168:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, T., T. Yamaguchi, K. Kato, M. Yoshizawa, Y. Nabeshima, S. Ohno, M. Hoshino, and K. Kaibuchi. 2005. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat. Cell Biol. 7:270–277. [DOI] [PubMed] [Google Scholar]
- Palazzo, A.F., C.H. Eng, D.D. Schlaepfer, E.E. Marcantonio, and G.G. Gundersen. 2004. Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science. 303:836–839. [DOI] [PubMed] [Google Scholar]
- Peränen, J., M. Rikkonen, M. Hyvönen, and L. Kääriäinen. 1996. T7 vectors with modified T7lac promoter for expression of proteins in Escherichia coli. Anal. Biochem. 236:371–373. [DOI] [PubMed] [Google Scholar]
- Perez, F., G.S. Diamantopoulos, R. Stalder, and T.E. Kreis. 1999. CLIP-170 highlights growing microtubule ends in vivo. Cell. 96:517–527. [DOI] [PubMed] [Google Scholar]
- Piehl, M., and L. Cassimeris. 2003. Organization and dynamics of growing microtubule plus ends during early mitosis. Mol. Biol. Cell. 14:916–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger, B., J. Al Bassam, L. Dehmelt, R.A. Milligan, and S. Halpain. 2004. MAP2c, but not tau, binds and bundles F-actin via its microtubule binding domain. Curr. Biol. 14:363–371. [DOI] [PubMed] [Google Scholar]
- Rogers, S.L., G.C. Rogers, D.J. Sharp, and R.D. Vale. 2002. Drosophila EB1 is important for proper assembly, dynamics, and positioning of the mitotic spindle. J. Cell Biol. 158:873–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, S.L., U. Wiedemann, U. Hacker, C. Turck, and R.D. Vale. 2004. Drosophila RhoGEF2 associates with microtubule plus ends in an EB1-dependent manner. Curr. Biol. 14:1827–1833. [DOI] [PubMed] [Google Scholar]
- Rosin-Arbesfeld, R., G. Ihrke, and M. Bienz. 2001. Actin-dependent membrane association of the APC tumour suppressor in polarized mammalian epithelial cells. EMBO J. 20:5929–5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, S.H., T. Cheng, L.Y. Jan, and Y.N. Jan. 2004. APC and GSK-3β are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr. Biol. 14:2025–2032. [DOI] [PubMed] [Google Scholar]
- Sini, P., A. Cannas, A.J. Koleske, P.P. Di Fiore, and G. Scita. 2004. Abl-dependent tyrosine phosphorylation of Sos-1 mediates growth-factor-induced Rac activation. Nat. Cell Biol. 6:268–274. [DOI] [PubMed] [Google Scholar]
- Tirnauer, J.S., S. Grego, E.D. Salmon, and T.J. Mitchison. 2002. EB1-microtubule interactions in Xenopus egg extracts: role of EB1 in microtubule stabilization and mechanisms of targeting to microtubules. Mol. Biol. Cell. 13:3614–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi, N., P. Marsh, R.G. Goold, A. Wood-Kaczmar, and P.R. Gordon-Weeks. 2005. Glycogen synthase kinase-3β phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J. Cell Sci. 118:993–1005. [DOI] [PubMed] [Google Scholar]
- van Hennik, P.B., J.P. ten Klooster, J.R. Halstead, C. Voermans, E.C. Anthony, N. Divecha, and P.L. Hordijk. 2003. The C-terminal domain of Rac1 contains two motifs that control targeting and signaling specificity. J. Biol. Chem. 278:39166–39175. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer, C.M., and E.D. Salmon. 1997. Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J. Cell Biol. 139:417–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann, T., and C.M. Waterman-Storer. 2001. Cell motility: can Rho GTPases and microtubules point the way? J. Cell Sci. 114:3795–3803. [DOI] [PubMed] [Google Scholar]
- Wittmann, T., H. Boleti, C. Antony, E. Karsenti, and I. Vernos. 1998. Localization of the kinesin-like protein Xklp2 to spindle poles requires a leucine zipper, a microtubule-associated protein, and dynein. J. Cell Biol. 143:673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann, T., G.M. Bokoch, and C.M. Waterman-Storer. 2003. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J. Cell Biol. 161:845–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann, T., G.M. Bokoch, and C.M. Waterman-Storer. 2004. a. Regulation of microtubule destabilizing activity of Op18/stathmin downstream of Rac1. J. Biol. Chem. 279:6196–6203. [DOI] [PubMed] [Google Scholar]
- Wittmann, T., R. Littlefield, and C.M. Waterman-Storer. 2004b. Fluorescent speckle microscopy of cytoskeletal dynamics in living cells. Live Cell Imaging: A Laboratory Manual. D.L. Spector and R.D. Goldman, editors. Cold Spring Harbor Press, New York. 187–204.
- Yin, H., L. You, D. Pasqualone, K.M. Kopski, and T.C. Huffaker. 2002. Stu1p is physically associated with β-tubulin and is required for structural integrity of the mitotic spindle. Mol. Biol. Cell. 13:1881–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, F.Q., J. Zhou, S. Dedhar, Y.H. Wu, and W.D. Snider. 2004. NGF-induced axon growth is mediated by localized inactivation of GSK-3β and functions of the microtubule plus end binding protein APC. Neuron. 42:897–912. [DOI] [PubMed] [Google Scholar]
- Zumbrunn, J., K. Kinoshita, A.A. Hyman, and I.S. Nathke. 2001. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3β phosphorylation. Curr. Biol. 11:44–49. [DOI] [PubMed] [Google Scholar]