

Abstract
The stress-activated protein kinase p38 and nitric oxide (NO) are proposed downstream effectors of excitotoxic cell death. Although the postsynaptic density protein PSD95 can recruit the calcium-dependent neuronal NO synthase (nNOS) to the mouth of the calcium-permeable NMDA receptor, and depletion of PSD95 inhibits excitotoxicity, the possibility that selective uncoupling of nNOS from PSD95 might be neuroprotective is unexplored. The relationship between excitotoxic stress–generated NO and activation of p38, and the significance of the PSD95–nNOS interaction to p38 activation also remain unclear. We find that NOS inhibitors reduce both glutamate-induced p38 activation and the resulting neuronal death, whereas NO donor has effects consistent with NO as an upstream regulator of p38 in glutamate-induced cell death. Experiments using a panel of decoy constructs targeting the PSD95–nNOS interaction suggest that this interaction and subsequent NO production are critical for glutamate-induced p38 activation and the ensuing cell death, and demonstrate that the PSD95–nNOS interface provides a genuine possibility for design of neuroprotective drugs with increased selectivity.
Introduction
Glutamate is an essential mediator of excitotoxicity, which is a form of neuronal death that can occur in a variety of brain regions subsequent to ischemic insult or other neurodegenerative conditions. Evidence from knockout mice and other models demonstrates the contributions of neuronal nitric oxide synthase (nNOS) and nitric oxide (NO) to glutamate-induced neuronal death (Huang et al., 1994; Dawson et al., 1996). The stress-activated protein kinase p38 is activated within minutes of glutamate receptor activation, and contributes to glutamate-induced neurotoxicity (Kawasaki et al., 1997; Cao et al., 2004). However, the relationship between NO production and p38 in cell death is unclear, as only delayed p38 activation has been observed upon application of NO donors to neuronal cells (Lin et al., 2001; Bossy-Wetzel et al., 2004). The postsynaptic density protein PSD95 tethers calcium-dependent nNOS to the mouths of NMDA receptor channels; this selective colocalization is believed to underlie the source specificity hypothesis, which states that calcium influx through NMDA receptors is especially neurotoxic (Aarts and Tymianski, 2003). Thus, complete ablation of PSD95 with antisense and dissociation of the entire PSD95 molecule from the NMDA receptor with PDZ1–2 decoy constructs are neuroprotective in ischemia models (Sattler et al., 1999; Aarts et al., 2002). Although these results are encouraging, PSD95 is known to link a large number of molecules to the NMDA receptor via its various domains; therefore, PSD95 dissociation/ablation will disrupt additional functions of the molecule. This disruption may be manifested as side effects. Indeed, it is unclear which of PSD95's functions is significant for the neuroprotection in these reports. The manner in which PSD95 mediates interaction of NMDA receptors with nNOS is partly understood. The PDZ1 domain of PSD95 can interact with the COOH terminus of the NMDA receptor, while PDZ2 is free to bind the NH2-terminal region of nNOS (Niethammer et al., 1996; Christopherson et al., 1999). Both the nNOS PDZ domain and the adjacent β finger sequence are implicated in this interaction (Brenman et al., 1996a; Christopherson et al., 1999; Tochio et al., 2000a).
The possible protective value of the more selective approach, targeting the PSD95–nNOS interaction itself, has yet to be examined. In this paper, we initially establish that glutamate-induced p38 activation and the resulting death of cerebellar granule neurons involve NO. Thus, nNOS inhibitors prevent the rapid glutamate-induced p38 activation and p38-dependent death. The p38 activation is transient and rapidly followed by pyknosis. Consistent with this, neuroprotection by p38 inhibitor is obtained only when the inhibitor is added before, and not after, the peak of p38 activation. Consistent with a role for NO in glutamate-induced cell death, p38 activation and pyknosis induced by NO donors are as rapid as when they are induced by glutamate.
Subsequently, we developed a decoy construct based on nNOS that we could show binds to the PDZ2 domain of PSD95. This construct prevented p38 activation and neuronal death induced by glutamate, but not those induced by NO donor. This suggests that the decoy construct indeed prevents p38 activation and pyknosis upstream of NO synthesis. Similarly, expression of the free PSD95–PDZ2 domain, which we demonstrate interacts with the NH2 terminus of nNOS, also inhibits pyknosis. We conclude that development of competitor sequences selectively disrupting only the PSD95–nNOS interface may have value as a neuroprotective strategy in excitotoxicity.
Results
NO contributes to excitotoxic neuronal cell death (Huang et al., 1994; Dawson et al., 1996), which can result in neuronal deficits in a variety of brain regions after stroke or the development other neurodegenerative conditions. Recent evidence suggests an essential role for the stress-activated protein kinase p38 in excitotoxic neuronal cell death in cultured cerebellar neurons (Kawasaki et al., 1997; Cao et al., 2004) and forebrain and hippocampal neurons (Legos et al., 2002), and in vivo in retinal neurons (Manabe and Lipton, 2003) and cerebral ischemia (Legos et al., 2001). NO species are known to activate p38 in nonneuronal cells, but this reportedly depends on a TAB1-mediated p38 autophosphorylation mechanism (Ge et al., 2002) that is not required for glutamate-evoked neuronal p38 activation (unpublished data). Information on the effect of NO on neuronal p38 is limited—in cerebellar granule neurons, an activation of p38 after a 3-h incubation with NO donor has been reported (Lin et al., 2001). Because glutamate activates p38 within 2 min and apoptosis is complete within 3 h in these cells (Cao et al., 2004), the significance of this result has been unclear. To consider the possible relationship between NO and p38, we first investigated in more detail the time course of p38 activation and its requirement for the cell death process. Addition of glutamate to cerebellar granule neuron cultures leads to a transient but strong activation of p38 at 5 min, with little elevation above basal level at 30 min and no detectable increase for the next 3 h (Fig. 1 A). Pyknosis is an early indicator of this form of cell death, preceding loss of membrane integrity by several hours (Cao et al., 2004). Cell death, assessed by pyknosis, is already detectable 60 min after glutamate addition and is complete within 3 h (Fig. 1 B). SB203580, at a concentration that selectively inhibits p38 in intact cerebellar granule neurons (Coffey et al., 2000, 2002; Cao et al., 2004), is known to prevent this pyknosis. Adding the inhibitor 30 min before the addition of glutamate strongly prevents the pyknosis, but if the inhibitor is added 30 min after glutamate exposure, the pyknosis is indistinguishable from that in controls (Fig. 1 C). These data suggest that the early transient increase of p38 activity is important for glutamate-induced pyknosis.
Figure 1.
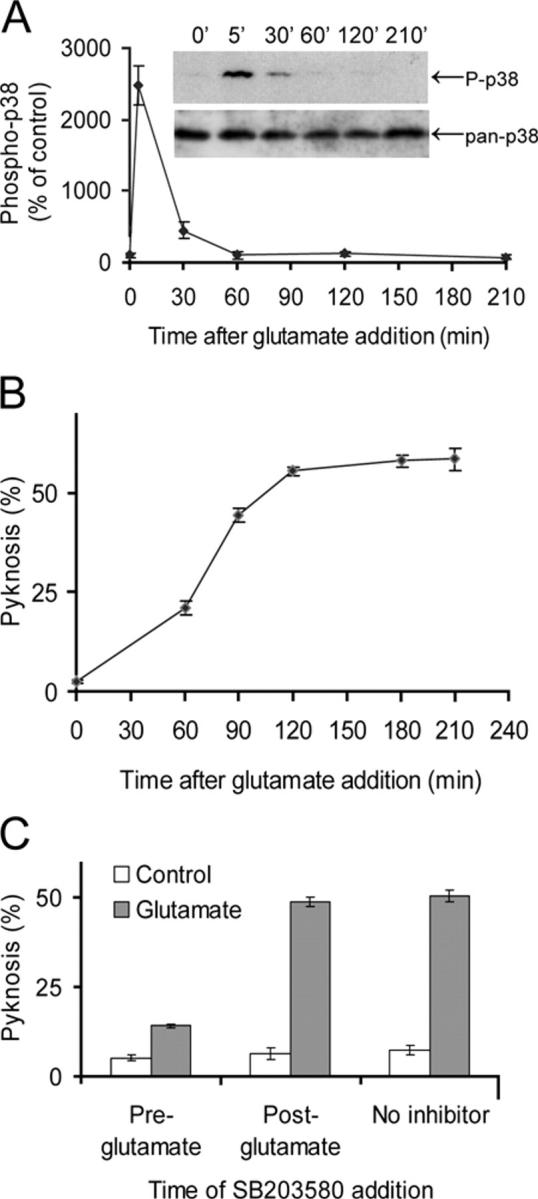
Glutamate-induced neuronal death requires the early phase of p38 activation. (A) Phospho-p38 levels in cerebellar granule neuron extracts prepared at times indicated after exposure to 50 μM glutamate. Phosphorylated p38 levels increase rapidly and fall to basal levels from 60 min after glutamate exposure. The pan-p38 blot indicates equal loading of samples. Means ± range are shown (n = 2). (B) Pyknosis of cerebellar granule neurons was assessed at times indicated after the start of a 30-min glutamate exposure. The pyknosis is complete 2–3 h after exposure to 50 μM glutamate. Means ± SEM are shown (n = 3). (C) Pyknosis was assessed 3 h after a 30-min glutamate exposure, in the presence of 1 μM SB203580, to specifically inhibit p38, added either 30 min before the start or immediately after the end of the 30-min glutamate exposure. Only pretreatment with inhibitor prevented the pyknosis. Means ± SEM are shown (n = 3).
We subsequently examined the effect of nNOS inhibitors on p38 activation 5 min after glutamate addition, when phospho-p38 levels are at their highest. 7-Nitroindazole, which does not discriminate between NOS isoforms (with selectivity ratios of 0.9–1.4-fold; for review see Alderton et al., 2001), and N-ω-propyl-l-arginine, which selectively targets nNOS (Zhang et al., 1997), both substantially and significantly reduced the glutamate-induced rapid increase in p38 activation loop phosphorylation observed at 5 min (Fig. 2, A and B). After this, the glutamate-induced pyknosis was also significantly diminished, as expected (Fig. 2, C and D). Both of these inhibitors are competitive with arginine, and thus the effects of the inhibitors are completely reversed by the presence of excess arginine in the culture medium (Fig. 2).
Figure 2.

Inhibitors of NOS and nNOS reduce glutamate-induced p38 activation and pyknosis. (A) Immunoblot revealing phospho-p38 levels in cerebellar granule neuron extracts prepared at times indicated, in the presence of carrier (DMSO) or the pan-NOS– and nNOS-specific inhibitors 7-Nitroindazole (3 μM; 7-NI) and N-ω-propyl-l-arginine (1 μM; Nw-PLA), respectively. These inhibitors are competitive with arginine, thus arginine was either absent, to allow the inhibitors to block the enzyme, or present, to prevent them from blocking it. Glutamate induces rapid p38 activation; this response is reduced by pan-NOS– and nNOS-specific inhibitors, but only when arginine is absent. The pan-p38 blot indicates equal loading of samples. (B) Replicates of data as in A were normalized and means ± SEM are shown (n = 3). Asterisks indicate significant difference of inhibitor-treated samples from control by paired t test (P < 0.002, or better, in all cases). (C) Neurons were treated with 50 μM glutamate in the presence or absence of NOS inhibitors and in the absence or presence of arginine, as shown (conditions with arginine and inhibitors, but without glutamate, were not tested). Cells were fixed and DNA stained with Hoechst 33342, and the inhibitors reduced glutamate-induced pyknosis. (D) Replicates of data as in C are shown as means ± SEM (n = 6). Asterisk indicates significant difference of inhibitor treated samples from control by paired t test (P < 0.002, or better, in all cases).
If NO species are genuinely involved in glutamate-induced p38 activation, then NO should, like glutamate, activate p38 within minutes of addition. Therefore, an NO donor was added to cerebellar granule neuron cultures and immunoblotting with phospho-p38 antibody was performed on cell lysates prepared at times after addition of NO donor, as shown (Fig. 3). We used the NONOate diethylamine/NO adduct (Dea/NO), which degrades with a t 1/2 of 2.1 min (at 37°C), to release NO. Dea/NO at 250 or 10 μM induced a substantial increase in phospho-p38 level (Fig. 3, A [top and middle] and B). The lower concentration is only threefold greater than the amount used in a recent study to supply a physiologically relevant amount of NO sufficient to enhance long-term potentiation without effects on basal synaptic transmission (Bon and Garthwaite, 2003). The activation of p38 by Dea/NO appears to occur by a direct effect on the neurons and not by indirect stimulation of glutamate release, because the NMDA receptor antagonist MK801 does not prevent it (unpublished data). It has been suggested that peroxynitrite (ONOO−) may mediate the neurotoxic actions of NO. Therefore, we also tested this, at a concentration reported to activate p38 in 293 cells (Ge et al., 2002); once again, a rapid p38 activation loop phosphorylation was detected (Fig. 3, A [bottom] and B). If concentrations of NO donor sufficient to activate p38 are of relevance to p38-mediated death by glutamate, then the donors should induce rapid pyknosis in a manner similar to that of glutamate. Pyknosis of cells was measured between 30 and 180 min after addition of donor, revealing that Dea/NO indeed induces a rapid pyknosis dependent on the amount of donor added (Fig. 3 C). The pan-caspase inhibitor zVAD-fmk failed to inhibit p38-dependent pyknosis induced by glutamate (Cao et al., 2004). Pyknosis induced by NO and ONOO− is similar in that zVAD-fmk fails to prevent it as well (unpublished data).
Figure 3.
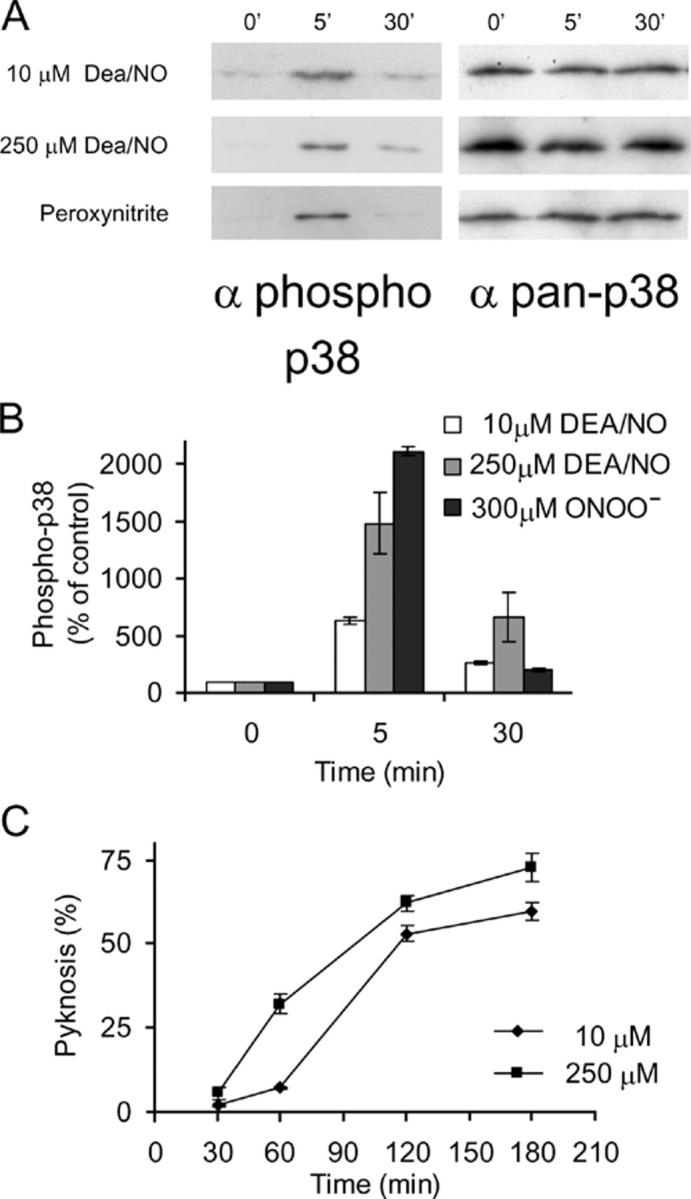
NO species activate neuronal p38 and induce rapid pyknosis. (A) Application of NO donors, as shown, to neurons increases levels of phospho-p38, with a time course similar to that induced by glutamate. The pan-p38 blot indicates equal loading of samples. (B) Replicates of the data in A were quantified and means ± SEM are shown (n = 3–4). (C) Pyknosis of cerebellar granule nuclei was assessed at the times indicated, after application of NO donor Dea/NO at the concentrations shown. Means ± range are shown (n = 2).
Based on the experiments just described, we concluded that glutamate-induced activation of p38 and the subsequent death of cerebellar granule neurons require activity of nNOS and can be reproduced with NO donors. PSD95 ablation/dissociation has been shown to be neuroprotective (Sattler et al., 1999; Aarts et al., 2002), but as described in the Introduction, it may have additional effects. Because hypotheses exist concerning the mechanism by which this protein mediates coupling of NMDA receptors to nNOS and the consequent sensitization of the enzyme to glutamate-mediated calcium influx, we developed a construct expected to bind PSD95 in a manner identical to, and therefore competitive with, that of endogenous nNOS. The domain structures of full length nNOS and the NH2-terminal fragment we used are shown in Fig. 4 A. This NH2-terminal fragment contains the nNOS PDZ domain and the adjacent β finger, both of which are required for binding PSD95-PDZ2 (Christopherson et al., 1999; Tochio et al., 2000a), and we therefore named it nNOS-PBD (PSD95-binding domain). Transfection with GFP and GST fusion constructs leads to expression of a protein running at ∼70 kD (Fig. 4 B and not depicted). To investigate whether this construct is able to selectively bind PSD95-PDZ2, we cotransfected GST-tagged nNOS-PBD into COS7 cells with GFP-tagged PSD95-PDZ1, PSD95-PDZ2, or PSD95-PDZ3, or GFP alone. Pull-down of nNOS-PBD with immobilized glutathione revealed that a selective interaction had formed within intact cells with PDZ2 but not PDZ1, PDZ3, or unfused GFP (Fig. 4 C), and that this interaction was sufficiently stable to be detected after cell lysis and multiple washing steps.
Figure 4.

nNOS-PBD selectively and stably interacts with the PSD95-PDZ2 domain. (A) Domain map of nNOSα (1,433 aa in length). The PDZ and β finger (βƒ) domains have been reported to interact with the NMDA receptor scaffold PSD95, leading to the hypothesis that this interaction may confer on NMDA receptors an increased propensity to produce NO. The synthetic construct nNOS-PBD (PSD95-binding domain; amino acids 1–300), which incorporates all sequences reported to be required for PSD95 interaction, is also shown. (B) The epitope-tagged construct GFP-nNOS-PBD expresses a single species of the expected migration in 293 cells. The positions of calibrated molecular mass markers are shown on the left. (C) The selectivity and stability of the interaction between PSD95-PDZ domains and nNOS was monitored by coprecipitation of the PDZ domains with GST-tagged nNOS-PBD, using glutathione immobilized on beads. COS7 cells were cotransfected with GST-nNOS-PBD and GFP-tagged PSD95-PDZ1, PSD95-PDZ2, or PSD95-PDZ3, or empty GFP vector (GFP), as indicated. Only PDZ2 was revealed by anti-GFP immunoblotting of the washed beads (pull-down, bottom). The input blot (top) shows that equal amounts of GFP fusion proteins were expressed in the different cell lysates, and the anti-GST pull-down blot (middle) shows that equal amounts of GST-nNOS were captured from the lysates by the beads.
The purpose of producing the nNOS-PBD construct was to prevent glutamate-induced activation of p38 and the resulting cell death. Therefore, we investigated whether the construct was capable of doing this. Cerebellar neuron cultures were cotransfected with empty vector or nNOS-PBD together with GST-tagged p38. Thus, we were able to selectively recover p38 from transfected neurons with glutathione immobilized on beads. In the presence of empty vector, the recovered p38 exhibited a large increase in activation loop phosphorylation in response to glutamate. However, the presence of cotransfected nNOS-PBD substantially reduced the p38 activation (Fig. 5, A and B). Subsequently, neurons were transfected with nNOS-PBD or empty vector together with GFP marker plasmid so that the transfected cells could be identified. Glutamate induced pyknosis in ∼50% of neurons in the presence of empty vector, but the presence of nNOS-PBD greatly reduced this response (Fig. 5, C and D).
Figure 5.

nNOS-PBD inhibits glutamate-evoked activation of p38α and subsequent pyknosis. (A) Neurons were cotransfected with p38α and nNOS-PBD or empty vector (pCMV), as indicated. Cells were stimulated with glutamate or control, p38α was collected from the transfected cell population, and activation was detected with phospho-specific antibodies. Glutamate activated transfected p38α, and the presence of nNOS-PBD reduced this activation. Immunodetection of total p38 with pan-p38 antibody demonstrates equal loading. (B) Replicates of the data in A were quantified and means ± SEM are shown (n = 3). Asterisk indicates significant difference from empty vector–transfected glutamate-treated samples by paired t test (P < 0.01). (C) Neurons were transfected with nNOS-PBD (PBD) or empty vector (pCMV), together with GFP as a transfection marker, and treated with control or for 30 min with 50 μM glutamate. After 3 h, cells were fixed and DNA stained with Hoechst 33342, to allow assessment of pyknosis. GFP and Hoechst images are shown, as indicated. The nNOS-PBD reduced the glutamate-induced pyknosis. Transfected neurons with normal nuclei are indicated by arrows, and transfected neurons with pyknotic nuclei are indicated by arrowheads. (D) Replicates of data as in C are shown as means ± SEM (n = 3). Asterisk indicates significant difference from empty vector–transfected glutamate-treated samples by paired t test (P < 0.001).
Next, we considered whether nNOS-PBD inhibits glutamate-induced p38 activation and cell death by acting upstream or downstream of NO. Neurons were challenged with NO donor as in Fig. 2, and p38 from transfected cells showed a large increase in phosphorylation that was not prevented by cotransfection with PBD (Fig. 6, A and B). NO donor–induced pyknosis was also not prevented by transfection with nNOS-PBD (Fig. 6 C).
Figure 6.
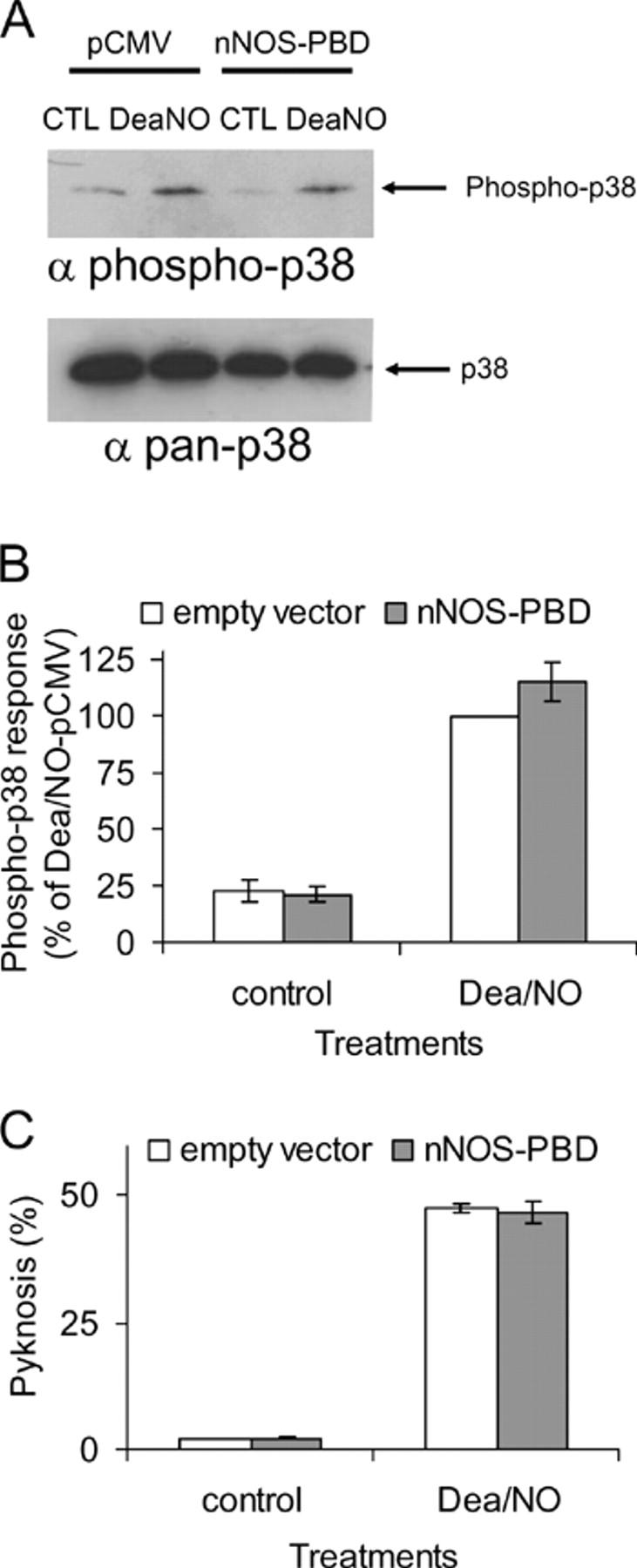
nNOS-PBD does not prevent NO-evoked activation of p38α or subsequent pyknosis. (A) Neurons were cotransfected with p38α and nNOS-PBD or empty vector (pCMV), as indicated. Cells were stimulated with 10 μM Dea/NO as NO donor (as described in the Fig. 3 legend), or with control, p38α was collected from the transfected cell population, and activation was detected with phospho-specific antibodies. Dea/NO activated transfected p38α, but the presence of nNOS-PBD did not reduce this activation. Immunodetection of total p38 with pan-p38 antibody demonstrates equal loading. (B) Replicates of the data in A were quantified and means ± SEM are shown (n = 3) (C) Neurons were transfected with nNOS-PBD (PBD) or empty vector (pCMV), together with GFP as a transfection marker, as described in the Fig. 5 (C and D) legend, and then treated with Dea/NO. The nNOS-PBD did not affect the Dea/NO-induced pyknosis. Means ± SEM (n = 3) are shown.
The nNOS-PBD construct was designed to selectively interfere with the NMDA receptor–PSD95 complex. Thus, it was important to evaluate whether it perturbed the general properties of the NMDA receptors, other than nNOS-dependent p38 activation. Patch-clamp recordings of whole-cell currents induced by the rapid application of NMDA applied via a U-tube showed that cells transfected with either nNOS-PBD or vector control were indistinguishable (Fig. 7 A). We detected no significant differences in induced current amplitude (Fig. 7 B, left) or time to peak current (Fig. 7 B, right). Capacitance was also unchanged (unpublished data), which suggests that no gross alterations in cell structure were induced. NMDA receptor activity leads to calcium influx into the cell. However, the actual changes in cytoplasmic free calcium also depend on the calcium-handling machinery of the cell. We measured the calcium response of cells transfected with nNOS-PBD or vector control by cotransfecting a fluorescence resonance energy transfer (FRET)–based calcium reporter “precocious cameleon,” or YC2.12 (Nagai et al., 2002). Once again, the nNOS-PBD– and vector-transfected cells were indistinguishable (Fig. 7 C).
Figure 7.

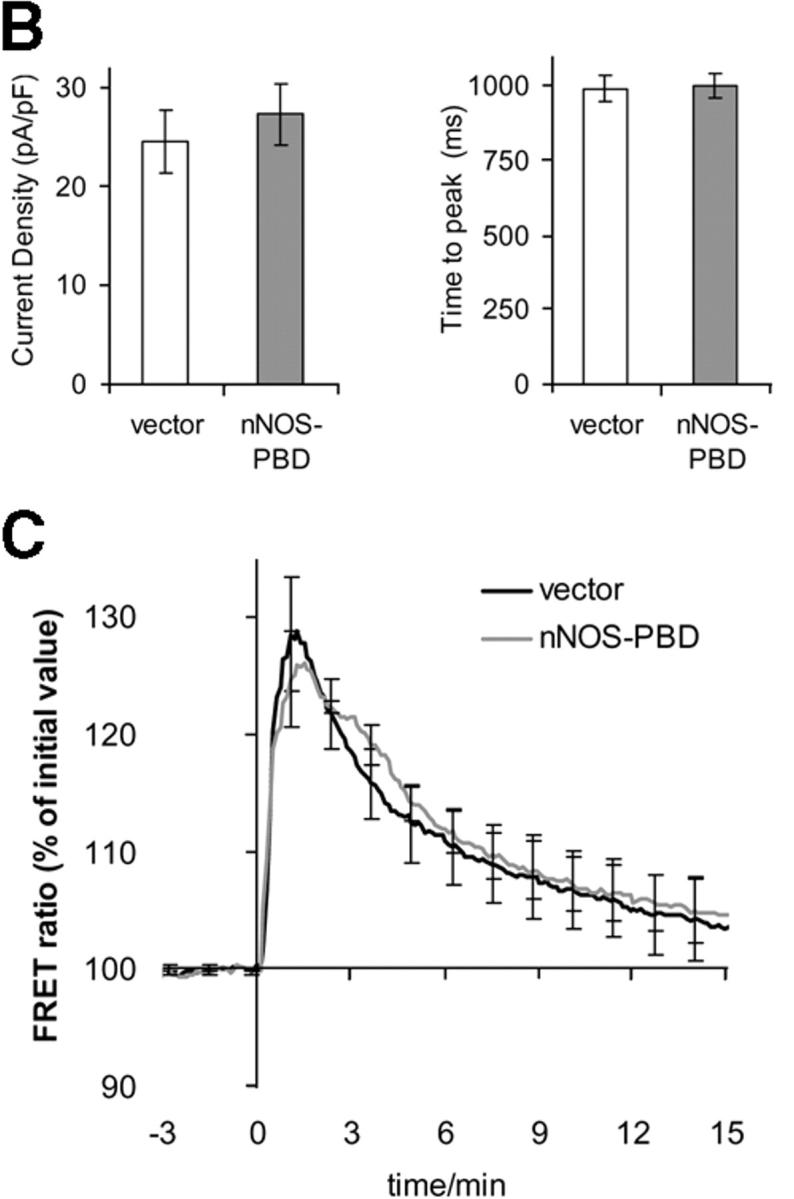
nNOS-PBD does not perturb NMDA receptor electrophysiological characteristics or NMDA-induced calcium response. (A) Whole-cell currents recorded from neurons transfected with GFP-tagged nNOS-PBD or pEGFP-C1 vector as control, after the rapid (U-tube) application of 200 μM NMDA, where indicated. To isolate NMDA receptor–gated currents, the U-tube solution contained tetrodotoxin, bicuculline, and NBQX, together with d-serine to saturate the glycine site, in addition to NMDA. (B) Peak current density (left) and time to peak current (right) were calculated from replicates, and means ± SEM are shown. (C) Neurons were cotransfected with FRET-based calcium probe YC2.12, together with either nonfluorescent pEBG-nNOS-PBD or pEBG vector control, as indicated. Cells were excited with 440 nm and images were acquired alternately at 480 nm (CFP emission) and 530 nm (raw FRET signal). 100 μM NMDA was added at time 0. Data are expressed as FRET ratio, i.e., raw FRET signal/CFP emission signal, which increases as calcium levels rise, bringing the fluorophores closer to one another and thereby increasing the FRET signal and quenching the CFP.
Together, these data support the proposal that the nNOS-PBD construct reduces glutamate-induced p38 activation and pyknosis by acting upstream of NO production, without causing general perturbation of either the electrophysiological characteristics of the NMDA receptor or the downstream signaling pathways. This suggests that a protein sequence that interacts with PSD95 in the same way as nNOS may be sufficient to confer significant neuroprotection via inhibition of glutamate-evoked p38 activation. Conversely, it would be expected that the PSD95-PDZ2 domain, which binds nNOS sequences (Fig. 4 C), would also be capable of conferring neuroprotection, whereas PDZ3 would not. PDZ1 may also be expected to be neuroprotective via interaction with the COOH termini of NMDA receptor subunits, but this interaction can be anticipated to dissociate PSD95 from the receptor complex and, therefore, to nonspecifically affect all aspects of NMDA receptor–PSD95 function. It is possible that the free PDZ2 domain may also act in this way, as PDZ2 is able to interact in vitro and in yeast with COOH-terminal peptide sequences derived from several NMDA receptor subunits (Niethammer et al., 1996), but those NMDA receptors that associate via PSD95 to nNOS may do so only via NMDA receptor interaction with PDZ1, because PDZ2 mediates the interaction with nNOS (Christopherson et al., 1999). We investigated the ability of glutamate to evoke pyknosis of cells transfected with the constructs shown in Fig. 4 C; i.e., either PSD95-PDZ1, PSD95-PDZ2, PSD95-PDZ3, or empty epitope vector (unfused GFP). Both PDZ2 and PDZ1 conferred neuroprotection, but PDZ3 failed to protect (Fig. 8).
Figure 8.

Free PDZ1 and PDZ2 domains inhibit glutamate-induced death, but PDZ3 has no effect. Neurons were transfected with the GFP-tagged PSD95-PDZ domains described in the Fig. 4 C legend, or empty vector, and then treated with glutamate or not treated. Pyknosis of transfected cells was assessed as described in the Fig. 5 (C and D) legend. Replicates were quantitated and means ± SEM are shown (n = 3). Asterisk indicates a significant difference between cells transfected with GFP-PDZ1 or GFP-PDZ2, compared with empty vector pCMV, by paired t test (P < 0.005, or better).
Discussion
Excitotoxic neuronal death is the consequence of overstimulation of glutamate receptors, which leads to excessive calcium influx. Excitotoxicity is believed to contribute to disorders such as ischemia, traumatic brain injury, Parkinson's disease, Huntingdon's disease, Alzheimer's disease, and amyotrophic lateral sclerosis. The calcium influx causes induction of NO production by the nNOS that is activated by binding of calcium–calmodulin (for review see Alderton et al., 2001). As a result, several different NO species may be produced, and these have been reported to have either neuroprotective or neurotoxic consequences (for review see Nelson et al., 2003). In spite of this complexity, there is evidence that nNOS contributes to ischemic cell death. For example, targeted deletion of the second exon of nNOS that contains the α isoform start codon eliminates expression of the nNOSα isoform without affecting β and γ isoforms. The resulting nNOSαΔ/Δ mice exhibit a reduced sensitivity to ischemic neuronal death (Huang et al., 1994) and reduced NMDA-induced death of cortical cultures (Dawson et al., 1996). In addition, small molecule nNOS inhibitors have neuroprotective actions (for review see Chabrier et al., 1999). Several mechanisms, including activation of poly(ADP-ribose) polymerase, inhibition of cytochrome c oxidase and other mitochondrial proteins, release of free cytoplasmic zinc, and activation of Trp channels, have been suggested to contribute to NO-induced neurodegeneration (Brown and Borutaite, 2002; Aarts et al., 2003; Bossy-Wetzel et al., 2004; for review see Yu et al., 2003). However, it is not clear whether these mechanisms are independent, interacting, or whether they form part of a single neurodegenerative pathway, and to what extent p38 is involved in these processes.
Thus, inhibition of nNOS can be expected to reduce excitotoxicity. However, this alone does not imply that PSD95–nNOS interaction is necessary or important for neurotoxicity, nor whether p38 might mediate a neurotoxic consequence of this interaction. The source specificity hypothesis states that calcium influx through NMDA receptors is especially neurotoxic (Aarts and Tymianski, 2003; Hardingham and Bading, 2003). This has been interpreted as a consequence of PSD95-mediated coupling of NMDA receptors and nNOS. The expectation that disrupting PSD95–nNOS interaction would have neuroprotective benefit is based on the validity of these hypotheses and assumptions. The mechanism of PSD95–nNOS interaction has been well characterized, and atomic detail is available from nuclear magnetic resonance structure and molecular modeling studies (Tochio et al., 2000b). Such data may aid possible development of small molecule compounds targeting this interaction. It is of interest to note that, even in the absence of small molecule inhibitors, the use of cell-permeable peptides has recently proven to be a realistic approach to reducing cell death in animal models of excitotoxicity (Aarts et al., 2002; Borsello et al., 2003). However, the possible neuroprotective value of targeting the PSD95–nNOS interface has been unexplored. Although ablating PSD95 or uncoupling it from the NMDA receptor provides substantial neuroprotection in an ischemia model (Sattler et al., 1999; Aarts et al., 2002), PSD95 is a multidomain protein believed to be a central mediator of assembly of the postsynaptic density complex, consisting of a vast array of signaling and structural molecules coupled to glutamate receptors in these specialized regions of neuronal cells (Husi et al., 2000). Thus, complete removal of PSD95 can be anticipated to have consequences beyond the dissociation of nNOS from the NMDA receptor (Aarts and Tymianski, 2003), thereby bringing with it the potential for undesirable side effects. The neuroprotective effects of PSD95 ablation or dissociation in the ischemia model may be the result of uncoupling of nNOS from glutamate-evoked calcium influx, but this remains unclear (Aarts and Tymianski, 2003). Further investigation of this issue is required before any firm conclusions can be drawn, and the source specificity hypothesis itself remains controversial (Hardingham and Bading, 2003). Any evidence that suggests a neuroprotective value of disrupting the PSD95–nNOS interface may have therapeutic importance, as it would increase the number of possible drug targets that could be considered for diseases involving excitotoxicity. It is anticipated that an improved clinical outcome, an increased primary effect, and fewer side effects might be obtained by aiming at more than one target in disease-causing signaling pathways. Furthermore, agents that disrupt the interaction between nNOS and PSD95 possess increased specificity, compared with small molecule catalytic site inhibitors, for two reasons: (1) only the α isoform of nNOS can interact with PSD95, whereas the catalytic domains are identical in the other isoforms (for review see Alderton et al., 2001), which will be unaffected by the aforementioned strategy; and (2) only the functions of the α isoform that are dependent on its interaction with PSD95, e.g., coupling to calcium influx through NMDA receptor, will be inhibited, as the nNOSα enzyme will not otherwise be affected.
In this study, we considered whether the PSD95–nNOS interface might be a suitable target for neuroprotective agents and whether the p38 pathway might mediate PSD95–nNOS interaction–dependent cell death. We chose a model of glutamate-induced neuronal death that we have characterized in some detail (Cao et al., 2004). Excitotoxicity can affect a variety of different brain regions and contribute to several neurodegenerative conditions. The morphological and biochemical features of degenerating neurons suggest that multiple mechanisms may underlie this form of cell death, even within a single brain region under a single stress, such as in ischemic cerebral cortex (Fukuda et al., 1999; Didenko et al., 2002). More effective neuroprotection will result from an understanding of different forms of neuronal cell death, which requires the use of multiple model systems. Glutamate-induced death in the cerebellar granule neuron model is NMDA receptor dependent and has properties (Cao et al., 2004; for review see Yu et al., 2003) reported to be associated with excitotoxic death in several other systems, including cultured cortical neurons and the ischemic brain. These properties include lumpy chromatin condensation (Sohn et al., 1998; Fukuda et al., 1999; Didenko et al., 2002), caspase independence (Didenko et al., 2002; Yu et al., 2002), insensitivity to inhibitors of transcription and translation (Csernansky et al., 1994; Lobner and Choi, 1996; Gwag et al., 1997), sensitivity to p38 inhibition (Legos et al., 2001, 2002), involvement of NO or nNOS (Huang et al., 1994; Dawson et al., 1996), and sensitivity to poly(ADP-ribose) polymerase inhibition (for review see Yu et al., 2003). Thus, the cerebellar granule neuron model may be particularly useful for studying forms of excitotoxic cell death with these properties.
Here, we demonstrated that the p38 activation and cell death in glutamate-treated cerebellar granule neurons were both sensitive to inhibitors of NOS and nNOS. Subsequently, we demonstrated that NO donor was capable of activating p38 and cell death in this model in a manner strikingly similar to the way glutamate induces death. This is consistent with the proposal that glutamate-induced cell death in this model involves generation of NO, a predictable but necessary prerequisite for this model to be useful for considering the possible neuroprotective value of targeting the PSD95–nNOS interaction. It is important to note that an additional prerequisite is the validity of the source specificity hypothesis, for if neurotoxicity were dependent merely on calcium load and not on localization, then nNOS would continue to contribute to neurotoxicity even if no longer physically associated with the NMDA receptor. nNOS activity also appeared to be necessary (and sufficient) for the activation of p38 that is critical for glutamate-evoked cell death (Fig. 9 A). Therefore, any neuroprotection that is the result of disrupting the PSD95–nNOS interface should inhibit glutamate-evoked p38 activation as well as cell death.
Figure 9.

A hypothetical model proposed to explain the disruption of neuronal death signaling revealed by the constructs investigated. (A) In an untransfected neuron, PSD95 is reportedly able to bring nNOS in close proximity to the calcium pore of the NMDA receptor. As a result, glutamate induces activation of nNOS and, subsequently, of p38. (B) The presence of nNOS-PBD in transfected cells (in excess of endogenous PSD95) competes with and prevents interaction of endogenous nNOS with PSD95, leading to a reduced ability of glutamate to activate p38 and pyknosis, which supports the source specificity hypothesis. (C) However, the presence of nNOS-PBD has no effect on the responses to addition of exogenous NO as this bypasses the requirement for PSD95–nNOS interaction. (D) Related scenarios in the presence of transfected PDZ1, 2 or 3. PDZ1 is reportedly capable of interacting with the NMDA receptor not nNOS, but this too reduces the ability of glutamate to induce pyknosis. PDZ2 is more effective, mimicking nNOS-PDZ2 in its actions, whereas PDZ3 reportedly interacts neither with NMDA receptor nor with nNOS, which explains its failure to prevent glutamate-induced pyknosis.
The NH2-terminal region of nNOSα we used, nNOS-PBD, selectively and stably bound PDZ2, as expected (Fig. 4). Expression of this construct in neurons inhibited p38α activation and reduced the subsequent cell death induced by glutamate treatment. This suggests that nNOS-PBD binds to a target in neuronal cells that is necessary for glutamate-induced p38 activation and subsequent cell death. The normal electrophysiological characteristics of the NMDA receptors and the cells' calcium response to NMDA were unaffected, which suggests that the action of nNOS-PBD was specific to the NMDA receptor–p38 pathway. The simplest explanation is that it is PDZ2 of PSD95 (or related molecules such as PSD93; Brenman et al., 1996b) that is affected in these experiments, leading to inhibition of the PSD95–nNOS interaction (Fig. 9 B). Because only a small amount of nNOS immunostaining colocalized with PSD95 immunostaining in these cells (unpublished data) and microscopy does not have sufficient resolution to discern disruption of the physical interaction between the molecules, it was not possible for us to directly visualize this effect. Instead, we reasoned that if the nNOS-PBD acts by preventing interaction of PSD95 (or a related molecule) with nNOS, thereby preventing NO production and the downstream consequences, then it should be possible to bypass this effect by exogenously supplying NO, which we had already demonstrated induced cell death in a manner very similar to that induced by glutamate. Indeed, transfection with nNOS-PBD had no effect on NO-induced activation of p38 or the subsequent neuronal death (Fig. 9 C). We sought additional evidence for the role of this interaction by generating expression of PDZ2 to compete with endogenous PSD95 in the nNOS interaction, or of PDZ3 as a control. PDZ2 again inhibited glutamate-induced neuronal cell death, whereas PDZ3, which is not known to have any importance in glutamate-induced neuronal cell death, had no effect (Fig. 9 D, middle and right). PDZ1 was also somewhat protective. This is not surprising, because PDZ1 can interact with the COOH termini of subunits of the NMDA receptor complex (while, simultaneously, PDZ2 can bind nNOS; Christopherson et al., 1999), thereby dissociating the entire PSD95 molecule from the NMDA receptors (Aarts and Tymianski, 2003; Fig. 9 D, left).
In conclusion, these results suggest that the interaction between nNOS and PSD95 (or a related molecule such as PSD93) is important for glutamate-induced activation of p38α stress-activated protein kinase and the ensuing cell death, and that the nNOS–PDZ2 interface is a target suitable for neuroprotective drug design efforts. It can be anticipated that, compared with agents that dissociate PSD95 from NMDA receptors, such reagents will have less effect on the NMDA receptor–PSD95 interaction and thus will produce considerably lower disruption of the delicate and highly complex postsynaptic density apparatus.
Materials and methods
Antibodies and plasmids
Mouse anti-GFP (CLONTECH Laboratories, Inc.), rabbit anti–phospho-p38 (New England Biolabs, Inc.) and mouse anti–pan-p38 (Transduction Labs) were used with secondary reagents from various sources (Santa Cruz Biotechnology, Inc. and UBI). The coding sequence for the NH2-terminal 300 amino acids of nNOS was prepared by PCR-based methods from plasmid pPIC-ZA-nNOS (a gift from Bernt Mayer, Karl-Franzens-Universitat, Graz, Graz, Austria) and inserted in-frame with the GFP coding sequence of the plasmid pEGFP-C1 (CLONTECH Laboratories, Inc.) to make GFP-nNOS-PBD. pEBG-nNOS-PBD was prepared by inserting the same sequence into the plasmid pEBG (a gift from Bruce Mayer, University of Connecticut Health Center, Farmington, CT) in-frame with the GST coding sequence. PDZ domains from PSD95 were PCR amplified from the plasmid pCMV-PSD95 (a gift from Y. Hata, Osaka University Medical School, Suita, Osaka, Japan). PDZ1 (aa 60–155), PDZ2 (aa 155–249), and PDZ3 (aa 302–402) were subsequently inserted in-frame into the plasmid pEGFP-C1. pEBG-p38α was a gift from Bruce Mayer. pCMV was a gift from S. van den Heuvel (Massachusetts General Hospital, Boston, MA).
Cell culture and glutamate and NO donor treatment
Primary cultures of cerebellar granule neurons were prepared and maintained as described previously (Courtney et al., 1997). Neurons used were cultured for 7–9 d in vitro (DIV). Glutamate treatment was performed as described previously (Cao et al., 2004). Cells were briefly rinsed in Mg-free Locke's buffer (154 mM NaCl, 5.6 mM KCl, 3.6 mM NaHCO3, 1.3 mM CaCl2, 5.6 mM d-glucose, and 5 mM Hepes, pH 7.4) and placed in the same buffer with glutamate (50 μM) for 30 min, or as shown in the legend of Fig. 1; 10 μM glycine was routinely included with glutamate because it is an essential coagonist for the NMDA receptor. Subsequently, cells were rinsed in Locke's buffer with 1 mM MgCl2, and conditioned medium was replaced for the times indicated in the legends of Fig. 1; Fig. 3 C; Fig. 5, C and D; Fig. 6 C; and Fig. 8. NO treatment was performed by adding 10 or 250 μM NO donor Dea/NO (DeaNONOate) or 300 μM ONOO− to the cell culture media directly, and these conditions were maintained throughout the whole experiment. The concentration of ONOO− was determined by measuring its absorbance at 302 nm (ɛ = 1,670 M−1 cm−1). The concentration of ONOO− was measured for every experiment, just before it was added to the samples. Pharmacological agents (3 μM 7-Nitroindazole, 1 μM N-ω-propyl-l-arginine, and 1 μM SB203580) were added 60 min before glutamate treatment, and the agents were also present in all media in which the cells were subsequently incubated. The NOS inhibitors are competitive with arginine; therefore, cells were placed in arginine-free conditions during the period of incubation with these inhibitors (or incubation with carrier, in the case of controls). 1 mM arginine was added to the preincubation and stimulation solutions, where indicated in the legend of Fig. 2.
Immunoblotting
Immunoblotting was performed as described previously (Coffey et al., 2002) by treating cells as indicated (see legends of Fig. 1 A; Fig. 2, A and B; Fig. 3, A and B; and Fig. 4 B), rinsing rapidly in ice-cold PBS, lysing in 1× Laemmli buffer, boiling, clearing, resolving by SDS-PAGE, and performing electrotransfer. Western blotting was performed according to standard protocols, and blots were developed using ECL reagents according to the manufacturer's instructions.
Cell death assay
After treatment as described in the Cell culture and glutamate and NO donor treatment section (typically after 3 h or as indicated in the legends of Figs. 1 B and 3 C), cells were stained with Hoechst 33342, fixed, and scored on the basis of nuclear morphology. A pyknotic nucleus was considered to indicate death of the cell.
Pyknosis assay for transfected neurons
Cerebellar granule cultures at 6–7 DIV were transfected as described previously (Coffey et al., 2000). They were cotransfected with GFP marker plasmids and either empty vector (pCMV), GFP-nNOS-PBD, GFP-PSD95-PDZ1, GFP-PSD95-PDZ2, or GFP-PSD95-PDZ3. When tested, cotransfection efficiency was almost 100% (Coffey et al., 2002; Hongisto et al., 2003). 24 h after transfection, cells were treated with or without glutamate or NO donor Dea/NO as described in the Cell culture and glutamate and NO donor treatment section. 3 h after stimulation, cells were fixed with 4% PFA, rinsed with ice-cold PBS, and stained with Hoechst 33342. For transfected neurons, fluorescence image fields of GFP emission using 450–490 nm of excitation light and a 20× air objective were taken to locate transfected cells, and the corresponding image of Hoechst fluorescence was examined to determine whether transfected cells had pyknotic nuclei. Four evenly spaced fields were counted per coverslip. Imaging of DNA dyes and transfected fluorescent proteins was performed with a cooled CCD (model KX85; Apogee) under control of MicroCCD software (Diffraction Limited) and a microscope (model IX70; Olympus) with a 20× air objective (0.4 NA) and appropriate filter cubes.
Assay for activation of transfected p38α
7 d after plating in 35-mm dishes, cerebellar granule neurons were cotransfected with EBG-p38α and either empty vector (pCMV) or GFP-nNOS-PBD. 24 h after transfection, cells were treated as described in the legends of Figs. 5 and 6 for 5 min, rinsed once with ice-cold PBS, and lysed in 500 μl of lysis buffer (20 mM Hepes, pH 7.4; 2 mM EGTA; 50 mM β-glycerophosphate; 1 mM DTT; 1 mM Na3VO4; 1% Triton X-100; 10% glycerol; 50 mM NaF; 1 mM benzamidine; 1 μg/ml aprotinin, leupeptin, and pepstatin; and 100 μg/ml PMSF). Homogenized and precleared supernatants were incubated with 10 μl (bed volume) prewashed S-hexylglutathione agarose beads for 3 h at 4°C. Beads were spun out and washed three times in lysis buffer, and then the drained beads were boiled in 40 μl 1× Laemmli sample buffer, for immunoblotting.
Pull-down assay for PSD95–nNOS interaction
1 d after plating in 10-cm dishes, COS7 cell cultures were cotransfected with EBG-nNOS-PBD and either GFP-PDZ1, GFP-PDZ2, GFP-PDZ3, or GFP-C1. After 48 h of transfection, cells were rinsed once with ice-cold PBS and lysed in 800 μl of low-salt buffer (20 mM Na2 β-glycerophosphate; 30 mM NaF; 2 mM EDTA; 2 mM Na4P2O5; 1 mM DTT; 10 μg/ml aprotinin, leupeptin, and pepstatin A; 0.5 mM AEBSF; and 0.5% igepal). Homogenized and precleared supernatants were rotated for 3 h at 4°C with 15 μl (bed volume) S-hexylglutathione agarose beads preequilibrated in low-salt buffer. Beads were spun out and washed three times in the same buffer, and then the aspirated pellet was boiled in 50 μl 1× Laemmli sample buffer for immunoblotting.
Electrophysiological recordings
Whole-cell currents were recorded from single cultured cerebellar granule cells (at 7 DIV) and transfected with either pEGFP-C1 (empty vector) or the PSD95-PDZ2–binding region of nNOS (pEGFP-nNOS) using an amplifier (Axopatch 200B; Axon Instruments, Inc.). Recorded membrane currents were digitized using a Digidata 1320A interface (Axon Instruments, Inc.) and analyzed using pCLAMP software. Patch pipettes were pulled from thin-wall borosilicate glass (1.5-mm outer diameter and 1.17-mm inner diameter; Clarke Electromedical) and fire polished to give a final resistance of ∼5 MΩ when filled. The pipette-filling solution contained (mM): 145 potassium gluconate, 10 Hepes, 5 EGTA, 5 MgCl2, 5 Na2ATP, and 0.2 GTP; adjusted to pH 7.2. The extracellular solution contained (mM): 145 NaCl, 5 KCl, 1 CaCl2, 5 Hepes, 5 glucose, and 20 sucrose; adjusted to pH 7.4. NMDA receptor currents were elicited at a holding potential of −60 mV by the rapid (U-tube) application of 200 μM NMDA in extracellular solution containing 20 μM d-serine, 50 μM bicuculline, 1 μM tetrodotoxin, and 5 μM NBQX.
FRET-based imaging of cytoplasmic free calcium
Cerebellar granule neurons were cultured on 10-mm square glass coverslips and cotransfected at 6–7 DIV with the precocious cameleon YC2.12 (a gift from A. Miyawaki, RIKEN Brain Research Institute, Wako, Japan; Nagai et al., 2002) and pEBG vector or pEBG-nNOS-PBD, as indicated in the legend of Fig. 7 C. This calcium probe is based on the Venus YFP variant that is most resistant to changes in pH and chloride, and matures quickly, thereby avoiding artifactually low FRET caused by retarded maturation of the acceptor fluorophore. The following day, the coverslips were washed once in Locke's buffer with 1 mM MgCl2 and twice more in Locke's buffer without MgCl2, and then placed in 1 ml of Locke's buffer without MgCl2 in a chamber on a microscope (model IX70; Olympus). Cells were illuminated with a mercury lamp, a neutral density filter, and a 440 nm/21 nm excitation filter. CFP and YFP emission image pairs were acquired (with a 2-s integration time per channel and 13 s between image pairs) through a 455-nm dichroic mirror and 480 nm/30 nm and 530 nm/26 nm filters in a filter wheel (CVI laser; Apogee Instruments, Inc.) mounted parfocally in front of a cooled CCD (model KX85; Apogee Instruments, Inc.). The acquired CFP emission images represent CFP signals quenched by FRET, and are referred to hereafter as CC images. The acquired Venus YFP emission images represent raw FRET signals, and are referred to hereafter as CY images. As calcium rises, it causes the probe to fold to a more compact conformation, leading to increased CY signal and quenching the CC signal. The ratio of background-corrected CY and CC signals was calculated and is directly related to calcium changes. NMDA and glycine (at final concentrations of 100 μM and 10 μM, respectively) were added at time 0. FRET time courses (CY/CC ratio) were calculated from the acquired image series with image analysis software developed by the authors (Lindqvist et al., 1995) that had been modified to permit the processing of these image datasets as just described. Background-corrected fluorescence ratio time courses were normalized to the average prestimulation CY/CC ratio values for each cell, the means of cells within each field were calculated, and the data are presented in Fig. 7 C as means ± SEM of independent experiments.
Acknowledgments
We thank Bruce Mayer, Bernt Mayer, Yutaka Hata, Atsushi Miyawaki, and Sander van den Heuvel for providing plasmids used in this study. We thank Eleanor Coffey for advice on the manuscript and John Challis and Richard Evans for their help and advice in setting up the NMDA receptor current recordings.
This work was funded by the Academy of Finland (grants 72446, 78232, 203520, and 206903), the Magnus Ehrnrooths Foundation, and the University of Kuopio. M.J. Courtney is an Academy of Finland researcher.
Abbreviations used in this paper: Dea, diethylamine; DIV, days in vitro; FRET, fluorescence resonance energy transfer; NO, nitric oxide; nNOS, neuronal NOS; NOS, NO synthase; ONOO−, peroxynitrite; PBD, PSD95-binding domain.
References
- Aarts, M., Y. Liu, L. Liu, S. Besshoh, M. Arundine, J.W. Gurd, Y.T. Wang, M.W. Salter, and M. Tymianski. 2002. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 298:846–850. [DOI] [PubMed] [Google Scholar]
- Aarts, M.M., and M. Tymianski. 2003. Novel treatment of excitotoxicity: targeted disruption of intracellular signaling from glutamate receptors. Biochem. Pharmacol. 66:877–886. [DOI] [PubMed] [Google Scholar]
- Aarts, M., K. Iihara, W.L. Wei, Z.G. Xiong, M. Arundine, W. Cerwinski, J.F. MacDonald, and M. Tymianski. 2003. A key role for TRPM7 channels in anoxic neuronal death. Cell. 115:863–877. [DOI] [PubMed] [Google Scholar]
- Alderton, W.K., C.E. Cooper, and R.G. Knowles. 2001. Nitric oxide synthases: structure, function and inhibition. Biochem. J. 357:593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bon, C.L., and J. Garthwaite. 2003. On the role of nitric oxide in hippocampal long-term potentiation. J. Neurosci. 23:1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel, E., M.V. Talantova, W.D. Lee, M.N. Scholzke, A. Harrop, E. Mathews, T. Gotz, J. Han, M.H. Ellisman, G.A. Perkins, and S.A. Lipton. 2004. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 41:351–365. [DOI] [PubMed] [Google Scholar]
- Borsello, T., P.G. Clarke, L. Hirt, A. Vercelli, M. Repici, D.F. Schorderet, J. Bogousslavsky, and C. Bonny. 2003. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 9:1180–1186. [DOI] [PubMed] [Google Scholar]
- Brenman, J.E., D.S. Chao, S.H. Gee, A.W. McGee, S.E. Craven, D.R. Santillano, Z. Wu, F. Huang, H. Xia, M.F. Peters, et al. 1996. a. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 84:757–767. [DOI] [PubMed] [Google Scholar]
- Brenman, J.E., K.S. Christopherson, S.E. Craven, A.W. McGee, and D.S. Bredt. 1996. b. Cloning and characterization of postsynaptic density 93, a nitric oxide synthase interacting protein. J. Neurosci. 16:7407–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G.C., and V. Borutaite. 2002. Nitric oxide inhibition of mitochondrial respiration and its role in cell death. Free Radic. Biol. Med. 33:1440–1450. [DOI] [PubMed] [Google Scholar]
- Cao, J., M.M. Semenova, V.T. Solovyan, J. Han, E.T. Coffey, and M.J. Courtney. 2004. Distinct requirements for p38α and JNK stress-activated protein kinases in different forms of apoptotic neuronal death. J. Biol. Chem. 279:35903–35913. [DOI] [PubMed] [Google Scholar]
- Chabrier, P.E., C. Demerle-Pallardy, and M. Auguet. 1999. Nitric oxide synthases: targets for therapeutic strategies in neurological diseases. Cell. Mol. Life Sci. 55:1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson, K.S., B.J. Hillier, W.A. Lim, and D.S. Bredt. 1999. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 274:27467–27473. [DOI] [PubMed] [Google Scholar]
- Coffey, E.T., V. Hongisto, M. Dickens, R.J. Davis, and M.J. Courtney. 2000. Dual roles for JNK in developmental and stress responses in cerebellar granule neurons. J. Neurosci. 20:7602–7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey, E.T., G. Smiciene, V. Hongisto, J. Cao, S. Brecht, T. Herdegen, and M.J. Courtney. 2002. JNK2/3 is specifically activated by stress, mediating c-jun activation, in the presence of constitutive JNK1 activity in cerebellar neurons. J. Neurosci. 22:4335–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney, M.J., K.E. Åkerman, and E.T. Coffey. 1997. Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J. Neurosci. 17:4201–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csernansky, C.A., L.M. Canzoniero, S.L. Sensi, S.P. Yu, and D.W. Choi. 1994. Delayed application of aurintricarboxylic acid reduces glutamate-induced cortical neuronal injury. J. Neurosci. Res. 38:101–108. [DOI] [PubMed] [Google Scholar]
- Dawson, V.L., V.M. Kizushi, P.L. Huang, S.H. Snyder, and T.M. Dawson. 1996. Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase-deficient mice. J. Neurosci. 16:2479–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didenko, V.V., H. Ngo, C.L. Minchew, D.J. Boudreaux, M.A. Widmayer, and D.S. Baskin. 2002. Caspase-3-dependent and -independent apoptosis in focal brain ischemia. Mol. Med. 8:347–352. [PMC free article] [PubMed] [Google Scholar]
- Fukuda, T., H. Wang, H. Nakanishi, K. Yamamoto, and T. Kosaka. 1999. Novel non-apoptotic morphological changes in neurons of the mouse hippocampus following transient hypoxic-ischemia. Neurosci. Res. 33:49–55. [DOI] [PubMed] [Google Scholar]
- Ge, B., H. Gram, F. Di Padova, B. Huang, L. New, R.J. Ulevitch, Y. Luo, and J. Han. 2002. MAPKK-independent activation of p38α mediated by TAB1-dependent autophosphorylation of p38α. Science. 295:1291–1294. [DOI] [PubMed] [Google Scholar]
- Gwag, B.J., J.Y. Koh, J.A. DeMaro, H.S. Ying, M. Jacquin, and D.W. Choi. 1997. Slowly triggered excitotoxicity occurs by necrosis in cortical cultures. Neuroscience. 77:393–401. [DOI] [PubMed] [Google Scholar]
- Hardingham, G.E., and H. Bading. 2003. The yin and yang of NMDA receptor signalling. Trends Neurosci. 26:81–89. [DOI] [PubMed] [Google Scholar]
- Hongisto, V., N. Smeds, S. Brecht, T. Herdegen, M.J. Courtney, and E.T. Coffey. 2003. Lithium blocks the c-jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol. Cell. Biol. 23:6027–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Z., P.L. Huang, N. Panahian, T. Dalkara, M.C. Fishman, and M.A. Moskowitz. 1994. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 265:1883–1885. [DOI] [PubMed] [Google Scholar]
- Husi, H., M.A. Ward, J.S. Choudhary, W.P. Blackstock, and S.G. Grant. 2000. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 3:661–669. [DOI] [PubMed] [Google Scholar]
- Kawasaki, H., T. Morooka, S. Shimohama, J. Kimura, T. Hirano, Y. Gotoh, and E. Nishida. 1997. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J. Biol. Chem. 272:18518–18521. [DOI] [PubMed] [Google Scholar]
- Legos, J.J., J.A. Erhardt, R.F. White, S.C. Lenhard, S. Chandra, A.A. Parsons, R.F. Tuma, and F.C. Barone. 2001. SB 239063, a novel p38 inhibitor, attenuates early neuronal injury following ischemia. Brain Res. 892:70–77. [DOI] [PubMed] [Google Scholar]
- Legos, J.J., B. McLaughlin, S.D. Skaper, P.J. Strijbos, A.A. Parsons, E. Aizenman, G.A. Herin, F.C. Barone, and J.A. Erhardt. 2002. The selective p38 inhibitor SB-239063 protects primary neurons from mild to moderate excitotoxic injury. Eur. J. Pharmacol. 447:37–42. [DOI] [PubMed] [Google Scholar]
- Lin, S., Y. Zhang, R. Dodel, M.R. Farlow, S.M. Paul, and Y. Du. 2001. Minocycline blocks nitric oxide-induced neurotoxicity by inhibition p38 MAP kinase in rat cerebellar granule neurons. Neurosci. Lett. 315:61–64. [DOI] [PubMed] [Google Scholar]
- Lindqvist, C., C. Holmberg, C. Oetken, M. Courtney, A. Stahls, and K.E. Akerman. 1995. Rapid Ca2+ mobilization in single LGL cells upon interaction with K562 target cells–role of the CD18 and CD16 molecules. Cell. Immunol. 165:71–76. [DOI] [PubMed] [Google Scholar]
- Lobner, D., and D.W. Choi. 1996. Preincubation with protein synthesis inhibitors protects cortical neurons against oxygen-glucose deprivation-induced death. Neuroscience. 72:335–341. [DOI] [PubMed] [Google Scholar]
- Manabe, S., and S.A. Lipton. 2003. Divergent NMDA signals leading to proapoptotic and antiapoptotic pathways in the rat retina. Invest. Ophthalmol. Vis. Sci. 44:385–392. [DOI] [PubMed] [Google Scholar]
- Nagai, T., K. Ibata, E.S. Park, M. Kubota, K. Mikoshiba, and A. Miyawaki. 2002. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20:87–90. [DOI] [PubMed] [Google Scholar]
- Nelson, E.J., J. Connolly, and P. McArthur. 2003. Nitric oxide and S-nitrosylation: excitotoxic and cell signaling mechanism. Biol. Cell. 95:3–8. [DOI] [PubMed] [Google Scholar]
- Niethammer, M., E. Kim, and M. Sheng. 1996. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J. Neurosci. 16:2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler, R., Z. Xiong, W.Y. Lu, M. Hafner, J.F. MacDonald, and M. Tymianski. 1999. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 284:1845–1848. [DOI] [PubMed] [Google Scholar]
- Sohn, S., E.Y. Kim, and B.J. Gwag. 1998. Glutamate neurotoxicity in mouse cortical neurons: atypical necrosis with DNA ladders and chromatin condensation. Neurosci. Lett. 240:147–150. [DOI] [PubMed] [Google Scholar]
- Tochio, H., Y.K. Mok, Q. Zhang, H.M. Kan, D.S. Bredt, and M. Zhang. 2000. a. Formation of nNOS/PSD-95 PDZ dimer requires a preformed β-finger structure from the nNOS PDZ domain. J. Mol. Biol. 303:359–370. [DOI] [PubMed] [Google Scholar]
- Tochio, H., F. Hung, M. Li, D.S. Bredt, and M. Zhang. 2000. b. Solution structure and backbone dynamics of the second PDZ domain of postsynaptic density-95. J. Mol. Biol. 295:225–237. [DOI] [PubMed] [Google Scholar]
- Yu, S.W., H. Wang, M.F. Poitras, C. Coombs, W.J. Bowers, H.J. Federoff, G.G. Poirier, T.M. Dawson, and V.L. Dawson. 2002. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 297:259–263. [DOI] [PubMed] [Google Scholar]
- Yu, S.W., H. Wang, T.M. Dawson, and V.L. Dawson. 2003. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol. Dis. 14:303–317. [DOI] [PubMed] [Google Scholar]
- Zhang, H.Q., W. Fast, M.A. Marletta, P. Martasek, and R.B. Silverman. 1997. Potent and selective inhibition of neuronal nitric oxide synthase by N ω-propyl-L-arginine. J. Med. Chem. 40:3869–3870. [DOI] [PubMed] [Google Scholar]