

Abstract
We report the characterization of factor inhibiting activating transcription factor 4 (ATF4)–mediated transcription (FIAT), a leucine zipper nuclear protein. FIAT interacted with ATF4 to inhibit binding of ATF4 to DNA and block ATF4-mediated transcription of the osteocalcin gene in vitro. Transgenic mice overexpressing FIAT in osteoblasts also had reduced osteocalcin gene expression and decreased bone mineral density, bone volume, mineralized volume, trabecular thickness, trabecular number, and decreased rigidity of long bones. Mineral homeostasis, osteoclast number and activity, and osteoblast proliferation and apoptosis were unchanged in transgenics. Expression of osteoblastic differentiation markers was largely unaffected and type I collagen synthesis was unchanged. Mineral apposition rate was reduced in transgenic mice, suggesting that the lowered bone mass was due to a decline in osteoblast activity. This cell-autonomous decrease in osteoblast activity was confirmed by measuring reduced alkaline phosphatase activity and mineralization in primary osteoblast cultures. These results show that FIAT regulates bone mass accrual and establish FIAT as a novel transcriptional regulator of osteoblastic function.
Introduction
Bone formation is an important physiological process that regulates skeletal growth, bone remodeling, and fracture repair. This process is dependent on the actions of two cell types: the bone-forming osteoblasts and the bone-resorbing osteoclasts. The osteoblasts, of mesenchymal origin, are responsible for bone matrix protein deposition and subsequent mineralization in both intramembranous and endochondral bone formation (Erlebacher et al., 1995). Osteoclasts are cells of hematopoietic origin that are responsible for resorbing extracellular matrix (Boyle et al., 2003). The equilibrium between bone formation and resorption is tightly controlled, and imbalances between the two processes lead to bone disease (Harada and Rodan, 2003; Zelzer and Olsen, 2003).
Several transcription factors have been identified as regulators of osteoblastic differentiation and function (Karsenty and Wagner, 2002), including Dlx5 (Miyama et al., 1999), Runx2/Cbfa1 (Ducy et al., 1997; Komori et al., 1997), and Osx (Nakashima et al., 2002). It is likely that some of these factors control the activity or expression of one another, forming defined pathways, as suggested by the Dlx5-dependent induction of Runx2/Cbfa1 and Osx in pluripotential cells (Lee et al., 2003a,b). Similarly, the basic helix-loop-helix transcription factors Twist-1 and -2 were recently shown to regulate osteoblast differentiation by interacting with Runx2/Cbfa1 to inhibit its activity (Bialek et al., 2004). It is thus becoming evident that complete understanding of the transcriptional control of osteoblastic differentiation and function will require further analysis of the cross talk and interactions between particular activators and/or repressors of gene transcription.
Several members of the basic domain-leucine zipper (bZip) family of transcription factors have also been shown to control osteoblast development or activity. In addition to the AP-1 family members Fra-1 (Jochum et al., 2000) and ΔFosB (Sabatakos et al., 2000), the bZip factor activating transcription factor 4 (ATF4) was also recently shown to regulate osteoblast biology. Yang et al. (2004) showed that ATF4 is a substrate of the RSK2 (ribosomal S6 kinase-2) kinase and regulates the onset of osteoblast differentiation, type I collagen synthesis, osteoblast-specific gene expression, and osteoblast terminal differentiation. ATF4 was shown to be the osteocalcin promoter binding factor Osf1 (Ducy and Karsenty, 1995; Schinke and Karsenty, 1999; Yang et al., 2004) and to regulate osteocalcin gene transcription in a RSK2-dependent manner (Yang et al., 2004). Mice deficient for ATF4 are runted (Tanaka et al., 1998; Hettmann et al., 2000; Masuoka and Townes, 2002) and harbor low bone mass (Yang et al., 2004). ATF4 can form homodimers (Hai and Curran, 1991; Vallejo et al., 1993) but can also heterodimerize with a variety of partners (Hai and Curran, 1991; Chevray and Nathans, 1992; Vallejo et al., 1993). The dimerization partner appears to influence specificity of DNA binding (Vallejo et al., 1993) as well as transcriptional activity (Fawcett et al., 1999; Lim et al., 2000).
We report the cloning and characterization of factor-inhibiting ATF4-mediated transcription (FIAT), a 66-kD leucine zipper nuclear protein that interacts with ATF4 to inhibit binding of ATF4 to its cognate response element and blocks ATF4-mediated transcriptional activation of the osteocalcin gene promoter in vitro. Transgenic mice overexpressing FIAT under the control of the osteoblast-specific fragment of the α1(I) collagen promoter were generated to study the role of FIAT in bone-forming cells in vivo. These mice displayed an osteopenic phenotype accompanied by reduced osteocalcin gene transcription, decreased bone mineral density, trabecular volume, and bone rigidity. Although osteoblast proliferation and apoptosis were unaffected, osteoblast activity was shown to be reduced. However, type I collagen mRNA transcription and protein synthesis were unchanged. Thus, transgenic expression of FIAT mimics several but not all aspects of the phenotype of ATF4-deficient bone and suggests that FIAT inhibition of ATF4 activity may be one of the pathways through which FIAT affects osteoblast function.
Results
FIAT Interacts with ATF4
We identified FIAT in a yeast two-hybrid screen for proteins interacting with the αNAC transcriptional coactivator (Moreau et al., 1998; Yotov et al., 1998). The interaction of the partial FIAT cDNA with αNAC was confirmed in yeast, but we could not observe an interaction between the full-length FIAT and the coactivator in mammalian cells (unpublished data). Thus, the physiological relevance of the putative FIAT–αNAC interaction remains unclear and was not further pursued.
The full-length 1.8-kb FIAT cDNA translates into a 66-kD protein that localizes to the nucleus in calvarial osteoblasts (Fig. 1, A–C) and in ROS 17/2.8 osteoblastic cells (Majeska et al., 1980; not depicted). The FIAT protein (accession no. NP_060830, encoded by a gene mapping to Xp22.1, accession no. NM_018360) is predicted to form a long coiled-coil at its COOH terminus, a structure that favors protein–protein interactions. Moreover, computer modeling predicts the presence of putative leucine zippers within the FIAT protein (Fig. 1 A). While this manuscript was under revision, a protein identical to FIAT was characterized on the basis of its extended COOH-terminal coiled-coil and shown to interact with syntaxin family members (Nogami et al., 2004). Combined with the data presented herein, it appears that FIAT can interact with a variety of partners. Sorting out the physiological relevance of all interactions will require further studies. The FIAT mRNA is around 4.5 kb in length with an extended 3′-untranslated region (unpublished data) and is ubiquitously expressed (Nogami et al., 2004).
Figure 1.

FIAT protein. (A) Amino acid sequence of the human FIAT protein. The putative leucine zipper is shaded in gray. (B) Nuclear extracts from MC3T3-E1 osteoblastic cells at confluence (lane 1) or after 6 d of growth in the presence of ascorbic acid and β-glycerophosphate (lane 2) were probed with the anti-FIAT peptide antibody. The arrow identifies the 66-kD FIAT protein. (C) Calvarial osteoblasts from primary cultures were stained with the anti-FIAT antibody. The endogenous FIAT protein is detected in the nucleus of the cells.
The presence of a putative leucine zipper within the FIAT protein sequence (Fig. 1 A) prompted us to investigate whether FIAT could form homodimeric or heterodimeric interactions. Yeast two-hybrid assays using FIAT as both the bait and target molecules revealed that FIAT could not homodimerize (Fig. 2, A and B). In a search for putative heterodimerization partners, we used the FIAT bait to screen cDNA libraries from MC3T3-E1 osteoblastic cells (Sudo et al., 1983) or primary cultures of osteoblasts (Ecarot-Charrier et al., 1983) in the two-hybrid assay. Three independent clones encoding the ATF4 transcription factor were isolated from the MC3T3-E1 library, whereas two different clones were obtained from the primary osteoblasts library, thus identifying ATF4 as a target molecule (Fig. 2, A and B). To confirm this interaction, reciprocal coimmunoprecipitation assays were performed using ROS 17/2.8 osteoblastic cells. Immunoprecipitation of endogenous ATF4 coprecipitated endogenous FIAT (Fig. 2 C, left). Reciprocally, the ATF4 protein coimmunoprecipitated with endogenous FIAT (Fig. 2 C, right). The use of unrelated antibodies or protein A–Sepharose alone confirmed the specificity of the immunoprecipitation reactions (Fig. 2 C, lanes 1 and 2 on each panel). These results demonstrate that FIAT and ATF4 interact in mammalian osteoblasts.
Figure 2.

FIAT interacts with ATF4. (A and B) Yeast two-hybrid protein interaction assays. (A) Growth on two-minus selection media. (B) Growth on three-minus selection media. (A and B) position 1, FIAT “bait” + ATF4 “target;” position 2, negative control; position 3, positive control; position 4, FIAT “bait” + FIAT “target” without leucine zipper; position 5, FIAT “bait” + FIAT “target;” position 6, negative control. Note that on three-minus selection media only yeasts at position 1 (FIAT + ATF4) and position 3 (positive control) grow, demonstrating a positive interaction between FIAT and ATF4. The failure of yeasts to grow at positions 4 and 5 confirm that FIAT does not homodimerize. (C) Coimmunoprecipitation. Endogenous ATF4 was immunoprecipitated from ROS 17/2.8 osteoblastic cells using an anti-ATF4 antibody, and the immunoprecipitates were probed with the anti-FIAT antibody (left). Reciprocally, endogenous FIAT was immunoprecipitated from ROS 17/2.8 cells using the anti-FIAT antibody, and the immunoprecipitates were probed with an anti-ATF4 antibody (right). FIAT was coimmunoprecipitated with ATF4 (left, lane 3) and ATF4 was also coimmunoprecipitated with FIAT (right, lane 3). Protein A–Sepharose or unrelated antibodies failed to precipitate the target proteins (lanes 1–2).
FIAT represses ATF4 DNA binding and transcriptional activity
ATF4 was recently characterized as an important transcriptional regulator of osteoblast differentiation binding to the OSE1 site of the osteocalcin promoter to regulate osteocalcin gene transcription (Yang et al., 2004). ATF4 was shown to be a substrate for the RSK2 kinase, and phosphorylation by RSK2 enhanced the transcriptional activity of ATF4 (Yang et al., 2004). We thus examined whether or not the interaction of FIAT with ATF4 would impact on the DNA-binding and transcriptional activity of ATF4 and regulate osteocalcin gene transcription in mammalian cells. In the first series of experiments, COS-7 cells were cotransfected with a reporter construct in which canonical ATF4 binding sites were subcloned upstream of the thymidine kinase minimal promoter region and expression vectors for ATF4 and FIAT. The recombinant ATF4 protein strongly induced the transcription of the reporter gene, whereas FIAT by itself was without effect (Fig. 3 A). Coexpression of FIAT with ATF4 significantly inhibited ATF4-mediated transcription (Fig. 3 A). To test the impact of FIAT on the ATF4-mediated activation of osteocalcin gene transcription, osteoblastic MC3T3-E1 cells were transfected with a reporter construct in which six copies of the OSE-1 regulatory element from the osteocalcin gene promoter, which binds ATF4 (Yang et al., 2004), were introduced upstream of the adenovirus type 2 major late promoter fused to the luciferase reporter gene (vector OSE1-luc; Ducy and Karsenty, 1995). The cells also received expression vectors for ATF4, FIAT, and RSK2. As previously reported (Yang et al., 2004), ATF4 induced the transcription of the OSE1-luc reporter, and this effect was enhanced by RSK2 coexpression (Fig. 3 B). Increasing amounts of FIAT suppressed the transcriptional activity of ATF4, even in the presence of RSK2 (Fig. 3 B). A control reporter vector in which the OSE1 response element was mutated did not respond to ATF4 expression (Fig. 3 B, mutOSE1). FIAT by itself was without effect (Fig. 3 B). Similar results were obtained when MC3T3-E1 or COS-7 cells were transfected with a reporter construct under the control of the proximal 147-bp osteocalcin gene promoter (unpublished data). These experiments demonstrated that FIAT was able to inhibit ATF4-mediated transcription and that this inhibition was maintained even when ATF4 was activated by phosphorylation.
Figure 3.

FIAT represses ATF4-mediated transcription and binding of ATF4 to DNA. (A) COS-7 cells were transfected with a luciferase reporter gene under the control of three canonical ATF4 binding sites, together with expression vectors for human ATF4 and human FIAT. (B) MC3T3-E1 osteoblastic cells were transfected with a reporter construct in which the luciferase gene was regulated by six copies of the wild-type (OSE1-luc) or mutated (mut OSE1) OSE1 sequence from the mouse osteocalcin gene promoter. Cotransfected plasmids included expression vectors for ATF4, RSK2, or FIAT (2× signifies that twice the amount of plasmid was used). Reporter gene activity was measured with a luminometer 24 h after transfection. The activity measured in cells transfected with the reporter construct and empty expression vectors was arbitrarily ascribed a value of 1. FIAT inhibited ATF4-induced transcription from both templates (A and B), even in the presence of an expression vector for the RSK2 kinase (B). FIAT expression by itself does not affect transcription from the reporter templates. (C) EMSA with an OSE1 oligonucleotide probe, ROS 17/2.8 nuclear extract, and recombinant FIAT. The ATF4 binding complex was identified using specific anti-ATF4 antibodies (lanes 3–5). FIAT did not bind the probe but inhibited binding of ATF4 to the DNA.
To examine the mechanisms involved, we performed electrophoretic mobility shift assays (EMSAs) with nuclear extracts from ROS 17/2.8 osteoblastic cells and recombinant FIAT protein. The probe used was a 26-bp oligonucleotide corresponding to the OSE1 binding element and flanking nucleotides from the mouse osteocalcin promoter sequence. We observed the previously reported multiple retarded complexes between nuclear ROS 17/2.8 proteins and the probe (Ducy and Karsenty, 1995; Schinke and Karsenty, 1999; Yang et al., 2004), and the ATF4 binding complex was identified using specific inhibition of DNA binding by anti-ATF4 antibodies (Fig. 3 C, lanes 1–5). Recombinant FIAT protein did not bind the OSE1 probe (Fig. 3 C, lane 8), but dose-dependently inhibited the binding of ATF4 to the OSE1 element (Fig. 3 C, lanes 6 and 7). Together with the data presented in Fig. 2, we interpret these results to mean that FIAT heterodimerizes with nuclear ATF4 to prevent its binding to DNA and repress ATF4-mediated gene transcription.
Reduced bone mass in mice expressing a FIAT transgene
To study the impact of FIAT on osteoblast development in vivo, transgenic mice expressing a FIAT transgene were generated. The transgene contained the full-length 1.8-kb FIAT cDNA fused to the 2.3-kb α1(I) collagen promoter (Fig. 4 A). This promoter fragment is known to direct transgene expression specifically in osteoblasts but not in other type I collagen-producing cells (Rossert et al., 1995; Dacquin et al., 2002). Tissue specificity of transgene expression was assessed by RT-PCR, and results confirmed that the transgene was expressed only in bone but not in other tissues (Fig. 4 B). The level of transgene expression in bone tissue was comparable to endogenous FIAT gene expression (Fig. 4 C). Histological analysis of tibial sections obtained from 3-mo-old mice revealed an osteopenic phenotype in transgenic mice. The bone mass reduction was manifested as decreased number and size of trabeculae as well as disturbed secondary centers of ossification (Fig. 4 D). The same phenotype was observed in another strain of transgenic mice, confirming that it was not integration site dependent (unpublished data).
Figure 4.
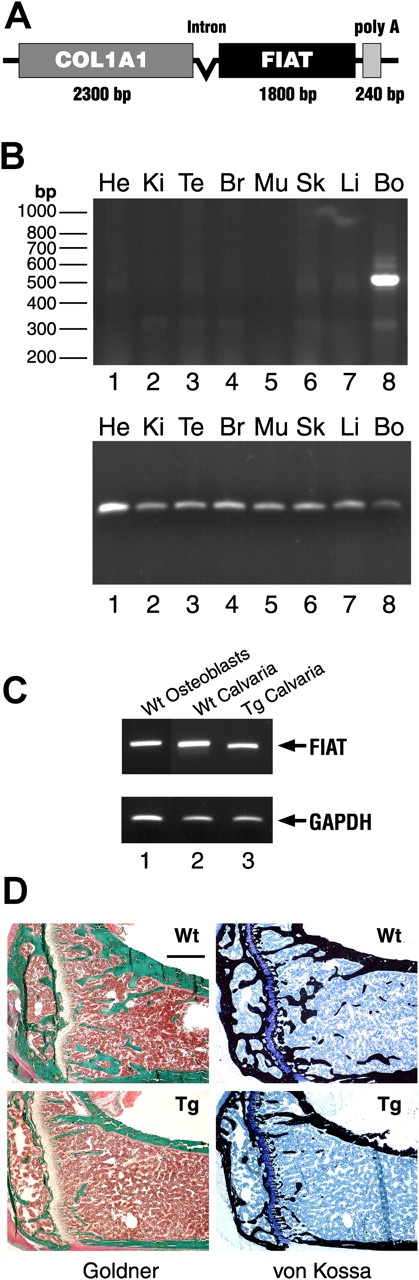
FIAT transgenic mice. (A) Schematic representation of the Col 1-FIAT transgene. Poly A, SV40 polyadenylation signal. (B) RT-PCR assay of transgene expression. Top panel shows specific FIAT transgene expression using primers from the FIAT sequence and the SV40 polyadenylation sequence. Bottom panel shows GAPDH expression. He, heart; Ki, kidney; Te, tendons; Br, brain; Mu, skeletal muscle; Sk, skin, Li, liver; Bo, bone. (C) Endogenous FIAT and transgene expression in osteoblasts and bone. RT-PCR with primers for endogenous FIAT (lanes 1 and 2) or primers specific for the transgene (lane 3). RNA was from osteoblast primary cultures (lane 1) or calvaria. Wt, wild type; Tg, transgenic. (D) Reduced bone mass in FIAT transgenic mice. Goldner (left) and von Kossa (right) stains of tibial sections from FIAT transgenic mice (bottom) and wild-type littermates (top) at 3 mo old. Note the smaller trabeculae and reduced secondary center of ossification in transgenic animals. Bar, 500 μM.
The osteopenic phenotype of the FIAT transgenic mice was further characterized using dual energy X-ray absorptiometry (DEXA), histomorphometry, and biomechanical analyses. DEXA scanning revealed a significant decrease (21%) in bone mineral density between transgenic animals and wild-type littermates (Fig. 5 A). Detailed histomorphometric measurements demonstrated a dramatic decrease in bone volume (70%), mineralized bone volume (45%), trabecular thickness (38%), and trabecular number (55%) (Fig. 5, B–E). Although the number of osteoblasts was not statistically different between transgenic animals and littermate controls (Fig. 5 G), measurements of double-labeled tetracycline tibial sections (Fig. 5, J and K) revealed a lowered mineral apposition rate (46%) and bone formation rate (52%) in the transgenics (Fig. 5, H and I). The same trends were observed in younger transgenic animals at 3 wk of age (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200412139/DC1). In 6-mo-old mice where bone formation rates are decreased, static histomorphometric parameters were similar between wild-type and transgenic littermates (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200412139/DC1).
Figure 5.
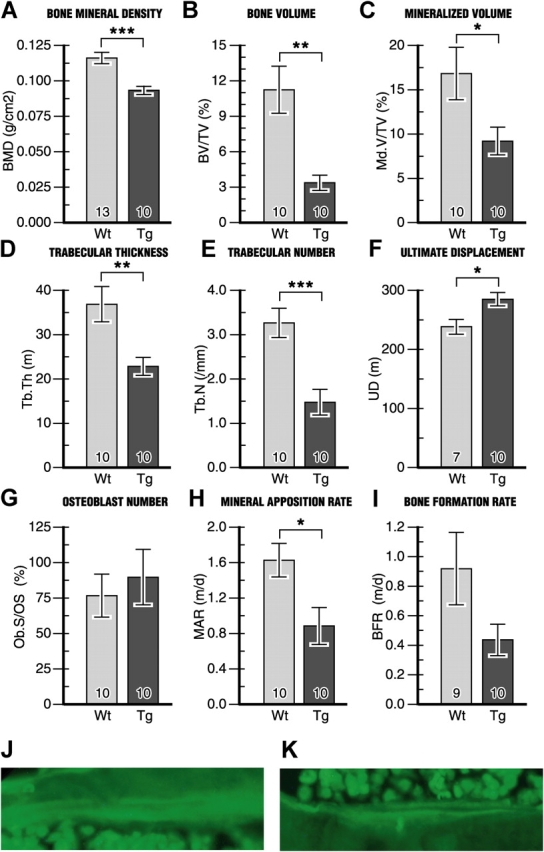
Histomorphometric analysis of bones from FIAT transgenic mice compared with wild-type littermate controls at 3 mo old reveals osteopenia. (A–I) The number of samples is indicated within each bar. (J and K) Double-labeling of bone surfaces with demeclocycline used to measure dynamic indices in H and I. Tg, transgenic; Wt, wild type. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To determine if the measured changes had any effect on the mechanical properties of bone, femurs were subjected to mechanical testing using the three-point bending method. Bones from transgenic animals exhibited a 20% increase in ultimate displacement compared with wild-type controls (Fig. 5 F), demonstrating that the reduced bone mineral density, lowered bone mass, and disrupted trabecular architecture resulted in a loss of rigidity in bones of transgenic animals.
FIAT impairs osteoblast activity
A decrease in bone mass can be secondary to endocrine dysfunction or result from defects in bone resorption or formation. Biochemical analysis of calcium, phosphate, and alkaline phosphatase levels in blood revealed no differences between blood parameters in wild type and transgenics (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200412139/DC1), ruling out perturbations in mineral homeostasis and abnormal turnover as the cause of the phenotype. Similarly, histomorphometric measurement of osteoclast number, TRAP staining, and assessment of osteoclastic activity by collagen cross-link assays showed similar results between the two genotypes (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200412139/DC1), eliminating any effect of the transgene on the number and activity of osteoclasts.
To determine if FIAT transgene expression altered osteoblast proliferation and/or apoptosis, osteoblasts were stained with PCNA to determine the rate of osteoblast proliferation, as well as by the TUNEL assay to evaluate apoptosis. These assays showed that FIAT did not affect the proliferation or the apoptotic rate of osteoblasts (Fig. 6), thus ruling out such mechanisms as causes of the osteopenic phenotype. Histomorphometric data had shown an attenuated mineral apposition rate and bone formation rate in transgenic mice (Fig. 5, H–K), hinting at the possibility of a defect in osteoblastic activity. When primary osteoblast cultures obtained from 7-d-old calvaria were analyzed for the production of alkaline phosphatase, a marker of osteoblastic differentiation and function, a statistically significant 34% decrease was observed in transgenic mice compared with wild type (Fig. 7 A). Similarly, alizarin red staining of mineralized nodules formed by primary osteoblast cultures exhibited a 70% reduction in transgenic animals compared with wild type (Fig. 7 B). Primary osteoblast cultures established from 3-mo-old bone marrow stromal cells also displayed a similar pattern (Fig. 7, C and D). The decrease in mineralization was readily evident in alizarin red–stained cultures (Fig. 7, E and F).
Figure 6.
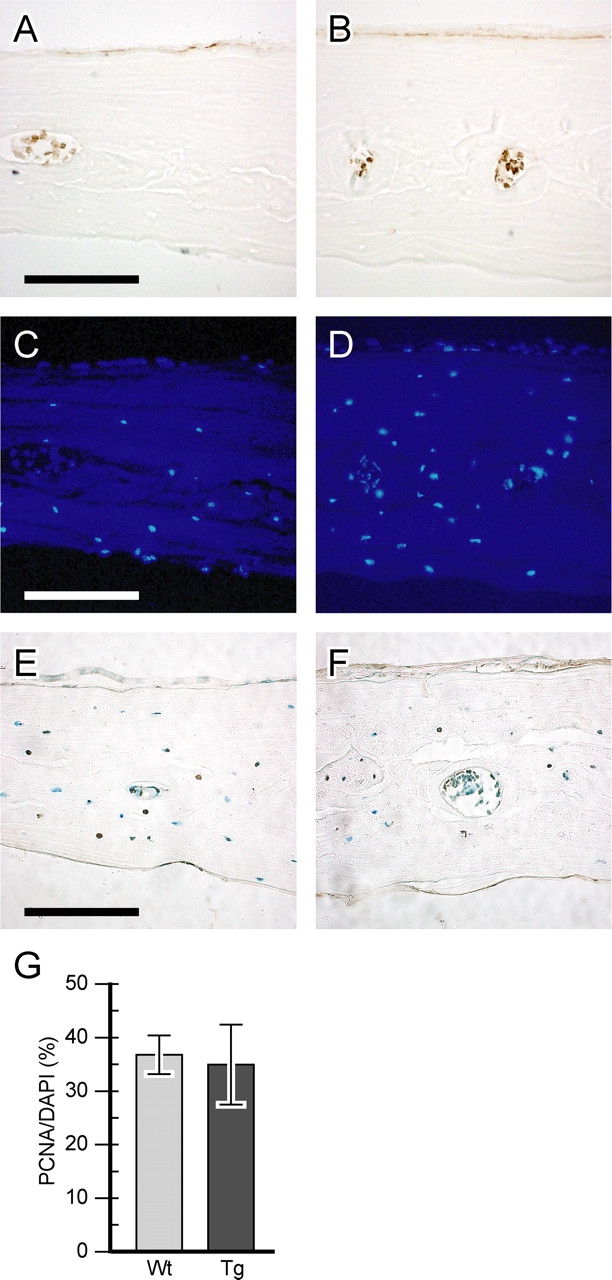
Normal osteoblast proliferation and survival in FIAT transgenic mice. Calvarial sections from wild-type (A and C) or transgenic (B and D) animals were stained for PCNA (A and B) and with a DAPI nuclear stain (C and D). Proliferation was graphed as PCNA-positive cells over DAPI-positive cells (G). Sections from wild-type (E) and transgenic (F) littermates were stained by TUNEL to assess apoptosis. Bars, 100 μM.
Figure 7.
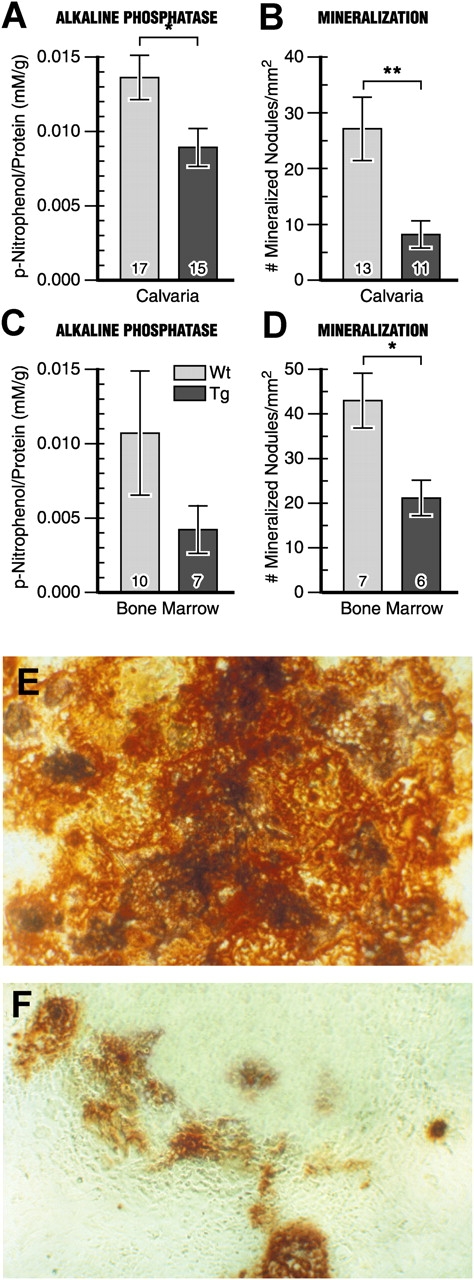
Reduced activity of FIAT transgenic osteoblasts. Primary cultures of osteoblasts were obtained from 6–8-d-old calvaria (A and B) or 3-mo-old bone marrow stromal cells (C and D). Cells were assayed for AP level after 7 d or stained with alizarin red after 14 d. Number of samples assayed is indicated within each bar. AP concentration was normalized by protein concentration per well (A and C). Dark red mineralized nodules were expressed per squared millimeter (B and D) and cells were photographed with bright-field microscopy (E, wild type; F, transgenic). *, P < 0.05; **, P < 0.01.
To evaluate the impact of FIAT transgene expression on the transcription of markers of osteoblast differentiation, RNA levels of bone markers such as Ocn, Fiat, Atf4, Runx2/Cbfa1, Osx, and Bsp, were quantitated by real-time RT-PCR (Fig. 8). FIAT transgene expression inhibited Ocn transcription (Fig. 8 A), as was observed in transient transfection assays (Fig. 3). Fiat expression was elevated in the transgenics because the real-time assay could detect transcripts from both the endogenous and transgenic Fiat alleles. Atf4, Runx2/Cbfa1, and Osx gene expression were unchanged, whereas Bsp mRNA levels were increased. Together, these data suggest that deregulated FIAT expression in bone cells caused an osteopenic phenotype due to a perturbation in osteoblastic activity, without much impact on differentiation.
Figure 8.

Expression of osteoblast expression markers. RNA was extracted from 3-mo-old calvaria, reverse-transcribed, and amplified with specific TaqMan probes using real-time PCR. Relative expression between wild-type (Wt) and transgenic (Tg) animals was calculated by the relative standard curve method and normalized to GAPDH. *, P < 0.05.
ATF4 controls the transcription of genes regulating the cellular import of amino acids (Harding et al., 2003). This was shown to affect the production of the major secreted protein of osteoblasts, type I collagen, and thus ATF4-deficient bones have less type I collagen in the matrix (Yang et al., 2004). We examined whether or not FIAT transgenic bones were similarly affected. van Gieson staining of tibial sections from wild-type and transgenic littermates did not reveal major changes in type I collagen content in the matrix between the two genotypes (Fig. 9, A and B). This finding was confirmed using quantification of col1A1 transcription by real-time RT-PCR (Fig. 9 C) and by measuring hydroxyproline content in bone tissue (Fig. 9 D). In addition, type I collagen synthesis from primary cultures of osteoblasts, in the presence or absence of nonessential amino acids, was unaffected by the expression of the FIAT transgene (Fig. 9 E). Thus, deregulated FIAT transgene expression mimics some, but not all, aspects of the phenotype of ATF4-deficient bone. Nevertheless, our results show that perturbing FIAT expression in osteoblasts impacts on bone mass accrual and strongly suggest that FIAT is a key determinant of osteoblast function.
Figure 9.
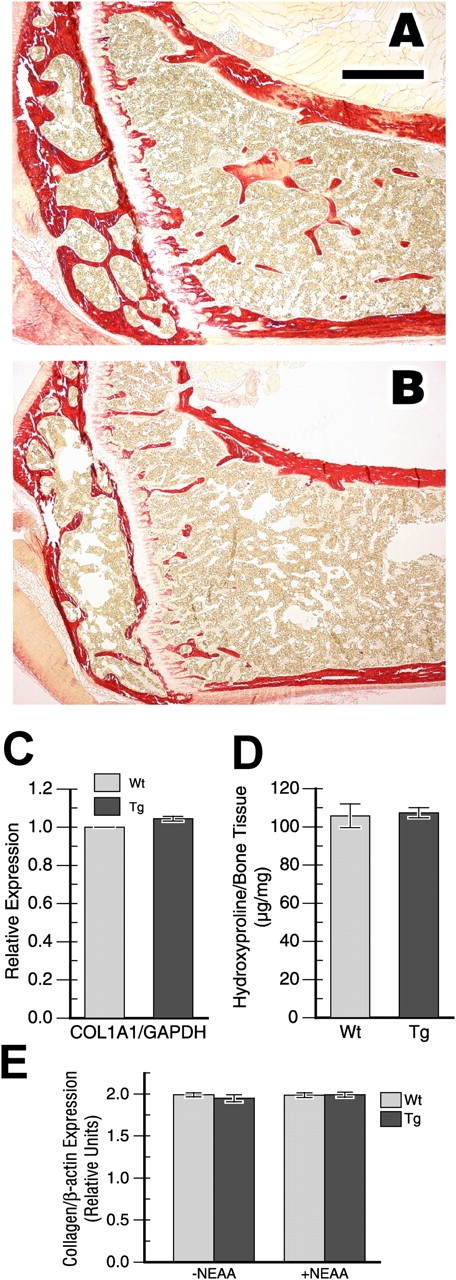
Type I collagen expression and synthesis in FIAT transgenic mice. Femoral sections from wild-type (A) or FIAT transgenic (B) littermates were stained with the van Gieson dye to examine type I collagen expression. Bar, 500 μM. (C) Calvarial Col1A1 mRNA levels were quantified by real-time PCR. (D) Collagen protein content in whole tibias from 3-mo-old mice was estimated using hydroxyproline quantification. (E) Type I collagen synthesis analysis. The histogram shows expression of [3H]proline–labeled α1 and α2 chains of type I collagen relative to β-actin expression (n = 9 independent primary osteoblast cultures from different litters for each bar). NEAA, nonessential amino acids; Wt, wild-type; Tg, transgenics.
Discussion
This study uncovers FIAT as a novel leucine zipper protein that regulates bone mass. Mice specifically expressing a FIAT transgene in osteoblasts exhibited an osteopenic phenotype characterized by reduced bone mineral density, bone volume, trabecular number, and bone rigidity. Furthermore, this bone mass reduction was due to defective osteoblastic function without changes in osteoblast proliferation or apoptosis. We hypothesize that inhibition of ATF4 transcriptional activity through heterodimer formation may be one of the molecular mechanisms by which FIAT controls bone mass.
FIAT transgene expression
The level of expression of the FIAT transgene was comparable to the expression level measured for the endogenous FIAT allele (Fig. 3 C), leading to a slight overexpression when quantified by real-time RT-PCR (Fig. 8 B). This translated into slightly increased FIAT protein expression in primary osteoblasts cultured from transgenic animals (unpublished data). We propose that the slight overexpression combined with the altered transcriptional regulation of FIAT expression conferred by the type I collagen promoter driving the transgene jointly impact on the osteoblast to affect its function.
FIAT regulates bone mass by affecting osteoblast activity
Bone formation is dependent on the equilibrium between bone resorption by osteoclasts and bone formation by osteoblasts (Karsenty and Wagner, 2002). Resorption is influenced by osteoclast number and activity. Our results showed that these parameters were not affected in FIAT transgenic mice. In contrast, bone formation is influenced by osteoblast number and activity. The number of osteoblasts is a function of cell differentiation, proliferation, and survival. We assessed differentiation using a battery of markers and RNA isolated from bones, primary calvarial cultures from newborn animals, and marrow stromal cell cultures of adult (3-mo-old) mice. Relative expression of all markers assessed was similar irrespective of the source of the RNA (unpublished data). Our results show no differences between the expression of most of the markers tested, except an increase in Bsp transcript levels and a decrease in Ocn gene transcription (further discussed in the following section). We interpret these results to mean that FIAT transgene expression has no major effect on osteoblast differentiation.
Similarly, osteoblast proliferation and survival (apoptosis) were unaffected by the FIAT transgene. However, dynamic histomorphometry revealed a reduction in mineral apposition rate and bone formation rate in transgenic mice, thus suggesting that decreased osteoblastic activity was responsible for the osteopenic phenotype. This hypothesis was confirmed by measuring reduced alkaline phosphatase activity and diminished mineralization by primary osteoblast cultures. These assays also proved that the effect of the transgene is cell-autonomous, as expected, because FIAT is not a secreted protein but resides in the nucleus.
Molecular mechanisms of FIAT repression of ATF4
The most plausible explanation for FIAT repression of ATF4-mediated transcription is that FIAT inhibits ATF4 by dimerizing with ATF4 through the leucine zipper region. Because FIAT does not have a readily identifiable DNA binding domain, we surmised that it could prevent ATF4 from binding to the DNA to activate transcription by forming inactive heterodimers. We have shown that this is the case using recombinant FIAT and nuclear extracts from osteoblastic ROS 17/2.8 cells (Fig. 3 C). Further structure-function analysis with deleted or site-specifically mutated ATF4 and FIAT recombinant proteins are underway to more precisely define the respective domains that mediate interactions between the two proteins. Other nonmutually exclusive possible mechanisms include FIAT interference with the interaction between ATF4 and other transcription factors or FIAT recruitment of corepressors to the chromatin. Additional experiments will be required to determine whether or not these mechanisms contribute to the FIAT repressor activity.
FIAT repressed osteocalcin gene transcription in cultured osteoblasts and in transgenic bone. The inhibition observed in bones from FIAT transgenic animals was not as pronounced as that measured in ATF4-deficient mice (Yang et al., 2004). The inhibition of ATF4 transcriptional activity by FIAT would be dependent on the relative expression of FIAT and ATF4 in transgenic animals, and it is possible that FIAT transgene expression was not high enough to block all ATF4-mediated transcription of the osteocalcin gene.
It is noteworthy that the inhibition of osteocalcin gene expression observed in this study correlated with a decrease in osteoblast function, whereas total osteocalcin gene ablation leads to an increase in osteoblast activity (Ducy et al., 1996). Although it is possible that this difference only reflects the relative degree of inhibition of osteocalcin expression, it is more likely that ablation of osteocalcin, a matrix constituent, leads to a different phenotype than the perturbation of a transcriptional regulatory cascade that, although it regulates the transcription of osteocalcin, impacts on other transcriptional events early in the differentiation program of the osteoblast.
Repression of ATF4-mediated transcription by FIAT is most likely responsible for the aspects of the FIAT transgenic mice phenotype that are a phenocopy of the ATF4-deficient animals, such as reduced osteocalcin transcription, low bone mass, and reduced osteoblast activity (Yang et al., 2004). Several differences exist between the two strains, however. As previously mentioned, transgenic FIAT expression did not have a major impact on the onset or extent of osteoblast differentiation. FIAT also did not affect type I collagen synthesis (Fig. 9). Type I collagen is the most abundant protein produced by osteoblasts and constitutes the majority of the mineralization scaffold in bone. ATF4 regulates amino acid import in several cell types (Harding et al., 2003), and the reduction in type I collagen synthesis in ATF4-deficient mice was associated with this function of the ATF4 protein (Yang et al., 2004). It remains possible that FIAT does not modulate this activity of the ATF4 protein and thus would not impact on type I collagen synthesis. Alternatively, our results could be due to the relatively low level of FIAT overexpression achieved in the transgenics. Additional studies (including structure-function analysis of the FIAT–ATF4 interaction and its impact on the transcription of other ATF4 target genes besides osteocalcin, FIAT RNA knock-down, or FIAT ablation by gene targeting) will be required to elucidate this point.
It also remains a formal possibility that FIAT may disturb bone formation by other mechanisms in addition to suppressing ATF4 transcriptional activity. FIAT could heterodimerize with additional bZip transcription factors, such as Fra-1 or ΔFosB, that have been shown to regulate bone mass accrual (Jochum et al., 2000; Sabatakos et al., 2000). Alternatively, FIAT could sequester dimerization partners away from bone mass–controlling bZip factors. A likely candidate remains c-Jun, a well established dimerization partner of Fos family members (Hartl et al., 2003).
At any rate, our results show that the transcriptional control of osteoblast activity is a tightly regulated phenomenon and confirm that protein–protein interactions play a critical role in the precise regulation of gene expression in bone-forming cells.
Materials and methods
Yeast two-hybrid
cDNA libraries from osteoblastic MC3T3-E1 cells or cultured primary osteoblasts were prepared in the pAD-GAL4 vector (Stratagene) using the HybriZAP Two-Hybrid cDNA Gigapack cloning kit and following the manufacturer's instruction. The αNac or FIAT baits were prepared by subcloning the respective cDNAs into the GAL4-DBD expression plasmid (Stratagene). The screen was performed according to the manufacturer's protocols, and interaction between molecules was identified in yeast that were grown in selective medium lacking three essential amino acids: TRP, LEU, and URA.
Western blotting, immunocytochemistry, and coimmunoprecipitation
A peptide corresponding to FIAT residues 111–125 was synthesized, coupled to ovalbumin, and used to raise rabbit polyclonal antibodies following standard protocols. The polyclonal antisera (1:1,500 dilution) was used to probe immunoblots of nuclear extracts from osteoblastic MC3T3-E1 cells. Anti–rabbit antibodies conjugated with HRP (1:25,000 dilution) were used as secondary antibodies and detected by ECL Western blotting detection reagents (Amersham Biosciences). The antibody was also used for indirect immunofluorescence. Primary cultures of osteoblasts were obtained from 6–8-d-old calvaria. In brief, calvaria were minced and digested sequentially (5 × 10 min) with αMEM containing 0.2% collagenase D (Roche) and 0.1% hyaluronidase (Roche). Fractions 3 to 5 were collected and plated in a 6-well plate with αMEM containing 10% FBS, 2% Glutamax, 1% penicillin, 1% streptomycin, and 1% Fungizone (Invitrogen). Cells attached for 7 d and were cultured in the presence of 10−8 M dexamethasone (Porter et al., 2003; Yang et al., 2003). The primary osteoblasts were fixed in 4% PFA and permeabilized with 0.2% Triton X-100. After blocking with 1% blocking reagent (Roche) supplemented with 0.2% Tween-20, the cells were incubated with the anti-FIAT antibody (1:200). The cells were incubated for 1–2 h at RT with a rhodamine-conjugated anti–rabbit IgG secondary antibody (dilution 1:500) to detect endogenous FIAT protein. Coverslips were mounted in Vectashield (with DAPI) mounting medium (Vector Laboratories).
For coimmunoprecipitation, 100–200 μg of nuclear proteins from ROS 17/2.8 osteoblastic cells in buffer D (20 mM Hepes-KOH, pH 7.9, 25% glycerol, 150 mM NaCl, 1.5 mM MgCl2, and 0.2 mM EDTA with inhibitors of proteases: 5 μg/ml leupeptin, aprotinin, pepstatin A, and 1 mM PMSF) were precleared with 50 μl of protein A–Sepharose slurry (Amersham Biosciences) for 1 h at 4°C with gentle rocking. After centrifugation, the cleared extract was incubated overnight at 4°C with gentle rocking, with or without 1–2 μg of specific or unrelated antibody, and with 50 μl of protein A–Sepharose slurry. The precipitates were washed with hypotonic buffer (10 mM Hepes-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, and protease inhibitors as above) and resuspended in 25 μl of PBS. A 10-μl aliquot was mixed with SDS-sample buffer, separated on 12% SDS-PAGE, and transferred to PVDF, and the blots were probed with anti-ATF4 antibodies (Santa Cruz Biotechnology, Inc.) or anti-FIAT antiserum, and then with anti–rabbit secondary antibody conjugated to HRP. Proteins were detected by chemifluorescence with ECL Plus Western blotting detection reagents (Amersham Biosciences).
Transfection experiments
COS-7 or MC3T3-E1 cells were plated at 1.5 × 105 cells/well in a 6-well plate and transfected with 400 ng of the ATF sites reporter (provided by T. Hai, Ohio State University, Columbus, OH; Liang and Hai, 1997) or 1 μg of the OSE1-luc reporter (Ducy and Karsenty, 1995), 400 ng of pCMV5ATF4, 25 ng of pK3HRSK2 (all gifts of G. Karsenty, Baylor College of Medicine, Houston, TX), 400 or 800 ng of pcDNA3.1/V5-HIS-TOPOFIAT, 50 ng of pSV6TKCAT (obtained from R. Tjian, University of California, Berkeley, Berkeley, CA), and a varying amount of pBluescript reporter vectors using Lipofectamine (Invitrogen) reagents according to the manufacturer's instructions. Luciferase assay and chloramphenicol acetyltransferase ELISA (Roche) were performed 24 h after transfection following the manufacturer's protocol. Data represent ratios of luciferase/chloramphenicol transferase activity and values are the means of three independent transfection experiments performed in duplicate.
EMSAs
Complementary oligonucleotides corresponding to the OSE1 binding site within the murine osteocalcin proximal promoter region (5′-CCTGCTCCTCCTGCTTACATCAGAGA-3′) were synthesized with an overhang, annealed, and labeled with [32P]-labeled dNTPs by Klenow fill-in using standard protocols (Ausubel et al., 1993).
The recombinant FIAT protein was purified using NEB's IMPACT system as described previously (Quelo et al., 2002). ROS17/2.8 nuclear extracts were prepared following the technique of Andrews and Faller (1991).
Recombinant protein (300 or 600 ng) or nuclear extracts (10 μg) were incubated for 30 min at 4°C in 20 μl of binding buffer (20 mM Hepes, pH 7.9, 60 mM KCl, 1 mM DTT, 1 mM EDTA, 100 ng of polydI-dC, and 12% glycerol). Labeled probe (10,000 dpm) was added to the binding reaction mixture. For supershift assays, anti-ATF4 or unrelated antibody (2–4 μg; Santa Cruz Biotechnology, Inc.) was added to the binding reaction for 30 min before the addition of the labeled probe. The bound mixtures were size-fractionated on a nondenaturing 5% polyacrylamide gel at 160 V for 100 min in 0.5× Tris-borate-EDTA buffer. The gel was subsequently dried and autoradiographed.
Generation of FIAT transgenic mice
FIAT transgenic mice were generated using a pCI (Promega)-based vector containing the osteoblast-specific 2.3-kb α1(I)collagen promoter (a gift from B. de Crombrughe, MD Anderson Cancer Center, Houston, TX; Rossert et al., 1995), an SV40 small intron, the 1.8-kb full-length FIAT cDNA, and the 240-bp SV40 polyadenylation signal. The linearized construct was injected at 1 μg/ml into fertilized eggs using standard methodology (Hogan et al., 1994). Founder animals were detected by Southern blot. Tissue specificity of transgene expression was examined by RT-PCR. The transgenic line was maintained by crossing with wild-type C57BL/6 mice and genotyped by PCR using FIAT primers (5′-ATCCATCAAAGCGCCATCAAAGCG-3′ and 5′-ACAAATAAAGCAATAGCATCACAA-3′) and Gapdh (glyceraldehydes-3-phosphate dehydrogenase) primers (5′-CACCATGGAGAAGGCCGGG-3′ and 5′-GACGGACACATTGGGGGTAG-3′). Mice were killed and analyzed at 3 mo old. Demeclocycline (Sigma-Aldrich) was injected at 30 ng/g of mice twice at 5-d intervals before sacrifice. At least 10 animals of each genotype were used for all experiments, and statistical significance was assessed by t test. Error bars represent SEM. All animal experimentation was approved by the Institutional Animal Care and Use Committee.
RT-PCR
RNA from primary osteoblasts or 3-mo-old calvaria was extracted with TRIzol (Invitrogen) following the manufacturer's instructions. 5 μg of RNA were reverse transcribed into cDNA with 500 ng of random primers, 25 mM dNTPs, 5 μl of 0.1 M DTT, 10 μl 5× first strand buffer, 3.5 μl RNAguard (Roche), and 2 μl M-MLV reverse transcriptase (Promega). Endogenous or transgenic Fiat and Gapdh genes were amplified by PCR using the primers described in the section Generation of FIAT transgenic mice.
Morphological analysis
Bone mineral density was determined by DEXA and data were analyzed by Lunar PIXImus software before mice were killed. Bone samples were fixed in 4% PFA, either embedded in methyl methacrylate, or decalcified and embedded in paraffin for future studies. For histological analysis, methyl methacrylate–embedded tibiae sections (5 μm) were stained with Goldner, Toluidine blue, and von Kossa reagents. Histomorphometric measurements were performed using Nova Prime software (BioQuant Image Analysis Corporation). Femurs preserved in PBS were first scanned with microcomputerized tomography (MicroCT) before performing three-point bending test at the Centre for Bone and Periodontal Research of McGill University.
Osteoblast proliferation and apoptotic experiments
Demineralized paraffin-embedded calvarial sections (6 μm) were used for all immunohistochemical experiments. Proliferation rate of osteoblasts was determined by the Proliferating Cell Nuclear Antigen (PCNA) Staining Kit (Zymed Laboratories) following the manufacturer's instructions. Sections were counterstained with DAPI (Vector Laboratories), and values were expressed as the number of proliferating osteoblasts over the number of DAPI-positive cells. Apoptotic rate of osteoblasts was assessed using the TUNEL-based in situ Cell Death Detection Kit, POD (Roche), followed by staining with DAB substrate (Vector Laboratories). Experiments were performed according to the manufacturers' instructions except that sections were unmasked with 0.1% trypsin and counterstained with methyl green.
Colorimetric determination of AP and alizarin red staining
Primary cultures of osteoblasts were obtained from 6–8-d-old calvaria or 3-mo-old bone marrow stromal cells. Osteoblasts from calvaria were prepared as described in the section Western blotting, immunocytochemistry, and coimmunoprecipitation. For osteoblasts from bone marrow stromal cells, epiphysis from both ends of femurs were cut off. Bone marrow cells were flushed out with a 22-gauge needle with the previous medium, resuspended, and plated at 106 cells per well in a 6-well plate. After cell attachment on day 4, medium was changed every 3 d, provided with additional 50 μg/ml ascorbic acid (Franceschi and Iyer, 1992), 10 mM β-glycerophosphate (Quarles et al., 1992), and 10−8 M dexamethasone (Porter et al., 2003; Yang et al., 2003). Cells were assayed for AP level after 7 d or stained with alizarin red after 14 d. Cells were trypsinized with 0.05% trypsin-EDTA and plated at 5 × 104 cells/well in two 12-well plates separately. For colorimetric determination of AP level, cells were fixed by 4% PFA and lysed. 1 mg/ml 4-nitrophenyl phosphate (Sigma-Aldrich) was added to each well and incubated for 30 min before reading at 405 nm. Protein concentration in cell lysate was determined by the Bradford assay. AP concentration was normalized by protein concentration per well. For alizarin red staining, cells were fixed with 4% PFA and stained with 0.5% alizarin red (Sigma-Aldrich) at pH 5.0. Dark red mineralized nodules were expressed per squared millimeter and cells were photographed with bright-field microscopy.
Real-time RT-PCR
RNA from 3-mo-old calvaria was extracted with TRIzol (Invitrogen) following the manufacturer's instructions. 5 μg of RNA were reverse transcribed into cDNA using the High Capacity cDNA Archive kit as per the manufacturer's recommendations (Applied Biosystems). Real-time PCR amplification was performed on a 7700 instrument (Applied Biosystems) using specific TaqMan assays for Osteocalcin (OCN), Fiat, Atf4, Runx2/Cbfa1, Osterix (Osx), Bone Sialoprotein (Bsp), or type I collagen (col1A1) and the TaqMan Universal PCR Master Mix (Applied Biosystems). Expression level of each mRNA was quantified by the relative standard curve method (User bulletin #2; ABI Prism 7700 Sequence Detection System) and normalized to Gapdh levels.
van Gieson staining
The van Gieson stain was prepared by dissolving 0.5 g of Sirius red F3B (Direct Red; Sigma-Aldrich) in 500 ml of saturated picric acid in water. Deparaffinized, rehydrated sections were stained for 1 h in van Gieson stain, rinsed in 0.5% acetic acid, dehydrated in three changes of 100% ethanol, cleared in xylene, and mounted.
Analysis of collagen content and synthesis
The collagen content of bones was analyzed as hydroxyproline content by the method of Burleigh et al. (1974). In brief, demineralized tibial bone (typically 2–5 mg) was hydrolyzed in 6 M HCl (40 μl/mg) at 110°C for 20 h. A 35-μl aliquot of the digest was neutralized by the addition of 35 μl of 6 M NaOH, and the solution was then diluted by the addition of 1 ml of water and clarified by the addition of activated charcoal resin. After centrifugation, a 20-μl aliquot of the supernatant was analyzed for hydroxyproline content after treatment with chloramine T reagent and color development with dimethylaminobenzaldehyde. Absorbance was measured at 560 nm and compared with hydroxyproline standards (0.1–5 μg in 20 μl). Collagen synthesis by osteoblasts was determined following the technique described by Yang et al. (2004) with the following modifications: wild-type and FIAT transgenic primary osteoblasts were labeled for 12 h with 50 μCi/ml of [3H]proline (Amersham Biosciences) in high-glucose DME (GIBCO BRL) supplemented with 2% dialyzed FBS, 2 mM Glutamax (GIBCO BRL), and 55 μM β-mercaptoethanol in the presence or absence of nonessential amino acids mix. Cells were homogenized in 500 μl of PBS with protease inhibitors (5 μg/ml leupeptin, aprotinin, pepstatin A, and 1 mM PMSF). 50 μl were used for protein quantification using the Bradford assay. A 200-μl additional fraction served for β-actin determination by Western blotting. The remaining 250 μl of cell homogenate were digested to collagen with 50 μg/ml of pepsin, precipitated, and resolved by SDS-PAGE on a 7% gel with 2 M urea as described previously (Yang et al., 2004). The wet gels were soaked in amplifying reagent (Amplify; Amersham Biosciences), dried, and exposed to Hyperfilm MP (Amersham Biosciences). The intensity of the signal was quantified using the GeneTools software (v. 2.11.03; Syngene USA) and normalized to β-actin expression.
Image acquisition
Micrographs in Figs. 1, 4, 5, 6, and 9 and in Fig. S4 were visualized on a microscope (model DM-R; Leica) with Fluotar objectives (magnification/NA: 5×/0.15; 10×/0.30; 20×/0.50; 40×/0.75). The micrographs in Fig. 7 were visualized on an inverted microscope (model DM-IL; Leica). For Fig. 7, images were acquired on film (160 ASA; Tungsten) with a camera (model MPS52; Wild Leitz) connected to a photoautomat (model MPS46; Wild Leitz), and the slides were digitalized with a scanner (model Super Coolscan 4000 ED; Nikon). For Fig. 2, the plates were photographed using a digital camera (model Coolpix 995; Nikon). All other images were acquired using a digital camera (model DC300F; Leica) with the IM50 image management software (Leica).
Image processing included whole image channel filtering to remove noise and whole image adjustment of brightness, contrast, color balance, and sharpening using Adobe Photoshop v. 5.5. Photoshop images were then flattened and imported into Adobe Illustrator v. 8.0 to build the final montages. A shadow was removed from the top center area of Fig. 7 F using the Photoshop cloning tool.
Online supplemental material
Four supplementary figures showing histomorphometric analysis of bones from FIAT transgenic mice and wild-type littermate controls at 3 wk (Fig. S1) and 6 mo (Fig. S2) old, blood biochemistry (Fig. S3), and osteoclast number, differentiation, and activity (Fig. S4) are available at http://www.jcb.org/cgi/content/full/jcb.200412139/DC1.
Acknowledgments
We thank Mia Esser and Louise Marineau for expert technical help with the transgenic animals, Sabrina Pacheco for genotyping the mice, and Alice Arabian for breeding colony management and technical help. Mark Lepik prepared the figures. Dr. Benoit de Crombrughe, Dr. Tsonwin Hai, and Dr. Robert Tjian provided plasmids. We are grateful to McGill University and the Genome Quebec Innovation Centre for allowing repeated usage of the real-time PCR instrument. Bone densitometry was performed at the Centre for Bone and Periodontal Research of McGill University. R. St-Arnaud is indebted to Dr. Gérard Karsenty and to Xiangli Yang for providing plasmids and sharing experimental protocols and unpublished data.
This work was supported by a grant from the Shriners of North America.
L. Verlinden's present address is Laboratorium voor experimentele geneeskunde en endocrinologie, Onderwijs en Navorsing, Universitaire Ziekenhuizen Gasthuisberg, B-3000 Leuven, Belgium.
Abbreviations used in this paper: ATF4, activating transcription factor 4; bZip, basic domain-leucine zipper; DEXA, dual energy X-ray absorptiometry; EMSA, electrophoretic mobility shift assay; FIAT, factor-inhibiting ATF4-mediated transcription.
References
- Andrews, N.C., and D.V. Faller. 1991. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 19:2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1993. Enzymatic manipulation of DNA and RNA. In Current Protocols in Molecular Biology. John Wiley and Sons, NY. 3.5.7–3.5.9.
- Bialek, P., B. Kern, X. Yang, M. Schrock, D. Sosic, N. Hong, H. Wu, K. Yu, D.M. Ornitz, E.N. Olson, et al. 2004. A twist code determines the onset of osteoblast differentiation. Dev. Cell. 6:423–435. [DOI] [PubMed] [Google Scholar]
- Boyle, W.J., W.S. Simonet, and D.L. Lacey. 2003. Osteoclast differentiation and activation. Nature. 423:337–342. [DOI] [PubMed] [Google Scholar]
- Burleigh, M.C., A.J. Barrett, and G.S. Lazarus. 1974. Cathepsin B1. A lysosomal enzyme that degrades native collagen. Biochem. J. 137:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevray, P.M., and D. Nathans. 1992. Protein interaction cloning in yeast: identification of mammalian proteins that react with the leucine zipper of Jun. Proc. Natl. Acad. Sci. USA. 89:5789–5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacquin, R., M. Starbuck, T. Schinke, and G. Karsenty. 2002. Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev. Dyn. 224:245–251. [DOI] [PubMed] [Google Scholar]
- Ducy, P., and G. Karsenty. 1995. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol. Cell. Biol. 15:1858–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy, P., C. Desbois, B. Boyce, G. Pinero, B. Story, C. Dunstan, E. Smith, J. Bonadio, S. Goldstein, C. Gundberg, et al. 1996. Increased bone formation in osteocalcin-deficient mice. Nature. 382:448–452. [DOI] [PubMed] [Google Scholar]
- Ducy, P., R. Zhang, V. Geoffroy, A.L. Ridall, and G. Karsenty. 1997. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 89:747–754. [DOI] [PubMed] [Google Scholar]
- Ecarot-Charrier, B., F.H. Glorieux, M. van der Rest, and G. Pereira. 1983. Osteoblasts isolated from mouse calvaria initiate matrix mineralization in culture. J. Cell Biol. 96:639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlebacher, A., E.H. Filvaroff, S.E. Gitelman, and R. Derynck. 1995. Toward a molecular understanding of skeletal development. Cell. 80:371–378. [DOI] [PubMed] [Google Scholar]
- Fawcett, T.W., J.L. Martindale, K.Z. Guyton, T. Hai, and N.J. Holbrook. 1999. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 339:135–141. [PMC free article] [PubMed] [Google Scholar]
- Franceschi, R.T., and B.S. Iyer. 1992. Relationship between collagen synthesis and expression of the osteoblast phenotype in MC3T3-E1 cells. J. Bone Miner. Res. 7:235–246. [DOI] [PubMed] [Google Scholar]
- Hai, T., and T. Curran. 1991. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. USA. 88:3720–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada, S., and G.A. Rodan. 2003. Control of osteoblast function and regulation of bone mass. Nature. 423:349–355. [DOI] [PubMed] [Google Scholar]
- Harding, H.P., Y. Zhang, H. Zeng, I. Novoa, P.D. Lu, M. Calfon, N. Sadri, C. Yun, B. Popko, R. Paules, et al. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 11:619–633. [DOI] [PubMed] [Google Scholar]
- Hartl, M., A.G. Bader, and K. Bister. 2003. Molecular targets of the oncogenic transcription factor jun. Curr. Cancer Drug Targets. 3:41–55. [DOI] [PubMed] [Google Scholar]
- Hettmann, T., K. Barton, and J.M. Leiden. 2000. Microphthalmia due to p53-mediated apoptosis of anterior lens epithelial cells in mice lacking the CREB-2 transcription factor. Dev. Biol. 222:110–123. [DOI] [PubMed] [Google Scholar]
- Hogan, B., R. Beddington, F. Costantini, and E. Lacy. 1994. Manipulating the Mouse Embryo. A Laboratory Manual, second edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 497 pp.
- Jochum, W., J.P. David, C. Elliott, A. Wutz, H. Plenk Jr., K. Matsuo, and E.F. Wagner. 2000. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat. Med. 6:980–984. [DOI] [PubMed] [Google Scholar]
- Karsenty, G., and E.F. Wagner. 2002. Reaching a genetic and molecular understanding of skeletal development. Dev. Cell. 2:389–406. [DOI] [PubMed] [Google Scholar]
- Komori, T., H. Yagi, S. Nomura, A. Yamaguchi, K. Sasaki, K. Deguchi, Y. Shimizu, R.T. Bronson, Y.H. Gao, M. Inada, et al. 1997. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 89:755–764. [DOI] [PubMed] [Google Scholar]
- Lee, M.H., Y.J. Kim, H.J. Kim, H.D. Park, A.R. Kang, H.M. Kyung, J.H. Sung, J.M. Wozney, and H.M. Ryoo. 2003. a. BMP-2-induced Runx2 expression is mediated by Dlx5, and TGF-beta 1 opposes the BMP-2-induced osteoblast differentiation by suppression of Dlx5 expression. J. Biol. Chem. 278:34387–34394. [DOI] [PubMed] [Google Scholar]
- Lee, M.H., T.G. Kwon, H.S. Park, J.M. Wozney, and H.M. Ryoo. 2003. b. BMP-2-induced Osterix expression is mediated by Dlx5 but is independent of Runx2. Biochem. Biophys. Res. Commun. 309:689–694. [DOI] [PubMed] [Google Scholar]
- Liang, G., and T. Hai. 1997. Characterization of human activating transcription factor 4, a transcriptional activator that interacts with multiple domains of cAMP-responsive element-binding protein (CREB)-binding protein. J. Biol. Chem. 272:24088–24095. [DOI] [PubMed] [Google Scholar]
- Lim, C., H. Sohn, Y. Gwack, and J. Choe. 2000. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) binds ATF4/CREB2 and inhibits its transcriptional activation activity. J. Gen. Virol. 81:2645–2652. [DOI] [PubMed] [Google Scholar]
- Majeska, R.J., S.B. Rodan, and G.A. Rodan. 1980. Parathyroid hormone-responsive clonal cell lines from rat osteosarcoma. Endocrinology. 107:1494–1503. [DOI] [PubMed] [Google Scholar]
- Masuoka, H.C., and T.M. Townes. 2002. Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood. 99:736–745. [DOI] [PubMed] [Google Scholar]
- Miyama, K., G. Yamada, T.S. Yamamoto, C. Takagi, K. Miyado, M. Sakai, N. Ueno, and H. Shibuya. 1999. A BMP-inducible gene, dlx5, regulates osteoblast differentiation and mesoderm induction. Dev. Biol. 208:123–133. [DOI] [PubMed] [Google Scholar]
- Moreau, A., W.V. Yotov, F.H. Glorieux, and R. St-Arnaud. 1998. Bone-specific expression of the alpha chain of the nascent polypeptide-associated complex, a coactivator potentiating c-Jun-mediated transcription. Mol. Cell. Biol. 18:1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima, K., X. Zhou, G. Kunkel, Z. Zhang, J.M. Deng, R.R. Behringer, and B. de Crombrugghe. 2002. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 108:17–29. [DOI] [PubMed] [Google Scholar]
- Nogami, S., S. Satoh, S. Tanaka-Nakadate, K. Yoshida, M. Nakano, A. Terano, and H. Shirataki. 2004. Identification and characterization of taxilin isoforms. Biochem. Biophys. Res. Commun. 319:936–943. [DOI] [PubMed] [Google Scholar]
- Porter, R.M., W.R. Huckle, and A.S. Goldstein. 2003. Effect of dexamethasone withdrawal on osteoblastic differentiation of bone marrow stromal cells. J. Cell. Biochem. 90:13–22. [DOI] [PubMed] [Google Scholar]
- Quarles, L.D., D.A. Yohay, L.W. Lever, R. Caton, and R.J. Wenstrup. 1992. Distinct proliferative and differentiated stages of murine MC3T3-E1 cells in culture: an in vitro model of osteoblast development. J. Bone Miner. Res. 7:683–692. [DOI] [PubMed] [Google Scholar]
- Quelo, I., M. Hurtubise, and R. St-Arnaud. 2002. alphaNAC requires an interaction with c-Jun to exert its transcriptional coactivation. Gene Expr. 10:255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossert, J., H. Eberspaecher, and B. de Crombrugghe. 1995. Separate cis-acting DNA elements of the mouse pro-α1(I) collagen promoter direct expression of reporter genes to different type I collagen-producing cells in transgenic mice. J. Cell Biol. 129:1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatakos, G., N.A. Sims, J. Chen, K. Aoki, M.B. Kelz, M. Amling, Y. Bouali, K. Mukhopadhyay, K. Ford, E.J. Nestler, and R. Baron. 2000. Overexpression of DeltaFosB transcription factor(s) increases bone formation and inhibits adipogenesis. Nat. Med. 6:985–990. [DOI] [PubMed] [Google Scholar]
- Schinke, T., and G. Karsenty. 1999. Characterization of Osf1, an osteoblast-specific transcription factor binding to a critical cis-acting element in the mouse osteocalcin promoters. J. Biol. Chem. 274:30182–30189. [DOI] [PubMed] [Google Scholar]
- Sudo, H., H.A. Kodama, Y. Amagai, S. Yamamoto, and S. Kasai. 1983. In vitro differentiation and calcification in a new clonal osteogenic cell line derived from newborn mouse calvaria. J. Cell Biol. 96:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, T., T. Tsujimura, K. Takeda, A. Sugihara, A. Maekawa, N. Terada, N. Yoshida, and S. Akira. 1998. Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes Cells. 3:801–810. [DOI] [PubMed] [Google Scholar]
- Vallejo, M., D. Ron, C.P. Miller, and J.F. Habener. 1993. C/ATF, a member of the activating transcription factor family of DNA-binding proteins, dimerizes with CAAT/enhancer-binding proteins and directs their binding to cAMP response elements. Proc. Natl. Acad. Sci. USA. 90:4679–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L., T. Tao, X. Wang, N. Du, W. Chen, S. Tao, Z. Wang, and L. Wu. 2003. Effects of dexamethasone on proliferation, differentiation and apoptosis of adult human osteoblasts in vitro. Chin. Med. J. (Engl.). 116:1357–1360. [PubMed] [Google Scholar]
- Yang, X., K. Matsuda, P. Bialek, S. Jacquot, H.C. Masuoka, T. Schinke, L. Li, S. Brancorsini, P. Sassone-Corsi, T.M. Townes, et al. 2004. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell. 117:387–398. [DOI] [PubMed] [Google Scholar]
- Yotov, W.V., A. Moreau, and R. St-Arnaud. 1998. The alpha chain of the nascent polypeptide-associated complex functions as a transcriptional coactivator. Mol. Cell. Biol. 18:1303–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelzer, E., and B.R. Olsen. 2003. The genetic basis for skeletal diseases. Nature. 423:343–348. [DOI] [PubMed] [Google Scholar]