

Abstract
Ca2+ is a highly versatile intracellular signal that regulates many different cellular processes, and cells have developed mechanisms to have exquisite control over Ca2+ signaling. Epidermal growth factor (EGF), which fails to mobilize intracellular Ca2+ when administrated alone, becomes capable of evoking [Ca2+]i increase and exocytosis after bradykinin (BK) stimulation in chromaffin cells. Here, we provide evidence that this sensitization process is coordinated by a macromolecular signaling complex comprised of inositol 1,4,5-trisphosphate receptor type I (IP3R1), cAMP-dependent protein kinase (PKA), EGF receptor (EGFR), and an A-kinase anchoring protein, yotiao. The IP3R complex functions as a focal point to promote Ca2+ release in two ways: (1) it facilitates PKA-dependent phosphorylation of IP3R1 in response to BK-induced elevation of cAMP, and (2) it couples the plasmalemmal EGFR with IP3R1 at the Ca2+ store located juxtaposed to the plasma membrane. Our study illustrates how the junctional membrane IP3R complex connects different signaling pathways to define the fidelity and specificity of Ca2+ signaling.
Introduction
Coordination of cellular functions resides in the ability of a cell to convert extracellular stimuli into appropriate responses (Delmas et al., 2002). These stimuli are relayed and processed by signaling pathways that are not organized in a linear fashion, but that instead display a complex network-like behavior with important cross talk between different signaling modules (Fivaz and Meyer, 2003). The question of how signaling specificity is achieved has therefore become central in the field of signaling research (Hur and Kim, 2002; Fivaz and Meyer, 2003).
Ca2+ is a highly versatile intracellular signal that regulates processes as diverse as fertilization, proliferation, apoptosis, secretion, and information processing in neuronal cells. It is, therefore, absolutely necessary for the spatio-temporal aspects of the Ca2+ signaling system to be carefully controlled (Berridge et al., 2003). Regulation of the Ca2+ release channel, inositol 1,4,5-trisphosphate receptor (IP3R) is suggested to be one of the mechanisms that cells have developed to tailor Ca2+ signaling. It has been postulated that the subcellular localization of IP3Rs, combined with their isoform-specific functions, provide a mechanism for defining Ca2+ signaling patterns (Takei et al., 1998; Thrower et al., 2001; Echevarria et al., 2003; Leite et al., 2003). To date, three mammalian IP3R subtypes have been identified. These receptor subtypes possess high homology (60–70%) in their primary structures and share basic properties, but interesting differences such as their IP3 sensitivity, subcellular distribution, and regulation by binding partners and kinases have been observed (Hagar et al., 1998; Miyakawa et al., 1999; Taylor et al., 2004). The subtype-specific roles of IP3Rs in various aspects of cell signaling and function are just beginning to be unraveled, and it might provide an important mechanism for coordinating Ca2+ signals within the cell (Leite et al., 2003; Hattori et al., 2004).
Exquisite modulation of Ca2+ signaling can also be achieved by the ability of IP3R to integrate signals from numerous signaling molecules and proteins including kinases and phosphatases (Patterson et al., 2004). IP3R can be phosphorylated by multiple kinases including cAMP-dependent protein kinase (PKA), cGMP-dependent protein kinase (PKG), PKC, Ca2+/CaM-dependent protein kinase II (CaMKII), and nonreceptor tyrosine kinases. A consistent pattern in phosphorylation of the IP3R is that these modifications alter the Ca2+ release properties of the channel. For some of the kinases, the scaffolding proteins that mediate recruitment to their site of action on the IP3R have been identified (Schlossmann et al., 2000; Berridge et al., 2003; Patterson et al., 2004; Tu et al., 2004). For example, IP3R is phosphorylated by tyrosine kinase Lyn, which results in increased activity in B cells. This phosphorylation event is facilitated by the B cell scaffold protein with ankyrin repeats (BANK) that links together Lyn, IP3R, and the B cell receptor (Yokoyama et al., 2002). Phosphorylation of IP3R type 1 (IP3R1) by PKA has been the most well-characterized of all the kinases that affect IP3Rs (Thrower et al., 2001), and studies have established that PKA phosphorylation activates IP3R1 by increasing its sensitivity to IP3 (Nakade et al., 1994; Tang et al., 2003). Interestingly, PKA has been found to copurify with IP3R1 in rat brain (DeSouza et al., 2002; Tu et al., 2004). Although the physiological relevance of IP3R association with PKA remains to be established, it is logical to assume that PKA phosphorylation of IP3R might be facilitated by an anchoring protein in a signaling complex to provide extreme precision in Ca2+ signaling.
It has been reported that EGF, which fails to mobilize intracellular Ca2+ when administrated alone, becomes capable of evoking [Ca2+]i increase and neurotransmitter release specifically after bradykinin (BK) stimulation in rat pheochromocytoma PC12 cells (Pandiella and Meldolesi, 1989; Hur et al., 2004). Here, we suggest a molecular mechanism for this sensitization process, providing evidence for a role of the IP3R1 signaling complex as a focal point for cross talk between different signaling pathways. Here, we demonstrate that the IP3R signaling complex couples the BK-induced cAMP–PKA pathway to the EGF-evoked Ca2+ release pathway via tethering PKA, EGF receptor (EGFR), and an anchoring protein, yotiao (AKAP9) to IP3R1. Our study showing colocalization of IP3R1 and EGFR located at closely apposing membranes of the intracellular Ca2+ store and the plasma membrane (PM), respectively, illustrates how interaction between signaling molecules located at membrane junctions provides the structural foundation for site- and context-specific Ca2+ signaling.
Results
Involvement of PKA in sensitization of the functionally silent EGF-induced Ca2+ signaling and exocytosis
EGF, which fails to evoke [Ca2+]i increase and exocytosis when administrated alone, becomes capable of triggering both responses when applied after BK-induced [Ca2+]i rise and exocytosis returns to the basal level in adrenal medullary chromaffin cells (Fig. 1). We previously showed that sensitization of the EGF-evoked [Ca2+]i rise was induced specifically by BK, but not by other Ca2+-elevating agents such as high K+, ionomycin, and UTP, an agonist of the G protein–coupled P2Y2 receptors (Hur et al., 2004). To decipher the specificity of BK among other Ca2+-elevating agents, we next sought to investigate which other factor(s) downstream of BK might be responsible for the sensitization process. Because BK activates the cAMP–PKA pathway as well as the PLC/Ca2+ pathway (Graness et al., 1997), we examined whether the cAMP–PKA pathway was involved. For this purpose, it was first confirmed that BK, but not high K+, ionomycin, and UTP, stimulated generation of cAMP (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200411034/DC1). As shown in Fig. 1, both EGF-evoked [Ca2+]i rise and exocytosis subsequent to BK stimulation were markedly blocked by an inhibitor of PKA, H89 in chromaffin cells. The inhibitory effect of H89 on the BK-induced sensitization of EGF-evoked [Ca2+]i rise was also observed in rat pheochromocytoma PC12 cells (Fig. 2 A), albeit to a lesser extent as compared with its effect in chromaffin cells. A competitive antagonist of PKA, RpcAMPS had a similar inhibitory effect on the EGF-induced [Ca2+]i rise subsequent to BK stimulation (Fig. 2 B), supporting the notion that PKA participates in regulation of the sensitization process. Furthermore, sensitization of the EGF-induced [Ca2+]i rise was markedly impaired in PKA-deficient PC12 cells as compared with wild-type PC12 cells (Fig. 2 C), confirming the involvement of PKA. To verify the specificity of H89, we tested whether H89 had any nonspecific inhibitory effects in PKA-deficient cells. It is important to note that EGF-induced [Ca2+]i rise subsequent to BK stimulation was not affected by H89 in PKA-deficient cells (Fig. 2 D), excluding its possible nonspecific inhibitory effects on the sensitization process.
Figure 1.

Sensitization of the functionally silent EGF-induced [Ca2 + ]i increase and exocytosis by BK in rat adrenal chromaffin cells. (A) Carbon fiber amperometry was used to detect exocytosis from single chromaffin cells in real time. Typical amperometric responses from adrenal chromaffin cells after addition of 20 nM EGF and/or 1 μM BK are shown. Involvement of PKA was examined by application of H89 (10 μM) before BK stimulation. (B) Normalized rate of exocytosis. Number of amperometric spikes over a 20-s recording period was divided by that over a 20-s control period. Each point represents a mean ± SEM value obtained from 15 control and 12 H89-treated cells from three separate experiments on different batches of cells. The normalized rate of exocytosis in the control period is assigned a value of 1.0 and indicated by a horizontal broken line. (C and D) Fura-2 intracellular Ca2+ imaging of chromaffin cells in response to 1 μM BK and/or 20 nM EGF stimulation. H89 (10 μM) was treated before BK stimulation as indicated. Representative Ca2+ transients are shown in C, and averaged Ca2+ traces from multiple cells are shown in D. At least three independent experiments were performed and cells from two coverslips were analyzed per experiment.
Figure 2.

Involvement of PKA in the sensitization process. (A) Fura-2–loaded PC12 cells (106) were pre-treated with either vehicle or H89 (10 μM), followed by BK, and then finally stimulated with EGF. Representative traces from more than three independent experiments are shown. (B) Fura-2–loaded PC12 cells were pre-treated with H89 (10 μM) or RpcAMPS (100 μM), followed by BK, and then finally stimulated with EGF. (C) EGF-induced [Ca2+]i increase subsequent to BK stimulation were compared in fura-2–loaded wild-type and PKA-deficient PC12 cells. (D) PKA-deficient cells were pretreated with vehicle, H89 (10 μM), or PP2 (10 μM), followed by BK, and then finally stimulated with EGF. (B–D) Peak amplitudes in the BK- and/or EGF-induced [Ca2+]i rise were measured. (E) PC12 cells were treated with vehicle, BK (2 min), EGF (200 pM, 2 min), or sequentially with BK (10 min) followed by EGF (200 pM, 2 min) in the presence or absence of H89 (10 μM) as indicated. Cell lysates were immunoprecipitated with the EGFR antibody, and the precipitates were immunoblotted with anti-phosphotyrosine antibody. Immunoblotting with the EGFR antibody was performed for normalization. EGFR phosphorylation was quantitated by densitometry and normalized. (F) PC12 cells were treated with vehicle, dibutyryl cAMP (1 mM), or BK for 3 min and tyrosine phosphorylated EGFRs were monitored as in E. (G) PC12 cells were stimulated as indicated and the level of IP3 was measured. BK/EGF denotes for cells stimulated with EGF (1 min) after BK (10 min) pretreatment. H89 (10 μM) or PP2 (10 μM) was pretreated for 3 min. The concentration of BK was 1 μM and EGF was 20 nM, unless otherwise stated. All error bars are mean ± SEM of a minimum of three experiments. **, P < 0.01.
To determine the target site at which PKA exerts its action in the sensitization process, we first examined the possible role of PKA in activation of EGFR and PLC. Tyrosine phosphorylation of the EGFR and phosphoinositide hydrolysis were monitored to indicate activities of the EGFR and PLC, respectively. As shown in Fig. 2 E, neither BK-induced transactivation of the EGFR (lanes 3 and 4) nor the potentiation effect of BK on the subsequent EGF-induced EGFR phosphorylation (lanes 5 and 6) was affected by H89. Besides, phosphorylation of EGFR was not induced either by dibutyryl cAMP, a membrane-permeable form of cAMP that activates PKA (Fig. 2 F), or forskolin, a direct activator of adenylyl cyclase (not depicted). EGF, which is unable of elicit phosphoinositide hydrolysis when administrated alone, gives rise to substantial amount of IP3 generation when applied after BK-induced IP3 generation has returned to the basal level. PLC activation elicited by application of EGF subsequent to BK stimulation was not inhibited by H89, whereas it was completely abrogated by a Src kinase inhibitor, PP2 (Fig. 2 G). Together, these results suggest that neither EGFR nor PLC is the molecular site at which PKA exerts its action in the sensitization process.
IP3R1 is required for the sensitization process
We have previously demonstrated that the EGF-induced [Ca2+]i rise sensitized by BK is attributed to PLC-dependent Ca2+ release from internal stores rather than Ca2+ influx across the PM (Hur et al., 2004). Therefore, another possible target of the sensitization could be the calcium release machinery. To address this possibility, we first examined the effects of 2-aminoethyl diphenylborate (2APB) and ryanodine on the sensitization process, to inhibit IP3R- and ryanodine receptor–mediated Ca2+ increase, respectively. Sensitization was completely blocked by 2APB but not ryanodine, suggesting that IP3R was responsible for the [Ca2+]i increase in the sensitization process (Fig. 3 A).
Figure 3.

Expression and subcellular localization of IP3R subtypes. (A) Effects of 2APB and ryanodine on the sensitization. Fura-2–loaded PC12 cells (106) were stimulated with BK (1 μM), followed by incubation with 2APB (75 μM) or ryanodine (1 μM) for 3 min, and then finally stimulated with EGF (20 nM). The peak amplitude of the EGF-induced [Ca2+]i rise was measured. Error bars are mean ± SEM of four separate experiments. **, P < 0.01. (B) In situ hybridization of the IP3R transcripts for each subtype in adrenal gland. Light micrographic images of adrenal gland sections with either antisense or sense IP3R RNA probes are shown. Bar, 400 μm. (C) Western blot analysis of adrenal medulla, PC12 cells, and CHO-K1 cells with antibodies against IP3R subtypes. (D and E) Immunoconfocal microscopy images showing colocalization of IP3R1 and flotillin-1 in adrenal chromaffin cells. Cells were triple labeled with propidium iodide (PI, red), anti–flotillin-1 antibody (blue), and with either anti-IP3R1 or anti-IP3R3 antibody (green). Immunoconfocal microscopy and postacquisition processing of images were performed as described in Materials and methods. (F) IP3R1 and PKA are present in DRMs. Extracts from adrenal medulla were fractionated by sucrose density gradient centrifugation, and each fraction was analyzed by immunoblotting with the indicated antibodies.
Next, we examined expression of IP3R subtypes in the adrenal medulla and PC12 cells by in situ hybridization and Western blot analysis. IP3R1 and IP3R3 were predominantly expressed in the adrenal medulla and PC12 cells (Fig. 3, B and C). Because the sensitization process occurred in and required the integrity of cholesterol-rich, detergent-resistant membrane (DRM) microdomain (so-called lipid raft; Hur et al., 2004), we then examined localization of the IP3R subtypes versus flotillin-1, which is known as a marker of lipid raft. IP3R1, but not IP3R3, was detected in the DRM fraction (Fig. 3 F). Consistently, immunoconfocal microscopy analysis revealed that flotillin-1 preferentially colocalized with IP3R1 (Fig. 3, D and E).
To determine the IP3R subtype responsible for the sensitization process, we performed knockdown studies using siRNA against IP3R1 and IP3R3. siRNA designed for each receptor subtype specifically knocked down the target IP3R subtype without affecting expression of another (Fig. 4 A). As shown in Fig. 4 B, the peak height in the BK-induced Ca2+ increase was reduced in cells transfected with siRNA against either IP3R1 or IP3R3, suggesting that both IP3R1 and IP3R3 were responsible for the BK-induced [Ca2+]i rise. Interestingly, time to reach the peak value was also slightly delayed in cells transfected with siRNA against IP3R1, whereas knockdown of IP3R3 resulted in reduction of the amplitude without altering the kinetics of Ca2+ rise (Fig. 4 B). Importantly, EGF-induced [Ca2+]i rise subsequent to BK stimulation was nearly eliminated in cells transfected with siRNA against IP3R1 but not IP3R3, demonstrating that IP3R1 was selectively involved in sensitization of the EGF-induced Ca2+ increase. Consistent with the Ca2+ response, amperometric analysis in single cells revealed that EGF-evoked exocytosis subsequent to BK stimulation was also nearly abolished in IP3R1 siRNA-transfected cells, demonstrating the requirement of IP3R1 in the sensitization process (Fig. 4 C).
Figure 4.
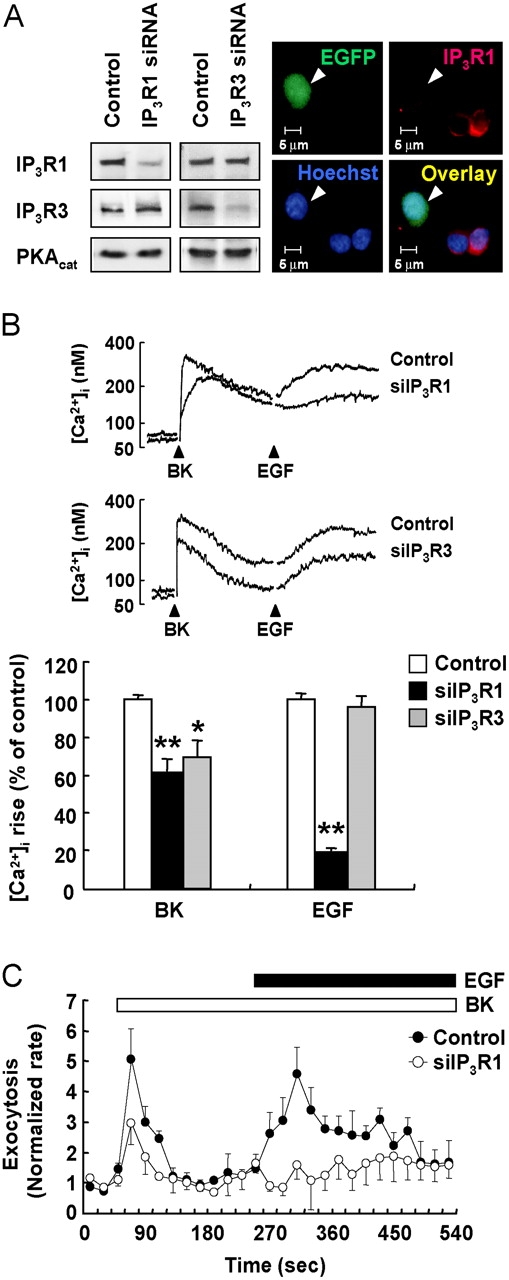
IP3R1 is required in the sensitization process. (A) PC12 cells were transfected with siRNA against IP3R1 or IP3R3 as indicated. pEGFP was cotransfected as a marker. Effects of siRNA were confirmed by Western blot analysis and immunocytochemistry. (Left) Representative immunoblots after transfection with siRNA against IP3R1 or IP3R3. (Right) Typical images after transfection with IP3R1 siRNA. (B) Cells transfected with siRNA as indicated were loaded with fura-2 for Ca2+ measurements. Cells were treated with 1 μM BK, followed by 20 nM EGF. Typical Ca2+ transients are presented, and the peak heights in the BK- and EGF-induced [Ca2+]i increase were measured. *, P < 0.05; **, P < 0.01. (C) Carbon fiber amperometry was used to detect exocytosis from single cells in real time. Control or IP3R1 siRNA-transfected PC12 cells were treated with 1 μM BK followed by 20 nM EGF as indicated. The rate of exocytosis was normalized as in Fig. 1 B. Each point presented is a mean ± SEM of three experiments.
EGFR, PKA, and yotiao (AKAP9) comprise a macromolecular signaling complex with IP3R1
Because both PKA and IP3R1 were detected in the DRM fraction (Fig. 3 F), we examined whether IP3R1 and PKA were part of a signaling complex. Immunoprecipitation with an antibody against the catalytic subunit of PKA revealed an endogenous signaling complex composed of IP3R1, EGFR, and yotiao, which belongs to the superfamily of A-kinase anchoring protein (AKAP; Fig. 5 A). Yotiao selectively coimmunoprecipitated with IP3R1, but not IP3R3 (Fig. 5 B). Components of the IP3R1 complex were also detected in immunoprecipitates of the EGFR (Fig. 5 C) and IP3R1, and the complex formation was not altered by BK stimulation (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200411034/DC1). Collectively, these results show that yotiao, PKA, and EGFR comprise a macromolecular signaling complex with IP3R1, and suggest a model in which a physically coupled complex of PKA-yotiao-IP3R1-EGFR forms before BK stimulation.
Figure 5.
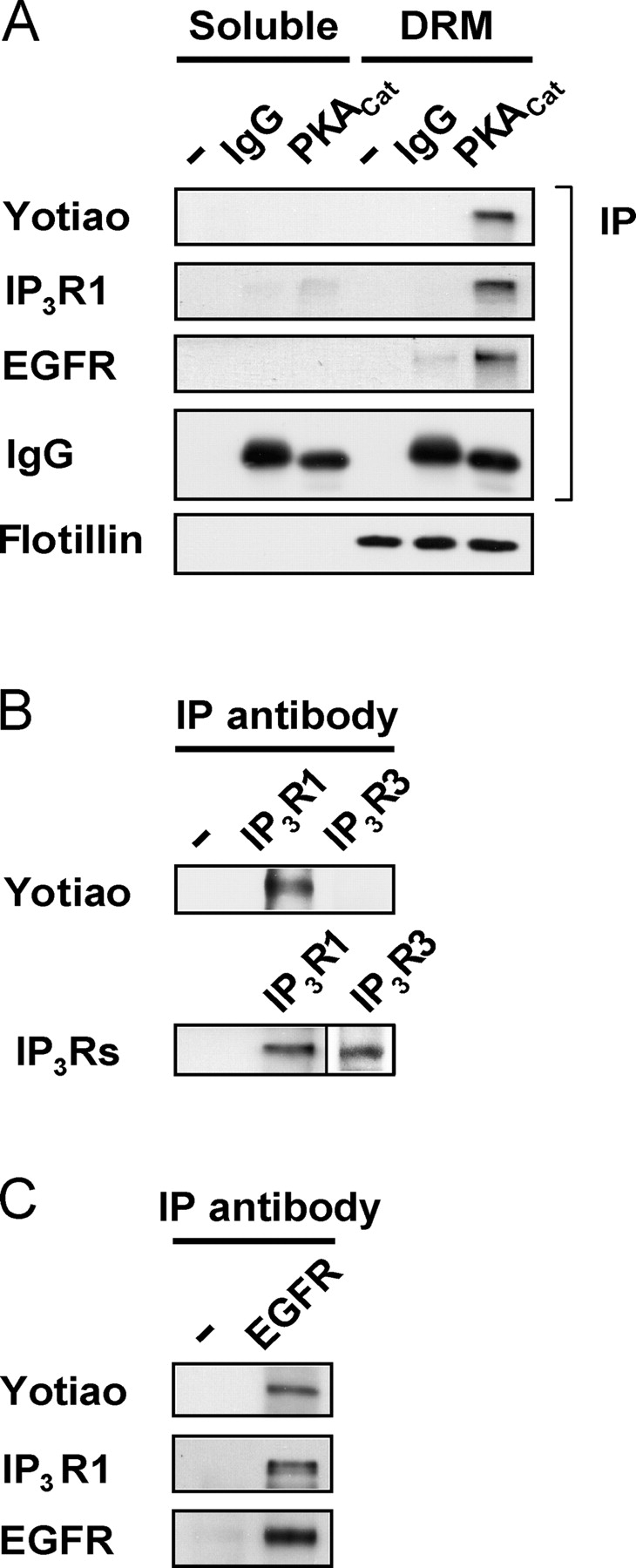
Yotiao (AKAP9), PKA, and EGFR comprise a macromolecular signaling complex with IP3R1. (A) Association of yotiao, IP3R1, and EGFR with PKA in the DRM fraction. PC12 cell lysates were fractionated by sucrose density gradient centrifugation. DRM and soluble fractions were pooled and subjected to immunoprecipitation with an antibody against the catalytic subunit of PKA (PKACatα) or IgG. Precipitates were analyzed by blotting with the indicated antibodies. (B) Yotiao coimmunoprecipitates with IP3R1, but not IP3R3. PC12 cell lysates were immunoprecipitated with an antibody against IP3R1 or IP3R3. Precipitates were analyzed by blotting with anti-yotiao antibody. (C) PC12 cell lysates were immunoprecipitated with IgG or antibodies against EGFR. Precipitates were analyzed by blotting with the indicated antibodies. Primary antibody was omitted in lanes marked “−”. Immunoblots presented are representative of at least three independent experiments.
Images from confocal microscopy analysis revealed that IP3R1 and EGFR exhibited distinct but overlapping subcellular distributions, and significant colocalization of IP3R1 with EGFR was observed at the cell periphery (Fig. S1). To provide further confirmation for the colocalization and to examine the subcellular locations of the signaling complex at the ultrastructural level, we performed immunogold EM (Fig. 6). The gold particles labeling IP3R1 molecules were detected specifically at the membranes of the secretory granules and the ER as well as at the PM. EGFRs were primarily shown to be at the PM though some of them were also found in the ER. These EGFRs appear to be the ones that are synthesized by ribosomes in the ER. Importantly, immunogold EM clearly revealed colocalization of IP3R1 and EGFR located at closely apposing membranes of the intracellular Ca2+ store and the PM, respectively (Fig. 6 and Table I).
Figure 6.
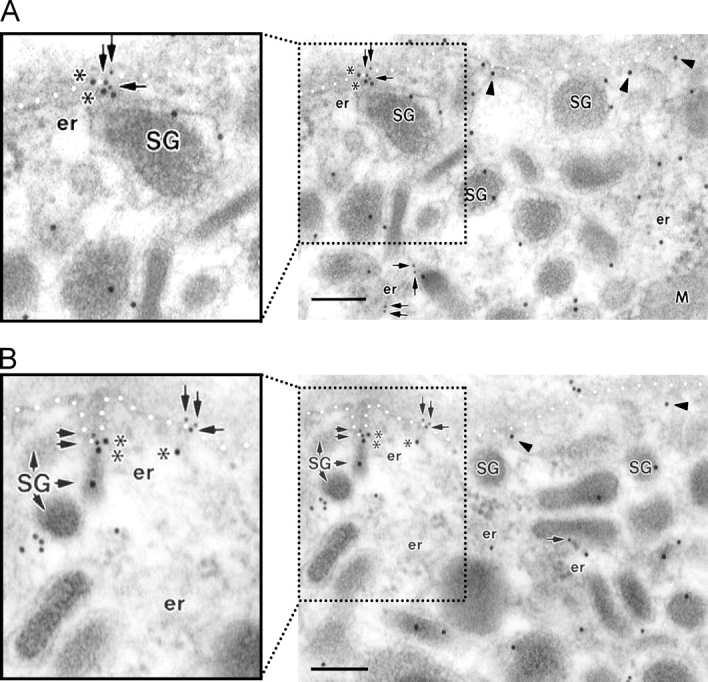
Immunogold EM showing colocalization of EGFR and IP3R1 in adrenal chromaffin cells. A and B show representative images from different preparations. Rat adrenal medullary chromaffin cells were double immunolabeled for the IP3R1 (15 nm gold) and EGFR (10 nm gold) with the affinity purified IP3R1-specific anti–rabbit antibody and anti–sheep EGFR antibody, respectively, as described in Online supplemental material. The gold particles labeling IP3Rs are localized at the membranes of the secretory granules (SG), the ER, as well as at the PM. IP3Rs at the ER juxtaposed to the PM are marked with arrowheads. Note that there are no nonspecific IP3R1-labeling gold particles in mitochondria (M). The EGFRs (PM and ER, arrows) are primarily localized at the PM though it also appears in the ER. The IP3R1 molecules that colocalize with the EGFR are marked with asterisks. The boundary of the PM is denoted with white dots for clarity. Bars, 200 nm.
Table I.
Distribution of distances between EGFR at the PM and IP 3 R1 at Ca 2+ stores in rat adrenal medullary chromaffin cellsa
| Distanceb | <15 nm | 15–50 nm | >50 nm |
|---|---|---|---|
| Labeling percentage | 38% | 27% | 35% |
Images from four different tissue preparations labeled with gold particles for both EGFR and IP3R1 were analyzed. The length of total PM measured was 49.04 μm.
IP3R1 and EGFR were considered to be colocalized when the distance between the gold particles labeling both receptors was within the length of a single gold particle.
IP3R1 is phosphorylated upon BK stimulation in a PKA-dependent manner
Because BK activated the cAMP–PKA pathway and PKA coimmunoprecipitated with IP3R1, we investigated whether IP3R1 was phosphorylated by BK in a PKA-dependent manner. For this purpose, we immunoprecipitated IP3R1 and performed Western blot analysis by using an antibody designed to recognize substrates phosphorylated by PKA. This antibody recognizes phospho-serine/threonine residues that are preceded by arginine at the −3 position (RXXpS/T), comprising the recognition site of PKA. This antibody has been used to demonstrate PKA-mediated phosphorylation of IP3R2 (Bruce et al., 2002) and IP3R3 (Straub et al., 2002). As shown in Fig. 7 A, BK induced PKA-dependent phosphorylation of IP3R1, an effect which was completely blocked by H89.
Figure 7.

Phosphorylation of IP3R1 upon application of BK. (A) BK-induced phosphorylation of IP3R1 occurs in a PKA-dependent manner. (Top) PC12 cells were treated with vehicle or BK (1 μM, 5 min) in the presence or absence of H89 (10 μM) as indicated. Lysates were immunoprecipitated with IP3R1 antibody, and Western blot analysis of the precipitates was performed by using an antibody designed to recognize substrates phosphorylated by PKA. (Bottom) PC12 cells treated with vehicle (NT) or BK were immunoprecipitated with the phospho-(serine/threonine) PKA substrate antibody, and Western blot analysis of the precipitates was performed by using an antibody against IP3R1. (B) Time course of the BK induced phosphorylation of IP3R1. 1 μM BK was applied for the indicated time periods and cell lysates were subjected to immunoprecipitation with antibodies against IP3R1. Phosphorylation of IP3R1 was detected as described in A.
In our previous study, we showed that BK induced a long-lasting sensitization: the peak height in the EGF-induced [Ca2+]i rise reached its maximum at 5–10 min after BK application and the level was sustained up to 1 h (Hur et al., 2004). To investigate the possible relationship between the sensitization process and the BK-induced phosphorylation of IP3R1, we examined the time course of phosphorylation of IP3R1 after BK application. As shown in Fig. 7 B, BK-induced phosphorylation of IP3R1 was persistent, whereas phosphorylation of EGFR was rather transient (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200411034/DC1). Interestingly, the pattern of BK-induced phosphorylation of IP3R1 correlated with the time course of BK-mediated sensitization of the calcium response.
AKAP signaling complex is required for the PKA-dependent phosphorylation of IP3R1 by BK and promotes the sensitization process
AKAPs position PKA at defined subcellular locations within a cell (Bauman and Scott, 2002). A defining characteristic of AKAPs is a conserved sequence that forms a binding site for the regulatory subunit of PKA. A consensus PKA anchoring motif called “AKAP-in silico (AKAP-IS)” was recently derived from a comprehensive analysis of AKAPs' sequences and bioinformatic design of an optimal binding peptide. AKAP-IS binds PKA with subnanomolar affinity and is an effective antagonist of PKA anchoring inside cells (Alto et al., 2003). To delineate a role of the AKAP signaling complex in regulation of the BK-induced sensitization process, we transfected expression constructs encoding AKAP-IS (AMAQIEYLAKQIVDNAIQQAKA) or the scrambled sequence (AMAQDVEIQLKAAYNQKLIAIA) as a control. Transfection of AKAP-IS disrupted the association of IP3R1, EGFR, and yotiao with PKA (Fig. 8 A). Importantly, AKAP-IS effectively blocked the PKA-dependent phosphorylation of IP3R1 in response to BK stimulation (Fig. 8 B). Furthermore, sensitization of the EGF-induced [Ca2+]i increase was substantially impaired in cells transfected with AKAP-IS as compared with those transfected with the scrambled sequence (Fig. 8 C). Because the AKAP-IS peptide would disrupt association of PKA with all AKAPs, we performed knockdown studies using siRNA against yotiao to confirm that AKAP9 is involved in the sensitization process. As shown in Fig. 8 D, the EGF-induced [Ca2+]i rise subsequent to BK stimulation was significantly blocked by transfection with yotiao siRNA, providing convincing evidence for involvement of AKAP9 in sensitization of the EGF response. Together, these results demonstrate that the AKAP9 signaling complex is essential for establishment of the IP3R1 signaling complex and regulation of the PKA-dependent IP3R phosphorylation in response to BK stimulation.
Figure 8.
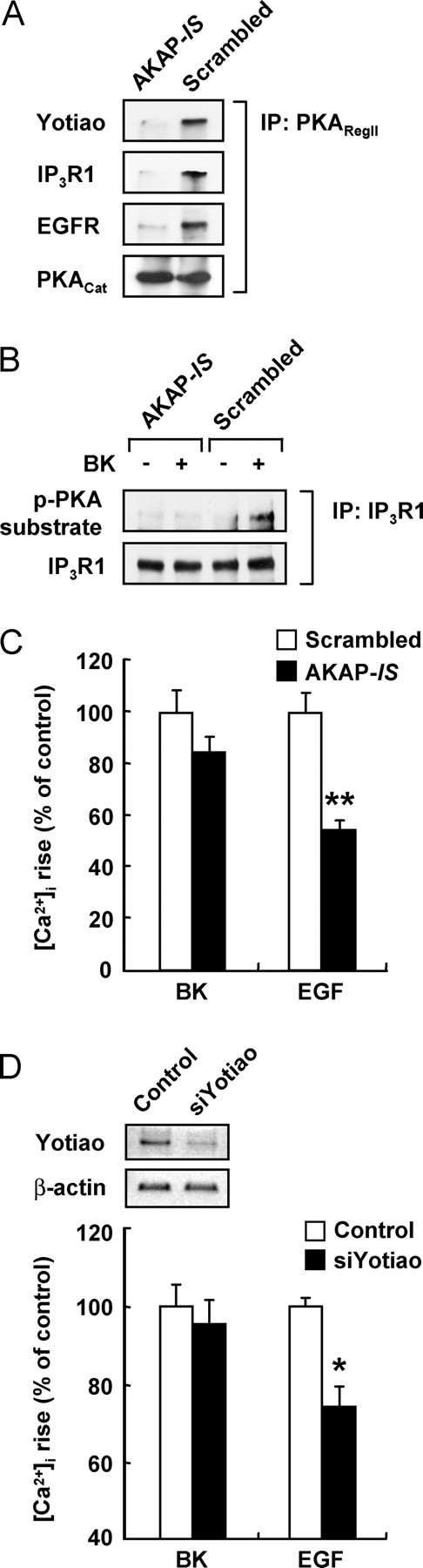
AKAP signaling complex facilitates BK-induced phosphorylation of IP3R1 and the sensitization process. (A) Lysates of PC12 cells transfected with either AKAP-IS or the scrambled sequence were immunoprecipitated with an antibody against the regulatory subunit (RIIα) of PKA, and precipitates were analyzed by blotting with the indicated antibodies. (B) Anchored pool of PKA is required for BK-induced phosphorylation of IP3R1. PC12 cells transfected with either AKAP-IS or the scrambled sequence were treated with vehicle or BK, and PKA-dependent phosphorylation of IP3R1 was examined as in Fig. 7. (C) Sensitization is facilitated by the AKAP signaling complex. PC12 cells transfected with either AKAP-IS or the scrambled sequence were loaded with fura-2 for [Ca2+]i measurements. (D) Involvement of yotiao in the sensitization process. (Top) Expression of yotiao mRNA in the control and yotiao siRNA-transfected cells was analyzed by RT-PCR as described in Materials and methods. Results from RT-PCR performed with β-actin primers are presented as a control to verify that equal amount of cDNA was used in each reaction. Inverted imaged are shown. (Bottom) Control and yotiao siRNA-transfected cells were loaded with fura-2 for [Ca2+]i measurements. (C and D) Cells were stimulated with BK (1 μM, 5 min) followed by EGF (20 nM), and the peak heights in the BK- and EGF-induced [Ca2+]i increase were measured. Error bars are mean ± SEM of three experiments. *, P < 0.05; **, P < 0.01.
Discussion
Stimulation of the G protein–coupled BK B2 receptor results in activation of signaling pathways that ultimately sensitize the functionally silent Ca2+ signaling triggered by EGFR, a prototypic member of the superfamily of receptor tyrosine kinase (RTK). This study suggests a structural and functional role of the IP3R complex in the regulation of cross talk between signaling pathways mediated by two different classes of cell surface receptors. Here, we demonstrate that the sensitization process is coordinated by a macromolecular signaling complex comprised of IP3R1, PKA, EGFR, and an anchoring protein, yotiao. This IP3R signaling complex functions to facilitate Ca2+ release via two important strategies: (1) it couples IP3R1 with EGFR located at closely apposing membranes of the Ca2+ store and the PM, respectively, to promote Ca2+ release from intracellular stores upon receptor activation at the PM, and (2) facilitates a physical association between PKA and IP3R1 to increase the sensitivity of IP3R1 via PKA-dependent phosphorylation.
An important strategy for ensuring specificity in Ca2+ signaling is to have signaling components organized into discrete microdomains. A previous study on muscarinic M1 receptors and BK B2 receptors provided functional evidence that the specificity and sensitivity of IP3-mediated Ca2+ signaling is determined by the spatial proximity of membrane receptors and IP3Rs (Delmas et al., 2002). They suggested that discrete microdomains enabled BK but not muscarinic stimulation to activate IP3R, although both receptors were equally efficient at activating PLC. These findings lead to the question of how the proximity between IP3Rs and plasmalemmal receptors is established. Emerging evidence reveals a privileged communication between the PM and the intracellular Ca2+ store. The ER network reaches from the nuclear envelope to the cell periphery and forms junctional contacts with the PM, in which the two apposed membranes are separated by a small gap. At present, what causes a particular portion of the PM to form a junction is not fully understood. It has been suggested that the junctional PM might have a specific lipid composition, such as those of lipid rafts that function as platforms for signaling molecules (Johenning and Ehrlich, 2002; Poburko et al., 2004). Alternatively, recent studies have shed new light on the multiprotein signaling complexes that mediate the communication (Tu et al., 1998; Xiao et al., 1998; Yuan et al., 2003; Treves et al., 2004). For example, Homer proteins have been shown to form a physical tether cross-linking metabotropic glutamate receptors (mGluRs) with IP3Rs (Tu et al., 1998; Xiao et al., 1998) as well as regulating interactions between TRPC calcium channels and IP3Rs (Yuan et al., 2003). Here, we provide biochemical evidence for a physical association of IP3R1 with EGFR (Fig. 5) and show colocalization of IP3R1 and EGFR located at closely apposing membranes of the intracellular Ca2+ store and the PM, respectively (Fig. 6). EGFR might be tethered to the complex by directly binding to the anchoring protein AKAP9 as shown by the interaction between AKAP150 and the M-type potassium channel, KCNQ2 (Hoshi et al., 2003). Alternatively, it may require another adaptor that functions as a bridge between AKAP9 and EGFR, as illustrated by recruitment of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor or the N-methyl-d-aspartic acid (NMDA) receptor to the AKAP79/150 complexes via Stargazin, synapse-associated protein-97, or postsynaptic density protein-95 (Chen et al., 2000; Colledge et al., 2000). Further studies are required to determine how EGFR binds to the IP3R complex. Interestingly, secretory granules were also located in close proximity to the colocalization sites. It is likely that the sensitization of Ca2+ signaling and exocytosis occurs at organellar junctions where the Ca2+ store, secretory granule, and the PM are closely juxtaposed. It seems that the IP3R complex organize microdomains of closely juxtaposed Ca2+ store and the PM, influencing local Ca2+ concentration and possibly stimulating the subsequent vesicle secretion in an effort to enhance local and rapid control mechanisms as well as to selectively transduce and integrate signals.
Our results highlight the sophisticated degree of regulation in order to achieve signaling specificity in receptor cross talk. We suggest that the initially silent EGF-induced Ca2+ signaling is acquired in response to BK stimulation via orchestration of at least two different signaling pathways, i.e., sensitization of the EGFR and the Ca2+ release machinery, IP3R, via Src and PKA, respectively. In our previous study, we demonstrated that Src-dependent activation of EGFR was required in the sensitization process. However, that activation of EGFR cannot entirely explain how BK induces sensitization of the Ca2+ signaling pathway triggered by EGF, because sensitization could not be induced by other agents that had been reported to activate the EGFR. Moreover, EGFR-mediated activation of the MAPK pathway was not affected by BK stimulation (Hur et al., 2004), suggesting that an additional regulatory mechanism was involved specifically in sensitization of the Ca2+ signaling pathway. Here, we propose that the sensitization process is facilitated by the IP3R complex that functions to coordinate Ca2+ release, enabling IP3R1 to respond accurately to intracellular messages.
A number of biochemical studies analyzed PKA phosphorylation of IP3R1 (Danoff et al., 1991; Nakade et al., 1994; Wojcikiewicz and Luo, 1998; Thrower et al., 2001; Patterson et al., 2004). PKA phosphorylates purified IP3R1 at two known sites, serine 1589 and 1755. The neuronal type IP3R1 is preferentially phosphorylated at Ser1755, whereas the peripheral IP3R1 is more heavily phosphorylated at Ser1589 (Thrower et al., 2001; Patterson et al., 2004). Despite the wealth of biochemical information, there has been little evidence so far, demonstrating PKA-dependent phosphorylation of IP3R1 in response to endogenous agonist stimulation in intact cells. Another important, unresolved question is how PKA, which has access to a range of substrates in intact cells, can be specifically implicated in the regulation of IP3R1. Here, we provide an explanation for both questions by suggesting a role for the AKAP signaling complex in the BK-induced phosphorylation of IP3R1, which functions to facilitate the sensitization process. AKAPs are a functionally defined group of proteins that bind PKA, providing discrete localization within a cell to allow for the spatially and temporally controlled phosphorylation of target proteins in response to local increases in cAMP (Carnegie and Scott, 2003). Yotiao (AKAP9) belongs to a class of AKAP, and it functions to anchor PKA to NMDA receptors in the brain, thereby contributing to modulation of current flow through NMDA receptors in response to the cAMP-dependent pathway (Westphal et al., 1999). Recently, Tu et al. (2004) provided interesting biochemical evidence demonstrating that yotiao and IP3R1 associate in a complex in rat brain and addressed that future experiments would be needed to establish the physiological relevance of this association. By investigation of dopamine-induced Ca2+ oscillations in striatal medium spiny neurons, they proposed a model in which the yotiao complex plays a pivotal role in cross talk between cAMP and Ca2+ signaling pathways (Tang and Bezprozvanny, 2004). Our study shows that PKA-dependent phosphorylation of IP3R1 occurs in response to BK stimulation in PC12 cells, an event which is regulated by a macromolecular signaling complex comprised of yotiao, PKA, IP3R1, and EGFR. We provide evidence that this IP3R signaling complex couples the BK-induced cAMP–PKA pathway to the EGF-evoked Ca2+ signaling pathway. Together with the analysis of Ca2+ signaling in dopaminergic pathways (Tang and Bezprozvanny, 2004), these results demonstrate the importance of IP3R signaling complex in the intricate interplay between cAMP and Ca2+ signaling pathways.
The adrenal gland is a part of the sympathetic nervous system, and adrenal chromaffin cells synthesize, store, and release catecholamine hormones. Emerging evidence suggests that hyperactivity of catecholaminergic systems is responsible for hyperarousal symptoms in stress disorders, such as posttraumatic stress disorder (Carrasco and Van de Kar, 2003; O'Donnell et al., 2004). Interestingly, several lines of experimental evidence point to a connection between these disorders and various inflammatory diseases (Tucker et al., 2004), supporting the concept that malfunction of the sympathetic nervous system is associated with dysregulation of the immune system, increasing the risk for stress-related medical illnesses. It is thus plausible to postulate that when the BK signaling, a representative proinflammatory signal, is up-regulated in response to activation of plasma-derived inflammatory system, this would enhance the level of secretion of catecholamines via activating adrenal chromaffin cells, not only by directly acting on the BK receptor, but also by inducing sensitization of the EGF-induced neurotransmitter release. EGF is a conventional mitogenic factor that stimulates the proliferation of epithelial cells and fibroblasts, but many studies showed that EGF also enhances the differentiation, maturation, and survival of various types of neuronal cells (Yamada et al., 1997; Wong and Guillaud, 2004). Results from our present study imply that EGF can also function as a neuromodulator, in addition to functioning as a neurotrophic factor. Considering that catecholamines secreted from the adrenal gland have diverse physiological roles, receptor-regulatory mechanisms for exocytosis in chromaffin cells would provide valuable information about therapeutical approaches for the treatment of stress-related disorders.
In conclusion, our study highlights the sophisticated degree of regulatory mechanisms that work in concert to render specificity in receptor cross talk, and emphasizes a role of the IP3R macromolecular signaling complex as a focal point for the reception, propagation, and integration of distinct intracellular signals. We suggest that the functionally silent EGF-induced Ca2+ signaling is acquired in response to BK stimulation via regulation of at least two components of the EGF signaling system that are mechanistically distinct (Fig. 9). First, BK triggers Src-dependent phosphorylation of EGFR, an event which results in enhanced recruitment and activation of PLCγ1 (Hur et al., 2004). Second, BK induces PKA-dependent phosphorylation of IP3R1, increasing its responsiveness to IP3. Assembling IP3R1, PKA, yotiao, and EGFR into a signaling complex provides a mechanism to facilitate not only phosphorylation of IP3R1 but also stimulus-dependent control of function. Our study illustrates how a junctional membrane IP3R complex contributes to expedite selective and efficient signal propagation, providing a previously unknown mechanism for defining the fidelity and specificity of Ca2+ signaling.
Figure 9.
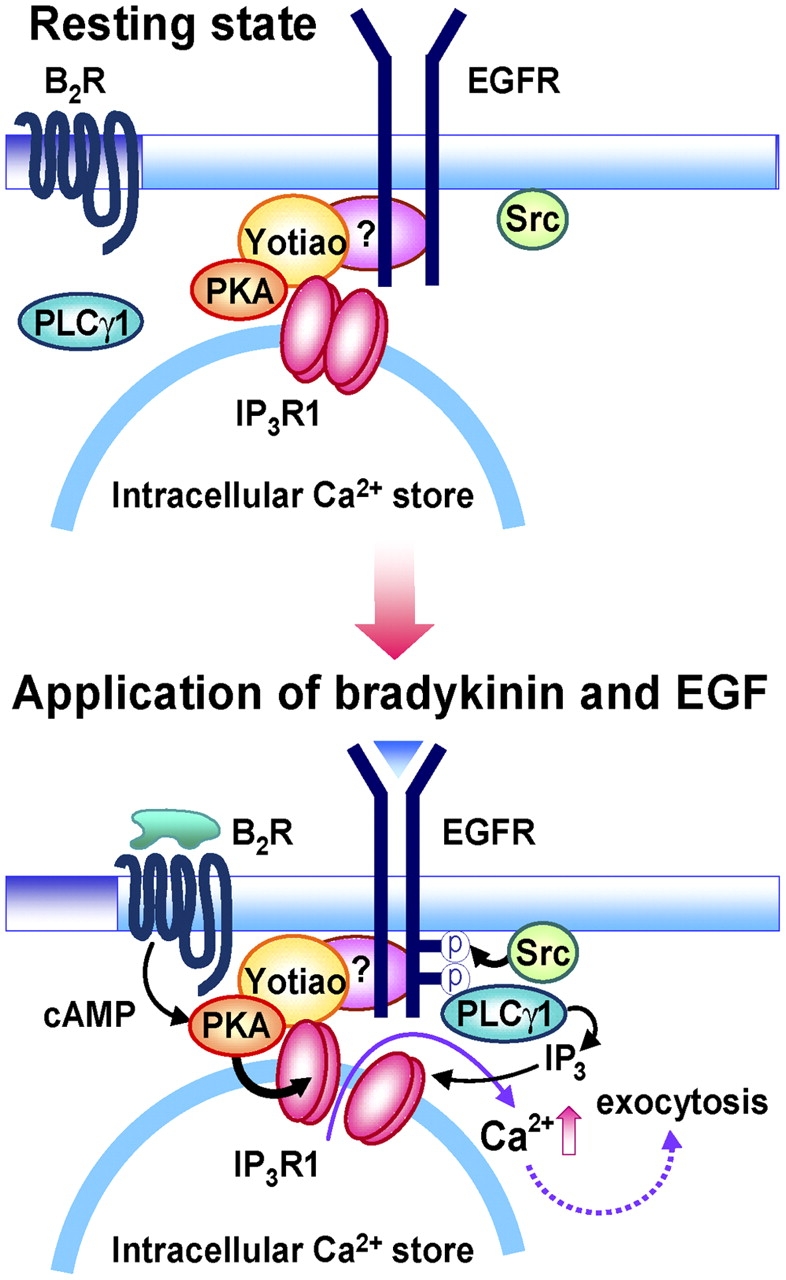
Suggested model for the sensitization of EGF-induced Ca2 + release and exocytosis by BK. The functionally silent EGF-induced Ca2+ signaling is acquired in response to BK stimulation via regulation of at least two components in the EGF signaling system, i.e., EGFR and IP3R1. First, BK triggers Src-dependent phosphorylation of EGFR, which results in enhanced recruitment and activation of PLCγ1, thereby allowing for generation of IP3. Second, BK induces PKA-dependent phosphorylation of IP3R1, increasing its responsiveness to IP3. IP3 generated by PLC activation becomes capable of triggering Ca2+ release, which leads to neurotransmitter release.
Materials and methods
Antibodies
Rabbit pAbs against IP3R subtypes were raised using peptides specific to the terminal 10–13 aa of type 1 (HPPHMNVNPQQPA), type 2 (SNTPHENHHMPPA), and type 3 (FVDVQNCMSR). The IP3R antibodies were affinity purified and the specificity was confirmed (Yoo et al., 2001). Other antibodies used were anti-phosphotyrosine (4G10) and anti-EGFR antibodies, which were purchased from Upstate Biotechnology; anti–flotillin-1 antibody, which was purchased from BD Transduction Laboratories; antibodies against the catalytic (PKACatα) and regulatory subunits of PKA (PKARegIIα), which were purchased from Santa Cruz Biotechnology, Inc.; phospho-serine/threonine PKA substrate antibody, which was purchased from Cell Signaling Technology; and yotiao antibody, which was purchased from Zymed Laboratories.
Isolation and primary cultures of rat adrenal medullary chromaffin cells
Primary cultures of rat adrenal chromaffin cells were prepared as we described in detail elsewhere (Hur et al., 2004).
Cell culture
Wild-type and PKA-deficient PC12 cells were grown in RPMI 1640 supplemented with 10% BCS and 5% HS. PKA-deficient PC12 cells stably expressing mutant regulatory subunits of PKA that are deficient in cAMP binding were previously described (Ginty et al., 1991; Cheng et al., 2002), and were provided by Y. Chern (Institute of Biomedical Sciences, Taiwan, China). Cells were incubated in serum-free medium overnight before use.
Amperometric measurement
Electrophysiological recording conditions were as we described in detail elsewhere (Kim et al., 2000). For data acquisition and analysis, pClamp 8 software (Axon Instruments, Inc.) and IGOR software (WaveMetrics, Inc.) were used.
Measurement of [Ca2+]i and its imaging
The level of [Ca2+]i was measured as we previously described (Hur et al., 2004). For Ca2+ imaging experiments, a monochromator-based spectrophotofluorimetric system (DeltaScan Illumination System; PTI) was used. Cells were loaded with 4 μM fura-2/AM (Molecular Probes) in the presence of sulphinpyrazone (250 μM). The coverslips were mounted on a stage of a microscope (model Eclipse TE300; Nikon) and cells were intermittently excited at 340 and 380 nm. Intensified charge-coupled device video camera (IC-200; PTI) was used to collect information. The fluorescence signal at 510 nm was collected, and the ratio of fluorescence at the two excitation wavelengths was calculated by Imagemaster software (v. 1.4; PTI). Areas on the same coverslips without cells were recorded as background images.
Measurement of IP3
Concentration of IP3 in cells was determined by competition assay with [3H]IP3 as we described in detail elsewhere (Kim et al., 1999).
RNA interference
siRNA duplexes targeting IP3R1 (5′-UGUACCAGGUGCUCUUGAC-3′ and 5′-GCACCAGCAGCUACAACUA-3′), IP3R3 (5′-AACACCAGCUGGAAGAUCAAC-3′ and 5′-AAGAAGUUCCGAGAUUGCCUC-3′), and yotiao (5′-AAGCAGACACAUGGUACGAUU-3′, 5′-AACGACGAGCUUCAUAAGAUA-3′, and 5′-AAAAUUCGAAGCCGCCAUUAA-3′) were purchased from Dharmacon. Transfection with siRNA pools was performed by electroporation and down-regulation of IP3Rs was confirmed by immunofluorescence and Western blot analysis. Effect of yotiao siRNA was confirmed by quantitative RT-PCR. For quantitative RT-PCR, cellular RNA was extracted from control and yotiao siRNA-expressing cells and 2 μg of total RNA was reverse-transcribed into cDNA by M-MuLV reverse transcriptase (Roche Molecular Biochemicals). Primers used in PCR were as follows: yotiao forward 5′-CACGAGGCTGAGATTACAAA-3′ and reverse 5′-TCATTTCATTTTGCAAGCCG-3′; β-actin forward 5′-TGTCACCAACTGGGACGATA-3′ and reverse 5′-GGGGTGTTGAAGGTCTCAAA-3′.
Immunoprecipitation and immunoblotting
Immunoblot analysis was performed as we previously described (Hur et al., 2004). For immunoprecipitation, samples were lysed using a hypotonic buffer (10 mM Tris/HCl, pH 7.5, 10 mM NaCl, 0.5 mM EGTA, 1 mM EDTA, 1% NP-40, and 0.25% sodium deoxycholate) containing inhibitors of proteases and phosphatases. After sonication, samples were cleared of insoluble debris by centrifugation at 15,000 g. Anti-EGFR, anti-PKACat, anti-PKARegII, or affinity-purified anti-IP3R1 antibodies coupled to protein A– or G–Sepharose beads were incubated with solubilized lysates. MBS buffer (25 mM Mes, 150 mM NaCl, pH 6.5) containing 0.5% Triton X-100 was used for immunoprecipitation with the DRM fractions. Isolation of the DRM microdomains was performed as we described in detail elsewhere (Hur et al., 2004). The beads were washed four times with the hypotonic or MBS buffer described above. Bound proteins were resolved by SDS-PAGE and analyzed by Western blotting. Signals were detected with an ECL detection system (Neuronex Co.).
Confocal microscopy and image analysis
Adrenal chromaffin cells fixed with 4% PFA were incubated with blocking solution (2.5% BSA and 5% HS in PBS) for 30 min at 4°C. Slides were incubated overnight at 4°C with anti–flotillin-1 and anti-IP3R1 or anti-IP3R3 antibody as indicated. After washing, samples were incubated with Cy3- (Jackson ImmunoResearch Laboratories) and FITC-conjugated secondary antibody (Santa Cruz Biotechnology, Inc.) for 1 h at RT. After washing, samples were treated with RNase (2 μg/ml) for 30 min at RT, followed by a 15-min incubation with 1 μg/ml propidium iodide. Slides were mounted and visualized by a confocal laser scanning microscopy (LSM 510 Meta; Carl Zeiss MicroImaging, Inc.). Postacquisition processing of images was performed with Adobe Photoshop 7.0 software.
Statistical analysis
All traces and immunoblots presented are representative of at least three separate experiments. All quantitative data are presented as means ± SEM of a minimum of three experiments. Comparisons between two groups were analyzed via t test, and values of P < 0.05 were considered to be significant.
Online supplemental material
Table S1 shows that BK induces generation of cAMP in PC12 cells. Confocal images showing distribution of EGFR and IP3R1 and colocalization of these receptors at the cell periphery in (A) vehicle- and (B) BK-treated cells are presented in Fig. S1. In Fig. S1, C shows that components of the IP3R1 complex were also detected in immunoprecipitates of the IP3R1, and the complex formation was not altered by BK stimulation. Fig. S2 shows the time course of BK induced-phosphorylation of EGFR. Methods for measurement of cAMP, immunogold EM and in situ hybridization are described in Online supplemental methods, available at http://www.jcb.org/cgi/content/full/jcb.200411034/DC1.
Acknowledgments
Authors are grateful to Ms. Yun-Hee Kim for confocal microscopy instrumentation and Dr. Byoung Dae Lee for critical discussions.
This work was supported by the National Research Laboratory Program (2000-N-NL-01-C-150), the Brain Neurobiology Research Program (M10412000088-04N1200-08810), the System Bio-Dynamics National Research Center of the Ministry of Science and Technology, and the Brain Korea 21 Program of the Ministry of Education.
Abbreviations used in this paper: 2APB, 2-aminoethyl diphenylborate; AKAP, A-kinase anchoring protein; AKAP-IS, AKAP-in silico; BK, bradykinin; DRM, detergent-resistant membrane; EGFR, EGF receptor; IP3R, inositol 1,4,5-trisphosphate receptor; IP3R1, IP3R type 1; NMDA, N-methyl-d-aspartic acid; PKA, cAMP-dependent protein kinase; PM, plasma membrane.
References
- Alto, N.M., S.H. Soderling, N. Hoshi, L.K. Langeberg, R. Fayos, P.A. Jennings, and J.D. Scott. 2003. Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proc. Natl. Acad. Sci. USA. 100:4445–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman, A.L., and J.D. Scott. 2002. Kinase- and phosphatase-anchoring proteins: harnessing the dynamic duo. Nat. Cell Biol. 4:E203–E206. [DOI] [PubMed] [Google Scholar]
- Berridge, M.J., M.D. Bootman, and H.L. Roderick. 2003. Calcium signaling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4:517–529. [DOI] [PubMed] [Google Scholar]
- Bruce, J.I., T.J. Shuttleworth, D.R. Giovannucci, and D.I. Yule. 2002. Phosphorylation of inositol 1,4,5-trisphosphate receptors in parotid acinar cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. J. Biol. Chem. 277:1340–1348. [DOI] [PubMed] [Google Scholar]
- Carnegie, G.K., and J.D. Scott. 2003. A-kinase anchoring proteins and neuronal signaling mechanisms. Genes Dev. 17:1557–1568. [DOI] [PubMed] [Google Scholar]
- Carrasco, G.A., and L.D. Van de Kar. 2003. Neuroendocrine pharmacology of stress. Eur. J. Pharmacol. 463:235–272. [DOI] [PubMed] [Google Scholar]
- Chen, L., D.M. Chetkovich, R.S. Petralia, N.T. Sweeney, Y. Kawasaki, R.J. Wenthold, D.S. Bredt, and R.A. Nicoll. 2000. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 408:936–943. [DOI] [PubMed] [Google Scholar]
- Cheng, H.C., H.M. Shih, and Y. Chern. 2002. Essential role of cAMP-response element-binding protein activation by A2A adenosine receptors in rescuing the nerve growth factor-induced neurite outgrowth impaired by blockage of the MAPK cascade. J. Biol. Chem. 277:33930–33942. [DOI] [PubMed] [Google Scholar]
- Colledge, M., R.A. Dean, G.K. Scott, L.K. Langeberg, R.L. Huganir, and J.D. Scott. 2000. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 27:107–119. [DOI] [PubMed] [Google Scholar]
- Danoff, S.K., C.D. Ferris, C. Donath, G.A. Fischer, S. Munemitsu, A. Ullrich, S.H. Snyder, and C.A. Ross. 1991. Inositol 1,4,5-trisphosphate receptors: distinct neuronal and nonneuronal forms derived by alternative splicing differ in phosphorylation. Proc. Natl. Acad. Sci. USA. 88:2951–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas, P., N. Wanaverbecq, F.C. Abogadie, M. Mistry, and D.A. Brown. 2002. Signaling microdomains define the specificity of receptor-mediated InsP(3) pathways in neurons. Neuron. 34:209–220. [DOI] [PubMed] [Google Scholar]
- DeSouza, N., S. Reiken, K. Ondrias, Y.M. Yang, S. Matkovich, and A.R. Marks. 2002. Protein kinase A and two phosphatases are components of the inositol 1,4,5-trisphosphate receptor macromolecular signaling complex. J. Biol. Chem. 277:39397–39400. [DOI] [PubMed] [Google Scholar]
- Echevarria, W., M.F. Leite, M.T. Guerra, W.R. Zipfel, and M.H. Nathanson. 2003. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat. Cell Biol. 5:440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fivaz, M., and T. Meyer. 2003. Specific localization and timing in neuronal signal transduction mediated by protein-lipid interactions. Neuron. 40:319–330. [DOI] [PubMed] [Google Scholar]
- Ginty, D.D., D. Glowacka, C. DeFranco, and J.A. Wagner. 1991. Nerve growth factor-induced neuronal differentiation after dominant repression of both type I and type II cAMP-dependent protein kinase activities. J. Biol. Chem. 266:15325–15333. [PubMed] [Google Scholar]
- Graness, A., A. Adomeit, B. Ludwig, W.D. Muller, R. Kaufmann, and C. Liebmann. 1997. Novel bradykinin signaling events in PC12 cells: stimulation of the cAMP pathway leads to cAMP-mediated translocation of protein kinase Cɛ. Biochem. J. 327:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar, R.E., A.D. Burgstahler, M.H. Nathanson, and B.E. Ehrlich. 1998. Type III InsP3 receptor channel stays open in the presence of increased calcium. Nature. 396:81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori, M., A.Z. Suzuki, T. Higo, H. Miyauchi, T. Michikawa, T. Nakamura, T. Inoue, and K. Mikoshiba. 2004. Distinct roles of inositol 1,4,5-trisphosphate receptor types 1 and 3 in Ca2+ signaling. J. Biol. Chem. 279:11967–11975. [DOI] [PubMed] [Google Scholar]
- Hoshi, N., J.S. Zhang, M. Omaki, T. Takeuchi, S. Yokoyama, N. Wanaverbecq, L.K. Langeberg, Y. Yoneda, J.D. Scott, D.A. Brown, and H. Higashida. 2003. AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat. Neurosci. 6:564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur, E.M., and K.T. Kim. 2002. G protein-coupled receptor signaling and cross-talk: achieving rapidity and specificity. Cell. Signal. 14:397–405. [DOI] [PubMed] [Google Scholar]
- Hur, E.M., Y.S. Park, B.D. Lee, I.H. Jang, H.S. Kim, T.D. Kim, P.G. Suh, S.H. Ryu, and K.T. Kim. 2004. Sensitization of epidermal growth factor-induced signaling by bradykinin is mediated by c-Src. Implications for a role of lipid microdomains. J. Biol. Chem. 279:5852–5860. [DOI] [PubMed] [Google Scholar]
- Johenning, F.W., and B.E. Ehrlich. 2002. Signaling microdomains: InsP(3) receptor localization takes on new meaning. Neuron. 34:173–175. [DOI] [PubMed] [Google Scholar]
- Kim, K.T., D.S. Koh, and B. Hille. 2000. Loading of oxidizable transmitters into secretory vesicles permits carbon-fiber amperometry. J. Neurosci. 20:RC101–RC105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y.H., T.J. Park, Y.H. Lee, K.J. Baek, P.G. Suh, S.H. Ryu, and K.T. Kim. 1999. Phospholipase C-δ1 is activated by capacitative calcium entry that follows phospholipase C-β activation upon bradykinin stimulation. J. Biol. Chem. 274:26127–26134. [DOI] [PubMed] [Google Scholar]
- Leite, M.F., E.C. Thrower, W. Echevarria, P. Koulen, K. Hirata, A.M. Bennett, B.E. Ehrlich, and M.H. Nathanson. 2003. Nuclear and cytosolic calcium are regulated independently. Proc. Natl. Acad. Sci. USA. 100:2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa, T., A. Maeda, T. Yamazawa, K. Hirose, T. Kurosaki, and M. Iino. 1999. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 18:1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakade, S., S.K. Rhee, H. Hamanaka, and K. Mikoshiba. 1994. cAMP-dependent phosphorylation of an immunoaffinity-purified homotetrameric inositol 1,4,5-trisphosphate receptor (type I) increases Ca2+ flux in reconstituted lipid vesicles. J. Biol. Chem. 269:6735–6742. [PubMed] [Google Scholar]
- O'Donnell, T., K.M. Hegadoren, and N.C. Coupland. 2004. Noradrenergic mechanisms in the pathophysiology of post-traumatic stress disorder. Neuropsychobiology. 50:273–283. [DOI] [PubMed] [Google Scholar]
- Pandiella, A., and J. Meldolesi. 1989. Reinforcement of signal generation at B2 bradykinin receptors by insulin, epidermal growth factors, and other growth factors. J. Biol. Chem. 264:3122–3130. [PubMed] [Google Scholar]
- Patterson, R.L., D. Boehning, and S.H. Snyder. 2004. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu. Rev. Biochem. 73:437–465. [DOI] [PubMed] [Google Scholar]
- Poburko, D., K.H. Kuo, J. Dai, C.H. Lee, and C. van Breemen. 2004. Organellar junctions promote targeted Ca2+ signaling in smooth muscle: why two membranes are better than one. Trends Pharmacol. Sci. 25:8–15. [DOI] [PubMed] [Google Scholar]
- Schlossmann, J., A. Ammendola, K. Ashman, X. Zong, A. Huber, G. Neubauer, G.X. Wang, H.D. Allescher, M. Korth, M. Wilm, et al. 2000. Regulation of intracellular calcium by a signaling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature. 404:197–201. [DOI] [PubMed] [Google Scholar]
- Straub, S.V., D.R. Giovannucci, J.I. Bruce, and D.I. Yule. 2002. A role for phosphorylation of inositol 1,4,5-trisphosphate receptors in defining calcium signals induced by Peptide agonists in pancreatic acinar cells. J. Biol. Chem. 277:31949–31956. [DOI] [PubMed] [Google Scholar]
- Takei, K., R.M. Shin, T. Inoue, K. Kato, and K. Mikoshiba. 1998. Regulation of nerve growth mediated by inositol 1,4,5-trisphosphate receptors in growth cones. Science. 282:1705–1708. [DOI] [PubMed] [Google Scholar]
- Tang, T.S., and I. Bezprozvanny. 2004. Dopamine receptor-mediated Ca2+ signaling in striatal medium spiny neurons. J. Biol. Chem. 279:42082–42094. [DOI] [PubMed] [Google Scholar]
- Tang, T.S., H. Tu, Z. Wang, and I. Bezprozvanny. 2003. Modulation of type 1 inositol 1,4,5-trisphosphate receptor function by protein kinase A and protein phosphatase 1α. J. Neurosci. 23:403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, C.W., P.C. da Fonseca, and E.P. Morris. 2004. IP3 receptors: the search for structure. Trends Biochem. Sci. 29:210–219. [DOI] [PubMed] [Google Scholar]
- Thrower, E.C., R.E. Hagar, and B.E. Ehrlich. 2001. Regulation of Ins(1,4,5)P3 receptor isoforms by endogenous modulators. Trends Pharmacol. Sci. 22:580–586. [DOI] [PubMed] [Google Scholar]
- Treves, S., C. Franzini-Armstrong, L. Moccagatta, C. Arnoult, C. Grasso, A. Schrum, S. Ducreux, M.X. Zhu, K. Mikoshiba, T. Girard, et al. 2004. Junctate is a key element in calcium entry induced by activation of InsP3 receptors and/or calcium store depletion. J. Cell Biol. 166:537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, H., T.S. Tang, Z. Wang, and I. Bezprozvanny. 2004. Association of type 1 inositol 1,4,5-trisphosphate receptor with AKAP9 (Yotiao) and protein kinase A. J. Biol. Chem. 279:19375–19382. [DOI] [PubMed] [Google Scholar]
- Tu, J.C., B. Xiao, J.P. Yuan, A.A. Lanahan, K. Leoffert, M. Li, D.J. Linden, and P.F. Worley. 1998. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 21:717–726. [DOI] [PubMed] [Google Scholar]
- Tucker, P., W.D. Ruwe, B. Masters, D.E. Parker, A. Hossain, R.P. Trautman, and D.B. Wyatt. 2004. Neuroimmune and cortisol changes in selective serotonin reuptake inhibitor and placebo treatment of chronic posttraumatic stress disorder. Biol. Psychiatry. 56:121–128. [DOI] [PubMed] [Google Scholar]
- Westphal, R.S., S.J. Tavalin, J.W. Lin, N.M. Alto, I.D. Fraser, L.K. Langeberg, M. Sheng, and J.D. Scott. 1999. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 285:93–96. [DOI] [PubMed] [Google Scholar]
- Wojcikiewicz, R.J., and S.G. Luo. 1998. Phosphorylation of inositol 1,4,5-trisphosphate receptors by cAMP-dependent protein kinase. Type I, II, and III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cells. J. Biol. Chem. 273:5670–5677. [DOI] [PubMed] [Google Scholar]
- Wong, R.W., and L. Guillaud. 2004. The role of epidermal growth factor and its receptors in mammalian CNS. Cytokine Growth Factor Rev. 15:147–156. [DOI] [PubMed] [Google Scholar]
- Xiao, B., J.C. Tu, R.S. Petralia, J.P. Yuan, A. Doan, C.D. Breder, A. Ruggiero, A.A. Lanahan, R.J. Wenthold, and P.F. Worley. 1998. Homer regulates the association of group 1 metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron. 21:707–716. [DOI] [PubMed] [Google Scholar]
- Yamada, M., T. Ikeuchi, and H. Hatanaka. 1997. The neurotrophic action and signalling of epidermal growth factor. Prog. Neurobiol. 51:19–37. [DOI] [PubMed] [Google Scholar]
- Yokoyama, K., I.H. Su Ih, T. Tezuka, T. Yasuda, K. Mikoshiba, A. Tarakhovsky, and T. Yamamoto. 2002. BANK regulates BCR-induced calcium mobilization by promoting tyrosine phosphorylation of IP3 receptor. EMBO J. 21:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo, S.H., Y.S. Oh, M.K. Kang, Y.H. Huh, S.H. So, H.S. Park, and H.Y. Park. 2001. Localization of three types of the inositol 1,4,5-trisphosphate receptor/Ca2+ channel in the secretory granules and coupling with the Ca2+ storage proteins chromogranins A and B. J. Biol. Chem. 276:45806–45812. [DOI] [PubMed] [Google Scholar]
- Yuan, J.P., K. Kiselyov, D.M. Shin, J. Chen, N. Shcheynikov, S.H. Kang, M.H. Dehoff, M.K. Schwarz, P.H. Seeburg, S. Muallem, and P.F. Worley. 2003. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell. 114:777–789. [DOI] [PubMed] [Google Scholar]