

Abstract
Vascular endothelial growth factor (VEGF) is a critical mediator of blood vessel formation during development and in pathological conditions. In this study, we demonstrate that VEGF bioavailability is regulated extracellularly by matrix metalloproteinases (MMPs) through intramolecular processing. Specifically, we show that a subset of MMPs can cleave matrix-bound isoforms of VEGF, releasing soluble fragments. We have mapped the region of MMP processing, have generated recombinant forms that mimic MMP-cleaved and MMP-resistant VEGF, and have explored their biological impact in tumors. Although all forms induced similar VEGF receptor 2 phosphorylation levels, the angiogenic outcomes were distinct. MMP-cleaved VEGF promoted the capillary dilation of existent vessels but mediated a marginal neovascular response within the tumor. In contrast, MMP-resistant VEGF supported extensive growth of thin vessels with multiple and frequent branch points. Our findings support the view that matrix-bound VEGF and nontethered VEGF provide different signaling outcomes. These findings reveal a novel aspect in the regulation of extracellular VEGF that holds significance for vascular patterning.
Introduction
Vascular patterning during development is guided by precise spatial cues, multiple and sequentially regulated growth factors, and guidance signals (Darland and D'Amore, 2001; Rossant and Hirashima, 2003). The result is a highly organized and hierarchical vascular array that is typical of nonpathological adult tissues. In contrast, vessel formation in tumors appears much less programmed and is improvised from a limited array of stimulatory factors (Carmeliet and Jain, 2000; McDonald and Choyke, 2003). Key among these is VEGF (Ferrara, 2000, 2002; Dvorak, 2002).
VEGF signaling is essential for the specification, morphogenesis, differentiation, and homeostasis of vessels (Gerber et al., 1999, 2002; Helmlinger et al., 2000; Damert et al., 2002; Ferrara et al., 2003). Furthermore, this signaling pathway is an integral component of pathological angiogenesis during tumor expansion (Inoue et al., 2002). In fact, the blockade of VEGF production or of VEGF receptor 2 (VEGFR2) phosphorylation results in the suppression of vascular growth and in the concomitant reduction of tumor mass and metastasis (Kim et al., 1993; Ferrara et al., 2004). Unlike most mammalian genes, the inactivation of only one allele results in embryonic lethality at mid-gestation from severe cardiovascular defects (Carmeliet et al., 1996; Ferrara et al., 1996). Interestingly, an organ-specific, twofold increase of VEGF can also lead to lethality (Miquerol et al., 2000). Thus, alterations of VEGF levels translate into significant pathological effects on the vasculature and on the organism as a whole.
Although much emphasis has been placed on understanding the production and stability of VEGF mRNA, relatively little attention has been given to the study of the stability and processing of VEGF proteins themselves. Upon secretion, VEGF becomes bound to the ECM (Park et al., 1993) and is widely viewed to act in a paracrine fashion. The interaction of VEGF with matrix proteins is mediated through the carboxy-terminal region, also known as a heparin-binding or ECM-binding domain (Houck et al., 1991, 1992). The regulation of VEGF in the extracellular environment has been implicated in the angiogenic switch, facilitating the transition from hyperplastic to malignant tumor formation (Bergers et al., 2000).
VEGF is encoded by a single gene that is located on chromosome 6p (Vincenti et al., 1996). The coding region spans 14 kb and contains eight exons. Through alternative splicing, a single pre-mRNA molecule can generate five isoforms (Robinson and Stringer, 2001). These vary in length from 121 to 206 aa (e.g., 121, 145, 165, 189, and 206) and differ by the presence or absence of sequences located in exons 6 and 7. Exon 8 is common to all isoforms (Tischer et al., 1991). Exons 6 and 7 have been shown to encode the ECM-binding domain of the protein. This domain is able to bind heparin sulfate proteoglycans and other matrix proteins (Houck et al., 1992; Poltorak et al., 1997) and is thought to be responsible for the sequestration of VEGF within the matrix. Thus, the matrix constitutes a reservoir for the growth factor, which becomes liberated by matrix breakdown via extracellular enzymes such as heparinases and plasmin (Houck et al., 1992; Plouet et al., 1997). Matrix metalloproteinases (MMPs) have also been implicated in the regulation of VEGF bioavailability from extracellular stores. However, a direct mechanism for their effects has remained elusive (Bergers et al., 2000; Rodriguez-Manzaneque et al., 2001). One possibility is that the release of VEGF occurs through the degradation of matrix proteins by MMPs, which is similar to the effects of heparinases and plasmin.
An alternative view is that VEGF is released by direct, intramolecular processing events. Here, we demonstrate that a subset of MMPs can cleave VEGF, releasing the receptor-binding domain from the matrix-binding motif of the protein. Further exploration into the biological significance of this processing event revealed that matrix-bound and soluble VEGF have alternative outcomes on vascular morphogenesis.
Results
VEGF is processed by a subset of MMPs
To test the hypothesis that VEGF could be a substrate of MMPs, we biotinylated the growth factor and performed a series of in vitro incubations with purified enzymes (Fig. 1 A). VEGF164 was cleaved by MMP3, 7, 9, and 19, releasing a 16-kD fragment. Two additional MMPs (1 and 16) also released the fragment, but less effectively. The presence of heparin aided processing by MMP3, but hindered the cleavage of VEGF by MMP9 (Fig. 1 A). Glycosylation was not required for proteolysis because nonglycosylated VEGF164 was cleaved in a similar manner (Fig. 1 B). Fragments could be detected 5 min after exposure to MMP3, as indicated by time course analysis. The cleavage product was stable, and it accumulated over time with concomitant reduction in the levels of intact growth factor (Fig. 1 B). Dose–response analysis also indicated that the cleavage event occurred in at least two stages. An intermediate species of a higher molecular mass was produced first, and a 13-kD stable fragment (equivalent to the 16-kD fragment from glycosylated growth factor) was subsequently generated, suggesting the presence of at least two proximal cleavage sites (Fig. 1 C). Interestingly, digestion was found to be optimal at slightly acidic pH (6.9–7.2; Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200409115/DC1). In addition, we showed that both matrix-bound isoforms (VEGF164 and VEGF188) were targeted by MMPs, which generated identical molecular mass fragments (Fig. 1 D).
Figure 1.

VEGF-A is cleaved by a subset of MMPs. (A) Biotinylated mVEGF164 was incubated with the indicated MMPs. The digestion products were resolved in tricine gels and were detected by avidin-HRP. (right) The presence of heparin was tested to evaluate its effect in VEGF processing by MMP3 and MMP9. (B and C) mVEGF164 was incubated with MMP3 at different time points (B) and molar ratios (C) as indicated. VEGF cleavage was visualized by SDS-PAGE followed by silver staining. In these experiments, nonglycosylated mVEGF164 was used, hence the smaller size. (D) Both mVEGF164 and mVEGF188 were incubated with MMP3. VEGF cleavage was examined with immunoblots probed with an amino-terminal antibody (463; see Fig. 3 A). Glycosylated mVEGF164 was used for A and D, and nonglycosylated mVEGF164 was used for B and C. Note the faster mobility in B and C. 44 kD, glycosylated mVEGF164 dimer; 22 kD, glycosylated mVEGF164 monomer; 19 kD, nonglycosylated mVEGF164 monomer; 16 kD, glycosylated mVEGF164-cleaved monomer fragment; 13 kD, nonglycosylated mVEGF164-cleaved monomer fragment; 6 kD, mVEGF164-cleaved monomer fragment. V, VEGF; M, MMP3; VM, VEGF + MMP3; ADAMTS, a disintegrin and metalloproteinase domain with thrombospondin repeats.
Proteolytic cleavage was inhibited by purified tissue inhibitors of MPs (TIMP)1 and TIMP2 (both of which are endogenous inhibitors of MMP3), but was not inhibited by aprotinin, a serine protease inhibitor (Fig. 2 A). Similarly, digestion by MMP9 was blocked by TIMP3 (Fig. 2 B). Plasmin was previously shown to cleave VEGF189 and release this isoform from the matrix in a dose-dependent manner (Park et al., 1993; Keck et al., 1997). Fig. 2 A shows that cleavage by plasmin was different from that generated by MMPs. Plasmin produced a smaller and less stable fragment than the one resulting from MMP exposure (Fig. 2 A). Furthermore, cleavage of VEGF by MMP3 also occurred when the growth factor was anchored by heparin. Bound growth factor that was exposed to MMP3 resulted in the release of a soluble 16-kD species (supernatant), whereas the 6-kD polypeptide remained bound (Fig. 2 C).
Figure 2.

Characterization of VEGF intramolecular cleavage in vitro and in vivo. (A) mVEGF164 was incubated with MMP3 or with plasmin in the presence of specific inhibitors. V, VEGF; M, MMP3; P, plasmin; T1, TIMP1; T2, TIMP2; A, aprotinin. (B) Cleavage by MMP9 and MMP3 was tested in the presence of heparin and TIMP3. (C) Heparin-bound, biotinylated mVEGF164 was exposed to MMP3, and soluble and bound fractions were evaluated by immunoblotting. Glycosylated mVEGF164 was used for A–C. 44 kD, glycosylated mVEGF164 dimer; 22 kD, glycosylated mVEGF164 monomer; 16 and 6 kD, glycosylated mVEGF164-cleaved monomer fragments. (D) VEGF fragments were detected in human ascites fluid from ovarian cancer in immunoblots with antibodies 375 (left) and 463 (right). The same blot was probed with both antibodies sequentially. (E) Serum levels of VEGF (intact and cleaved) in mice bearing mVEGF164 xenografts were measured by sequential “sandwich” ELISA with antibodies that recognize the receptor-binding domain and carboxy-terminal end of VEGF (amino acid coded by exon 8).
To ascertain whether this proteolytic event occurred in vivo, we used human ascites fluid from patients with ovarian cancer (collected without patient identifiers, under Institutional Review Board guidelines). Ascites fluid was collected in the presence of a cocktail of proteinase inhibitors to block cleavage events originating after isolation. Initial fractionation of VEGF was performed by affinity chromatography on a polyclonal anti-VEGF antibody column. The antibody interacted with multiple epitopes that were distributed throughout the entire VEGF protein, maximizing the retention of all isoforms and fragments. Eluted fractions were combined and were further evaluated with epitope-specific antibodies. A carboxy-terminal antibody (generated against residues coded by exon 8; see Fig. 3 A) recognized VEGF189, VEGF165, and VEGF121 on immunoblots but could not detect any cleaved fragments (Fig. 2 D). This result is consistent with the fact that the residues coded by exon 8 are removed after VEGF digestion by MMPs (see Fig. 3, A and B). In contrast, the amino-terminal antibody (see Fig. 3 A) was able to detect the intact isoforms (VEGF165 and VEGF121) in addition to a group of fragments ranging from 13 to 16 kD in molecular mass (Fig. 2 D). The fact that only the amino-terminal antibody was able to detect the smaller fragments is consistent with the occurrence of proteolysis in the carboxy-terminal region of VEGF. Both plasmin and MMP3 were also detected in the unfractionated ascites fluid (unpublished data).
Figure 3.

Localization of cleavage sites. (A) Structure of mVEGF164 and localization of cleavage sites. Cleavage sites were determined by Edman sequencing, MALDI-TOF MS, μLC/MSn, and μLC/MS/MS of 16 kD and 6 kD. 463 denotes antibody specific to the amino terminus (N-ter) of VEGF; 375 to the carboxy terminus (C-ter) of VEGF. (B) Glycosylated mVEGF164 was incubated with MMP3 or with plasmin at the following molar ratios: 8:1, 4:1, 2:1, and 1:1 (VEGF/proteinase). VEGF cleavage was examined by immunoblotting with antibodies 463 and 375. V, VEGF; 22 kD, glycosylated mVEGF164 monomer. (C, top) Schematic structure of mVEGF164, mVEGF113, and mVEGFΔ108–118 are shown. (bottom, left) Conditioned media from HEK 293T stable clones were examined by immunoblotting with antibody 463. (bottom, right) VEGFR2 phosphorylation was induced in PAE-VEGFR2 cells by 100 ng/ml VEGF. Antibodies against phosphotyrosine (PTyr) and VEGFR2 were used to examine phosphorylation of VEGFR2. (D) VEGFR2 phosphorylation was induced by different concentrations of VEGF as indicated. (E) Mesh-CAM assays were used for the evaluation of VEGF activity on E10 chicken embryos. The first panel illustrates the assay: polymerized, vitrogen-containing growth factor is placed onto CAM, and the angiogenic response (neovessels) is assessed independently of the preexisting CAM vessels. Each pellet contained 1 μg/mesh VEGF. Evaluation was determined 24 h after the application of pellets to CAM surface. Numbers below are the vessel mean/mm2 (±SD) obtained from the evaluation of four independent experiments. Bar, 100 μm. (F) Qualitative representation of angiogenesis in mice in response to a Matrigel plug containing different VEGF forms. (E and F) VEGF113 showed large, dilated vessels (brackets). In contrast, VEGFΔ108–118 showed significant sprouting of thin vessels (arrows). Indicated below is the average vascular density/mm2 (±SD) from four independent experiments. Vessel diameters shown in brackets are as follows: mVEGF164 = 15 μm; mVEGF113 = 109 μm (large) and 16 μm (small). Bar, 100 μm.
To further explore whether VEGF was cleaved in vivo, we devised a dual ELISA system that allowed the distinction between cleaved and intact VEGF based on the selective removal of amino acids that are coded by exon 8. Using this system, we determined that, in fact, 82–90% of circulating VEGF had been processed, as it lacked the carboxy-terminal domain (Fig. 2 E).
Processing of VEGF severs the receptor-binding domain from the extracellular, matrix-binding motif
Next, we mapped the cleavage sites in VEGF using matrix-assisted laser desorption time-of-flight (MALDI-TOF) mass spectrometry (MS), capillary and microcapillary nano–liquid chromatography MS (μLC/MSn), and microcapillary reverse-phase HPLC nano-electrospray tandem MS (μLC/MS/MS). Consistent with electrophoretic mobility evaluations, we found that proteolytic processing takes place in sequential steps. Initial cleavage occurs at residues 135, 120, and finally at residue 113 (Fig. 3 A). The events result in the dissociation of the receptor-binding motif from the ECM-binding domain. This proteolytic processing is different from the one mediated by plasmin (Fig. 3 B). Plasmin has been reported to cleave VEGF at position 110 (Park et al., 1993). Indeed, this fragment exhibits a faster mobility (13 kD) than the MMP3 fragment (16 kD; Fig. 3 B). Furthermore, plasmin proteolyses the small carboxy-terminal, 9-kD fragment to completion (as we have been unable to detect this fragment with carboxy-terminal antibodies), in contrast to its presence in samples that are exposed to MMP3 (6 kD; Fig. 3 B). Unlike digestion with MMP3, plasmin-produced fragments did not accumulate over time, and levels of the full-length growth factor became reduced, which is an indication of progressive degradation (Fig. S1 B).
MMP-cleaved VEGF fragments are able to phosphorylate VEGFR2 and induce angiogenesis
To ascertain whether the fragments were able to elicit receptor activation and angiogenesis, we generated a construct that mimicked the MMP-cleaved VEGF protein (VEGF113) and also developed an MMP-resistant VEGF form (VEGFΔ108–118; Fig. 3 C). The latter consisted of an 11-aa deletion between residues 108–118 (Fig. 3 C). These constructs were used to generate recombinant protein in human embryonic kidney (HEK) 293T cells and were tested for their susceptibility to MMP3 (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200409115/DC1). Even though the MMP-resistant VEGF contains aa 120 and 135, which are known to participate in the digestion of VEGF164, it appears that the removal of 108–118 alters the conformation of the growth factor so that aa 120 and 135 are no longer accessible sites for digestion.
Subsequently, porcine aortic endothelial (PAE) cells expressing VEGFR2 (PAE-VEGFR2) were exposed to various purified VEGF forms to evaluate receptor phosphorylation. MMP-cleaved and MMP-resistant VEGF forms were similarly capable of phosphorylating VEGFR2 (Fig. 3 D). Furthermore, dose–response analysis indicated that the kinetics of phosphorylation were similar to those displayed by wild-type VEGF164 (when isolated in an identical manner). Phosphorylation was detected at 50 ng and peaked at 1 μg when each one of the three protein forms was used (Fig. 3 D). More importantly, all forms were able to stimulate angiogenesis in the chorioallantoic membrane (CAM) assay (Fig. 3 E). In this assay, the neovessels are distinguished from the existent vascular network of CAM. Angiogenic growth is stimulated against gravity by growth factors that are embedded in a polymerized matrix (vitrogen), which is supported by a nylon mesh and placed onto CAM. The angiogenic response is noted when new vessels invade this acellular matrix, which is subsequently removed from CAM for visualization and quantitation. As seen in Fig. 3 E, a vitrogen plug with equivalent levels of the growth factor was placed onto the vascularized CAM. After 24 h, CAM was perfused, stained with rhodamine lectin, and the collagen plug was removed for evaluation under confocal microscopy. Interestingly, although all forms elicited an angiogenic response, VEGF113 showed fused, enlarged channels (Fig. 3 E, brackets); in contrast, MMP-resistant VEGF induced significant sprouting, resulting in thin, interconnected vessels (Fig. 3 E, arrows).
The isoforms were also tested in the Matrigel plug assay in mice. Each form was mixed with Matrigel and was injected subcutaneously; evaluation was performed after 7 d. Once again, all forms elicited an angiogenic response; however, the morphology of the vessels was significantly different (Fig. 3 F). Although VEGF164 displayed tortuous vessels of similar size to the adjacent vessels in the mouse, VEGF113 showed remarkably enlarged vessels (Fig. 3 F, brackets). In contrast, Matrigel that contained MMP-resistant VEGF demonstrated thin vessels with multiple branch points (Fig. 3 F, arrows).
Further exploration of biological effects that are mediated by the different VEGF forms was performed in tumor xenograft assays. T47D human carcinoma cell lines were transfected with expression vectors that coded for wild-type, MMP-cleaved, and MMP-resistant VEGF. Clones expressing similar levels of VEGF among the different constructs were selected for initial comparison (Fig. 4 A). Interestingly, we noted high molecular mass bands in the immunoblots from tumor lysates, a finding that was reproducible with several antibodies and was suggestive of macromolecular aggregates that included the growth factor. Transfected cells did not show differences in growth (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200409115/DC1). We also evaluated VEGF serum levels in animals bearing the four tumor types. In spite of similar VEGF levels within the tumor, the MMP-cleaved form (VEGF113) showed a 10–12-fold increase in the concentration of circulating growth factor (Fig. 4 B). Interestingly, tumors expressing VEGF113 grew poorly, were macroscopically pale, and demonstrated slow growth kinetics. In contrast, tumors expressing MMP-resistant VEGF displayed faster growth kinetics in comparison with control and with tumors expressing VEGF164 (Fig. 4 C). Together, these findings indicate that matrix-bound VEGF is more efficient in supporting a more functional angiogenic response than soluble VEGF. The results also revealed that circulating levels of VEGF are not necessarily direct indicators of tumor progression, as they did not correlate with tumor size or with vascular density. Similar findings were obtained with other clones and with a different human tumor cell line (HT1080).
Figure 4.
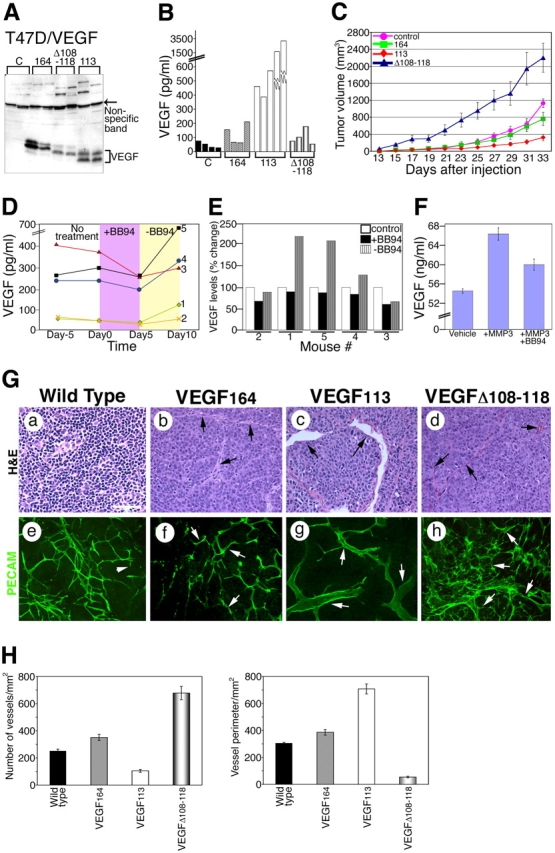
Induction of tumor growth in the presence of mVEGF 164 , mVEGF113, and mVEGF Δ 108–118. (A) Tumor lysates were immunoblotted with antibody 463. VEGF is indicated by a bracket. A nonspecific band (arrow) illustrates relative loading levels. Above this band, high molecular mass proteins were also immunoreactive. (B) Serum levels of VEGF in the different groups of mice bearing xenografts. (C) Tumor growth kinetics. Estimated volume was calculated using the following formula: volume (mm3) = 4/3π(0.5 × w)2 × (0.5 × l), where w is the width and l is the length. Results are mean ± SD. n = 8 for control, 164, and Δ108–118; n = 13 for 113. (D) Effect of treatment with MMP inhibitor BB94 on circulating serum levels of VEGF (n = 5). Each line corresponds to one mouse. VEGF measurements were performed 5 d before treatment (Day −5), immediately before treatment (Day 0), 5 d after treatment (Day 5), and 5 d after BB94 withdrawal (Day 10). (E) Percent change in VEGF levels from day 1 (1 d before the treatment with BB94). (F) VEGF levels in conditioned media of MMP3-treated and nontreated tumor explant cultures. Explants were incubated in the presence of vehicle, MMP3, and MMP3 + BB94. Levels of VEGF were determined by ELISA. (G) Microscopic analyses of tumor sections by hematoxylin and eosin (H&E) and platelet/endothelial cell adhesion molecule 1 (PECAM) staining. A representative tumor section of each mouse group is shown. Arrows point to vessels in b–h. Arrows in e–h indicate PECAM-positive vessels. Bar, 100 μm. (H) Quantitation of vascular density and size. (C, F, and H) Error bars are ±SD.
The relevance of MMPs to the levels of circulating VEGF was explored by treating mice carrying VEGF164-expressing tumors (size range 50–500 mm3) with the broad spectrum MMP inhibitor (BB94). Circulating VEGF levels were determined at four time points: (a) 5 d before treatment; (b) immediately before injection with BB94; (c) on the last day of the treatment; and (d) 5 d after BB94 withdrawal. VEGF levels decreased significantly at the end of 5 d of BB94 treatment in all mice (Fig. 4 D). The withdrawal of treatment was associated with a remarkable increase in serum VEGF levels (Fig. 4, D and E). We also isolated VEGF164 tumor explants and determined if exposure to MMP3 in the culture altered the concentration of VEGF in the conditioned media. The results shown in Fig. 4 F indicate that a relatively short exposure to active MMP3 results in the selective release of VEGF from tumor explants.
Vascular sprouting and branching is favored by matrix-bound VEGF, whereas vascular hyperplasia is induced by soluble VEGF
A detailed analysis of the tumor vessels revealed contrasting differences between the various VEGF forms. VEGF113-expressing tumors exhibited low vascular density and large size vessels. In contrast, MMP-resistant VEGF tumors were significantly more vascularized. Vessels from these tumors were generally thinner and displayed multiple branched points (Fig. 4, G and H). These findings were surprising, yet consistent amongst different clones and tumor cells that were used for transfections (T47D and HT1080). Vascular density, but not vascular volume, correlated with tumor size.
Vessels from areas adjacent to the tumor were also affected. In particular, vessels near the tumor-expressing VEGF113 displayed engorged and fragile vessels (Fig. 5). Vessels in the proximity of MMP-resistant VEGF tumors were also altered, but did not show significant hyperplasia. Instead, we noted that vessels from MMP-resistant tumors consisted of tortuous, thin capillaries, higher vascular density, and groups of capillary tufts that were reminiscent of the glomeruli bodies (Pettersson et al., 2000). Fig. 5 shows a detailed evaluation of vessel size that was observed in regions immediately adjacent (0.5 cm) and more peripheral (1 cm) to the tumor. Quantitations of the vessel diameter showed that vessels from VEGF113 were dilated and were more heterogeneous in size than vessels from tumors expressing other VEGF forms.
Figure 5.
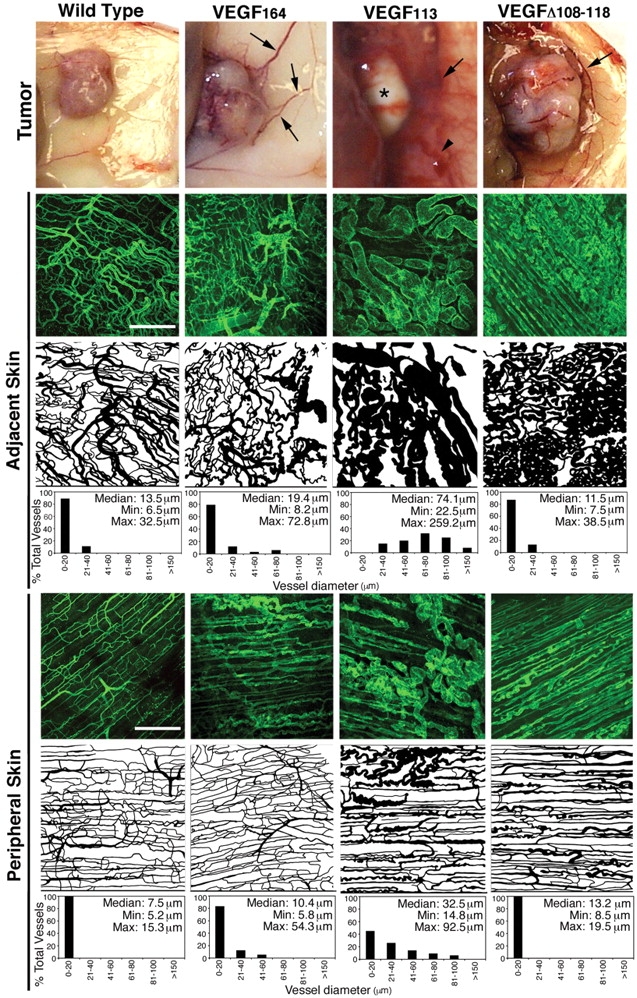
Effect of VEGF in vessels adjacent to tumors. Macroscopic appearance of tumor and tumor-surrounding skin is shown. Arrows point to large vessels. Arrowhead in VEGF113 denotes bleeding/fragility. Asterisk in VEGF113 shows the pale aspect of those tumors. Skin sections were stained with PECAM, and the vessel patterns from similar areas were traced and shown. These computer-generated images were used for quantitation of vessel diameter, as represented in the histograms (n = 8). Bars, 300 μm.
To further dissect the effect of these VEGF forms in capillary formation, we evaluated their effects in vitro. Purified growth factors at identical concentrations were included in fibrinogen/fibronectin gels before polymerization. Sepharose beads coated with PAE-VEGFR2 cells were mixed with the matrix as it polymerized and were observed consecutively for 3 d (Fig. 6 A). Wild-type VEGF164 elicited the growth of endothelial cells as sheets (Fig. 6 A, arrowheads) and induced capillary morphogenesis (Fig. 6 A, arrows). Interestingly, VEGF113 only induced the proliferation of endothelial cells as sheets. This contrasted with the strong morphogenic events that were mediated by MMP-resistant VEGF (Δ108–118). In the presence of this mutated growth factor, endothelial cells organized into cords, and phalloidin staining of the actin cytoskeleton demonstrated protrusion of multiple filopodia. Although visual inspection suggested that VEGF113 mediated a stronger proliferative response, a detailed quantitation of endothelial cell proliferation in all three cases did not reveal differences that were statistically significant (Fig. S3 B). These responses were mediated by VEGFR2. PAE cells that were not expressing VEGFR2 did not respond to the growth factor (Fig. 6 B). The effects were dependent on the presence of VEGF, as PAE-VEGFR2 cells did not respond in a similar manner when serum was used instead of VEGF (Fig. 6 B).
Figure 6.
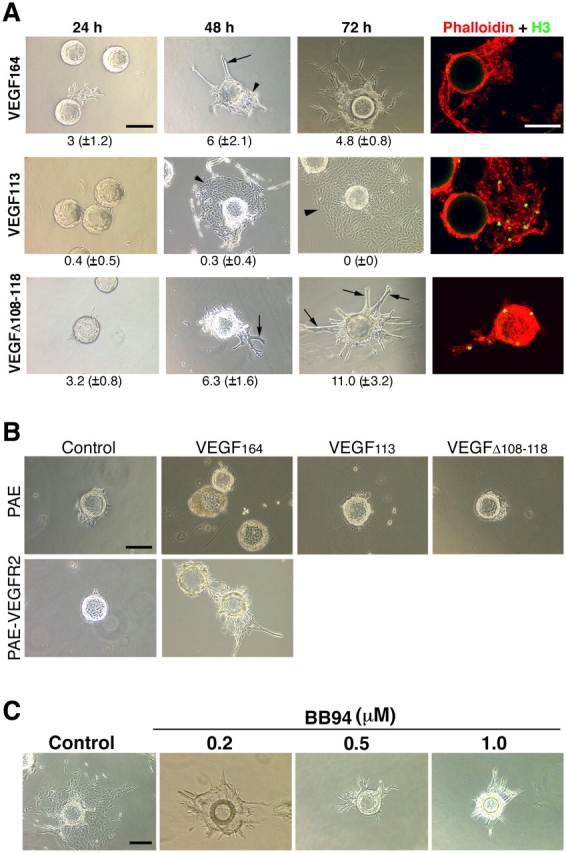
Morphogenic effects of different VEGF forms in vitro. In vitro angiogenesis assays were performed using Sepharose beads coated with PAE cells expressing VEGFR2 (PAE-VEGFR2). Endothelial-coated beads were embedded in fibrin/fibronectin gels containing different growth factors. (A) PAE-VEGFR2 cells formed cordlike structures in response to mVEGFΔ108–118 (arrows) and formed sheetlike structures when exposed to mVEGF113 (arrowheads). A mixture of both is seen in the presence of VEGF164. In the last column, simultaneous staining of cells with AlexaFluor546-conjugated phalloidin and with the antibody to phosphohistone H3 showed proliferation elicited by the different VEGF forms. Numbers below each panel correspond to the mean (±SD) of sprouts per bead. A total of five individual experiments were used for quantification. (B, top) In the absence of VEGFR2, PAE cells did not respond to VEGF113 or to mVEGFΔ108–118. (bottom) PAE-VEGFR2–coated beads were incubated with DME containing serum alone or in the presence of VEGF164. (C) Incorporation of increasing levels of BB94 showed a correlation between cord formation and MMP activity. Bars, 150 μm.
Together, the results indicate that in the case of VEGF164, the local, discrete digestion of VEGF is likely to occur as endothelial cells migrate and grow as sheets, whereas in the absence of such digestion, VEGF would mediate the organization of cords. To test this more directly, we included the broad-spectrum MMP inhibitor BB94 in the matrix assays and in the culture media. As predicted, we found that the number of cords was increased depending on the concentration of the MMP inhibitor (Fig. 6 C).
We next sought to determine whether VEGF was cleaved as cells invaded the matrix. For these experiments, we generated a peptide comprising the MMP cleavage sites that were identified in VEGF164 and a mutant peptide that was not cleaved by MMP3 (Fig. 7 A). Both peptides were conjugated to coumarin (fluorescence) at the amino terminus and to a 2,4-dinitrofluorobenzene (DNP; quencher) at the carboxy terminus to create a fluorescent peptide indicator of MMP activity. When intact, the peptide indicator did not emit a signal. Upon cleavage by MMP3, a strong fluorescence signal was recorded (Fig. 7 A). Inclusion of the wild-type peptide in fibrinogen gel assays revealed discrete areas of fluorescence in proximity to, or on the surface of, cells that invaded the gel, indicating that cleavage of the peptide had occurred (Fig. 7 B). No cleavage was detected when BB94 was included in the gel. Similarly, the mutant peptide lacking the cleavage site did not show a signal. Together, these results suggest that MMP cleavage of VEGF occurs in discrete, extracellular microdomains and results in distinct signaling outcomes. These in vitro findings are consistent with the effect of the different VEGF forms in tumors. Hyperplasia would be analogous to proliferation of cells as sheets (cleaved VEGF), whereas the invasion and morphogenesis of cords is analogous to active sprouting angiogenesis (uncleaved, matrix-tethered VEGF).
Figure 7.

Visualization of VEGF cleavage in vitro. (A) Coumarin-conjugated VEGF peptide and mutant control. Graph on the right shows proteinase-dependent emission of fluorescence. (B) When embedded in the fibrin/fibronectin gels, fluorescence is detected in areas adjacent to migrating endothelial cells (arrows). The inhibitor BB94 blocked the effect, and the mutant peptide did not emit fluorescence under similar conditions. Bar, 150 μm.
Discussion
We investigated the potential role of MMPs in the generation of bioactive VEGF from extracellular stores. In the process, we found that VEGF can be cleaved intramolecularly by a subset of MMPs. Processing results in the release of the receptor-binding domain from the ECM-binding motif that is present in the majority of VEGF-A isoforms. Furthermore, we noted that the state of free versus bound VEGF dictates whether a vascular network will undergo dilation/increased vessel size or will initiate active sprouting events, thus promoting two different modes of vascular expansion. The first requires the proliferation of cells as sheets, whereas the second entails the initial sensing of the environment by active filopodia extension, migration/invasion, and subsequent proliferation (Fig. 8). Overall, our findings support the view that matrix-bound and nontethered VEGF provide different signaling outcomes even though they act through the same cell surface receptor (VEGFR2).
Figure 8.

Model for the effect of MMPs in matrix-bound VEGF. (top) In the presence of a specific subset of MMPs, VEGF is cleaved (yellow) and is released from the matrix. This soluble VEGF induces vascular dilation in the existent vessels. (bottom) In the absence of MMPs, matrix-bound VEGF isoforms remain bound and induce sprouting angiogenesis events with little vascular dilation. The model implies that regulation of VEGF availability can be regulated by splicing events and/or extracellularly by the relative levels of MMPs.
The role of multiple VEGF-A isoforms, which modulates the interaction of VEGF with the matrix, has remained elusive. Using tumor cells with low levels of endogenous VEGF, Grunstein et al. (2000) introduced VEGF120, 164, and 188 isoforms individually and tested their effects in tumor angiogenesis. Although the approach did not eliminate endogenous VEGF from the invading stroma, the authors noted substantial differences in the vascularization of the tumors. In particular, VEGF188 displayed an extensive hypervascular response in comparison with the other isoforms (Grunstein et al., 2000). Subsequently, in a series of experiments, Ruhrberg et al. (2002) developed isoform-specific VEGF mice, revealing that VEGF-A isoforms play a critical role in the guidance of vascular patterning and in vessel diameter during development (Ruhrberg et al., 2002). Thus, mice expressing VEGF120 displayed lower vascular density, poor sprouting, and poor filopodia extension. In contrast, the sole expression of VEGF188 leads to increased multidirectional filopodia extension, higher vascular density, and disorganized patterning. Interestingly, mice that were heterozygous for both isoforms showed a “normalized” vasculature, suggesting that matrix association plays a role in vascular density and patterning.
Overall, the findings presented in this study confirm and extend the results observed with isoform-specific, VEGF-A transgenic animals. The introduction of a soluble VEGF-A isoform (VEGF113) resulted in poor vascular density and vessels that were larger than their wild-type counterparts. In contrast, the expression of MMP-resistant VEGF led to excessive and rather disorganized branching. Together, these studies indicate the existence of two modes for the regulation of VEGF interaction with ECM: (a) alternative splicing or (b) proteolytic processing. The second mode would directly depend on microenvironmental levels of specific MMPs/plasmins. Given the preponderance of MMPs during pathological conditions, we would predict that extracellular processing is likely to be a frequent regulatory event during tissue repair, inflammation, and in cancer (Coussens and Werb, 2002). In fact, the relative levels and availability of MMPs can offer an explanation for the heterogeneous nature of vessels in different tumors. Thus, discrete levels of MMPs could have an impact on VEGF signaling, resulting in alterations in vascular density, vessel diameter, and patterning through the direct alteration in the status of bound versus soluble VEGF. The concept that MMPs regulate the local distribution of VEGF has also been noted in a model of retinopathy in mice (Gerhardt, H., and C. Betsholtz, personal communication). Their findings provide evidence that MMP activity is linked to a cascade of events that regulate VEGF and vascular patterning.
Our results do not negate the possibility that the proteolysis of matrix molecules might also lead to the release of VEGF from extracellular stores. However, these events deserve a more detailed investigation, as does the nature of the interactions between VEGF and specific matrix molecules. Also, it is important to stress that several MMPs were found to mediate VEGF processing. 4 out of 13 MMPs tested were effective in severing VEGF from the matrix via specific intramolecular cleavage. Thus, it is likely that these effects are highly redundant in vivo.
MMPs play a significant role in matrix remodeling, enabling migration, and the establishment of new capillary beds (Heissig et al., 2003). Understanding the interplay between these molecules and the reciprocal effects on the tumor vasculature has been a major effort in the field. Recent findings that the inactivation of some MMPs by homologous recombination facilitates tumor expansion has propelled a reinterpretation of the initial protumorigenic view of MMPs in cancer (Overall and Lopez-Otin, 2002; Balbin et al., 2003). Similarly, the assumptions that MMPs were, in general, proangiogenic has been challenged by findings that some MMPs could also suppress neovascularization (Pozzi et al., 2000) by the production of angiogenesis inhibitors via proteolysis and other mechanisms (O'Reilly et al., 1999; Hamano et al., 2003). Our data also supports the view that MMPs act as sophisticated modulators rather than as simple inducers or suppressors. Understanding the discrete temporal control of MMPs and their spectrum of substrates and modulators will be essential to make strides toward novel therapeutic strategies that target MMPs in tumors.
The intramolecular cleavage of growth factors has been implicated in activation, in the alteration of biological properties, and in growth factor degradation (Fuentes-Prior et al., 2000; Junttila et al., 2000; McQuibban et al., 2000, 2002; Bergsten et al., 2001; Borrell-Pages et al., 2003; Nanba et al., 2003). In the case of the VEGF family, both VEGF-C and -D require extracellular cleavage for activation (McColl et al., 2003). Although VEGF-A165 is not proteolytically activated, it has been proposed that the longer isoforms (189 and 206) are hindered from inducing the activation of VEGF receptors and that cleavage events are required for the induction of mitogenic activities in endothelial cells (Houck et al., 1991).
In the process of elucidating the contribution of proteinases to the release of VEGF, we also confirm earlier reports that matrix-bound VEGF is active (Park et al., 1993; Poltorak et al., 1997; Hutchings et al., 2003). However, we note that the outcome of receptor activation mediated by soluble and bound VEGF was different. Although both forms were similarly capable of phosphorylating VEGFR2, they elicited different cellular responses by this same receptor. MMP-resistant/matrix-bound VEGF induced filopodia extension, discrete invasion of the stroma, and facilitated cell–cell associations that were consistent with tube formation; i.e., sprouting angiogenesis. In contrast, cleaved VEGF resulted in the proliferation of cells as sheets and in the broad invasion of the stroma, which is consistent with vascular hyperplasia. These findings could offer a handle on how to dissect the wealth of signaling pathways that are activated by VEGF. This growth factor has been implicated in the induction of permeability, proliferation, migration, differentiation, and morphogenesis. Although it is clear that all these signaling outcomes do not occur simultaneously, it has been difficult to ascertain the hierarchy of the effects and when/how one response prevails over others.
Angiogenesis is a complex, multifactorial phenomenon involving signals from endothelial cells and from the host tissue. Although the key players in this process have been identified, and their specific roles are being elucidated individually, a concrete understanding of the process will only come from the comprehensive acknowledgment that they act as a whole in a series of dynamically reciprocal interactions that modulate signaling outcomes.
Materials and methods
Cells and reagents
HEK 293T cells, breast tumor-derived T47D cells, and HT1080 cells were purchased from American Type Culture Collection and were grown in DME supplemented with 10% FCS. PAE and PAE-VEGFR2 (provided by Gera Neufeld, Technicon, Israel) were grown in Ham's F12 medium. Catalytically active forms of MMP2, 3, 7, 8, 9, MT1-MMP (MMP14), MT3-MMP (MMP16), ADAMTS4, and plasmin were purchased from Calbiochem. The activity of MMPs was tested against known substrates before use. Catalytically active forms of MMP13, 19, and 26 were provided by Carlos Lopez-Otin (University of Oviedo, Oviedo, Spain). Human VEGF165 was provided by the National Cancer Institute's Biological Resources Branch (Rockville, MD). mVEGF164 was purchased from R&D Systems, and nonglycosylated mVEGF164 was purchased from Chemicon. mVEGF-A cDNA isoforms 164, 188, and 120 were gifts from Patricia A. D'Amore (The Schepens Eye Research Institute, Boston, MA). Human VEGF165 expression vector was provided by Kevin Claffey (University of Connecticut, Storrs, CT). Antibodies to phosphotyrosine and VEGFR2 were previously described (Luque et al., 2003). Epitope-specific VEGF antibodies were provided by Donald Senger (Beth Israel Deaconess Medical Center, Boston, MA; Sioussat et al., 1993). Matrigel matrix was purchased from BD Biosciences. BB94 was provided by Gerry Weinmaster (University of California, Los Angeles, [UCLA], Los Angeles, CA).
Digestion of VEGF
Unless specified, VEGF was incubated with proteinases in incubation buffer (50 mM Tris-Cl, pH 7.45, 150 mM NaCl, 10 mM CaCl2, and 1 μm ZnCl2) at a 4:1 molar ratio (VEGF/proteinases) at 37°C for 4 h. To biotinylate mVEGF164, 5 μg VEGF was incubated with 1 mg/ml sulfo-N–hydroxy succinimido–biotin–long chain (Pierce Chemical Co.) overnight at 4°C, and the reaction mixture was dialyzed in 1× PBS to eliminate free biotin. For heparin-bound VEGF assays, plates were coated with 5 μg/ml heparin, washed, and incubated with biotinylated VEGF. Plates were then exposed to MMP3 or to buffer alone. Biotinylated VEGF reaction mixtures were analyzed by using 20% tricine-Tris gel, followed by avidin D coupled with HRP (Vector Laboratories), and were visualized by chemiluminescence. Whenever nonbiotinylated VEGF was used, products were either analyzed on silver-stained gel or were immunoblotted using anti-VEGF antibodies.
VEGF purification from ascites fluid
Ascites fluid was collected from ovarian cancer patients with the approval of the Institutional Review Board (a gift of Joseph Mortola and Michael Graubert, Beth Israel Deaconess Medical Center). No patient identifiers were retained. VEGF immunoaffinity column was prepared with polyclonal VEGF antibody (provided by Donald Senger). Ascites samples were loaded onto the column, were washed in binding buffer, and were eluted with NaCl2. The presence of VEGF in eluted fractions was assessed by immunoblotting with VEGF antibodies.
Amino-terminal amino acid sequencing and MS analysis
MMP3-cleaved mVEGF164 was separated by 20% tricine-Tris gel electrophoresis and was transferred onto polyvinylidene difluoride membranes by using 3-[cyclohexylamino]-1-propanesulfonic acid buffer. The resulting 16- and 6-kD VEGF fragments were amino-terminally sequenced using a microsequenator (model 477A; Applied Biosystems), which was a service provided by William Lane (Harvard Microchemistry Facility, Cambridge, MA). For MS and microsequencing analyses, nonglycosylated mVEGF164 was incubated with catalytically active MMP3, and 13-kD fragments were analyzed by using MALDI-TOF MS, μLC/MSn, and μLC/MS/MS on a quadrupole ion trap mass spectrometer (model Finnigan LCQ DECA; Harvard Microchemistry Facility).
Construction of stable cell lines and purification of proteins
mVEGF-A cDNA isoforms 120, 164, and 188 were cloned into pcDNA3.1 expression vectors (Invitrogen). For the construction of mVEGF113, PCR was performed by using primer P1 (5′-CAAGCGCGCAAGAGAGCGGG-3′) and primer P2 (5′-TCACCGCCTTGGCTTGTCACATC-3′). Reverse primer P3 (5′-TCTCCGCCTTGGCTTGTCACATC-3′) was used for the construction of mVEGF113his. The resulting product was subcloned into pCR-Blunt II-TOPO vectors (Invitrogen), and the insert was substituted into the analogous site in pcDNA3.1 plasmids. For the construction of mVEGFΔ108–118, sequential PCR was performed. Primer sets P1 and P4 (5′-GCTCTGAACATTTCTTTGGTCTGCATTC-3′), P5 (5′-GACCAAAGAAATGTTCAGAGCGGAGAAAGC-3′), and P2 were used for the initial PCR. The resulting fragments were then amplified in a second PCR step by using primers P1 and P2. The final product was subcloned into pcDNA3.1 plasmids as described above.
HEK 293T, T47D, and HT1080 cells were transfected with 1 μg p-cytomegalo virus-puro and 10 μg pcDNA3.1 or with expression vectors using calcium phosphate. Selection was performed 48 h after transfection with 1.5 μg/ml puromycin. All stable clones were evaluated for proliferation rates (Fig. S2). Recombinant mVEGF164 and mVEGFΔ108–118 were purified from the conditioned media of stable clones by heparin affinity chromatography using fast protein liquid chromatography (Bio-Rad Laboratories). Recombinant mVEGF113his was purified by Co2+ chelate affinity chromatography.
Phosphorylation assays
VEGFR2 phosphorylation was determined by immunoblotting as described previously (Luque et al., 2003).
CAM assay
The effect of mVEGF164, mVEGF113, and mVEGFΔ108–118 on angiogenesis was evaluated as previously described (Vazquez et al., 1999). The collagen gel was supplemented with 1 μg VEGF, and the extent of angiogenic response was measured by the use of rhodamine-conjugated Lens culinaris (Vector Laboratories) injected into the circulation of CAM. Evaluation was performed on a confocal system (model MRC-1024Es; Bio-Rad Laboratories) equipped with a microscope (model E800; Nikon) and a krypton/argon laser (model 60-WL-DZ; Bio-Rad Laboratories).
In vivo Matrigel plug assay
6-wk-old female nude mice (Charles River Laboratories) were anesthesized with avertin, were subcutaneously injected with 250 μl of growth factor–reduced Matrigel (BD Biosciences), and were supplemented with either serum-free DME or with 1 μg VEGF. After 7 d, the mice were killed, and the angiogenic response was evaluated. Matrigel plugs were fixed in 1% paraformaldehyde, and vessels were detected with a rat anti–mouse CD31 antibody followed by Cy3-labeled goat anti–rat IgG antibody (both purchased from Jackson ImmunoResearch Laboratories). Evaluation was performed on a confocal microscope (model MRC-1024; Bio-Rad Laboratories).
Xenograft tumor assays
T47D and HT1080 cell lines were injected subcutaneously (5 × 106 cells) in 6-wk-old female nude mice (Charles River Laboratories). Tumor growth was monitored every other day. When tumors reached 1,500 mm3, mice were killed, and dissected tumors were solubilized in lysis buffer as described previously (Rodriguez-Manzaneque et al., 2001). A total of 42 mice were injected with T47D, 42 mice were injected with T47D-expressing mVEGF164, 47 mice were injected with T47D-expressing mVEGF113, and 45 mice were injected with T47D-expressing mVEGFΔ108–118. A total of 60 mice were injected with HT1080, HT1080-expressing mVEGF164, mVEGF113, and mVEGFΔ108–118 (15 mice each). Nude mice that were injected with T47D-expressing mVEGF164 were used for BB94 treatment. BB94 was homogenized in PBS, pH 7.0, containing 0.02% Triton X-100 and was administered as an emulsion by intraperitoneal injection. Mice (cohort of five animals per group) were treated daily with 30 mg/kg for a total of 5 d.
ELISA
To quantify levels of VEGF in the serum, blood samples were collected and allowed to clot overnight at 4°C. Samples were centrifuged at 2,000 g for 20 min. The serum was assayed by using a “sandwich” mouse VEGF-ELISA kit (Calbiochem) according to the manufacturer's instructions. To detect VEGF forms with intact carboxy termini, VEGF antibody 375 (raised against amino acids coded by exon 8) was immobilized onto the surface of the 96-well plates and was assayed by using sandwich ELISA. After normalization, a comparison of both ELISA assays revealed the levels of cleaved and uncleaved VEGF. To determine the VEGF levels in tumor explant cultures, tumors were collected and incubated in 200 μl DME containing 0.5% FBS, either in the presence or absence of MMP3 (1 ng/μl) or MMP3 (1 ng/μl) in the presence of BB94 (10 μM). After 24 h of incubation at 37°C, the conditioned medium was removed and assayed by using a sandwich mouse VEGF-ELISA kit (Calbiochem).
Immunohistochemical analysis
Tumors were collected, fixed in 1% paraformaldehyde, and sectioned at 200 μm using a vibratome (Ted Pella). The evaluation of skin near tumors was performed as a whole-mount preparation. Vessels were visualized with a rat anti–mouse CD31 antibody followed by Cy3-labeled goat anti–rat IgG antibody (both from Jackson ImmunoResearch Laboratories). The evaluation was performed on a confocal system (model MRC-1024; Bio-Rad Laboratories). For hematoxylin and eosin staining, tumors were fixed in 4% paraformaldehyde, were paraffin embedded, sectioned, and stained by members of the UCLA Tissue Core Laboratory.
Quantitation of angiogenesis
Quantitation of vascular density was performed from confocal immunostained images. Images were collected randomly (two images from each tumor, CAM, or Matrigel; four to six individual samples were evaluated in every case), and vascular density was determined with the aid of IMAGEPRO 4.0 software (Media Cybernetics).
In vitro angiogenesis assay
Cytodex beads (712,000 beads/ml; Sigma-Aldrich) were incubated with PAE or with PAE-VEGFR2 cells and were then embedded into fibrinogen/fibronectin gel (2.5 mg/ml; Sigma-Aldrich) containing 100 ng VEGF and 1 U/μl thrombin (Sigma-Aldrich). The extent of angiogenesis was evaluated on a fluorescent inverted microscope (model Diaphot 300; Nikon). To assess cell proliferation, cells were simultaneously stained with AlexaFluor546-conjugated phalloidin (Molecular Probes) and with the antibody to phosphohistone H3 (Cell Signaling).
Cleavage of fluorescence-labeled peptide
MMP3 was incubated with fluorescent substrate at 37°C for 30 min with different molar ratios. The fluorescent substrates (7-methoxycoumarin-4-yl) acetic acid–CRPKKDRTKPENHCEPCK (2,4-dinitrofluorobenzene)-CONH2 (wild-type peptide) and (7-methoxycoumarin-4-yl) acetic acid–CRPKKDKPKPENHCEPCK (2,4-dinitrofluorobenzene)-CONH2 (mutant peptide) were custom synthesized at SynPep Corporation. Fluorescence was detected by a Dual-Scanning Microplate Spectrofluorometer (model SPECTRAmax GEMINI; Spinco Biotech) with an excitation/emission wavelength of 365:440 nm.
Online supplemental material
Fig. S1 shows the effect of pH on the proteolytic processing of VEGF by MMP3. It also compares the stability of the proteolyzed VEGF fragments that are released by MMP3 and plasmin. Fig. S2 provides results as to the proteolytic sensitivity of VEGF mutant forms (113 and Δ108–118) to MMP3 and plasmin. Fig. S3 compares the growth rates of T47D mammary carcinoma cells transfected with different forms of VEGF and shows a proliferative index of endothelial cells in response to different VEGF forms. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200409115/DC1.
Acknowledgments
We would like to thank several members of the angiogenic community that contributed with reagents and constructive criticisms to this work. In particular, Dr. Donald Senger for the epitope-specific antibodies; Dr. Patricia D'Amore for the cDNAs for mVEGF188, 164, and 120; Dr. Kevin Claffey for human VEGF165 expression vector; Dr. Carlos Lopez-Otin for several noncommercially available MMPs; Dr. Ann Zovein and Dr. Timothy Lane for critical reading of the manuscript; Dr. Elisabetta Dejana, Dr. Kari Alitalo, Dr. Zena Werb, and Dr. Christer Betsholtz for highly stimulating discussions.
This work was supported by grants from the National Cancer Institute (RO1CA077420 and CA065624) and from the Susan Komen Breast Cancer Foundation (PDF100649). S. Lee is a fellow funded by the Department of Defense (grant DAMD17-02-1-0328).
Abbreviations used in this paper: CAM, chorioallantoic membrane; HEK, human embryonic kidney; MALDI-TOF MS, matrix-assisted laser desorption time-of-flight MS; MMP, matrix metalloproteinase; μLC/MSn, capillary and microcapillary nano–liquid chromatography MS; μLC/MS/MS, microcapillary reverse-phase HPLC nano-electrospray tandem MS; MS, mass spectrometry; PAE, porcine aortic endothelial; PAE-VEGFR2, PAE cells expressing VEGFR2; PECAM, platelet/endothelial cell adhesion molecule 1; TIMP, tissue inhibitor of MPs; VEGFR2, VEGF receptor 2.
References
- Balbin, M., A. Fueyo, A.M. Tester, A.M. Pendas, A.S. Pitiot, A. Astudillo, C.M. Overall, S.D. Shapiro, and C. Lopez-Otin. 2003. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat. Genet. 35:252–257. [DOI] [PubMed] [Google Scholar]
- Bergers, G., R. Brekken, G. McMahon, T.H. Vu, T. Itoh, K. Tamaki, K. Tanzawa, P. Thorpe, S. Itohara, Z. Werb, and D. Hanahan. 2000. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2:737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsten, E., M. Uutela, X. Li, K. Pietras, A. Ostman, C.H. Heldin, K. Alitalo, and U. Eriksson. 2001. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat. Cell Biol. 3:512–516. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages, M., F. Rojo, J. Albanell, J. Baselga, and J. Arribas. 2003. TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J. 22:1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet, P., and R.K. Jain. 2000. Angiogenesis in cancer and other diseases. Nature. 407:249–257. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P., V. Ferreira, G. Breier, S. Pollefeyt, L. Kieckens, M. Gertsenstein, M. Fahrig, A. Vandenhoeck, K. Harpal, C. Eberhardt, et al. 1996. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 380:435–439. [DOI] [PubMed] [Google Scholar]
- Coussens, L.M., and Z. Werb. 2002. Inflammation and cancer. Nature. 420:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damert, A., L. Miquerol, M. Gertsenstein, W. Risau, and A. Nagy. 2002. Insufficient VEGFA activity in yolk sac endoderm compromises haematopoietic and endothelial differentiation. Development. 129:1881–1892. [DOI] [PubMed] [Google Scholar]
- Darland, D.C., and P.A. D'Amore. 2001. Cell-cell interactions in vascular development. Curr. Top. Dev. Biol. 52:107–149. [DOI] [PubMed] [Google Scholar]
- Dvorak, H.F. 2002. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 20:4368–4380. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. 2000. VEGF: an update on biological and therapeutic aspects. Curr. Opin. Biotechnol. 11:617–624. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. 2002. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer. 2:795–803. [DOI] [PubMed] [Google Scholar]
- Ferrara, N., K. Carver-Moore, H. Chen, M. Dowd, L. Lu, K.S. O'Shea, L. Powell-Braxton, K.J. Hillan, and M.W. Moore. 1996. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 380:439–442. [DOI] [PubMed] [Google Scholar]
- Ferrara, N., H.P. Gerber, and J. LeCouter. 2003. The biology of VEGF and its receptors. Nat. Med. 9:669–676. [DOI] [PubMed] [Google Scholar]
- Ferrara, N., K.J. Hillan, H.P. Gerber, and W. Novotny. 2004. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 3:391–400. [DOI] [PubMed] [Google Scholar]
- Fuentes-Prior, P., Y. Iwanaga, R. Huber, R. Pagila, G. Rumennik, M. Seto, J. Morser, D.R. Light, and W. Bode. 2000. Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature. 404:518–525. [DOI] [PubMed] [Google Scholar]
- Gerber, H.P., K.J. Hillan, A.M. Ryan, J. Kowalski, G.A. Keller, L. Rangell, B.D. Wright, F. Radtke, M. Aguet, and N. Ferrara. 1999. VEGF is required for growth and survival in neonatal mice. Development. 126:1149–1159. [DOI] [PubMed] [Google Scholar]
- Gerber, H.P., A.K. Malik, G.P. Solar, D. Sherman, X.H. Liang, G. Meng, K. Hong, J.C. Marsters, and N. Ferrara. 2002. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature. 417:954–958. [DOI] [PubMed] [Google Scholar]
- Grunstein, J., J.J. Masbad, R. Hickey, F. Giordano, and R.S. Johnson. 2000. Isoforms of vascular endothelial growth factor act in a coordinate fashion To recruit and expand tumor vasculature. Mol. Cell. Biol. 20:7282–7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamano, Y., M. Zeisberg, H. Sugimoto, J.C. Lively, Y. Maeshima, C. Yang, R.O. Hynes, Z. Werb, A. Sudhakar, and R. Kalluri. 2003. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 3:589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heissig, B., K. Hattori, M. Friedrich, S. Rafii, and Z. Werb. 2003. Angiogenesis: vascular remodeling of the extracellular matrix involves metalloproteinases. Curr. Opin. Hematol. 10:136–141. [DOI] [PubMed] [Google Scholar]
- Helmlinger, G., M. Endo, N. Ferrara, L. Hlatky, and R.K. Jain. 2000. Formation of endothelial cell networks. Nature. 405:139–141. [DOI] [PubMed] [Google Scholar]
- Houck, K.A., N. Ferrara, J. Winer, G. Cachianes, B. Li, and D.W. Leung. 1991. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol. Endocrinol. 5:1806–1814. [DOI] [PubMed] [Google Scholar]
- Houck, K.A., D.W. Leung, A.M. Rowland, J. Winer, and N. Ferrara. 1992. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 267:26031–26037. [PubMed] [Google Scholar]
- Hutchings, H., N. Ortega, and J. Plouet. 2003. Extracellular matrix-bound vascular endothelial growth factor promotes endothelial cell adhesion, migration, and survival through integrin ligation. FASEB J. 17:1520–1522. [DOI] [PubMed] [Google Scholar]
- Inoue, M., J.H. Hager, N. Ferrara, H.P. Gerber, and D. Hanahan. 2002. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell. 1:193–202. [DOI] [PubMed] [Google Scholar]
- Junttila, T.T., M. Sundvall, J.A. Maatta, and K. Elenius. 2000. Erbb4 and its isoforms: selective regulation of growth factor responses by naturally occurring receptor variants. Trends Cardiovasc. Med. 10:304–310. [DOI] [PubMed] [Google Scholar]
- Keck, R.G., L. Berleau, R. Harris, and B.A. Keyt. 1997. Disulfide structure of the heparin binding domain in vascular endothelial growth factor: characterization of posttranslational modifications in VEGF. Arch. Biochem. Biophys. 344:103–113. [DOI] [PubMed] [Google Scholar]
- Kim, K.J., B. Li, J. Winer, M. Armanini, N. Gillett, H.S. Phillips, and N. Ferrara. 1993. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 362:841–844. [DOI] [PubMed] [Google Scholar]
- Luque, A., D.R. Carpizo, and M.L. Iruela-Arispe. 2003. ADAMTS1/METH1 inhibits endothelial cell proliferation by direct binding and sequestration of VEGF165. J. Biol. Chem. 278:23656–23665. [DOI] [PubMed] [Google Scholar]
- McColl, B.K., M.E. Baldwin, S. Roufail, C. Freeman, R.L. Moritz, R.J. Simpson, K. Alitalo, S.A. Stacker, and M.G. Achen. 2003. Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J. Exp. Med. 198:863–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, D.M., and P.L. Choyke. 2003. Imaging of angiogenesis: from microscope to clinic. Nat. Med. 9:713–725. [DOI] [PubMed] [Google Scholar]
- McQuibban, G.A., J.H. Gong, E.M. Tam, C.A. McCulloch, I. Clark-Lewis, and C.M. Overall. 2000. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 289:1202–1206. [DOI] [PubMed] [Google Scholar]
- McQuibban, G.A., J.H. Gong, J.P. Wong, J.L. Wallace, I. Clark-Lewis, and C.M. Overall. 2002. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 100:1160–1167. [PubMed] [Google Scholar]
- Miquerol, L., B.L. Langille, and A. Nagy. 2000. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 127:3941–3946. [DOI] [PubMed] [Google Scholar]
- Nanba, D., A. Mammoto, K. Hashimoto, and S. Higashiyama. 2003. Proteolytic release of the carboxy-terminal fragment of proHB-EGF causes nuclear export of PLZF. J. Cell Biol. 163:489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly, M.S., D. Wiederschain, W.G. Stetler-Stevenson, J. Folkman, and M.A. Moses. 1999. Regulation of angiostatin production by matrix metalloproteinase-2 in a model of concomitant resistance. J. Biol. Chem. 274:29568–29571. [DOI] [PubMed] [Google Scholar]
- Overall, C.M., and C. Lopez-Otin. 2002. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer. 2:657–672. [DOI] [PubMed] [Google Scholar]
- Park, J.E., G.A. Keller, and N. Ferrara. 1993. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell. 4:1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson, A., J.A. Nagy, L.F. Brown, C. Sundberg, E. Morgan, S. Jungles, R. Carter, J.E. Krieger, E.J. Manseau, V.S. Harvey, et al. 2000. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab. Invest. 80:99–115. [DOI] [PubMed] [Google Scholar]
- Plouet, J., F. Moro, S. Bertagnolli, N. Coldeboeuf, H. Mazarguil, S. Clamens, and F. Bayard. 1997. Extracellular cleavage of the vascular endothelial growth factor 189-amino acid form by urokinase is required for its mitogenic effect. J. Biol. Chem. 272:13390–13396. [DOI] [PubMed] [Google Scholar]
- Poltorak, Z., T. Cohen, R. Sivan, Y. Kandelis, G. Spira, I. Vlodavsky, E. Keshet, and G. Neufeld. 1997. VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix. J. Biol. Chem. 272:7151–7158. [DOI] [PubMed] [Google Scholar]
- Pozzi, A., P.E. Moberg, L.A. Miles, S. Wagner, P. Soloway, and H.A. Gardner. 2000. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc. Natl. Acad. Sci. USA. 97:2202–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, C.J., and S.E. Stringer. 2001. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 114:853–865. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Manzaneque, J.C., T.F. Lane, M.A. Ortega, R.O. Hynes, J. Lawler, and M.L. Iruela-Arispe. 2001. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc. Natl. Acad. Sci. USA. 98:12485–12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant, J., and M. Hirashima. 2003. Vascular development and patterning: making the right choices. Curr. Opin. Genet. Dev. 13:408–412. [DOI] [PubMed] [Google Scholar]
- Ruhrberg, C., H. Gerhardt, M. Golding, R. Watson, S. Ioannidou, H. Fujisawa, C. Betsholtz, and D.T. Shima. 2002. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 16:2684–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sioussat, T.M., H.F. Dvorak, T.A. Brock, and D.R. Senger. 1993. Inhibition of vascular permeability factor (vascular endothelial growth factor) with antipeptide antibodies. Arch. Biochem. Biophys. 301:15–20. [DOI] [PubMed] [Google Scholar]
- Tischer, E., R. Mitchell, T. Hartman, M. Silva, D. Gospodarowicz, J.C. Fiddes, and J.A. Abraham. 1991. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 266:11947–11954. [PubMed] [Google Scholar]
- Vazquez, F., J.C. Rodriguez-Manzaneque, J.P. Lydon, D.P. Edwards, B.W. O'Malley, and M.L. Iruela-Arispe. 1999. Progesterone regulates proliferation of endothelial cells. J. Biol. Chem. 274:2185–2192. [DOI] [PubMed] [Google Scholar]
- Vincenti, V., C. Cassano, M. Rocchi, and G. Persico. 1996. Assignment of the vascular endothelial growth factor gene to human chromosome 6p21.3. Circulation. 93:1493–1495. [DOI] [PubMed] [Google Scholar]