

Abstract
Clostridial neurotoxins are bacterial endopeptidases that cleave the major SNARE proteins in peripheral motorneurons. Here, we show that disruption of synaptic architecture by botulinum neurotoxin C1 (BoNT/C) in central nervous system neurons activates distinct neurodegenerative programs in the axo-dendritic network and in the cell bodies. Neurites degenerate at an early stage by an active caspase-independent fragmentation characterized by segregation of energy competent mitochondria. Later, the cell body mitochondria release cytochrome c, which is followed by caspase activation, apoptotic nuclear condensation, loss of membrane potential, and, finally, cell swelling and lysis. Recognition and scavenging of dying processes by glia also precede the removal of apoptotic cell bodies, in line with a temporal and spatial segregation of different degenerative processes. Our results suggest that, in response to widespread synaptic damage, neurons first dismantle their connections and finally undergo apoptosis, when their spatial relationships are lost.
Introduction
Synapse remodelling is a physiological process used to refine neuronal circuits during development. Exuberant projections and axonal branches that have been directed toward inappropriate targets are usually removed by mechanisms, which seem to be evolutionary conserved (Bagri et al., 2003; Watts et al., 2003). Similar mechanisms are not confined to the developing brain, but are still present in the adult nervous system where they ensure structural plasticity of neurons in response to learning and memory (Bailey and Kandel, 1993; Lichtman and Colman, 2000). Local self-destruction programs regulate this fine refinement of neuronal circuits, thereby enabling a selective elimination of unused connections without affecting the remaining branches or the cell soma (Walsh and Lichtman, 2003). As often observed in nature, similar mechanisms seem to be active also in some pathological conditions where selective degeneration of injured axons can occur in response to local insults (Raff et al., 2002). Selective axonal degeneration in the peripheral nervous system has received considerable attention (Schaumburg et al., 1974; Coleman and Perry, 2002). Axotomy of peripheral nerves results in the degeneration of the distal portion of the axon by an active process termed Wallerian degeneration (Waller, 1850; Gillingwater and Ribchester, 2001). Alternatively, degeneration may begin in the distal axon extremities and progress backward to the soma by a “dying-back” process, as observed in many peripheral nerve diseases (Raff et al., 2002). The mechanisms leading to axonal degeneration and the final demise of the cell body are largely unknown, although there is compelling evidence that different execution systems operate in the axons and in the cell bodies during Wallerian degeneration (Finn et al., 2000; Raff et al., 2002). Interestingly, local apoptotic-like mechanisms may be recruited to dispose of synapses and dendritic spines, at least in in vitro models of injury (Davies et al., 1987; Ivins et al., 1998; Mattson et al., 1998).
Growing evidence suggests that disruption of synapse integrity or function may be even more relevant than neuronal loss in slow, central degenerative disorders such as Alzheimer's (AD), Huntington's (HD), and Parkinson's diseases. Loss of synapses is proportionally greater than the loss of neurons in AD patients (Davies et al., 1987), and a strong correlation exists between cognitive decline and loss of presynaptic terminals (Hatanpaa et al., 1999; Sze et al., 1997). A marked decrease in striatal volume, without changes in cell number, has also been reported in mouse models of HD (Mangiarini et al., 1996; Yamamoto et al., 2000). This suggests that the loss of neurites and synaptic failure may determine the HD phenotype. Interestingly, reduced synaptic connectivity without neuronal loss is also a relevant feature of neuropsychiatric diseases such as schizophrenia (McGlashan and Hoffman, 2000; Frankle et al., 2003; Gasic and Nicotera, 2003).
Widespread, but selective, synaptic damage can be elicited by using clostridial neurotoxins of the botulinum family. Botulinum neurotoxins (BoNTs) act inside neuronal synapses as metalloproteinases, which specifically cleave the major SNARE proteins (Blasi et al., 1993b; Schiavo et al., 1992a,b). In so doing, BoNTs block neurotransmitter release at the neuromuscular junction in vivo or in the cortical, hippocampal, and cerebellar granule neurons in vitro (Osen-Sand et al., 1996; Verderio et al., 1999; Foran et al., 2003).
Despite the accumulating evidence that the synapse, axon, and soma may represent separate degenerative compartments, it is still unclear whether synaptic damage may initiate neuronal death and whether this involves spatially and temporally distinct programs. To address these issues, we set up a model of axonal degeneration driven by BoNT/C-mediated synaptic damage in central neurons.
In cerebellar granule cells (CGCs), BoNT/C triggered two neurodegenerative events that were distinct in space and time: (1) an early neurite degeneration, which occurred independently of trophic stimulation and the activation of death-signaling kinases, and which did not involve the apoptosis-executing machinery (caspases); and (2) a late apoptotic demise of the cell bodies, which was prevented by kinase inhibition and involved cytochrome c release and caspase activation. The compartmentalization of the two degenerative events was emphasized by the selective recognition and removal of different regions in degenerating neurons. These data suggest that widespread synaptic damage caused by BoNT/C in central neurons can initiate different degenerative programs spatially and temporally that recapitulate many relevant features found in human neurodegenerative disorders.
Results
Synaptic disarray by BoNT/C causes CGC apoptosis
Once internalized by neurons, BoNT/C selectively proteolyzes syntaxin and SNAP-25, which are two of the major SNARE proteins responsible for neurotransmitter release. To determine whether BoNT/C was active, we examined the integrity of syntaxin and SNAP-25 in CGCs. 3 h after the addition of BoNT/C, the intensity of the syntaxin band on Western blot decreased progressively (Fig. 1 A). In agreement with recent findings (Foran et al., 2003), only a small amount of the syntaxin cleavage product was detected on gels, which is presumably caused by rapid degradation of the fragment. Also, SNAP-25 disappeared after treatment with the toxin, although with slower kinetics (Fig. 1 A). Thus, BoNT/C entered CGCs and acted as an intracellular specific protease in synapses.
Figure 1.
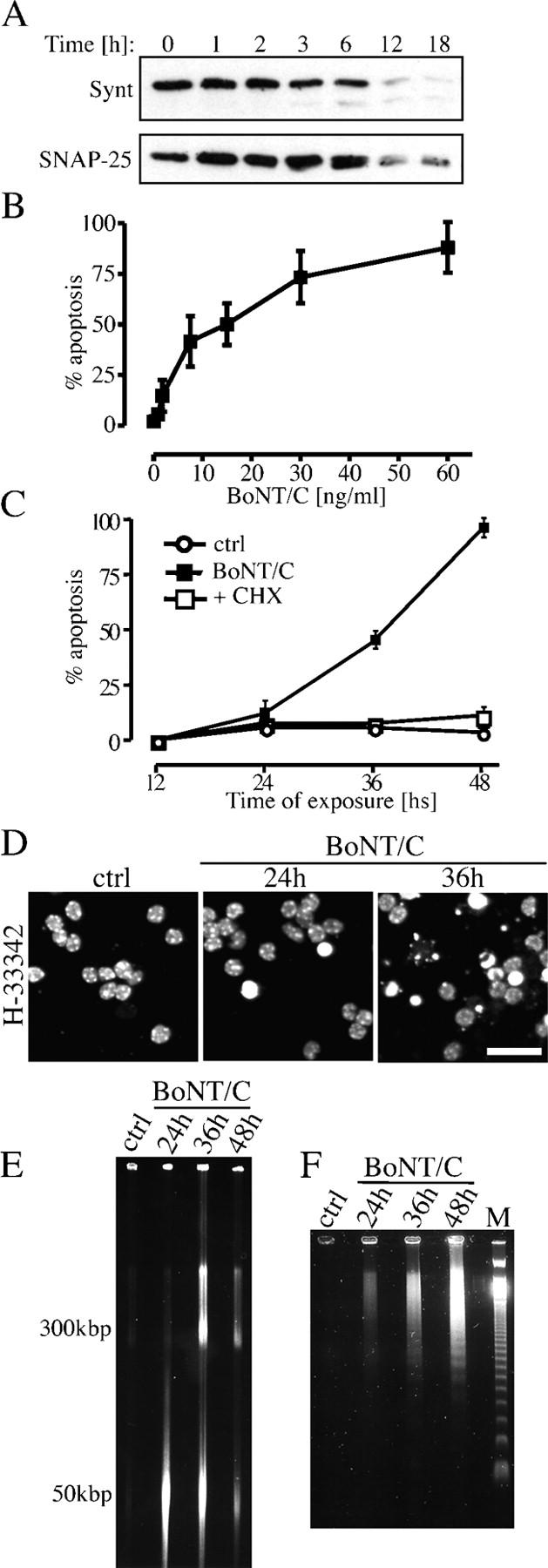
BoNT/C induces apoptosis in CGCs. (A) Time course of syntaxin and SNAP-25 cleavage analyzed by Western blot in CGC lysates. Cells were exposed to 20ng/ml BoNT/C for the time indicated. Concentration–response (B) and time course (C) of BoNT/C-induced cell death. In B, cell death was evaluated 30 h after treatment with the toxin, whereas in C, cells were exposed to 20 ng/ml BoNT/C in the absence or the presence of cycloheximide (CHX; 2 μM). Apoptosis is expressed as a percentage of condensed nuclei in cultures stained by H-33342 (D). Shown are HMM DNA fragmentation (E) and DNA laddering (F) in cultures exposed to 20 ng/ml BoNT/C for the indicated times. Data are means ± SD for triplicate determinations. Bar, 30 μm.
Next, we examined whether BoNT/C triggered death of CGCs. Cell death was concentration dependent in a range between 5 and 60 ng/ml BoNT/C and had obvious apoptotic features (Fig. 1 B). Nuclear condensation and fragmentation began only after 24 h, and became widespread by 36 h (Fig. 1, C and D). At these times, the plasma membrane of neuronal somata was still intact, as revealed by the retention of fluorescent calcein within cells (not depicted). Cell death was dependent on protein synthesis because it was completely prevented by coexposure to cycloheximide (Fig. 1 C). High molecular mass (HMM) DNA fragments were detected by field inversion gel electrophoresis (FIGE) after 24 h (Fig. 1 E), whereas oligonucleosomal DNA fragmentation was detected at much later time points (e.g., 36–48 h after exposure to BoNT/C; Fig. 1 F).
Cytochrome c was released from mitochondria within the cell body starting 24 h after exposure to BoNT/C, as shown by the loss of punctuate cytochrome c fluorescent staining and the appearance of a diffuse pattern within the cell soma and the nuclear region (Fig. 2 A). Caspase-3 was activated concomitantly, as shown by the increased DEVD-cleaving activity (Fig. 2 B) and by the formation of the 120-kD fragment of fodrin (Fig. 2 C). The broad spectrum caspase inhibitors zVAD-fluoromethylketone (fmk) and z-D-2,6-dichlorobenzoyloxymethylketone (cbk) blocked DEVDase activity (Fig. 2 B) as well as fodrin cleavage (Fig. 2 C). Accordingly, HMM and oligonucleosomal DNA fragmentation (Fig. 2, D and E, respectively), as well as the apoptotic morphological changes (Fig. 2 A and C), were prevented upon caspase inhibition. In contrast, z-VAD-fmk concentrations as high as 100 μM did not affect mitochondrial cytochrome c release (Fig. 2 A). Because, caspase-2 may not be effectively inhibited by zVAD-fmk (Nicholson, 1999), we used the more selective caspase-2 inhibitor zVDVAD-fmk (1 μM); zVDVAD-fmk reduced cell death only in part, suggesting, however, a possible role for caspase-2 in cell body apoptosis (Fig. 2 C).
Figure 2.
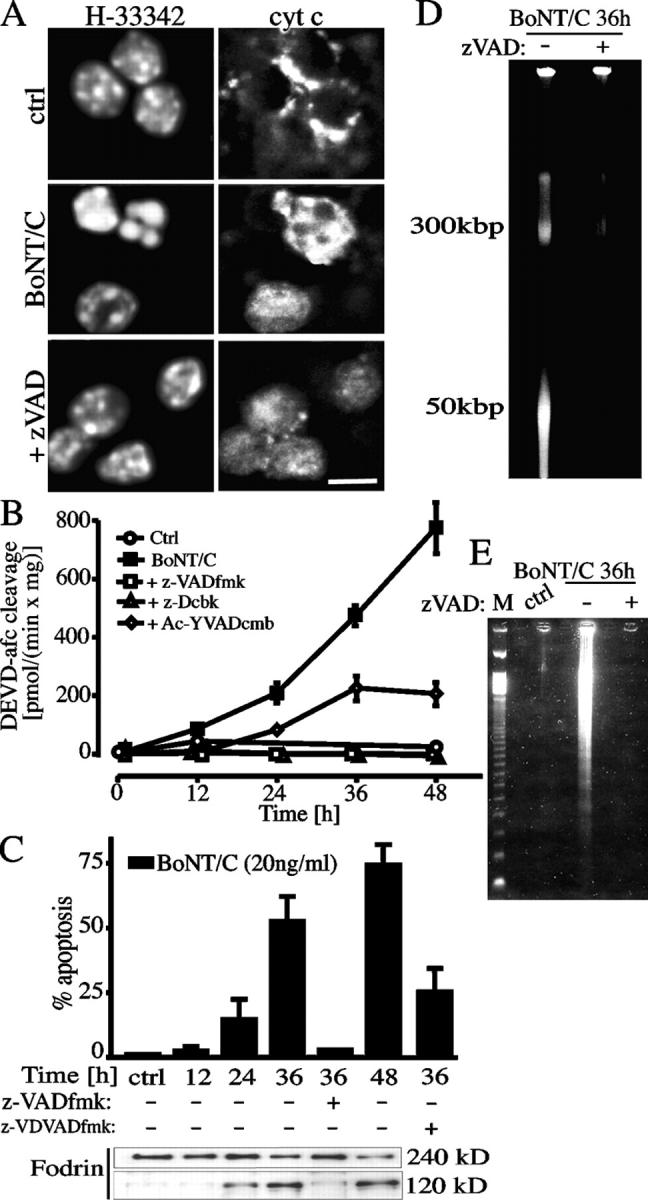
Apoptotic demise of the cell bodies is a caspase-dependent process. (A) Cytochrome c release analyzed by confocal microscopy at the level of the cell bodies in BoNT/C-treated CGCs. The release, denoted by the diffuse staining, was not prevented by caspase inhibition (e.g., by 100 μM zVAD-fmk). Cells were exposed to 20 ng/ml BoNT/C for 30 h. Shown are the time course of caspase activation measured by enzymatic assay (B) and fodrin cleavage (C) in CGC lysates exposed to 20 ng/ml BoNT/C for the indicated times. Caspase inhibition by 100 μM zVAD-fmk prevented the increase in DEVD-afc cleaving activity (B), fodrin cleavage and apoptosis (C), nuclear condensation (A), HMM DNA fragmentation (D), and DNA laddering (E). The caspase-2 inhibitor zVDVAD-fmk (1 μM) partially protected from apoptosis. Bar, 10 μm.
BoNT/C-induced neurodegeneration is not caused by a block of neurotransmitter release
CGC apoptosis might have been caused by BoNT/C-mediated block of neurotransmitter release that resulted in deprivation of essential cell stimulation/trophic support. To test this possibility, we first measured glutamate release stimulated by KCl in BoNT/C-treated neurons. Neurotransmitter release was fully blocked at toxin concentrations as low as 2.5 and 5 ng/ml. However, BoNT/C triggered only little apoptosis at those concentrations (Fig. 3 A).
Figure 3.
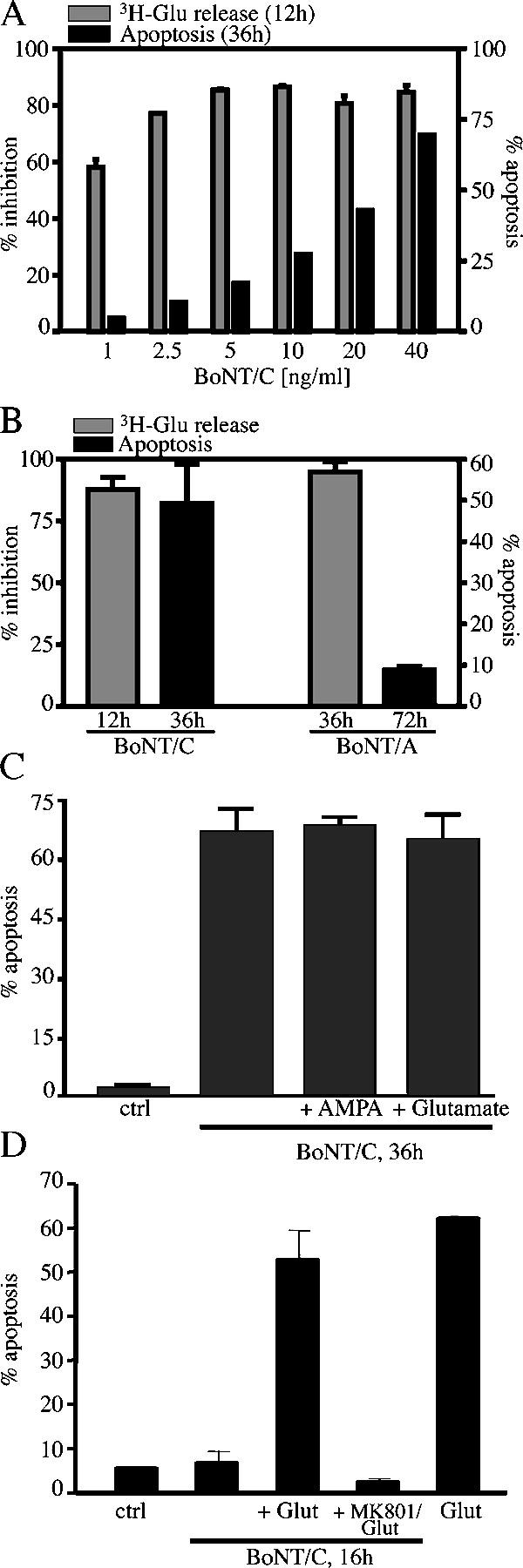
BoNT/C-induced apoptosis is independent of the block of neurotransmitter release and is not prevented by glutamatergic stimulation. (A) Dose–response of BoNT/C-induced block of neurotransmitter release and correlation with apoptosis. Neurotransmitter release was triggered by potassium depolarization in l-[G-3H]glutamine–loaded cultures. (B) Comparison of the block of neurotransmitter release with apoptosis induced by 20 ng/ml BoNT/C and 2 μg/ml BoNT/A. (C) CGCs were incubated with 20 ng/ml BoNT/C. After 12 h, 1 μM AMPA or 0.5 μM glutamate was added, and, after 36 h, apoptosis was evaluated by H-33342 staining. (D) CGCs were incubated with 20 ng/ml BoNT/C for 15 h. Then, they were exposed to 300 μM glutamate for 1 h, in the presence or in the absence of 2 μM MK801. Apoptosis was evaluated as a percentage of condensed nuclei. Data are means ± SD for triplicate determinations.
In a second set of experiments, CGCs were treated with BoNT/A. Whereas BoNT/C cleaves both syntaxin and SNAP-25, BoNT/A blocks neurotransmission by exclusively targeting SNAP-25 (Blasi et al., 1993a). In fact, treatment with BoNT/A caused an almost complete block of glutamate release. But, BoNT/A did not trigger neurite degeneration or apoptosis even up to 72 h (Fig. 3 B).
In adult neurons, physiological, glutamatergic stimulation is required for maintenance of postsynaptic structures (McKinney et al., 1999). Therefore, we exposed BoNT/C-treated neurons to a mild glutamatergic stimulation using either AMPA (0.1–10 μM) or glutamate (0.5–1 μM). Addition of either of the two agonists at the time of BoNT/C addition or 12 h after BoNT/C addition did not prevent apoptosis (Fig. 3 C). Nevertheless, neurons were still able to respond to glutamate stimulation because glutamate (300 μM for 1 h) triggered excitotoxicity in BoNT/C-treated cultures, which was prevented by blocking the NMDA receptor with 2 μM MK801 (Fig. 3 D). Together, these data show that loss of glutamatergic transmission was not the cause of CGC degeneration and that postsynaptic structures (i.e., NMDA receptors) remained operative in degenerating neurons.
Caspase-independent axo-dendritic degeneration precedes the apoptotic demise of neuronal bodies
Well before the onset of apoptosis and starting 8–12 h after addition of BoNT/C to CGCs, their dense axo-dendritic network underwent a progressive degeneration. Neurites displayed enlarged varicosities by 12 h (Fig. 4 A) and blebs in the form of pearl chainlike structures appeared along neurites (Fig. 4 B). Next, the axo-dendritic network underwent a widespread fragmentation. Notably, most of these fragmented neurites were still enclosed by an intact plasma membrane, as suggested by scanning electron microscopy (Fig. 4 B) and shown by their ability to retain the vital dye calcein-AM (see Fig. 6 B).
Figure 4.
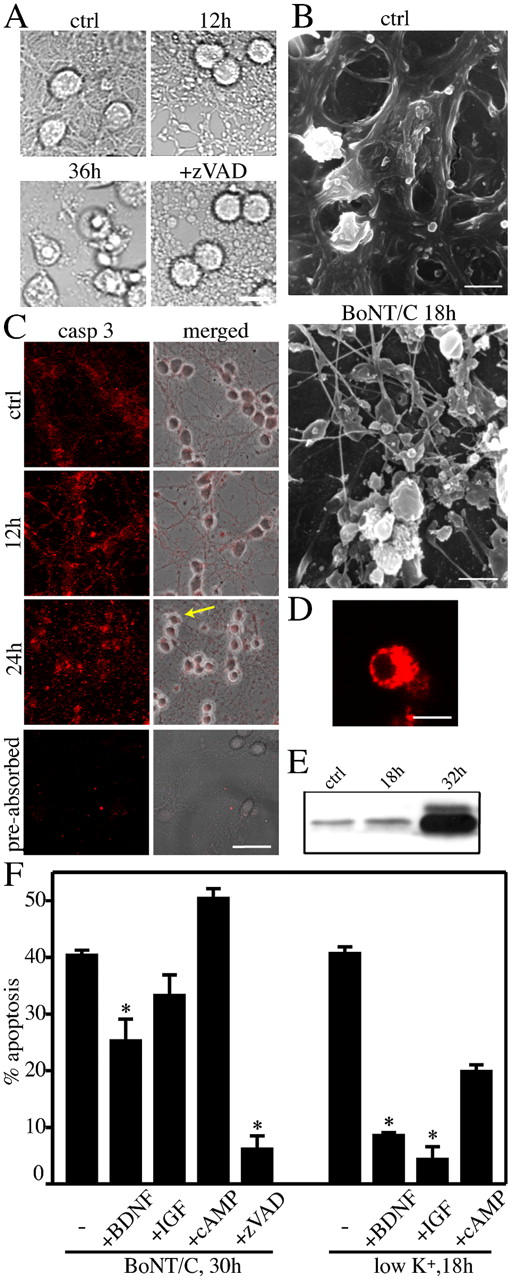
A caspase-independent axonal breakdown preceded the apoptotic demise of the neuronal bodies. Appearance of the axo-dendritic network in CGC cultures as shown by phase-contrast (A) and scanning electron microscopy (B). The disassembly of the neuronal projections in the presence of 20 ng/ml BoNT/C is noteworthy. The magnification used in B allows the visualization of the only neuronal network. The p17 fragment of caspase-3 was detected along the neuronal projections (C), as well as within the cell bodies (D), by confocal microscopy. Note the absence of staining when the antibody was preabsorbed with p20/p17 caspase-3. Fluorescence images in C represent single focal planes acquired at the level of the neurites. The arrow indicates the cell shown in D to illustrate the presence of processed caspase-3 at the level of the cell body at 24 h. The Western blot shows small amounts of caspase-3 processing in untreated neurons and 18 h after BoNT/C. Substantial activation is seen after 32 h, when most neurons have undergone apoptosis (E). Note that caspase inhibition by zVAD-fmk prevented nuclear condensation, but did not prevent the dismantling of the neurites (A). Neurotrophic factors (100 ng/ml BDNF and 200 ng/ml IGF-1) and cAMP (1 mM) did not prevent axonal degeneration (not depicted) and only BDNF partially protected from apoptosis (E). Neurotrophic factors efficacy was monitored in parallel cultures in which apoptosis was induced by K+ withdrawal. The percentage of apoptosis was evaluated by scoring condensed, H33342-positive nuclei. (F) Data are means ± SD for triplicate determinations; *, P < 0.005. Bars: (A) 10 μm; (B) 5 μm; (C) 30 μm; (D) 10 μm.
Figure 6.
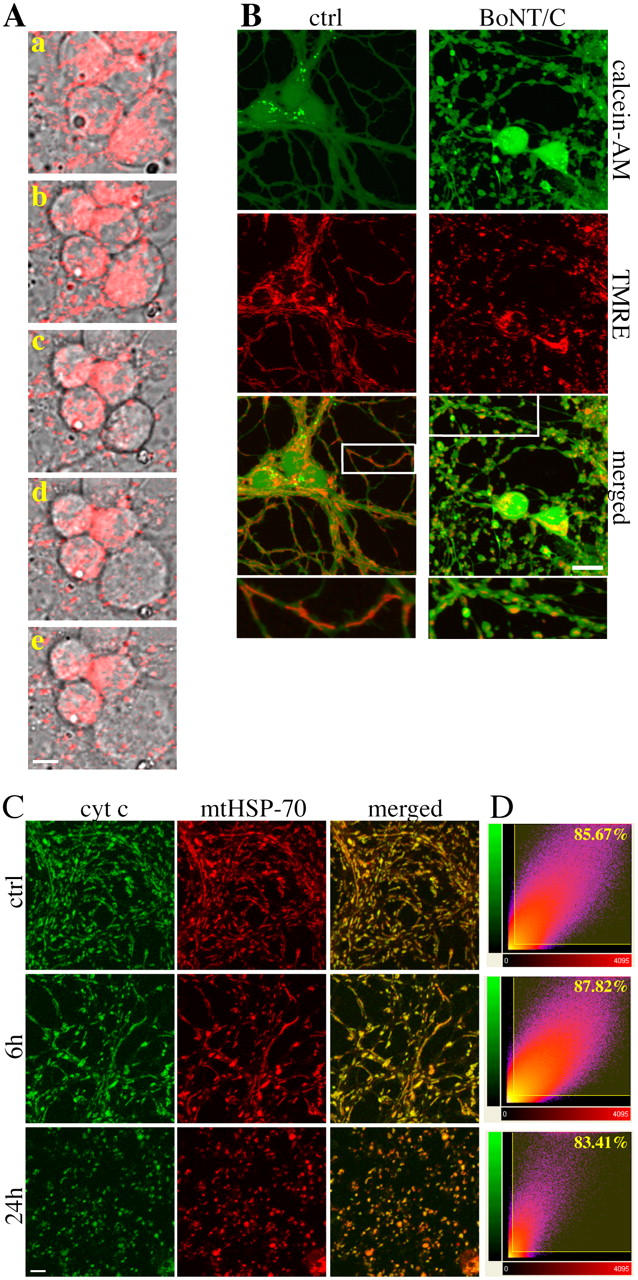
Mitochondrial integrity is retained during the early axonal degeneration, but it is lost in the late demise of the cell bodies. BoNT/C-treated CGCs were loaded with 20 nM TMRE and imaged by confocal microscopy (A and B). Time series imaging of mitochondria at the level of the cell bodies (A, a–e; time [h] a: 30; b: 37, c: 42, d: 42.5, and e: 43.5; Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200406126/DC1). (B) CGC treated with 20 ng/ml BoNT/C for 18 h were loaded with calcein-AM (green) and TMRE (red) and imaged by confocal microscopy. The bottom panels show the detail of the related insets. Note the presence of TMRE-positive mitochondria, which is indicative of a retained membrane potential in the degenerating neurites and the different mitochondrial morphology/distribution in BoNT/C-treated neurons versus control. (C and D) Analysis of cytochrome c release along the neurites in BoNT/C-treated cultures. CGCs were immunostained for cytochrome c and mtHSP-70. Confocal images were acquired from the same field (C) and colocalization quantified by ImarisColoc software (D). Scattergrams show the percentage of the green channel voxels (cytochrome c [cyt c]) that colocalize with the red channel ones (mtHSP-70). Bars: (A) 5 μm; (B) 10 μm; (C) 15 μm.
The breakdown of the neuronal projections was not prevented by z-VADfmk (Fig. 4 A) nor by zVDVAD-fmk (not depicted). Despite caspase activity being irrelevant for the dismantling of the neuronal network, proteolytic processing of caspase-3 was detected along the neurites when observed using an antibody specific for the large p17-20 fragment, which forms an essential part of the activated enzyme (Fig. 4 C). Notably, a small amount of positive staining for processed caspase-3 was detectable also in untreated neurons. To test whether this was caused by aspecific binding, the antibody was preadsorbed with processed caspase-3. As indicated in Fig. 4 C, preadsorption neutralized the positive immunostaining. Western blot analyses further confirmed the presence of the p20/p17 fragment in untreated neurons (Fig. 4 E), thus suggesting that a small amount of caspase-3 is normally processed in CGC cultures. Substantial amounts of processed caspase-3 were obviously detected in the cell bodies after 24 h (Fig. 4, C and D), which is consistent with the increase in caspase-3 activity (Fig. 2 B).
Because axo-dendritic degeneration caused by BoNT/C involved mechanisms different from those causing cell body apoptosis, we examined whether trophic factors known to be neuroprotective would differently modulate the two degenerative processes. We used growth factors (NGF, BDNF, and IGF-I) and cAMP, which is part of the intracellular signaling pathways of several neurotrophins (D'Mello et al., 1993; Gaiddon et al., 1996). NGF, IGF-I, and cAMP did not prevent apoptosis of neuronal somata (Fig. 4 F) nor did they protect from neurite fragmentation (not depicted). Similarly, replacement of the original medium with medium from parallel, untreated cultures did not protect from BoNT/C-induced neurodegeneration (unpublished data). Under the same conditions, 100 ng/ml BDNF reduced apoptosis, but did not prevent neurite degeneration (Fig. 4 F). These data further support the idea that two independent mechanisms are responsible for axo-dendritic degeneration and the demise of the cell bodies.
Neurites degenerate with cytoskeleton breakdown, but retain functional mitochondria
A general cytoskeletal disassembly occurred within the first 18 h after the exposure to BoNT/C. The dense actin network, which characterizes untreated cultures, became sparser, and the finest projections were lost; consequently, there was an overall loss of immunoreactivity (Fig. 5 A). Alterations of the microtubule network were also apparent. Tubulin-positive enlarged varicosities, or bleblike structures, became visible along the neurites 8 h after treatment and were evident on the remaining neurites by 20 h (Fig. 5, A and B). Interestingly, microtubule disassembly was also accompanied by abnormal phosphorylation of the tau protein. Phosphorylation of one of the tau KXGS motifs was detected 6 h after exposure to BoNT/C (Fig. 5 C) by using the AT-8–like antibody 11b directed against the phosphorylated epitope S202-205 that was detected in paired helical filaments-tau (Biernat et al., 1992; Goedert et al., 1992). Using a different set of antibodies (11b gave poor results with immunostaining), we then examined the appearance and localization of phosphorylated tau in degenerating neurons. After BoNT/C addition, the pattern of tau distribution changed from a very diffuse to a highly fluorescent, punctuate staining that was compatible with a disorganization/aggregation of tau filaments (Fig. 5 D).
Figure 5.
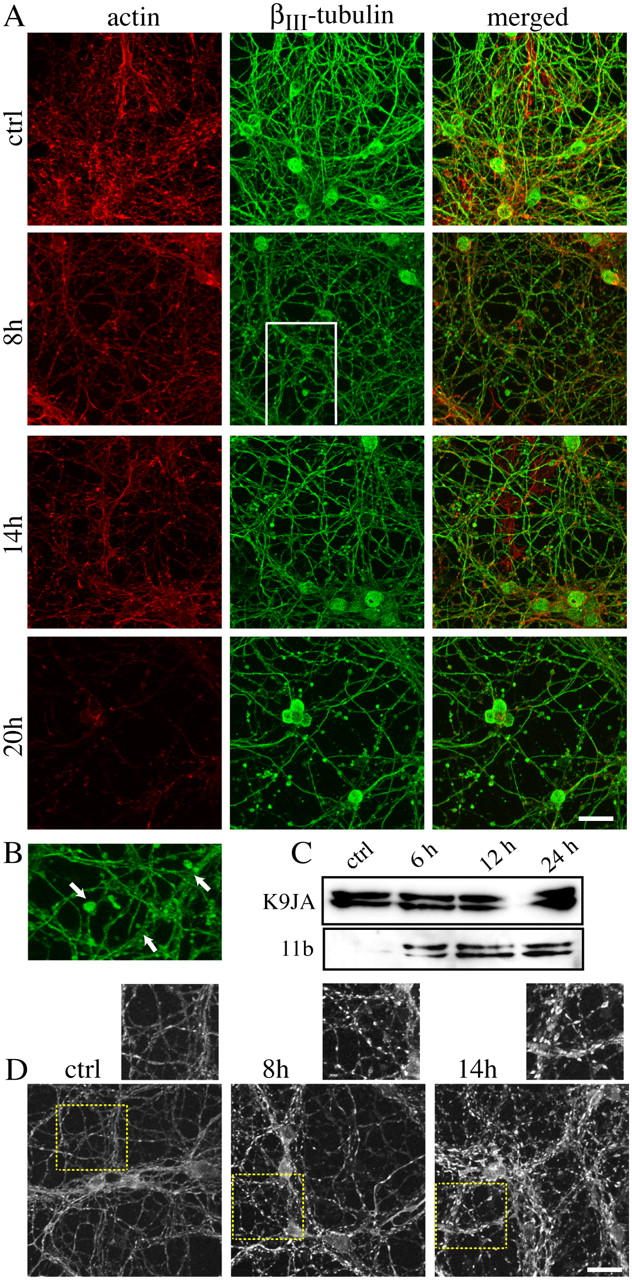
Cytoskeletal disassembly and abnormal tau phosphorylation accompanied the early degeneration of the neurites. (A) CGCs were exposed to 20 ng/ml BoNT/C for the times indicated and immunostained for actin and βIII-tubulin. Confocal images were acquired from the same field. The bulblike structures present at 8 h along the neurites are shown at higher magnification in B (arrows). (C) The total amount of tau protein was detected by the K9JA antibody, which recognizes tau independently of its phosphorylation. In the same cell extracts, abnormal tau phosphorylation was detected by using the S202/205 phosphospecific antibody 11b. Neurons were exposed to 20 ng/ml BoNT/C for the indicated times. (D) The pattern of tau distribution was determined by a different antibody (Innogenetics) and changed in cells exposed to 20 ng/ml BoNT/C. Images in A, B, and D are extended focus images derived by merging 50–60 focal planes acquired in the z dimension with a 63× oil 1.3 NA apochromat objective. Bars, 20 μm.
Thus, exposure to BoNT/C initiated an early cytoskeletal disarray at the level of the neuronal projections. Cytoskeletal alterations are intimately linked with changes in mitochondrial distribution along neuronal projections (Ebneth et al., 1998; Stamer et al., 2002). In BoNT/C-treated neurons, tetramethylrhodamine ethyl ester (TMRE)–loaded mitochondria along neurites initially (6 h) displayed higher longitudinal motility than those in control cells. By 12 h after the exposure to the toxin, mitochondria had lost their elongated shape as a result of shortening in a process morphologically analogous to mitochondrial fission (Bossy-Wetzel et al., 2003). Mitochondria progressively migrated into, and clumped in, discrete membrane-enclosed regions. Interestingly, blebs along the neurites always seemed to emerge around clustered mitochondria (unpublished data). Mitochondria in cell bodies clustered on one side opposite to the nucleus (Fig. 6 A and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200406126/DC1).
The membrane potential was remarkably retained in the mitochondria present along degenerating axo-dendritic projections, and, even at late time points, TMRE-positive mitochondria were still visible in most blebs and neurite fragments (Fig. 6 B). Mitochondrial integrity and membrane potential were initially retained also in the mitochondria clustering around the cell nucleus. However, shortly before nuclear condensation, cytochrome c was released from somatic mitochondria (Fig. 2 A) and the TMRE staining dissipated (Fig. 6 A and Video 1). In this case, the events occurred quite rapidly as shown in other cell types undergoing apoptosis (Goldstein et al., 2000). Soon after the loss of the mitochondrial membrane potential, cell bodies first shrunk, and then swelled and lysed (Fig. 6 A and Video 1).
To further confirm that mitochondria in fragmenting neurites retained structural integrity, we performed a double immunostaining for cytochrome c and the mitochondrial heat-shocked protein 70 (mtHSP-70). Colocalization was quantified using a point-spread function by using the ImarisColoc software. As shown in Fig. 6 (C and D), even at late time points, when the axo-dendritic network was severely compromised, cytochrome c and HSP-70 stainings remained colocalized in the fragmenting neurites. This corroborates the findings obtained with TMRE, and further suggests that mitochondrial functions are maintained long after the blebbing and the initial disintegration of the neurites (Fig. 6 B).
Involvement of upstream kinases in BoNT/C-mediated neurodegeneration
Although neurite degeneration and the death of the cell body are executed by different mechanisms, they may involve a common initiating lethal signal and simply diverge downstream before mitochondrial dysfunction. Because a lack of trophic and glutamatergic stimulation was excluded by the experiments discussed above (compare Fig. 3 D), we targeted some of the kinases implicated in neuronal death in both in vitro and in vivo models (Harper and LoGrasso, 2001; Cruz and Tsai, 2004; Jope and Johnson, 2004). These include the MAPK, GSK-3, and Cdk-5 signaling pathways. Because of the complexity of the MAPK cascade, we used CEP-1347, a synthetic inhibitor, which does not act on JNK (c-Jun NH2-terminal kinase) directly, but inhibits the upstream MLK (mixed lineage kinase). MLKs are upstream kinases that activate MAPKs including JNK, ERK1/2, ERK5, and p38 (Wang et al., 2004). The inhibition by 100 nM CEP-1347 delayed apoptosis induced by BoNT/C (Fig. 7 A), but it did not protect against the early degeneration of the neurites (Fig. 7 C). Similar results were obtained also with the Cdk-5 inhibitor roscovitine (10 μM) and with alsterpaullone (1 μM), which inhibits both GSK3 and Cdk-5 (Fig. 7 B). A more selective GSK-3 inhibitor (2 μM) had only a minor effect, although in combination with roscovitine it prevented cell death. These results suggest that both MAPKs and Cdk-5 have relevant roles in BoNT/C-triggered apoptosis. However, because none of the kinase inhibitors prevented neurite degeneration (Fig. 7 C), it is again apparent that neurite loss and apoptosis were triggered by entirely distinct signaling pathways.
Figure 7.
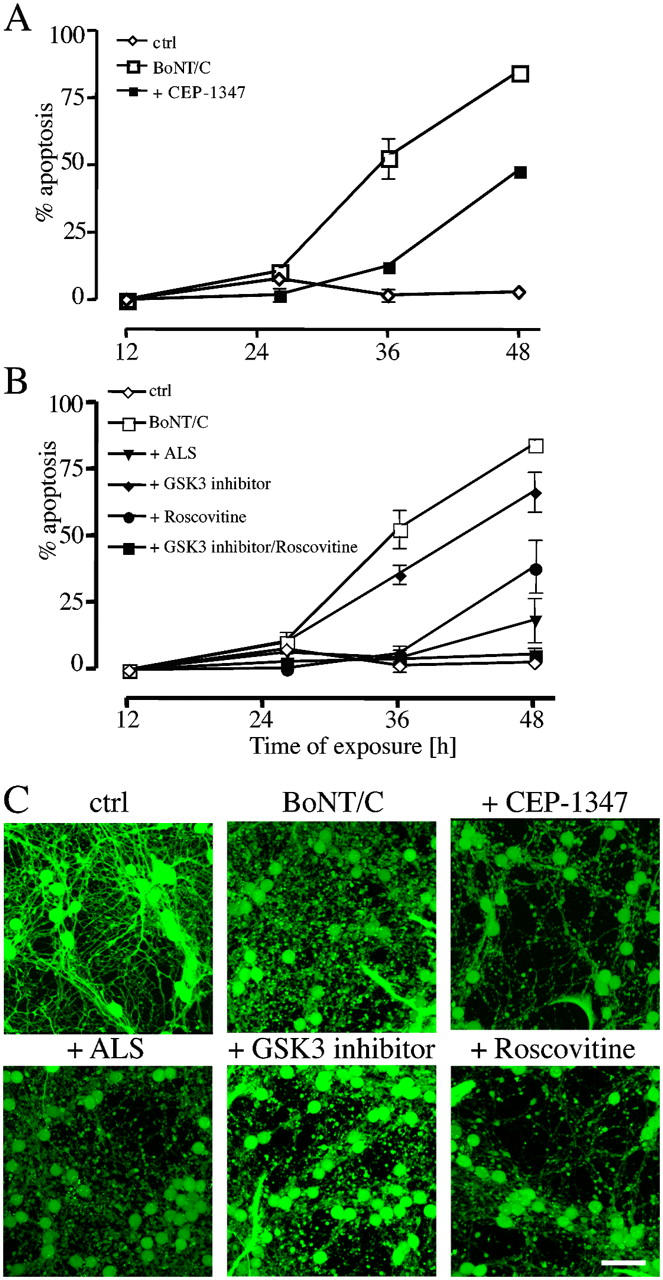
Effect of kinases inhibitors on BoNT/C-induced degeneration of the neurites and death of the cell bodies. Time course of BoNT/C-induced cell death in the presence of the MLK inhibitor CEP1347 (100 nM; A), GSK-3 inhibitor (2 μM), alsterpaullone, ALS (1 μM), and roscovitine, (10 μM; B). Apoptosis was evaluated as a percentage of condensed nuclei stained by H-33342. (C) CGCs were exposed to 20 ng/ml BoNT/C and different kinase inhibitors for 18 h and were loaded with calcein-AM before being imaged by confocal microscopy. Bar, 30 μm.
Recognition and phagocytosis of degenerating neurites precedes removal of apoptotic cell bodies
Finally, we reasoned that if different degeneration processes regulate loss of neuronal processes and apoptosis, signals for phagocytosis and scavenging should also differ in the two neuronal compartments. Glial cells actively scavenge neuronal debris or whole dying neurons (Bechmann and Nitsch, 1997a,b) and few glial cells (usually <5%) are present in CGC cultures. After exposure to BoNT/C, we noticed that the degenerating axo-dendritic network became less dense, and large areas in culture dishes were devoid of any remnants of neuronal neurites. Therefore, we monitored the progression of events in BoNT/C-treated CGC cultures over 48 h. As the degeneration and fragmentation of neuronal projections progressed, contaminant glial cells became highly mobile and began to probe the neuronal network by scavenging fragmented neurites and eventually cellular debris (Fig. 8 A and Videos 2 and 3, available at http://www.jcb.org/cgi/content/full/jcb.200406126/DC1). Most notably, scavenging was initially restricted to neuronal projections and did not affect the somata until they became clearly apoptotic. Therefore, we set out to determine whether a selective appearance of surface recognition molecules in degenerating neurites would trigger such selective phagocytosis. Cultures were loaded with Alexa 633–conjugated annexinV to recognize phosphatidylserine, which is one of the main signaling molecules in phagocytosis. Degenerating neurites, but not the cell bodies, displayed phosphatidylserine during the first 24 h (Fig. 8 B and Fig. S1). Somata undergoing apoptosis displayed phosphatidylserine later, and were then removed by resident glial cells. These data show that both recognition signals and phagocytosis occur by two temporally and spatially distinct steps.
Figure 8.
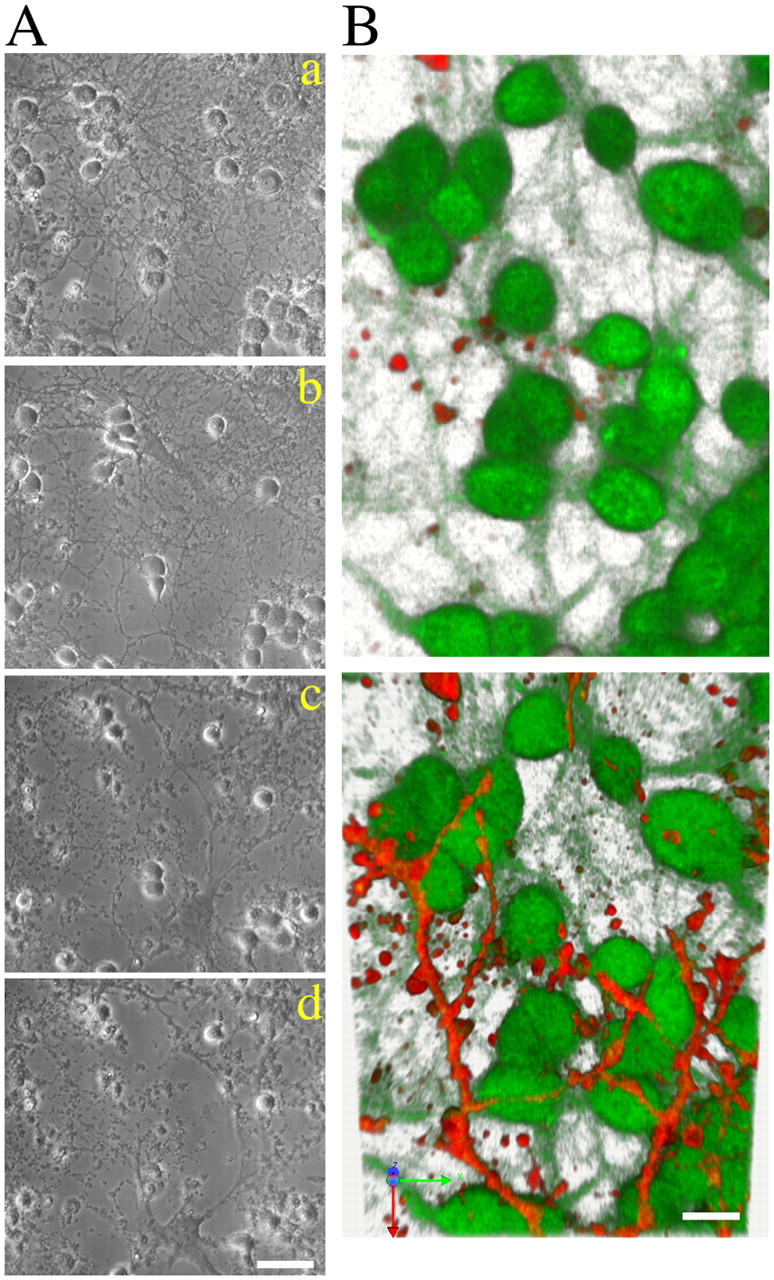
Selective phosphatidylserine exposure on degenerating neurites preluded their clearance by glial cells. BoNT/C-treated CGCs were followed for 48 h by conventional time-lapse microscopy (A; and Videos 2 and 3, available at http://www.jcb.org/cgi/content/full/jcb.200406126/DC1). As neurites degenerated and fragmented, a glial cell inhabiting the layer below the neuronal network became activated and begun clearing damaged projections. At a later stage (36–48 h), when cell bodies succumbed to apoptosis, the glial cell scavenged cell corpses (Videos 2 and 3, respectively). Images in A are extracted from Video 2 at the following times (h): a, 1; b, 23; c, 34; and d, 40. (B) BoNT/C-treated CGCs were loaded with Alexa 633–conjugated Annexin V (red) and calcein-AM (green) and imaged by time-lapse confocal microscopy (B and Fig. S1). The image in B and the rotation in Fig. S1 are three-dimensional renderings from multiple z-stacked images collected at 12 (top) and 18 h (bottom). The calcein stain is masked by the appearance of annexin positivity along the surface of neurites. Image stacks were collected by laser scanning microscopy and rendered using the Imaris software. Bars: (A) 25 μm; (B) 10 μm.
Discussion
Clostridial neurotoxins have long been used as a selective experimental tool for dissecting the molecular basis of synaptic transmission (Bagetta et al., 1990; Schiavo et al., 2000). Recently, their use has been extended to the therapeutic treatment of various human disturbances caused by excessive neurotransmitter release (Cordivari et al., 2004). Their specificity of action at the synapse is caused by the selective Zn2+ endopeptidase activity, which each serotype exerts against one of the three major SNARE proteins, syntaxin, SNAP-25, and VAMP/synaptobrevin. Among those three proteins, syntaxin is the only one being targeted by a single toxin serotype, BoNT/C (Schiavo et al., 1995). In mouse neurons, the cleavage is selective for syntaxin 1, which is localized on the presynaptic plasma membrane (Humeau et al., 2000; for review see Teng et al., 2001). In this study, we used clostridium neurotoxins as an experimental tool, to trigger synaptic damage in central nervous system neurons. BoNT/C triggered two spatially and temporally distinct programs, which mediate neurite degeneration and apoptosis. In CGCs, BoNT/C caused an early disassembly of the neuronal network characterized by progressive vesiculation and fragmentation (Fig. 4, A and B). This is in agreement with the synaptic terminal swelling and the neurite network vesiculation induced by BoNT/C in cortical, hippocampal, and spinal cord neurons (Osen-Sand et al., 1996; Williamson and Neale, 1998). The degeneration of the projections was independent of the loss of neurotransmitter release (Fig. 3), was insensitive to trophic stimulation (Fig. 4 F), and did not require protein synthesis. However, it was an active process as shown by the ability of the fragmented processes to retain plasma membrane integrity (Figs. 4 B and 6 B) and by the presence of functional mitochondria along the neurites (Fig. 6 B). On the contrary, the late demise of the cell body was accompanied by the loss of mitochondrial membrane potential and cytochrome c release, before the appearance of any nuclear change (Figs. 2 A and 6 A). Furthermore, although caspases contributed to the late apoptotic demise of the cell body (Fig. 2), caspase-independent mechanisms mediated the initial neurodegenerative events (Fig. 4 C). Caspase-independent axonal degeneration associated with caspase-dependent apoptosis of the cell body has been described in Wallerian degeneration of optic and sciatic nerve explants and in localized axonal degeneration induced by local neurotrophin deprivation in dorsal root ganglia (Finn et al., 2000). However, at difference with the predictions of these models, small amounts of processed caspase-3 were detected along the neuronal projections of BoNT/C-treated CGCs (Fig. 4 C), either reflecting an undefined housekeeping function of caspase-3 (Campbell and Holt, 2003; for review see Perfettini and Kroemer, 2003) or, reflecting a more effective antiapoptotic machinery that operates in the neuronal projections (Potts et al., 2003). The lack of any protective effect of caspase inhibitors on neurite degeneration (Fig. 4 A) suggests that caspase-3, although processed, is not active or relevant in dismantling CGN projections.
Neurodegeneration induced by BoNT/C in central neurons resembled axotomy in many of its features (e.g., role of caspases, cytoskeletal disarray, and phosphatidylserine exposure). However, we tested whether BoNT/C-induced neurodegeneration was attenuated or prevented in the Wallerian degeneration slow (Wld s) mice, spontaneous mutants in which axonal degeneration is greatly delayed, and found no protective effects (unpublished data). We also tested whether the ubiquitin–proteasome system was required in BoNT/C-triggered neurite degeneration as it is in the early stages of Wallerian degeneration (Zhai et al., 2003) or in axonal pruning (Watts et al., 2003). In our model, inhibition of the ubiquitin–proteasome system did not affect the degenerative events. Axodendritic fragmentation within the first 24 h was not prevented by short exposure to agents that block the proteasome system, nor was apoptosis (unpublished data). Together, this seems to suggest that also neurites, as well as the cell body, may activate distinct programs of self-destructions.
Although it is now accepted that cell death may involve distinct molecular mechanisms and take many manifestations different from classical apoptosis (Leist and Jaattela, 2001), the possibility of different death programs has seldom been examined for neurites, which play such an important role for the onset and manifestation of several pathologies. Neurodegeneration induced by BoNT/C in CGCs resembled several features also observed in neurodegenerative disease. A massive damage of the cytoskeleton underlay neurite disassembly. Both components of the cytoskeleton—microfilaments and microtubules—were affected. Whereas actin changes mainly consisted of a progressive, general loss, tubulin alterations were characterized by the appearance of typical rings along the neurites (Fig. 5, A and B). Similar cytoskeletal abnormalities have been observed in the early stage of Alzheimer's disease (Dickson et al., 1999) as well as in the aging brain (Vickers et al., 1996). In both cases, dystrophic neurites, which associated with β-amyloid plaque, showed bulb- and ringlike structures. These consisted mainly of neurofilaments, but also consisted of hyperphosphorylated tau proteins in the late stages of the disease. In our system, abnormal tau phosphorylation at the serine 202/205 residues was detected by AT-8–like antibodies during the early degeneration of neurite (Fig. 5 C). This was associated with a redistribution of tau along the neurites (Fig. 5 D).
Although we do not have firm evidence that the cytoskeletal disassembly and the tau alterations are the causes of the cell body apoptosis, there is ample evidence that activation and mislocalization of Cdk-5 and hyperphosphorylated tau can lead to apoptosis in other systems (Cheung and Ip, 2004). Because Cdk-5 inhibitors did not modify tau phosphorylation in BoNT/C-treated cultures (unpublished data), abnormal tau phosphorylation via Cdk-5 was not an essential requirement for apoptosis in this system. However, our data do not exclude that tau redistribution can play a fundamental role in both the neurite loss and the onset of apoptosis triggered by BoNT/C. An aberrant behavior of microtubules and microtubule-associated proteins can cause disturbances in axonal transport and impair axonal mitochondrial distribution in the early stages of Alzheimer's disease (Ebneth et al., 1998; Mandelkow and Mandelkow, 1998; Mandelkow et al., 2003). Clearly, our results with the kinase inhibitors eliminate the possibility that Cdk-5 or other upstream kinases are involved in the neurite loss. The selective effect of kinase inhibitors on cell body apoptosis places Cdk-5 and MAPK upstream of the demise of the cell body, but downstream of the initial neurodegenerative events after synaptic damage.
Interestingly, all the degenerative effects induced by BoNT/C were neurospecific, and the population of contaminant glial cells present in the culture was not affected. Similarly, BoNT/C did not induce any degenerative or hypertrophic effect in glial cultures (unpublished data). However, morphological changes indicative of a state of activation were detected in the glial cells present in BoNT/C-treated CGC cultures (unpublished data). A common feature of several neurodegenerative diseases is the abundance of reactive astrocytes and activated microglia in proximity to degenerating neurons. Astrocytes have been reported to accumulate and surround the area of amyloid deposition and undergo hypertrophy with the progression of neurite dystrophy (Mrak et al., 1996). This condition of reactive gliosis seems to be mimicked in BoNT/C-induced neurodegeneration, and therefore makes this model a valuable in vitro system for studying the interactions between injured neurons and glial cells. Moreover, glia seem to participate in the dismantling of degenerating neurites (Fig. 8 A and Videos 2 and 3), which exposed recognition molecules as phosphatidylserine, well before any sign of apoptosis was detected in neuronal somata (Fig. 8 B). The temporal and spatial segregation of the degenerative process, including the recognition and scavenging of dying neurites, may reflect a stereotyped response of neurons to injury, which may be relevant to the progression of slow degenerative diseases and aging. Because BoNT/C selectively cleaves synaptic proteins (Humeau et al., 2000; Schiavo et al., 2000), the finding that widespread disruption of synaptic architecture/function causes neurodegeneration supports the argument that prolonged synaptic destabilization may be sufficient to trigger neurodegenerative disease. Strikingly, the effects of BoNT/C on central neurons recapitulate several of the most relevant features found in human neurodegenerative disorders and in their animal models. Synaptic damage, cytoskeletal modifications, mitochondrial alterations, and apoptosis are featured in conditions such as Alzheimer's and Huntington's diseases, although the contribution of each of these processes to neurodegeneration and to disease manifestation remains unknown. In line with other toxin-related model systems that have played important roles in understanding neurodegenerative processes, the model described here opens the door to future exploration of the relationship between synaptic dysfunction, axonal damage and apoptosis in the CNS.
Materials and methods
Chemicals
Unless otherwise stated, all reagents were obtained from Sigma-Aldrich. BoNT/C and BoNT/A were initially provided by C. Montecucco (University of Padua, Padua, Italy), and were later obtained from Calbiochem. Caspase inhibitors zVAD-fmk, Ac-YVAD-chloromethylketone, and zD-cbk were obtained from Bachem. The caspase-2 inhibitor z-VD(OMe)VAD (OMe)-fmk was purchased from MP Biomedicals. The GSK-3 inhibitor was synthesized at H. Lundbeck A/S according to standard protocols. SYTOX, H-33342, calcein-AM, and TMRE were obtained from Molecular Probes. The caspase substrate, DEVD-aminotrifluoromethylcoumarine (afc) was obtained from Biomol. Solvents and inorganic salts were purchased from Merck or Riedel-de Haen.
Cell culture
Murine CGCs were isolated as described previously (Leist et al., 1997a). Dissociated neurons were plated on 100 μg/ml (250 μg/ml for glass surfaces) poly-l-lysine (molecular mass > 300 kD)–coated dishes at a density of ∼0.25 × 106 cells/cm2 (800,000 cells/ml; 500 μl/well, 24-well plate) and cultured in Eagle's basal medium (GIBCO BRL) supplemented with 10% heat-inactivated FCS, 20 mM KCl, 2 mM l-glutamine, penicillin–streptomycin, and 10 μM cytosine arabinoside (added 48 h after plating). Neurons were used without further medium changes after 6–7 d in vitro (DIV). The frequency of glial fibrillary acid protein-positive cells was <5%. The cultures were exposed to BoNT/C in their original medium. Stock solutions of clostridial neurotoxins were prepared in 150 mM NaCl, 10 mM Hepes, pH 7.4, containing 0.1% BSA.
For time-resolved imaging, cells were grown on ultrathin Lab-Tek chamber slides (Nunc) and exposed to 20 or 30 ng/ml BoNT/C at 6 DIV. 5 h after treatment, cells were loaded with either with 20 nM TMRE or with Alexa 633–conjugated annexin V (1:100) and 1 μM calcein-AM in the presence of 2 μM MK801 and 10 mM Hepes.
Viability assays
Apoptosis was quantified by staining neuronal cultures with 1 μg/ml H-33342 (membrane permeant, blue fluorescent chromatin stain) as described previously (Leist et al., 1997a). Apoptotic cells were characterized by scoring typically condensed nuclei. About 600–1,000 cells were counted in nine different fields in more than three different culture wells, and experiments were repeated in at least three different preparations. Mitochondrial function and integrity was assessed by the fluorescent, mitochondrial membrane potential (Δψ)–sensitive indicator TMRE (20 nM; λex = 568 nm and λem ≥ 590 nm).
HMM DNA fragmentation was quantified as described previously (Ankarcrona et al., 1995) by field inversion gel electrophoresis. About 1.3 × 106 cells (corresponding to two wells of a 12-well plate) were embedded into 40-μl agarose blocks. λ-DNA concatemers (n × 50 kbp) were used as molecular mass markers.
Oligonucleosomal DNA fragmentation (DNA laddering) was analyzed by conventional agarose gel electrophoresis (Wyllie, 1980; Thornberry, 1994). Gels (10% agarose) were stained with SyBRO green and photographed. DNA ladder (123-4; 182 bp) of the rat prolactin gene was used as a molecular mass marker.
Enzymatic assays
Caspase-3–like activity (measured by DEVD-afc cleavage) was assayed essentially as described previously (Thornberry, 1994; Leist et al., 1997b). CGCs were lysed in 25 mM Hepes, 5 mM MgCl2, 1 mM EGTA, 0.5% Triton X-100, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 μg/ml aprotinin, and 1 mM PEFA block, pH 7.5. The fluorimetric assay was performed in microtiter plates with a substrate concentration of 40 μM and a total protein amount of 5 μg. DEVD-afc cleavage was followed in the reaction buffer (50 mM Hepes, 10 mM DTT, 1% sucrose, and 0.1% CHAPS) over a period of 30 min at 37°C (λex = 390 nm and λem = 505 nm). Absolute activity was calibrated with afc standard solutions (0–2.5 μM). Triple measurements were performed for each sample.
Western blot analysis
CGCs were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris, 1% NP-40, 0.25% sodium deoxycholate, and 1 mM EGTA) supplemented with protease inhibitors (1 mM PMSF, 1 mM benzamidine, 1 mM iodoacetate, 1 mM iodoacetamide, 40 μM leupeptin, 10 μg/ml antipain, and 5 μg/ml pepstatin). For the detection of phosphorylated tau protein, the phosphatase inhibitor sodium orthovanadate (1 mM) was also added. Protein content was determined using the bicinchoninic acid method (Bio-Rad Laboratories).
Proteins were separated by SDS-PAGE with 15 μg protein/lane on 12% polyacrylamide gels for syntaxin, 5 μg protein/lane on 8% polyacrylamide gels for fodrin, 50 μg protein/lane on 8% polyacrylamide gels for tau protein, 100 μg protein/lane on 12% polyacrylamide gels for processed caspase-3, and then blotted onto nitrocellulose membranes (Amersham Biosciences) in a semi-dry blotter (Bio-Rad Laboratories). Blots were blocked in TNT (50 mM Tris, pH 8.0, 150 mM NaCl, and 0.05% Tween 20) containing 5% dried milk and incubated with either antisyntaxin mAb (1:10,000; clone 78.3; Synaptic Systems), anti–SNAP-25 mAb (1:10,000; clone 71.2; Synaptic Systems), antifodrin mAb (1:500; clone 1622; Chemicon International), anticleaved caspase-3 polyclonal antibody (1:1,000; Asp175; Cell Signaling), or anti-tau protein polyclonal antibodies (K9JA, 1:5, 000; 11b 1:2,000). Tau antibodies were a gift from Eva-Maria and Eckhard Mandelkow (Max-Planck Institute, Hamburg, Germany). Specifically stained bands were detected by chemiluminescence (ECL; Amersham Biosciences) using a polyclonal IgG horseradish peroxidase–coupled secondary antibody (1:1,000; BD Biosciences).
Determination of l-[G-3H]-glutamate release
Cells were exposed to BoNT/C as indicated in Fig. 3. To avoid excitotoxicity via the NMDA receptor, 2 μM MK801 (RBI; Biotrend Chemikalien GmbH) and 2 mM MgCl2 were added 30 min before the removal of the medium and were present throughout the loading, washing, and releasing steps of the assay. Neurons were loaded with 37 kBq/ml l-[G-3H]glutamine (Amersham Biosciences; spec. act., 1.96 TBq/mmol) in CSS25 (CSS with 25 mM KCl) for 30 min at 37°C and washed three times in the same buffer. During this incubation period, the labeled l-glutamine is taken up by the neurons, converted into glutamate by mitochondrial glutaminase and packed into synaptic vesicles (McMahon and Nicholls, 1990). After 30 min of equilibration in CSS5 (CSS with 5 mM KCl) at 37°C, l-glutamate release was measured after stimulation with 55 mM KCl or 10 μM veratridine for 4 min. Supernatants were removed and counted for tritium. Cells were then lysed in 0.3% Triton X-100/PBS for 30 min, and the residual accumulated l-[G-3H]glutamate was measured. Values were calculated as a percentage of the total cellular tritium content, the basal release was subtracted, and the control release was set to 100%.
Immunofluorescence
CGCs were grown on glass coverslips, fixed after the experiment with 4% PFA and permeabilized with 0.1% Triton X-100 in PBS. For the detection of active caspase, a rabbit polyclonal antibody (cleaved caspase-3, Asp175; Cell Signaling) was used. The specificity of the staining was tested by incubating the antibody with the p20/p17 fragment of caspase-3 in the presence of zVAD-fmk. To monitor cytoskeleton alterations, cells were incubated with the neurospecific mouse mAb anti–βIII-tubulin (Promega). As a secondary antibody, we used an Alexa (λex = 488 nm and λem = 512 nm)-coupled anti–mouse IgG (1:600, Molecular Probes). Tau protein was detected by using the mouse monoclonal AT8 antibody (Innogenetics). Actin was detected by Alexa 568–conjugated phalloidin (λex = 568 nm and λem = 603 nm). Cytochrome c and mtHSP-70 were detected via mouse mAbs (Cyt c: clone 6H2.B4 [PharMingen]; mtHSP-70: clone JG1 [Abcam]). For the double staining, antibodies for cytochrome c and HSP70 were labeled with Alexa Fluor 488 (λex = 495 nm and λem = 519 nm) and 546 (λex = 556 nm and λex = 573 nm), respectively, by using the Zenon mouse IgG labeling kit (Molecular Probes). Cultures were counterstained with 0.5 μg/ml H-33342.
Confocal microscopy
Confocal microscopy images were acquired using a laser scanning microscope (model LSM 510 Combi; Carl Zeiss MicroImaging, Inc.) equipped with visible and infrared Ti-Sa pulsed laser (Tsunami/Millenia Spectra-Physics). If not otherwise stated, all images were collected using a 40× oil 1.3 NA apochromat objective (Carl Zeiss MicroImaging, Inc.). Postacquisition processing was performed with Adobe Photoshop and Adobe Illustrator.
Time-resolved imaging
Images for time-resolved experiments were collected using either confocal (Figs. 6 A and 8 B, Video 1, and Fig. S1) or conventional (Fig. 8 A and Videos 2 and 3) microscopy. Cells were thermostated using a heated stage (Carl Zeiss MicroImaging, Inc.), and the temperature was kept constantly at 34°C (±1.5°C). Additionally, pH was maintained by perfusing cell with 5% CO2 using a CO2 controller (MicroImaging, Inc.). Images were collected with a time interval of 15 min. Settings were optimized in order to reduce photobleaching to a minimum, and laser stability was controlled using an internal photodiode monitor as reference.
Colocalization
Quantitative colocalization was performed using ImarisColoc software (Bitplane AG). Image stacks were collected using a laser scanning microscope. Image stacks were then loaded in Imaris, and quantitative colocalization was determined. Threshold for colocalization was automatically set using the automatic threshold function of the software. Intensity pairs of the two channels that exhibit no correlation (Pearson's correlation coefficient < 0) were excluded from the analysis. Once the first step was obtained, then the software calculated the percentage of voxels that colocalize.
Scanning electron microscopy
CGCs that were grown on glass coverslips were immersed in Karnovsky's fixative and ferrocyanide-reduced osmium tetroxide before being treated with 5% uranyl acetate and dehydrated with a series of alcohols. They were transferred to hexamethyldisilazane and air dried before being sputter coated with a 15-nm layer of gold and examined in an electron microscope (model 100CXII; JEOL) equipped with an ASID-4D scanning unit (JEOL). Postacquisition processing was performed with Adobe Photoshop and Adobe Illustrator.
Statistics
Experiments to determine cell viability or caspase activity were run as triplicates and repeated in three to eight cell preparations. Statistical significance was calculated on the original datasets using the t test. All Western blots and immunostainings were performed with samples from at least three independent cell preparations.
Online supplemental material
All videos were acquired as described in the Time-resolved imaging section, processed using QuickTimePro (Video 1) or Adobe Premiere Pro (Videos 2 and 3) and saved as Quicktime movies.
Fig. S1 shows the distribution of phosphatidylserine along neuronal projections in a three-dimensional reconstruction.Video 1 shows a dynamic recording of the mitochondrial membrane potential clustering around the cell nucleus of neurons exposed to 25 ng/ml BoNT/C for 33 h and almost completely deprived of connections. Video 2 shows the clearance of degenerating neurites by a glial cell. Video 3 shows the clearance of apoptotic neuronal bodies during the late phase of neurodegeneration. Online supplemental material is available http://www.jcb.org/cgi/content/full/jcb.200406126/DC1.
Acknowledgments
We thank Maria Azucena Guerra-Martin, Heike Neumann, Elvira Gawlitta-Gorka, Tim Smith, Anna Simpson, and Chris Guerin for their help. We are grateful to Professor C. Montecucco and Dr. Ornella Rossetto for initially providing BoNT/C and A and to Professors Eva-Maria and Eckhard Mandelkow for providing the tau antibodies K9JA and 11b.
This work was supported by a DFG grant (For324/2) and by the British Medical Research Council.
Abbreviations used in this paper: afc, aminotrifluoromethylcoumarine; BoNT/C/A, botulinum neurotoxin C1 or A; CGC, cerebellar granule cell; fmk, fluoromethylketone; HMM, high molecular mass; mtHSP-70, mitochondrial heat-shocked protein 70; TMRE, tetramethylrhodamine ethyl ester.
E. Fava's present address is Max-Planck Institute of Molecular Cell Biology and Genetics, 01307 Dresden, Germany.
References
- Ankarcrona, M., J.M. Dypbukt, E. Bonfoco, B. Zhivotovsky, S. Orrenius, S.A. Lipton, and P. Nicotera. 1995. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 15:961–973. [DOI] [PubMed] [Google Scholar]
- Bagetta, G., M.T. Corasaniti, G. Nistico, and N.G. Bowery. 1990. Behavioural and neuropathological effects produced by tetanus toxin injected into the hippocampus of rats. Neuropharmacology. 29:765–770. [DOI] [PubMed] [Google Scholar]
- Bagri, A., H.J. Cheng, A. Yaron, S.J. Pleasure, and M. Tessier-Lavigne. 2003. Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell. 113:285–299. [DOI] [PubMed] [Google Scholar]
- Bailey, C.H., and E.R. Kandel. 1993. Structural changes accompanying memory storage. Annu. Rev. Physiol. 55:397–426. [DOI] [PubMed] [Google Scholar]
- Bechmann, I., and R. Nitsch. 1997. a. Astrocytes and microglial cells incorporate degenerating fibers following entorhinal lesion: a light, confocal, and electron microscopical study using a phagocytosis-dependent labeling technique. Glia. 20:145–154. [DOI] [PubMed] [Google Scholar]
- Bechmann, I., and R. Nitsch. 1997. b. Identification of phagocytic glial cells after lesion-induced anterograde degeneration using double-fluorescence labeling: combination of axonal tracing and lectin or immunostaining. Histochem. Cell Biol. 107:391–397. [DOI] [PubMed] [Google Scholar]
- Biernat, J., E.M. Mandelkow, C. Schroter, B. Lichtenberg-Kraag, B. Steiner, B. Berling, H. Meyer, M. Mercken, A. Vandermeeren, M. Goedert, et al. 1992. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 11:1593–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi, J., E.R. Chapman, E. Link, T. Binz, S. Yamasaki, P. De Camilli, T.C. Sudhof, H. Niemann, and R. Jahn. 1993. a. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 365:160–163. [DOI] [PubMed] [Google Scholar]
- Blasi, J., E.R. Chapman, S. Yamasaki, T. Binz, H. Niemann, and R. Jahn. 1993. b. Botulinum neurotoxin C1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO J. 12:4821–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel, E., M.J. Barsoum, A. Godzik, R. Schwarzenbacher, and S.A. Lipton. 2003. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr. Opin. Cell Biol. 15:706–716. [DOI] [PubMed] [Google Scholar]
- Campbell, D.S., and C.E. Holt. 2003. Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron. 37:939–952. [DOI] [PubMed] [Google Scholar]
- Cheung, Z.H., and N.Y. Ip. 2004. Cdk5: mediator of neuronal death and survival. Neurosci. Lett. 361:47–51. [DOI] [PubMed] [Google Scholar]
- Coleman, M.P., and V.H. Perry. 2002. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 25:532–537. [DOI] [PubMed] [Google Scholar]
- Cordivari, C., V.P. Misra, S. Catania, and A.J. Lees. 2004. New therapeutic indications for botulinum toxins. Mov. Disord. 19:S157–S161. [DOI] [PubMed] [Google Scholar]
- Cruz, J.C., and L.H. Tsai. 2004. A Jekyll and Hyde kinase: roles for Cdk5 in brain development and disease. Curr. Opin. Neurobiol. 14:390–394. [DOI] [PubMed] [Google Scholar]
- D'Mello, S.R., C. Galli, T. Ciotti, and P. Calissano. 1993. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. USA. 90:10989–10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, C.A., D.M. Mann, P.Q. Sumpter, and P.O. Yates. 1987. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J. Neurol. Sci. 78:151–164. [DOI] [PubMed] [Google Scholar]
- Dickson, T.C., C.E. King, G.H. McCormack, and J.C. Vickers. 1999. Neurochemical diversity of dystrophic neurites in the early and late stages of Alzheimer's disease. Exp. Neurol. 156:100–110. [DOI] [PubMed] [Google Scholar]
- Ebneth, A., R. Godemann, K. Stamer, S. Illenberger, B. Trinczek, and E. Mandelkow. 1998. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J. Cell Biol. 143:777–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, J.T., M. Weil, F. Archer, R. Siman, A. Srinivasan, and M.C. Raff. 2000. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J. Neurosci. 20:1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foran, P.G., N. Mohammed, G.O. Lisk, S. Nagwaney, G.W. Lawrence, E. Johnson, L. Smith, K.R. Aoki, and J.O. Dolly. 2003. Evaluation of the therapeutic usefulness of botulinum neurotoxin B, C1, E, and F compared with the long lasting type A. Basis for distinct durations of inhibition of exocytosis in central neurons. J. Biol. Chem. 278:1363–1371. [DOI] [PubMed] [Google Scholar]
- Frankle, W.G., J. Lerma, and M. Laruelle. 2003. The synaptic hypothesis of schizophrenia. Neuron. 39:205–216. [DOI] [PubMed] [Google Scholar]
- Gaiddon, C., J.P. Loeffler, and Y. Larmet. 1996. Brain-derived neurotrophic factor stimulates AP-1 and cyclic AMP-responsive element dependent transcriptional activity in central nervous system neurons. J. Neurochem. 66:2279–2286. [DOI] [PubMed] [Google Scholar]
- Gasic, G.P., and P. Nicotera. 2003. To die or to sleep, perhaps to dream. Toxicol. Lett. 139:221–227. [DOI] [PubMed] [Google Scholar]
- Gillingwater, T.H., and R.R. Ribchester. 2001. Compartmental neurodegeneration and synaptic plasticity in the Wld(s) mutant mouse. J. Physiol. 534:627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert, M., M.G. Spillantini, N.J. Cairns, and R.A. Crowther. 1992. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 8:159–168. [DOI] [PubMed] [Google Scholar]
- Goldstein, J.C., N.J. Waterhouse, P. Juin, G.I. Evan, and D.R. Green. 2000. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2:156–162. [DOI] [PubMed] [Google Scholar]
- Harper, S.J., and P. LoGrasso. 2001. Signalling for survival and death in neurones: the role of stress-activated kinases, JNK and p38. Cell. Signal. 13:299–310. [DOI] [PubMed] [Google Scholar]
- Hatanpaa, K., K.R. Isaacs, T. Shirao, D.R. Brady, and S.I. Rapoport. 1999. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J. Neuropathol. Exp. Neurol. 58:637–643. [DOI] [PubMed] [Google Scholar]
- Humeau, Y., F. Doussau, N.J. Grant, and B. Poulain. 2000. How botulinum and tetanus neurotoxins block neurotransmitter release. Biochimie. 82:427–446. [DOI] [PubMed] [Google Scholar]
- Ivins, K.J., E.T. Bui, and C.W. Cotman. 1998. Beta-amyloid induces local neurite degeneration in cultured hippocampal neurons: evidence for neuritic apoptosis. Neurobiol. Dis. 5:365–378. [DOI] [PubMed] [Google Scholar]
- Jope, R.S., and G.V. Johnson. 2004. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29:95–102. [DOI] [PubMed] [Google Scholar]
- Leist, M., and M. Jaattela. 2001. Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2:589–598. [DOI] [PubMed] [Google Scholar]
- Leist, M., E. Fava, C. Montecucco, and P. Nicotera. 1997. a. Peroxynitrite and nitric oxide donors induce neuronal apoptosis by eliciting autocrine excitotoxicity. Eur. J. Neurosci. 9:1488–1498. [DOI] [PubMed] [Google Scholar]
- Leist, M., C. Volbracht, S. Kuhnle, E. Fava, E. Ferrando-May, and P. Nicotera. 1997. b. Caspase-mediated apoptosis in neuronal excitotoxicity triggered by nitric oxide. Mol. Med. 3:750–764. [PMC free article] [PubMed] [Google Scholar]
- Lichtman, J.W., and H. Colman. 2000. Synapse elimination and indelible memory. Neuron. 25:269–278. [DOI] [PubMed] [Google Scholar]
- Mandelkow, E.M., and E. Mandelkow. 1998. Tau in Alzheimer's disease. Trends Cell Biol. 8:425–427. [DOI] [PubMed] [Google Scholar]
- Mandelkow, E.M., K. Stamer, R. Vogel, E. Thies, and E. Mandelkow. 2003. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging. 24:1079–1085. [DOI] [PubMed] [Google Scholar]
- Mangiarini, L., K. Sathasivam, M. Seller, B. Cozens, A. Harper, C. Hetherington, M. Lawton, Y. Trottier, H. Lehrach, S.W. Davies, and G.P. Bates. 1996. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 87:493–506. [DOI] [PubMed] [Google Scholar]
- Mattson, M.P., J. Partin, and J.G. Begley. 1998. Amyloid beta-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 807:167–176. [DOI] [PubMed] [Google Scholar]
- McGlashan, T.H., and R.E. Hoffman. 2000. Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch. Gen. Psychiatry. 57:637–648. [DOI] [PubMed] [Google Scholar]
- McKinney, R.A., M. Capogna, R. Durr, B.H. Gahwiler, and S.M. Thompson. 1999. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat. Neurosci. 2:44–49. [DOI] [PubMed] [Google Scholar]
- McMahon, H.T., and D.G. Nicholls. 1990. Glutamine and aspartate loading of synaptosomes: a reevaluation of effects on calcium-dependent excitatory amino acid release. J. Neurochem. 54:373–380. [DOI] [PubMed] [Google Scholar]
- Mrak, R.E., J.G. Sheng, and W.S. Griffin. 1996. Correlation of astrocytic S100 beta expression with dystrophic neurites in amyloid plaques of Alzheimer's disease. J. Neuropathol. Exp. Neurol. 55:273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, D.W. 1999. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 6:1028–1042. [DOI] [PubMed] [Google Scholar]
- Osen-Sand, A., J.K. Staple, E. Naldi, G. Schiavo, O. Rossetto, S. Petitpierre, A. Malgaroli, C. Montecucco, and S. Catsicas. 1996. Common and distinct fusion proteins in axonal growth and transmitter release. J. Comp. Neurol. 367:222–234. [DOI] [PubMed] [Google Scholar]
- Perfettini, J.L., and G. Kroemer. 2003. Caspase activation is not death. Nat. Immunol. 4:308–310. [DOI] [PubMed] [Google Scholar]
- Potts, P.R., S. Singh, M. Knezek, C.B. Thompson, and M. Deshmukh. 2003. Critical function of endogenous XIAP in regulating caspase activation during sympathetic neuronal apoptosis. J. Cell Biol. 163:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff, M.C., A.V. Whitmore, and J.T. Finn. 2002. Axonal self-destruction and neurodegeneration. Science. 296:868–871. [DOI] [PubMed] [Google Scholar]
- Schaumburg, H.H., H.M. Wisniewski, and P.S. Spencer. 1974. Ultrastructural studies of the dying-back process. I. Peripheral nerve terminal and axon degeneration in systemic acrylamide intoxication. J. Neuropathol. Exp. Neurol. 33:260–284. [DOI] [PubMed] [Google Scholar]
- Schiavo, G., F. Benfenati, B. Poulain, O. Rossetto, P. Polverino de Laureto, B.R. DasGupta, and C. Montecucco. 1992. a. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 359:832-5. [DOI] [PubMed] [Google Scholar]
- Schiavo, G., B. Poulain, O. Rossetto, F. Benfenati, L. Tauc, and C. Montecucco. 1992. b. Tetanus toxin is a zinc protein and its inhibition of neurotransmitter release and protease activity depend on zinc. EMBO J. 11:3577–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo, G., C.C. Shone, M.K. Bennett, R.H. Scheller, and C. Montecucco. 1995. Botulinum neurotoxin type C cleaves a single Lys-Ala bond within the carboxyl-terminal region of syntaxins. J. Biol. Chem. 270:10566–10570. [DOI] [PubMed] [Google Scholar]
- Schiavo, G., M. Matteoli, and C. Montecucco. 2000. Neurotoxins affecting neuroexocytosis. Physiol. Rev. 80:717–766. [DOI] [PubMed] [Google Scholar]
- Stamer, K., R. Vogel, E. Thies, E. Mandelkow, and E.M. Mandelkow. 2002. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156:1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze, C.I., J.C. Troncoso, C. Kawas, P. Mouton, D.L. Price, and L.J. Martin. 1997. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J. Neuropathol. Exp. Neurol. 56:933–944. [DOI] [PubMed] [Google Scholar]
- Teng, F.Y., Y. Wang, and B.L. Tang. 2001. The syntaxins. Genome Biol. 2:reviews3012.1–3012.7. [DOI] [PMC free article] [PubMed]
- Thornberry, N.A. 1994. Interleukin-1 beta converting enzyme. Methods Enzymol. 244:615–631. [DOI] [PubMed] [Google Scholar]
- Verderio, C., S. Coco, O. Rossetto, C. Montecucco, and M. Matteoli. 1999. Internalization and proteolytic action of botulinum toxins in CNS neurons and astrocytes. J. Neurochem. 73:372–379. [DOI] [PubMed] [Google Scholar]
- Vickers, J.C., D. Chin, A.M. Edwards, V. Sampson, C. Harper, and J. Morrison. 1996. Dystrophic neurite formation associated with age-related beta amyloid deposition in the neocortex: clues to the genesis of neurofibrillary pathology. Exp. Neurol. 141:1–11. [DOI] [PubMed] [Google Scholar]
- Waller, A. 1850. Experiments on the section of glossopharyngeal and hypoglossal nerves of the frog and observations of the alternative produced therapy in the structure of their primitive fibers. Philos. Trans. R. Soc. London. 140:423–429. [Google Scholar]
- Walsh, M.K., and J.W. Lichtman. 2003. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 37:67–73. [DOI] [PubMed] [Google Scholar]
- Wang, L.H., C.G. Besirli, and E.M. Johnson Jr. 2004. Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu. Rev. Pharmacol. Toxicol. 44:451–474. [DOI] [PubMed] [Google Scholar]
- Watts, R.J., E.D. Hoopfer, and L. Luo. 2003. Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron. 38:871–885. [DOI] [PubMed] [Google Scholar]
- Williamson, L.C., and E.A. Neale. 1998. Syntaxin and 25-kDa synaptosomal-associated protein: differential effects of botulinum neurotoxins C1 and A on neuronal survival. J. Neurosci. Res. 52:569–583. [DOI] [PubMed] [Google Scholar]
- Wyllie, A.H. 1980. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 284:555–556. [DOI] [PubMed] [Google Scholar]
- Yamamoto, A., J.J. Lucas, and R. Hen. 2000. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 101:57–66. [DOI] [PubMed] [Google Scholar]
- Zhai, Q., J. Wang, A. Kim, Q. Liu, R. Watts, E. Hoopfer, T. Mitchison, L. Luo, and Z. He. 2003. Involvement of the ubiquitin-proteasome system in the early stages of Wallerian degeneration. Neuron. 39:217–225. [DOI] [PubMed] [Google Scholar]