

Abstract
A glutamic acid deletion (ΔE) in the AAA+ protein torsinA causes DYT1 dystonia. Although the majority of torsinA resides within the endoplasmic reticulum (ER), torsinA binds a substrate in the lumen of the nuclear envelope (NE), and the ΔE mutation enhances this interaction. Using a novel cell-based screen, we identify lamina-associated polypeptide 1 (LAP1) as a torsinA-interacting protein. LAP1 may be a torsinA substrate, as expression of the isolated lumenal domain of LAP1 inhibits the NE localization of “substrate trap” EQ-torsinA and EQ-torsinA coimmunoprecipitates with LAP1 to a greater extent than wild-type torsinA. Furthermore, we identify a novel transmembrane protein, lumenal domain like LAP1 (LULL1), which also appears to interact with torsinA. Interestingly, LULL1 resides in the main ER. Consequently, torsinA interacts directly or indirectly with a novel class of transmembrane proteins that are localized in different subdomains of the ER system, either or both of which may play a role in the pathogenesis of DYT1 dystonia.
Introduction
DYT1 dystonia is an autosomal dominant childhood-onset neurological disease characterized by prolonged involuntary twisting movements that reflect neuronal dysfunction rather than neurodegeneration (Fahn et al., 1987; Berardelli et al., 1998). The mechanism by which the pathogenic mutation in the AAA+ protein torsinA produces DYT1 dystonia is unknown (Ozelius et al., 1997). Because AAA+ proteins are chaperones that alter the conformation of substrates, the identity of substrate determines the biological pathway modulated by AAA+ protein function (Vale, 2000). For example, the role of the AAA protein NSF in neuronal function is best appreciated when one considers that it acts upon SNARE complexes.
TorsinA resides in the ER lumen, but several observations indicate that it interacts with a nuclear envelope (NE) substrate (for review see Gerace, 2004). In addition, disease-associated ΔE-torsinA accumulates abnormally in the NE, suggesting that NE dysfunction may contribute to disease pathogenesis (Goodchild and Dauer, 2004). Consequently, identifying a NE substrate of torsinA is likely to further our understanding of the molecular pathogenesis of DYT1 dystonia. Because torsinA is expected to alter the conformation of a NE lumenal protein, characterizing this interaction may also provide insight into the functional organization of the NE and the poorly understood roles of NE resident proteins and their associated genetic diseases.
Results and discussion
We have previously shown that, although wild-type (WT) torsinA is predominantly localized in the main ER, pathogenic ΔE-torsinA and a predicted “substrate trap” ATP hydrolysis-deficient EQ-torsinA concentrate in the NE (Fig. 1 A; Vale, 2000; Goodchild and Dauer, 2004). NE resident proteins typically concentrate in the nuclear membrane through a selective retention mechanism mediated by binding to the nuclear lamina (Burke and Stewart, 2002). Consequently, NE proteins are less mobile in the NE than in the ER membrane (Ellenberg et al., 1997). If torsinA interacts with a NE protein, it should therefore display similarly reduced mobility in the NE. We tested this concept by examining the mobility of torsinA using FRAP analysis of BHK21 cells transiently overexpressing GFPWT-, GFPΔE-, and GFPEQ-torsinA. At moderate expression levels, both GFPΔE- and GFPEQ-torsinA selectively localize in the NE (Fig. 1 B); these cells were used for NE FRAP measurements. Cells expressing higher levels of these proteins also contain fluorescence in the main ER (Fig. 1 D), allowing us to perform ER FRAP measurements. In the ER, all three forms of GFP-torsinA displayed a similar time course of fluorescence recovery (∼65% after 210 s; Fig. 1 E). In contrast, the NE fluorescence recovery of GFPΔE- and GFPEQ-torsinA was markedly slower than GFPWT-torsinA (Fig. 1 C). In the NE, only 50% of GFPΔE-torsinA and 40% of GFPEQ-torsinA fluorescence recovered within 330 s (Fig. 1 C), at which time 75% of GFPWT-torsinA fluorescence had returned. However, it is possible that contaminating fluorescence from ER GFPWT-torsinA may contribute to an overestimate of NE GFPWT-torsinA recovery.
Figure 1.
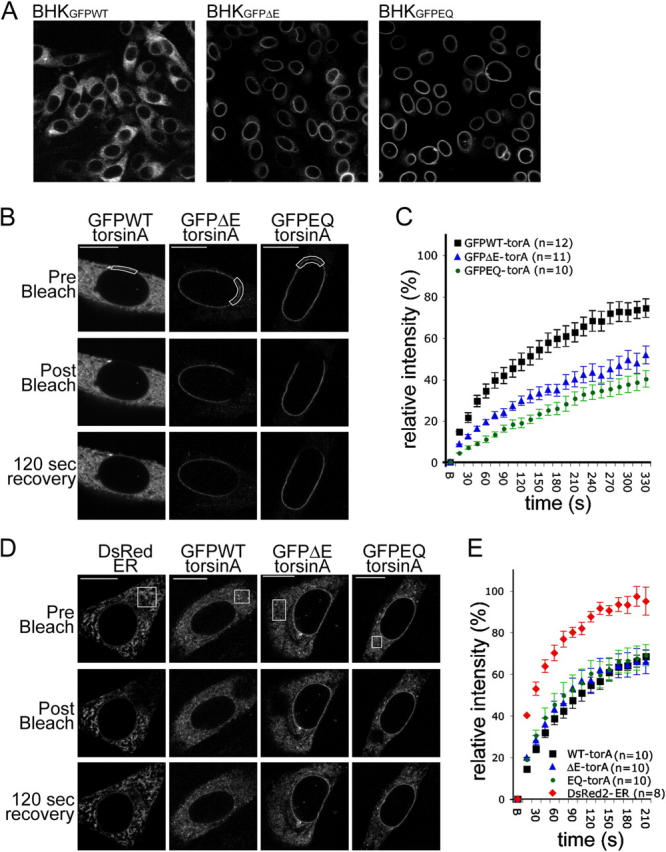
Pathogenic and substrate trap forms of torsinA display reduced mobility in the NE. (A) GFP immunolabeling of BHKGFPWT, BHKGFPΔE, and BHKGFPEQ stable cell lines. (B and D) GFP fluorescence of BHK21 cells transiently transfected with GFPWT-, GFPΔE-, or GFPEQ-torsinA and DsRed fluorescence of control cells transfected with DsRed2-ER (CLONTECH Laboratories, Inc.). Images show representative cells immediately before (top), immediately after (middle), and 120 s after (bottom) bleaching a ROI (boxed areas) in the NE (B) or ER (D). Bars, 10 μm. (C and E) Relative fluorescence intensity in the ROI as a function of time after photobleaching at time point “B” (B, bleach; see Materials and methods). Points show mean values and SEM.
The rate of GFPEQ-torsinA FRAP is slower than that of some well characterized transmembrane NE proteins (such as emerin), but is comparable to others (Ellenberg et al., 1997; Östlund et al., 1999; Daigle et al., 2001; Shimi et al., 2004). Because torsinA is restricted to the ER lumen/perinuclear space, it cannot bind to nuclear lamins. Therefore, these findings are consistent with the hypothesis that the NE accumulation of ΔE-torsinA is caused by an abnormal interaction with an immobilized transmembrane substrate. The rate of GFPΔE- and GFPEQ-torsinA fluorescence recovery is likely to be a function of (a) the degree to which its NE binding partner is immobilized and (b) the rate at which torsinA cycles on and off this partner. A higher rate of cycling might explain the faster recovery of GFPΔE-torsinA compared with GFPEQ-torsinA.
Lamina-associated polypeptide 1 (LAP1) is a torsinA binding protein
Based on the behavior of WT and mutant torsinA, we next sought to identify a torsinA NE binding partner. We developed a screening procedure based on the assumption that overexpressing a NE-localized torsinA substrate would increase the amount of torsinA in the NE, which is normally quite low. We selected candidate proteins that normally reside in the NE and contain a predicted lumenal domain that is conserved between mammalian species because these features indicate a potential functional role within the NE lumen. Cells stably expressing GFPWT-torsinA (BHKGFPWT; Fig. 1 A) were transfected with 18 candidate protein cDNAs in a reporter plasmid that coexpresses β-galactosidase (Table I and Fig. 2 A). Of all tested NE candidate proteins, only LAP1 recruited GFPWT-torsinA to the NE in a uniform perinuclear distribution reminiscent of substrate trap GFPEQ-torsinA (Table I and Fig. 2 A; compare transfected and untransfected cells). Occasionally, cells expressing high levels of lamin B receptor, LUMA, and Sun2 contained bright puncta of GFPWT-torsinA. These puncta were considered to be a nonspecific effect of gross overexpression because they were randomly located in the NE and ER. We further examined the LAP1 recruitment of GFPWT-torsinA by expressing myc-tagged LAP1 (myc-LAP1) in BHKGFPWT cells. As expected, cells expressing myc-LAP1 concentrated GFPWT-torsinA in the NE (Fig. 2 B), whereas the unrelated ER chaperone, protein disulphide isomerase (PDI), was unaltered (Fig. 2 C).
Table I. Effect of NE candidate proteins on GFPWT-torsinA.
| Candidate protein | Change in GFPWT-torsinA distribution |
|---|---|
| Emerin | None |
| gp210 | None |
| LAP1 | Increased NE labeling |
| LAP2β | None |
| Lamin B receptor | Puncta in ER/NE |
| LUMA | Puncta in ER/NE |
| MAN1 | None |
| Nesprin-1 | None |
| Nurim | None |
| NET3 | None |
| NET4 | None |
| NET26 | None |
| NET31 | None |
| NET39 | None |
| NET51 | None |
| Sun1 (Unc84A homologue) | None |
| Sun2 (Unc84B homologue) | Puncta in ER/NE |
| UNCL (Unc50 homologue) | None |
BHKGFPWT cells were transfected with NE resident proteins and transfected cells were identified by colabeling with antibodies against reporter proteins (see Fig. 2 A). The nuclear envelope transmembrane proteins (NET) are described by Schirmer et al. (2003) and UNCL by Fitzgerald et al. (2000). See Burke and Stewart (2002) for descriptions of other candidate proteins.
Figure 2.
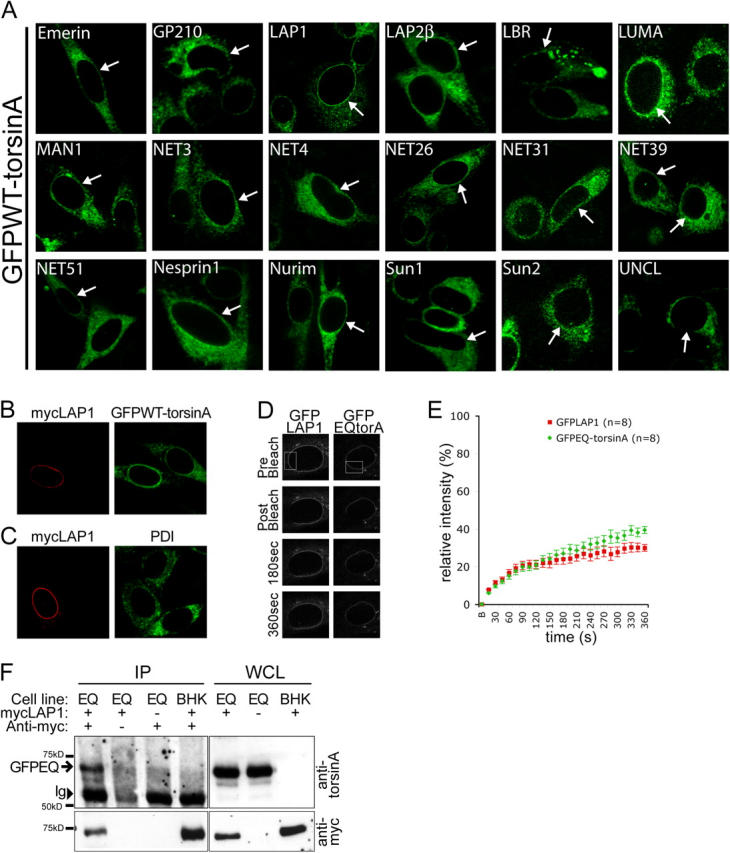
LAP1 recruits torsinA to the NE. (A) GFPWT-torsinA in BHKGFPWT cells transfected with different NE proteins. Arrows show transfected GFPWT-torsinA cells expressing candidate NE proteins, which were identified by colabeling for myc or β-galactosidase reporters (see online supplemental material for more details). (B) Immunolabeling of myc-LAP1–transfected BHKGFPWT cells with anti-myc and anti-GFP. (C) Immunolabeling of myc-LAP1–transfected BHK21 cells with anti-myc and anti-PDI. (D) GFP fluorescence of HeLa cells transiently transfected with GFPEQ-torsinA and GFP-LAP1 immediately before (prebleach), immediately after (postbleach), and at 180 and 360 s after photobleaching in the ROI (boxed area). (E) Relative fluorescence intensity in the ROI as a function of time after photobleaching. FRAP analysis was performed as described in Materials and methods. Points represent mean and SEM. (F) Coimmunoprecipitation of GFPEQ-torsinA with myc-LAP1. Immunoprecipitations with anti-myc antibody were performed with whole cell lysates (WCL) of BHKGFPEQ cells (Cell line: EQ) or BHK21 cells (Cell line: BHK) transfected with myc-LAP1. Immunoblots of WCL and immunoprecipitated proteins were probed with anti-torsinA (top panel) and anti-myc (bottom panel). GFPEQ-torsinA coimmunoprecipitates with myc-LAP1 from transfected BHKGFPEQ cells (position of GFPEQ-torsinA indicated by arrow) but not in the absence of anti-myc antibody (second lane), with mock-transfected BHKGFPEQ cells (third lane), or with myc-LAP1–transfected BHK21 cells (fourth lane). The position of immunoglobulin heavy chains is indicated (Ig, arrowhead).
To further assess LAP1 as a torsinA NE binding partner, we used FRAP to compare the mobility of GFP-LAP1 with GFPEQ-torsinA. We hypothesized that the FRAP of a torsinA binding partner should be equal to or less than that of substrate trap EQ-torsinA. Consistent with this notion, the rates of GFP-LAP1 and GFPEQ-torsinA NE FRAP are strikingly similar in the initial recovery period (Fig. 2 E). In later stages, GFP-LAP1 FRAP plateaus at ∼30% recovery, whereas GFPEQ-torsinA FRAP steadily increases, likely because AAA proteins containing the Walker B box E/Q mutation typically retain a low level of residual ATP hydrolysis activity (Whiteheart et al., 1994). Therefore, the comparative FRAP rates of GFPEQ-torsinA and GFP-LAP1 are consistent with LAP1 being a NE binding partner of torsinA. We also tested whether or not GFPEQ-torsinA coimmunoprecipitates with myc-LAP1. Anti-myc immunoprecipitations from lysates of BHKGFPEQ cells transfected with myc-LAP1 demonstrated that GFPEQ-torsinA coimmunoprecipitates with myc-LAP1 (Fig. 2 F), further supporting the idea that LAP1 and torsinA interact in the NE.
Next, we examined whether or not the lumenal domain of LAP1 is responsible for its interaction with torsinA, as predicted by our model. To explore this question, we tested if the isolated lumenal domain of LAP1 is capable of altering the perinuclear subcellular distribution of EQ-torsinA. We generated myc-tagged constructs containing the LAP1 lumenal domain with (myc-210LAP1) or without (myc-240LAP1) the transmembrane domain (Fig. 3 A; Kondo et al., 2002). As expected, these fragments fail to concentrate in the NE and instead localize in the main ER (Fig. 3 B, left). Expression of either LAP1 lumenal fragment produced a clear redistribution of GFPEQ-torsinA from the NE to the ER (Fig. 3 B). Myc-210LAP1 causes a similar redistribution of disease-associated GFPΔE-torsinA (Fig. 3 C), and in all instances we observed strong colocalization between labeling for GFP and myc (Fig. 3, B and C). The effect of the LAP1 lumenal domain was specific, as the lumenal domain of the nucleoporin gp210 (Wozniak and Blobel, 1992) did not alter the subcellular distribution of GFPEQ-torsinA (Fig. 3 B, bottom).
Figure 3.

TorsinA interacts with the conserved lumenal domain of LAP1. (A) Schematic illustration of LAP1 protein structure and the deletion mutants used in this study. (B) Immunofluorescent labeling of transfected BHKGFPEQ cells with anti-GFP and anti-myc antibodies. GFPEQ-torsinA is displaced by either LAP1 lumenal fragment but not by the lumenal fragment of gp210. (C) Immunofluorescent labeling of BHKGFPΔE cells transfected with myc-tagged 210LAP1 with anti-GFP and anti-myc antibodies.
These data indicate that LAP1 may be a NE-localized torsinA substrate. LAP1 was originally identified as the antigen recognized by a monoclonal antibody generated against purified rat liver nuclear envelopes (RL13). Three RL13 immunoreactive NE proteins were designated LAP1A, B, and C (with molecular masses of 75, 68, and 55 kD, respectively; Senior and Gerace, 1988). A single exon encodes the entire transmembrane and lumenal domains of LAP1 in rat, mouse, and human, suggesting that LAP1 isoforms vary only in their nucleoplasmic portion. Interestingly, the lumenal domain of human LAP1 is 86% identical to mouse LAP1, whereas the nucleoplasmic domains exhibit only 46% sequence identity. This comparison suggests that torsinA interacts with a domain of LAP1 that has a conserved role in the lumen of the NE.
Lumenal domain like LAP1 (LULL1) is a novel ER-localized LAP1 homologue
Because the LAP1 lumenal domain appears to be a torsinA-interacting motif, we searched for other proteins containing this domain by performing a BLAST search of the NCBI database. This search identified a single novel human cDNA (GenBank/EMBL/DDBJ accession no. NM_145034) encoding a protein with a lumenal domain like LAP1, which we named LULL1 (Fig. 4, A and B). The LULL1 gene encodes a protein containing a single predicted transmembrane domain and appears to have arisen from a gene duplication event because it is located adjacent to the LAP1 gene on human chromosome 1q24. cDNA clones also exist for rat and mouse forms of LULL1, and the LAP1 and LULL1 genes are also adjacent within these genomes. In contrast to the conserved lumenal domains of LAP1 and LULL1, there is significant divergence between the NH2-terminal regions of these proteins that extend outside of the secretory pathway (Fig. 4, A and B).
Figure 4.

LULL1 is an ER resident protein with homology to LAP1. (A) The percentage of amino acid sequence identity of predicted nucleoplasmic (LAP1), cytoplasmic (LULL1), and transmembrane and lumenal portions of human LULL1 and LAP1. (B) CLUSTAL W alignment of human LAP1 and LULL1 amino acid sequences. Asterisk indicates position with identical amino acid residues, colon indicates conserved amino acid residues, and period indicates semi-conserved amino acid residues. Predicted membrane spanning domains (determined with TMPred) are shaded and a conserved potential N-linked glycosylation site is boxed. (C) BHK21 cells transfected with myc-LULL1 were lysed in buffer with or without 1% Triton X-100 and centrifuged to separate lysates into soluble (S) and insoluble (P) fractions. Immunoblots of equal amounts of soluble and insoluble fractions were probed with anti-myc antibodies. (D) BHK21 cells transfected with myc-LULL1 and LULL1-myc were labeled with anti-myc and anti-PDI. (E) Immunoblotting of lysates from BHK21 cells transfected with myc-LAP1 or myc-LULL1 digested with PNGase F or endoglycosidase H and probed with anti-myc antibody.
To explore whether or not LULL1 interacts with torsinA, we isolated a human cDNA that matched the sequence of NM_145034. Transient transfection of BHK21 cells with myc-tagged LULL1 generates a protein of ∼75 kD that is insoluble in the absence of detergent but is solubilized by 1% Triton X-100, suggesting the presence of a membrane spanning domain (Fig. 4 C). When NH2- (myc-LULL1) or COOH-terminal (LULL1-myc)–tagged LULL1 were transfected into BHK21 or HeLa cells, they colocalized with PDI (Fig. 4 D and not depicted for HeLa cells), including in low expressing cells. Like torsinA, both LAP1 and LULL1 proteins are PNGaseF- and endoglycosidase H–sensitive glycoproteins, indicating that they are retained within the ER system (Fig. 4 E).
We transfected myc-LULL1 into BHKGFPEQ cells to determine if this ER-localized LAP1 homologue also interacts with torsinA. Consistent with this notion, myc-LULL1 produced a clear redistribution of GFPEQ-torsinA from the NE to the ER and there was strong colocalization between GFP and myc labeling in transfected cells (Fig. 5 A). We obtained similar results with a LULL1 fragment containing only the transmembrane and lumenal domains (208LULL1; Fig. 5 A), confirming that this domain is responsible for the effects observed with full-length LULL1. In addition, GFPEQ-torsinA coimmunoprecipitates with myc-LULL1 from lysates of myc-LULL1–transfected BHKGFPEQ cells (Fig. 5 B). Together, these results suggest that LULL1 interacts with torsinA in the main ER. Like torsinA, LAP1 and LULL1 mRNAs are widely expressed in both neural and nonneural tissue (Fig. 5 C), which is consistent with the hypothesis that these proteins may be physiologically relevant interactors of torsinA.
Figure 5.

TorsinA interacts with LULL1. (A) TorsinA interacts with the conserved lumenal domain of LULL1. Immunofluorescent labeling of transfected BHKGFPEQ cells with anti-GFP and anti-myc antibodies. Full-length LULL1 (top) and the LULL1 lumenal domain (bottom) recruit GFPEQ-torsinA to the ER. (B) TorsinA coimmunoprecipitates with myc-LULL1. Immunoprecipitations and immunoblotting were performed as in Fig. 2 F except that transfections were performed with myc-LULL1. Immunoglobulin heavy chains were not visible with the exposure time needed to visualize GFPEQ-torsinA. (C) RT-PCR of mouse LAP1 and LULL1 from whole tissue RNA. (D) Rabbit polyclonal antibodies against LAP1, LULL1, and torsinA similarly detect their respective antigens. BHK21 cells were transfected with myc-tagged mouse forms of LAP1, LULL1, and torsinA; and WCL was probed with anti-myc to confirm that similar amounts of transfected protein were loaded (top panel). Immunoblots were subsequently probed (bottom panel) with anti-LAP1, anti-LULL1, and anti-torsinA at concentrations that generated similar levels of immunoreactivity. Comparative images are from a simultaneous exposure of a single immunoblot. (E) Immunoblots of NIH-3T3 WCL probed with rabbit polyclonal antibodies. 15 μg of 1% SDS NIH-3T3 WCL were probed with rabbit polyclonal antibodies at the concentrations used in D. Images are from a simultaneous 2-s exposure of a single immunoblot. (F) NIH-3T3 cells transfected with GFPWT-torsinA (left) or GFPEQ-torsinA (right) and labeled with anti-GFP. (G) LAP1 and LULL1 interact more strongly with substrate trap EQ-torsinA. WCL were prepared from BHKGFPWT cells (Cell line: WT) and BHKGFPEQ cells (Cell line: EQ) transfected with myc-LAP1 (Tfct: LAP1) or myc-LULL1 (Tfct: LULL1). Proteins were immunoprecipitated from WCL with anti-myc antibody, eluted from protein G agarose beads and immunoblotted. Parallel control precipitations were performed in the absence of anti-myc antibody. Immunoblots of immunoprecipitated proteins and 2% of WCL were probed with anti-torsinA and anti-myc. WCL from BHKGFPWT cells contained more GFP-torsinA, myc-LAP1, and myc-LULL1 than BHKGFPEQ cells because this was necessary to visualize coprecipitated GFPWT-torsinA. The position of the 65-kD GFP-torsinA is indicated by an arrow (top). Neither myc-LAP1, myc-LULL1, or GFP-torsinA proteins were immunoprecipitated in the absence of anti-myc antibody.
Next, we sought to understand why, if torsinA interactors exist in both the NE and ER, substrate trap EQ-torsinA appears to localize exclusively to the NE. One important technical consideration is that the much smaller volume of the NE, compared with the ER, makes torsinA far easier to detect in the NE when subcellular localization is assessed by fluorescence microscopy. In addition, the relative steady-state levels of torsinA and its interactors will influence the subcellular localization of torsinA. To assess the relative steady-state levels of these proteins, we used rabbit polyclonal antibodies raised against the mouse forms of LAP1, LULL1, or torsinA that similarly detect their respective antigens (Fig. 5 D). In NIH-3T3 lysate, these antibodies recognize proteins of the appropriate molecular masses, including the three previously described isoforms of LAP1 (Fig. 5 E; Senior and Gerace, 1988). The relative intensity of anti-LAP1 and anti-LULL1 immunoreactivity suggests that there is significantly more LAP1 than LULL1 in NIH-3T3 fibroblasts (Fig. 5 E). Thus, GFPEQ-torsinA may localize to the NE in these cells (Fig. 5 F) because there is far more NE binding partner (LAP1).
To examine whether or not LAP1 and LULL1 may be torsinA substrates, we compared the interaction of these proteins with WT- and EQ-torsinA. Because AAA+ proteins typically form high affinity interactions with substrate when bound to ATP (Vale, 2000), substrates of torsinA will bind more tightly to EQ-torsinA than the WT protein. We tested if this was the case for LAP1 and LULL1 by performing immunoprecipitations on lysates from LAP1- or LULL1-transfected BHKGFPWT and BHKGFPEQ cell lines. GFPEQ-torsinA readily immunoprecipitated with either LAP1 or LULL1. However, to detect the association of GFPWT-torsinA with LAP1 or LULL1, it was necessary to perform immunoprecipitations from a much greater amount of protein lysate (Fig. 5 G, WCL). These data suggest strongly, but do not prove, that LAP1 and LULL1 are substrates of torsinA; an adaptor protein could mediate the interaction between torsinA and LAP1 or LULL1.
Several lines of evidence indicate that torsinA has a role in the NE (Gerace, 2004). We demonstrate that this function of torsinA may involve an interaction with LAP1, and that the behavior of LAP1 is consistent with that of a torsinA substrate (i.e., it is more tightly associated with EQ- than WT-torsinA). Although the functional role of LAP1 is poorly understood, it is known to bind A- and B-type lamins (Senior and Gerace, 1988; Foisner and Gerace, 1993; Martin et al., 1995). This suggests that alterations in torsinA function may affect the nuclear lamina, raising the possibility that DYT1 dystonia shares molecular abnormalities with diseases that result from laminA mutations (Burke and Stewart, 2002; De Sandre-Giovannoli et al., 2003). The fact that alterations in both lamin A and torsinA function lead to NE morphologic abnormalities is consistent with this notion (Sullivan et al., 1999; Naismith et al., 2004).
We also identify a novel ER protein, LULL1, that interacts with torsinA through a region conserved with the LAP1 lumenal domain; this protein also behaves like a torsinA substrate. The striking homology between the LAP1 and LULL1 lumenal domains suggests that they are similarly modified by the AAA+ chaperone activity of torsinA. In addition, LAP1 and LULL1 share other features. They both contain a single membrane-spanning domain and their nucleoplasmic (LAP1) and cytoplasmic (LULL1) regions are similarly sized. Consequently, these proteins may be engaged in similar roles in the NE and ER and contribute to a biological process that is common to both compartments. In light of the mechanism of AAA+ protein function, our data suggest that alterations in LAP1 or LULL1 activity may therefore participate in the pathogenesis of DYT1 dystonia.
Materials and methods
Cell culture
BHK21, NIH-3T3, and HeLa cell lines were cultured using standard conditions (American Type Culture Collection). The generation and characterization of BHKGFPWT, BHKGFPΔE, and BHKGFPEQ cells has been described previously (Goodchild and Dauer, 2004). All cell transfections were performed using Lipofectamine Plus (Invitrogen) according to the manufacturer's instructions.
FRAP
The day after transfection, cells were trypsinized and replated at 10–20% confluence in collagen-coated chambered coverglasses (LabTekII) in Dulbecco's minimum essential medium media containing 1% FBS. Immediately before imaging, this media was replaced with media containing 10 mM Hepes buffer, pH 7.5. Imaging and photobleaching were performed using a Plan NEOFLUAR 100×/1.30 oil objective on an inverted confocal microscope (model LSM510 Meta; Carl Zeiss MicroImaging, Inc.). Cells transfected with GFP fusion proteins were imaged with 488-nm light, DsRed with 516-nm light, using 2% laser power and a pin hole of 1 airy unit. After two imaging scans, a selected area of the ER or NE (region of interest [ROI]) was bleached using maximal laser power for 20 iterations, and then the photobleached cell was imaged at 15-s intervals for 3–6 min. Collected images were analyzed in Adobe Photoshop to calculate the mean fluorescence intensity in the ROI as a function of time after photobleaching. To correct for whole cell photobleaching caused by the bleaching pulse and image capture, fluorescence intensity was also measured in an unbleached area (UA) at all time points and a fractional correction calculated as UAt=n/UAPB. ROI fluorescence intensity at each time point was corrected by these values and then normalized so that the prebleach fluorescence level equaled 100 and immediate post-bleach level was zero.
Immunolabeling
Immunofluorescence labeling was performed on cells 48 h after transfection using methanol-fixed cells grown on collagen-coated glass coverslips (Carolina Scientific). Coverslips were blocked for 1 h at RT in block solution (PBS, 0.25% Triton X-100 and 10% normal donkey serum), incubated overnight at 4°C in primary antibodies diluted in block solution. The next day coverslips were washed, incubated with secondary antibodies (diluted in block solution), and washed in PBS before mounting using Vectashield Mounting Media with DAPI (Vector Laboratories). In double labeling experiments, GFP was detected with FITC- and myc with Texas red–conjugated secondary antibodies to minimize the possibility of “bleed through.” Digital Images were acquired using a laser scanning confocal microscope (model LSM510 Meta; Carl Zeiss MicroImaging, Inc.). FITC and Texas red images were acquired successively and figures were prepared in Adobe Photoshop.
Antibodies
Antibodies used were as follows: affinity purified rabbit polyclonal anti-torsinA raised against residues 319–332 of mouse torsinA (a gift from B. Lauring, Columbia University, New York, NY), rabbit polyclonal anti-LAP1 raised against residues 463–478 of mouse LAP1 (Covance Research Products), and rabbit polyclonal anti-LULL1 raised against residues 107–120 of mouse LULL1 (Covance Research Products). Other antibodies were rabbit anti-GFP 1:1,000 (AbCam), mouse anti–β galactosidase 1:100 (Sigma-Aldrich), mouse anti-myc 1:500 (CLONTECH Laboratories, Inc.), rabbit anti-myc 1:100 (Sigma-Aldrich), and mouse anti-PDI 1:100 (StressGen Biotechnologies). All secondary antibodies were raised in donkey from Jackson ImmunoResearch Laboratories.
Immunoprecipitation
Immunoprecipitations were performed at 24 h after transfection using mouse monoclonal anti-myc (CLONTECH Laboratories, Inc.) and Agarose Protein G immunoprecipitation kit (Roche) according to the manufacturers' instructions, except that buffers contained only Igepal CA630.
LULL1 characterization
The solubility of LULL1 was investigated by lysing myc-tagged LULL1-transfected BHK21 cells in buffer (50 mM Tris-HCl, pH 7.5, and protease inhibitors) with and without 1% Triton X-100. Homogenates were incubated on ice for 20 min and centrifuged at 20,000 g for 15 min to separate supernatant and pellet fractions. Pellets were solubilized by heating to 95°C in 180 μl of 1× lamaeli sample buffer. Supernatants were also brought to a 180-μl volume and 1× concentration. Equal volumes of pellet and supernatant fractions were used in SDS-PAGE. The glycosylation state of myc-tagged LAP1 and LULL1 was examined by digesting lysates from transfected BHK21 cells with PNGaseF or Endoglycosidase H (New England Biolabs, Inc.) according to the manufacturer's instructions.
RT-PCR
Total RNA was prepared from mouse tissues using Trizol reagent (Invitrogen), and cDNA was generated from 1 μg of total RNA with oligo dT primers and SuperScript III (Invitrogen), all according to the manufacturer's instructions. Normalization of samples was performed by increasing or decreasing the amount of template cDNA dependent on amplification efficiency determined using GAPDH primers. Primers were designed to amplify cDNA regions that spanned at least one intron boundary, along with the lumenal domain of LAP1 and LULL1, to prevent amplification from genomic DNA.
Online supplemental material
Details of plasmid construction for the candidate cDNA screen, primer details, and the generation of LAP1 and LULL1 fusion and truncated constructs is contained in the online supplemental material. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200411026/DC1.
Acknowledgments
We thank Howard Worman for cDNA clones and helpful comments on the manuscript. We also thank Steve Sturley and Jon Graff for helpful comments on the manuscript, Brett Lauring for generously supplying torsinA antibody, and Jamie Twaite and Connie Kim for superb technical assistance.
This work was supported by the Dystonia Medical Research and Bachmann-Strauss Foundations, the March of Dimes, the Parkinson's Disease Foundation, and The National Institute of Neurological Disorders and Stroke (grant NS050528-01A1).
Abbreviations used in this paper: LAP1, lamina-associated polypeptide 1; LULL1, lumenal domain like LAP1; NE, nuclear envelope; PDI, protein disulphide isomerase; ROI, region of interest; WCL, whole cell lysate; WT, wild-type.
References
- Berardelli, A., J.C. Rothwell, M. Hallett, P.D. Thompson, M. Manfredi, and C.D. Marsden. 1998. The pathophysiology of primary dystonia. Brain. 121:1195–1212. [DOI] [PubMed] [Google Scholar]
- Burke, B., and C.L. Stewart. 2002. Life at the edge: the nuclear envelope and human disease. Nat. Rev. Mol. Cell Biol. 3:575–585. [DOI] [PubMed] [Google Scholar]
- Daigle, N., J. Beaudouin, L. Hartnell, G. Imreh, E. Hallberg, J. Lippincott-Schwartz, and J. Ellenberg. 2001. Nuclear pore complexes form immobile networks and have a very low turnover in live mammalian cells. J. Cell Biol. 154:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli, A., R. Bernard, P. Cau, C. Navarro, J. Amiel, I. Boccaccio, S. Lyonnet, C.L. Stewart, A. Munnich, M. Le Merrer, and N. Levy. 2003. Lamin a truncation in Hutchinson-Gilford progeria. Science. 300:2055. [DOI] [PubMed] [Google Scholar]
- Ellenberg, J., E.D. Siggia, J.E. Moreira, C.L. Smith, J.F. Presley, H.J. Worman, and J. Lippincott-Schwartz. 1997. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 138:1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn, S., C.D. Marsden, and D.B. Calne. 1987. Classification and investigation of dystonia. Movement Disorders 2. C.D. Marsden and S. Fahn, editors. Butterworths, London. 332–358.
- Fitzgerald, J., D. Kennedy, N. Viseshakul, B.N. Cohen, J. Mattick, J.F. Bateman, and J.R. Forsayeth. 2000. UNCL, the mammalian homologue of UNC-50, is an inner nuclear membrane RNA-binding protein. Brain Res. 877:110–123. [DOI] [PubMed] [Google Scholar]
- Foisner, R., and L. Gerace. 1993. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell. 73:1267–1279. [DOI] [PubMed] [Google Scholar]
- Gerace, L. 2004. TorsinA and torsion dystonia: Unraveling the architecture of the nuclear envelope. Proc. Natl. Acad. Sci. USA. 101:8839–8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild, R.E., and W.T. Dauer. 2004. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc. Natl. Acad. Sci. USA. 101:847–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, Y., J. Kondoh, D. Hayashi, T. Ban, M. Takagi, Y. Kamei, L. Tsuji, J. Kim, and Y. Yoneda. 2002. Molecular cloning of one isotype of human lamina-associated polypeptide 1s and a topological analysis using its deletion mutants. Biochem. Biophys. Res. Commun. 294:770–778. [DOI] [PubMed] [Google Scholar]
- Martin, L., C. Crimaudo, and L. Gerace. 1995. cDNA cloning and characterization of lamina-associated polypeptide 1C (LAP1C), an integral protein of the inner nuclear membrane. J. Biol. Chem. 270:8822–8828. [DOI] [PubMed] [Google Scholar]
- Naismith, T.V., J.E. Heuser, X.O. Breakefield, and P.I. Hanson. 2004. TorsinA in the nuclear envelope. Proc. Natl. Acad. Sci. USA. 101:7612–7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Östlund, C., J. Ellenberg, E. Hallberg, J. Lippincott-Schwartz, and H.J. Worman. 1999. Intracellular trafficking of emerin, the Emery-Dreifuss muscular dystrophy protein. J. Cell Sci. 112:1709–1719. [DOI] [PubMed] [Google Scholar]
- Ozelius, L.J., J.W. Hewett, C.E. Page, S.B. Bressman, P.L. Kramer, C. Shalish, D. de Leon, M.F. Brin, D. Raymond, D.P. Corey, et al. 1997. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 17:40–48. [DOI] [PubMed] [Google Scholar]
- Schirmer, E.C., L. Florens, T. Guan, J.R. Yates III, and L. Gerace. 2003. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science. 301:1380–1382. [DOI] [PubMed] [Google Scholar]
- Senior, A., and L. Gerace. 1988. Integral membrane proteins specific to the inner nuclear membrane and associated with the nuclear lamina. J. Cell Biol. 107:2029–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimi, T., T. Koujin, M. Segura-Totten, K.L. Wilson, T. Haraguchi, and Y. Hiraoka. 2004. Dynamic interaction between BAF and emerin revealed by FRAP, FLIP, and FRET analyses in living HeLa cells. J. Struct. Biol. 147:31–41. [DOI] [PubMed] [Google Scholar]
- Sullivan, T., D. Escalante-Alcalde, H. Bhatt, M. Anver, N. Bhat, K. Nagashima, C.L. Stewart, and B. Burke. 1999. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147:913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale, R.D. 2000. AAA proteins. Lords of the ring. J. Cell Biol. 150:F13–F19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteheart, S.W., K. Rossnagel, S.A. Buhrow, M. Brunner, R. Jaenicke, and J.E. Rothman. 1994. N-ethylmaleimide–sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J. Cell Biol. 126:945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak, R.W., and G. Blobel. 1992. The single transmembrane segment of gp210 is sufficient for sorting to the pore membrane domain of the nuclear envelope. J. Cell Biol. 119:1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]