

Abstract
Changes in O2 tension can significantly impact cell survival, yet the mechanisms underlying these effects are not well understood. Here, we report that maintaining sympathetic neurons under low O2 inhibits apoptosis caused by NGF deprivation. Low O2 exposure blocked cytochrome c release after NGF withdrawal, in part by suppressing the up-regulation of BIMEL. Forced BIMEL expression removed the block to cytochrome c release but did not prevent protection by low O2. Exposing neurons to low O2 also activated hypoxia-inducible factor (HIF) and expression of a stabilized form of HIF-1α (HIF-1αPP→AG) inhibited cell death in normoxic, NGF-deprived cells. Targeted deletion of HIF-1α partially suppressed the protective effect of low O2, whereas deletion of HIF-1α combined with forced BIMEL expression completely reversed the ability of low O2 to inhibit cell death. These data suggest a new model for how O2 tension can influence apoptotic events that underlie trophic factor deprivation–induced cell death.
Introduction
The availability of neurotrophic factors plays an important role in determining whether developing neurons live or die as they innervate their target tissues (Burek and Oppenheim, 1996). A well-characterized model for studying trophic factor dependency involves withdrawing NGF from dissociated sympathetic neurons maintained in cell culture (Martin et al., 1988; Deckwerth and Johnson, 1993; Edwards and Tolkovsky, 1994). NGF withdrawal activates the intrinsic apoptotic pathway characterized by release of cytochrome c from mitochondria and subsequent activation of caspases (Deshmukh and Johnson, 1998; Neame et al., 1998). Cytochrome c release is coordinated by members of the BCL-2 family, most notably BAX and BIMEL (Deckwerth et al., 1998; Putcha et al., 2001; Whitfield et al., 2001), which in turn are regulated by c-Jun NH2-terminal kinases (JNKs; Harris and Johnson, 2001; Whitfield et al., 2001; Lei and Davis, 2003; Putcha et al., 2003).
A number of reports have shown that reducing O2 to as low as 1% can increase cell survival in vitro (Yun et al., 1997). Nonetheless, cells maintained in culture are typically exposed to O2 levels that far exceed those measured in vivo. For example, whereas normal tissue O2 levels range from 1 to 5% in the adult mammalian brain (Erecinska and Silver, 2001), dissociated neurons are routinely maintained in an atmosphere of 5% CO2 and 95% air, equivalent to ∼20% O2. Although reducing O2 tension can impact a wide range of biochemical processes (Bickler and Donohoe, 2002), a particularly well-characterized response recently implicated in neuronal cell survival involves activation of the transcription factor hypoxia-inducible factor (HIF; Halterman et al., 1999; Zaman et al., 1999; Piret et al., 2002; Soucek et al., 2003).
There are three isoforms of HIF, each consisting of a common β subunit and a unique α subunit; of these, HIF-1α is the best characterized (Semenza, 1999). When O2 is not limiting, HIF-1α readily associates with the von Hippel Lindau protein, part of an E3 ubiquitin ligase, and is rapidly polyubiquitinated and degraded by the proteasome. The interaction between HIF-1α and von Hippel Lindau protein is mediated by the O2-dependent hydroxylation of two proline residues in HIF-1α (Ivan et al., 2001; Jaakkola et al., 2001; Yu et al., 2001). The enzymes that catalyze this reaction are members of the egg-laying nine (EGLN) family of prolyl hydroxylases (Bruick and McKnight, 2001; Epstein et al., 2001). In hypoxic cells, EGLN prolyl hydroxylase activity is reduced leading to stabilization of HIF-1α and transactivation of a large and diverse group of HIF-responsive genes (for review see Safran and Kaelin, 2003).
Here, we show that exposing sympathetic neurons to low O2 during NGF deprivation significantly reduces their rate of cell death. Induction of BIMEL and loss of mitochondrial cytochrome c were both suppressed in NGF-deprived neurons exposed to low O2. Forced expression of BIMEL restored cytochrome c release but did not reverse the protective effect of low O2, suggesting that additional mechanisms were important for inhibiting cell death. Results from several experiments implicated HIF as a potential mediator of the neuroprotective effect of low O2. This was confirmed by microinjection experiments combining targeted deletion of HIF-1α with ectopic expression of BIMEL in the same neurons. These results provide a new model for how O2 tension influences the apoptotic events that underlie trophic factor deprivation–induced death.
Results
Reducing O2 inhibits death caused by NGF deprivation
To assess whether reducing O2 tension affects the survival of sympathetic neurons, dissociated neurons that had been maintained in vitro for 5 d under standard culture conditions (i.e., 5% CO2 and 95% air, equal to 20% O2) were refed with fresh NGF-containing media or deprived of NGF and immediately placed in incubators equilibrated to 20%, 2%, or 1% O2. Survival was assessed after staining cells with the DNA-binding dye Hoechst 33,342 and examining neuronal nuclei for evidence of chromatin condensation. Exposing NGF-maintained neurons to 1–2% O2 for up to 4 d had no effect on their survival (unpublished data). On the other hand, low O2 exposure markedly increased the survival of NGF-deprived neurons, with the greatest effect seen at 1% O2 (Fig. 1, A and B). Under these conditions, 75% of neurons exhibited normal nuclear morphology as late as 48 h after NGF deprivation. Although slightly smaller, the cell soma also resembled those of NGF-maintained neurons, whereas the neuronal processes appeared less well protected. Based on these initial observations, we speculated that reducing O2 might interfere with certain aspects of the cell death pathway known to be activated during trophic factor deprivation.
Figure 1.
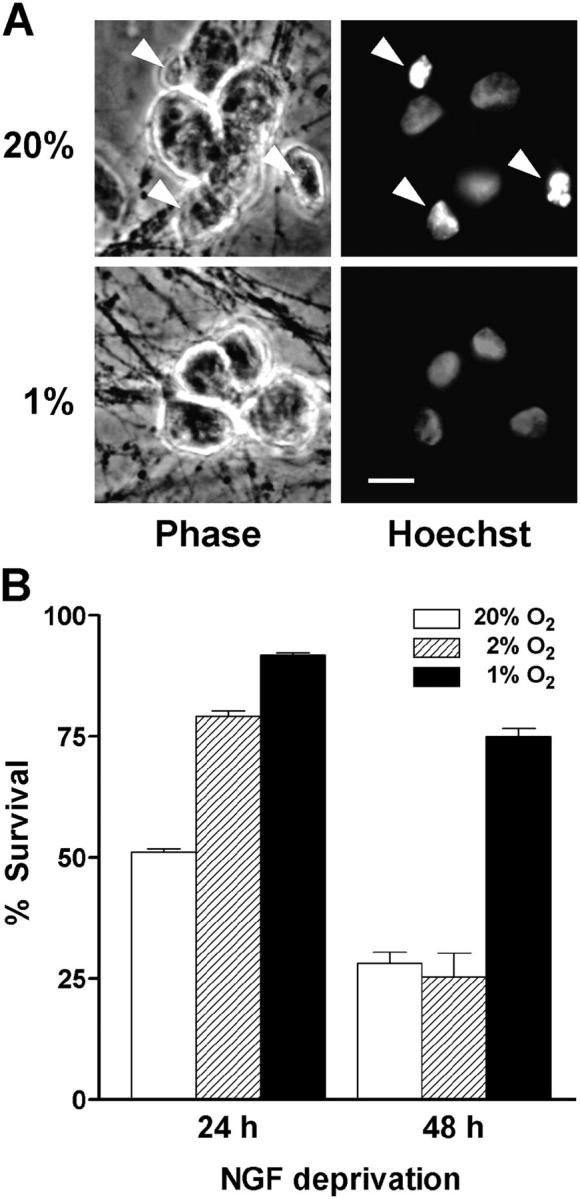
Low O 2 delays cell death caused by NGF deprivation. Sympathetic neurons were either deprived of NGF or refed with fresh NGF-containing media and immediately transferred to 20%, 2%, or 1% O2 incubators. At the end of the treatment, the cells were immediately placed in fixative and stained with Hoechst 33,342. (A) Phase-contrast and epifluorescence images show a single field of view for each condition. Cultures were exposed to either 20% or 1% O2 and deprived of NGF for 24 h. Note the presence of several apoptotic nuclei in cultures exposed to 20% O2 (arrowheads) but not in those at 1% O2. Bar, 15 μm. (B) Cells were scored for viability as described in Materials and methods. Results represent the percentage of cells having uniformly stained chromatin, phase-bright cell bodies, and clearly discernible nuclear membranes (mean ± SEM, n = 3).
Low O2 inhibits release of cytochrome c from mitochondria
Using immunofluorescence, we investigated the effect of low O2 on the localization of cytochrome c after NGF withdrawal. Neurons maintained with NGF at 20% O2 have a punctate, cytoplasmic distribution of cytochrome c immunofluorescence, indicative of its mitochondrial localization (Fig. 2, A and B). As seen by others (Deshmukh and Johnson, 1998; Neame et al., 1998), withdrawing NGF from these neurons results in an almost complete loss of punctate cytochrome c immunofluorescence. In contrast, the majority of neurons exposed to 1% O2 during NGF deprivation retained punctate cytochrome c immunofluorescence similar to NGF-maintained neurons.
Figure 2.

Low O 2 inhibits the loss of cytochrome c from mitochondria. Neurons were deprived of NGF or refed with fresh NGF-containing media and cultured at 20% or 1% O2 for 24 h. (A and B) Immunofluorescence was performed using an anti–cytochrome c antibody and cells were counterstained with Hoechst 33,342. Phase-contrast and epifluorescence images show a single field of view for each condition. Panels 1–4, 20% O2; panels 5–8, 1% O2. Bar, 20 μm. The percentage of cells with punctate cytochrome c (mean ± SEM) was determined in three independent experiments. (C) Whole cell lysates from neurons treated as described above were separated by 15% SDS-PAGE and immunoblotted with anti–cytochrome c antibody. NS represents a nonspecific protein detected by the primary antibody (similar levels of this protein confirm equal loading of the gel). NGF deprivation resulted in a 73 ± 9% decrease in cytochrome c levels in neurons exposed to 20% O2 compared with a 30 ± 3% decrease in neurons exposed to 1% O2 (percent decreases are the mean ± SEM from three independent experiments).
We also examined the effect of low O2 and NGF withdrawal on cytochrome c protein levels detected by immunoblotting whole cell lysates. In a previous study, immunoblots of mitochondrial and cytosolic fractions prepared from NGF-deprived neurons revealed the expected decrease in mitochondrial cytochrome c, but not an accumulation of cytochrome c in the cytosolic fractions (Putcha et al., 2000). Consequently, immunoblotting cytochrome c from whole cell lysates yielded the same results as analyzing subcellular fractions, a procedure that requires obtaining neurons from many more animals. As shown in Fig. 2 C, total cytochrome c levels decreased in neurons deprived of NGF and exposed to 20% O2. However, there was no change in the level of cytochrome c in the NGF-deprived cells exposed to 1% O2. Thus, these data together with the immunofluorescence results described above indicate that the neuroprotective effect of reduced O2 lies upstream to the loss of mitochondrial cytochrome c.
Induction of BIMEL after NGF withdrawal is suppressed by low O2
Because the BCL-2 family proteins BAX and BIMEL are critical for cytochrome c release after NGF withdrawal, we compared their protein levels before and after NGF withdrawal in neurons cultured under standard and reduced O2. We also analyzed expression of the pro-survival protein BCL-XL and for changes in the phosphorylation of c-Jun. Phosphorylation of c-Jun increases in neurons deprived of NGF under standard culture conditions (Ham et al., 1995; Virdee et al., 1997) and we observed an essentially identical increase in neurons exposed to low O2 (Fig. 3 A). BAX and BCL-XL protein levels remained unchanged after 20 h of NGF withdrawal, regardless of O2 tension. BIMEL protein levels, on the other hand, increased an average of fourfold when neurons were deprived of NGF under standard O2 conditions; but when NGF deprivation was performed at 1% O2, a much smaller increase in BIMEL was observed (Fig. 3, A and B; also see Fig. 4 B). This effect appeared to be specific because low O2 did not inhibit c-Jun phosphorylation or decrease the expression of any of the other proteins examined.
Figure 3.
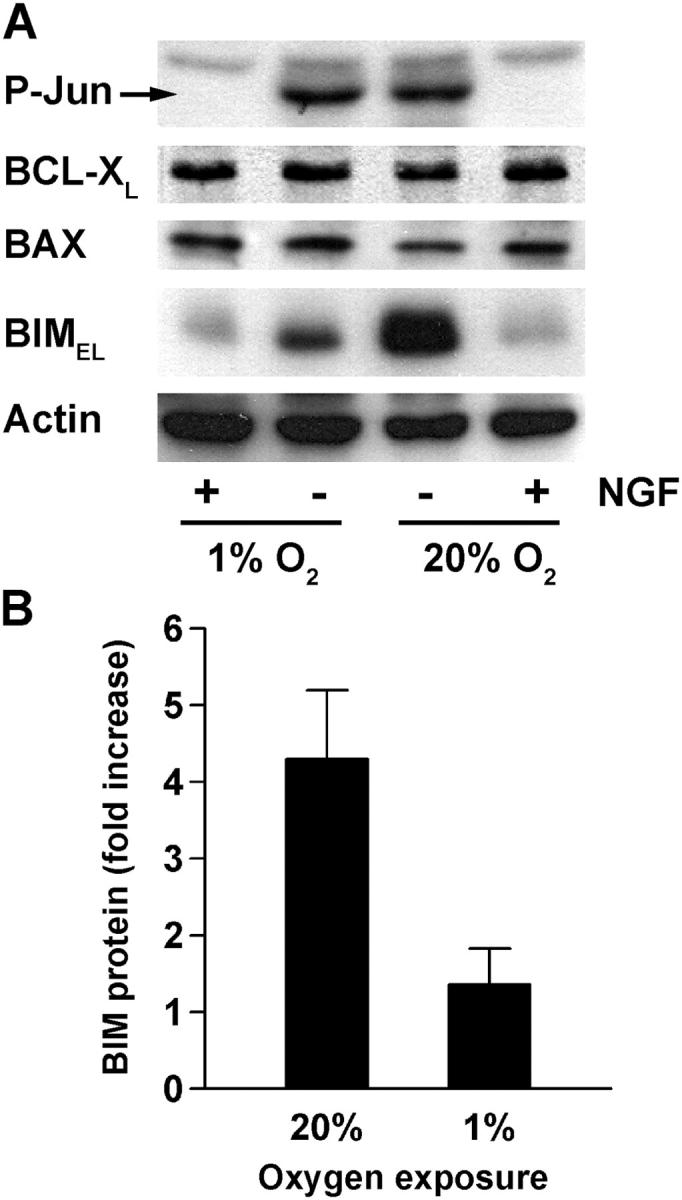
BIM EL induction during NGF deprivation is inhibited by exposure to low O2. (A) Neurons were treated as indicated and after 20 h whole cell lysates were prepared and immunoblotted with anti–phospho-c-Jun (Ser-63), anti BCL-XL, anti-BAX, anti-BIM, and anti-actin antibodies. (B) BIMEL protein levels were quantified using immunoblots from three independent experiments. The results (mean fold increase ± SEM) represent the ratio of BIMEL protein in NGF-deprived neurons to that in NGF-maintained neurons at either 20% or 1% O2.
Recent studies have suggested that phosphorylation of BIMEL by extracellular signal-regulated kinases (ERKs) is involved in targeting BIMEL to the proteasome (Biswas and Greene, 2002; Ley et al., 2003). We considered the possibility that a similar mechanism might underlie the suppression of BIMEL expression by low O2. Exposing control NGF-maintained neurons to 1% O2 for 20 h resulted in a small increase in phosphorylated ERK1/2 (Fig. 4 A, top). Nonetheless, phosphorylated ERK1/2 levels decreased similarly during NGF deprivation regardless of O2 tension (Fig. 4 A), suggesting that reducing O2 was not sufficient to stimulate ERK activation in sympathetic neurons in the absence of NGF signaling.
Figure 4.

Low O2 affects only some of the pathways involved in the induction of BIMEL after NGF withdrawal. (A) Neurons were treated with or without NGF and exposed to 20% or 1% O2 for the indicated times. Whole cell lysates were prepared and immunoblotted using anti–phospho-ERK1/2 or anti-ERK1/2 antibody. (B) Neurons were kept in the presence of NGF or deprived of NGF for 20 h at either 20% or 1% O2. Proteasome inhibitor (MG132, 50 μM) was added to one set of the NGF-deprived cultures. Whole cell lysates were immunoblotted with anti-BIM and anti-actin antibodies. The concentration of MG132 used was sufficient to inhibit proteasome activity as evidenced by its ability to stabilize HIF-1α (not depicted). (C) mRNA levels for BIMEL and cyclophilin were analyzed by semi-quantitative RT-PCR using total RNA prepared from neurons treated in the presence or absence of NGF for 20 h at 20% or 1% O2. BIMEL mRNA levels increased 4.2-fold during NGF deprivation in neurons exposed to 20% O2 compared with 1.9-fold in neurons exposed to 1% O2 (normalized to cyclophilin, n = 2). (D) Whole cell lysates were prepared from neurons treated with or without NGF and exposed to 1% O2 for the indicated times. The lysates were then immunoblotted with anti–phospho-c-Jun (Ser-63) and anti–phospho-Akt (Ser-473) antibodies. Arrows indicate the positions of the phosphorylated proteins. (E) Whole cell extracts prepared from cells treated in the presence or absence of NGF for 20 h at 20% or 1% O2 were immunoblotted with anti–phospho-FKHRL1 (Thr-32), anti–phospho-Akt, and anti–Akt antibodies. Similar results were seen in at least three independent experiments.
If low O2 exposure were to trigger proteasome-mediated destabilization of BIMEL, then inhibiting the proteasome might be expected to restore BIMEL levels to those found in normoxic, NGF-deprived neurons. To test this possibility, neurons were deprived of NGF under 20% or 1% O2 in the presence or absence of the proteasome inhibitor MG132 and then analyzed by immunoblotting for changes in BIMEL expression. As shown above, NGF deprivation resulted in increased BIMEL levels only in normoxic neurons (Fig. 4 B, compare second lane with fifth lane). Treatment with MG132 did little to reverse the suppression of BIMEL in neurons exposed to low O2 (Fig. 4 B, compare fifth lane with sixth lane). Together, these results indicate that the defect in BIMEL induction during NGF deprivation performed at low O2 is not due to ERK-dependent targeting of BIMEL to the proteasome.
Similar to BIMEL protein, the increase in BIMEL mRNA during NGF deprivation was also suppressed by low O2 (Fig. 4 C). Both c-Jun and the FOXO family member FKHRL1 have been implicated in up-regulating BIMEL transcription in cells deprived of NGF (Putcha et al., 2001; Harris and Johnson, 2001; Whitfield et al., 2001; Gilley et al., 2003). c-Jun is activated relatively soon after NGF withdrawal in part through JNK-dependent phosphorylation of Ser-63 (Virdee et al., 1997; Eilers et al., 1998). Activation of FKHRL1 after NGF withdrawal involves the dephosphorylation of sites originally phosphorylated by Akt protein kinase (Zheng et al., 2002; Gilley et al., 2003). Thus, FKHRL1 activation typically coincides with inactivation of Akt (Burgering and Kops, 2002).
To test whether the JNK/c-Jun and Akt/FKHRL1 pathways might be altered in neurons kept under different O2 conditions, we immunoblotted c-Jun and Akt using antibodies specific for their phosphorylated and activated forms. In neurons maintained at 20% O2, the amount of phosphorylated c-Jun increased within 5 h of NGF removal and remained elevated for at least 20 h. Conversely, phosphorylated Akt levels decreased within 5 h of NGF deprivation and remained low (unpublished data). These same changes in c-Jun and Akt phosphorylation occurred when the experiment was performed on neurons exposed to 1% O2 (Fig. 4 D), suggesting that the reduced O2 tension used in these studies is unlikely to impact either c-Jun activation or Akt inactivation after NGF withdrawal.
We also examined whether low O2 might directly influence FKHRL1 by altering its phosphorylation at Thr-32, a site targeted by Akt (Brunet et al., 1999). As shown previously (Gilley et al., 2003), neurons deprived of NGF under standard O2 conditions had reduced levels of Thr-32–phosphorylated FKHRL1 compared with neurons maintained with NGF (Fig. 4 E). Unexpectedly, the amount of FKHRL1 phosphorylated at this site did not decrease when NGF was withdrawn from neurons exposed to 1% O2, despite the concurrent decrease in Akt phosphorylation. These results suggest that activation of FKHRL1 in response to NGF withdrawal might be inhibited in neurons exposed to reduced O2. Such a scenario, if corroborated, could help explain the suppression of BIMEL mRNA expression that occurs under these conditions (Gilley et al., 2003).
Ectopic BIMEL reverses the ability of low O2 to block cytochrome c release but not cell death
The results described above suggest a model in which low O2 inhibits cell death by suppressing BIMEL induction, which in turn inhibits the release of cytochrome c from mitochondria and ultimately cell death. As a test of this idea, we infected neurons with a control adenovirus expressing EGFP or one expressing a BIMEL/EGFP fusion protein. The next day, the cells were deprived of NGF and switched to a 1% O2 environment for an additional 24 h, after which the cells were examined by immunofluorescence for cytochrome c localization. Parallel cultures were deprived of NGF for 48 h to assess the effects of BIMEL expression on cell survival. The majority of control EGFP-expressing neurons exposed to low O2 during NGF deprivation retained intense, punctate cytochrome c immunofluorescence (Fig. 5, A and B), similar to the results shown with uninfected neurons in Fig. 2. In contrast, very few of the NGF-deprived neurons exposed to low O2 and expressing BIMEL/EGFP retained punctate cytochrome c labeling. Despite their apparent lack of mitochondrial-localized cytochrome c, the majority of cells expressing BIMEL/EGFP remained healthy 48 h after withdrawing NGF (Fig. 5 C). Thus, whereas forced expression of BIMEL can overcome the block to cytochrome c release, there must be additional processes induced by low O2 (apparently downstream or independent of cytochrome c release) that can sustain cell survival in the absence of NGF.
Figure 5.

BIMEL expression promotes loss of mitochondrial cytochrome c but does not decrease survival in NGF-deprived neurons exposed to low O2. Neurons were infected with Ad-BIMEL/EGFP or Ad-EGFP adenoviruses (MOI = 100). The next day, NGF was withdrawn and the cells were immediately transferred to 1% O2. (A and B) After 24 h of NGF deprivation, the cells were fixed and analyzed for immunofluorescence using anti–cytochrome c antibody. The three panels for each treatment are of the same field of view. Bar, 15 μm. The percentage of EGFP-expressing, BIMEL/EGFP-expressing, and control (uninfected) cells showing punctate cytochrome c immunofluorescence was determined (means ± SEM, n = 3). Arrows in A point to a neuron expressing BIMEL-EGFP (top), its Hoechst-stained nucleus (middle), and its lack of punctate cytochrome c immunofluorescence (bottom). (C) Neurons were treated as described above except that NGF deprivation was continued for 48 h. The cells were then stained with Hoechst dye and scored for viability (mean ± SEM, n = 3). Except in the case of uninfected cells, only neurons positive for BIMEL/EGFP or EGFP expression were scored in B and C.
Low O2 exposure results in increased HIF activity. Because BIMEL expression failed to reverse the neuroprotective effect of low O2, we wondered if activation of HIF might contribute to the enhanced survival seen under these conditions. In initial experiments, we infected neurons using an adenovirus that expresses a luciferase reporter gene under the control of four tandem hypoxia response elements (Ad-HRE-luciferase). Not surprisingly, NGF-maintained neurons exposed to 1% O2 had increased HIF reporter gene activity compared with neurons kept under standard cell culture conditions (Fig. 6 A). NGF-deprived neurons exposed to 1% O2 displayed a similar elevation in HRE-luciferase activity, demonstrating that low O2-mediated activation of HIF in these neurons can occur in the absence of NGF signaling. HIF-1α protein levels also increased under the same conditions suggesting that at least part of the increase in HIF activity is due to HIF-1 (Fig. 6 B).
Figure 6.
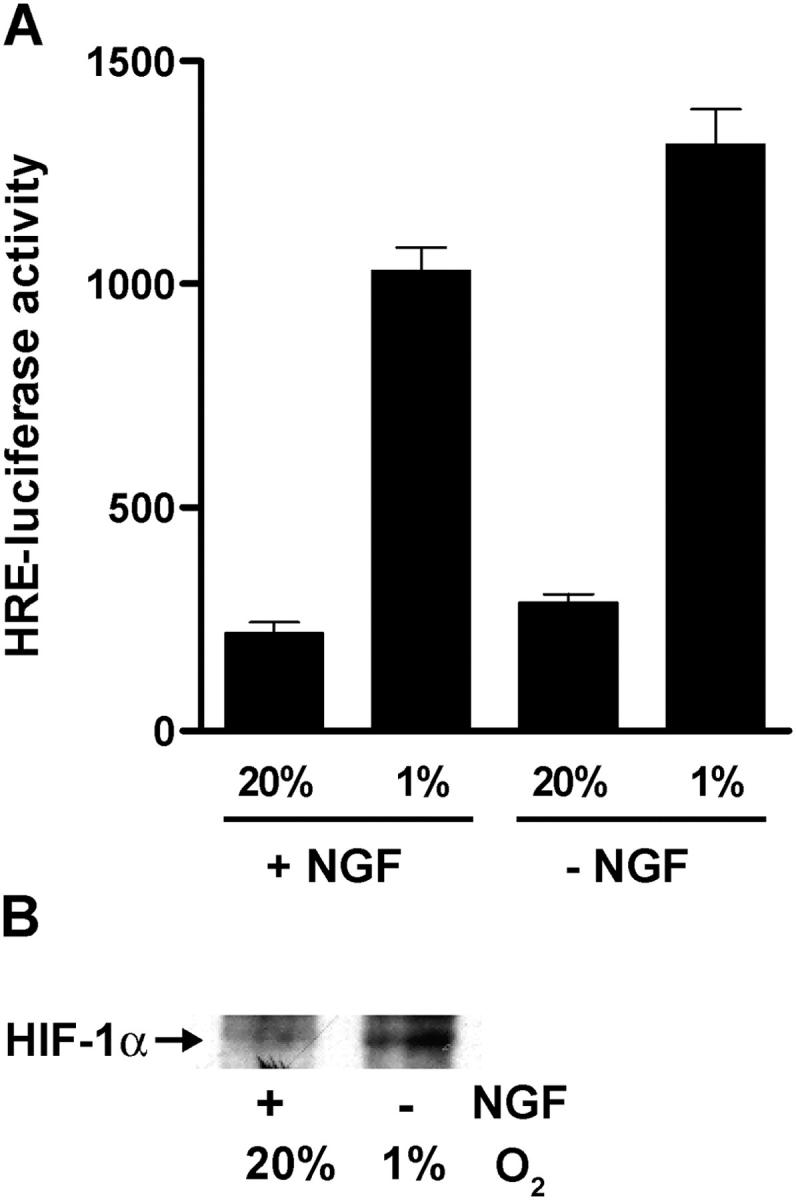
Low O2 exposure activates HIF in the presence or absence of NGF. (A) Neurons were infected with Ad-HRE-luciferase (MOI = 10) and 48 h later either continued in the presence of NGF or deprived of NGF for an additional 12 h at 20% or 1% O2. Cells were then lysed and relative luciferase activities were determined (mean ± SEM of triplicate wells; similar results were obtained in a second independent experiment). (B) Neurons were either deprived of NGF and incubated at 1% O2 or kept in the presence of NGF and cultured at 20% O2. After 20 h, HIF-1α protein expression was analyzed by immunoprecipitation followed by immunoblotting with an anti–HIF-1α antibody.
Stabilized HIF-1αPP→AG inhibits NGF deprivation–induced death
Having found that low O2 activates endogenous HIF in the neurons, we wondered whether stabilization of HIF in the absence of low O2 would be sufficient to inhibit trophic factor deprivation–induced death. A stabilized form of HIF-1α (HIF-1αPP→AG) was created by mutating the two proline residues that target HIF-1α for degradation to alanine and glycine. Transfection experiments in COS-7 cells done in the presence and absence of coexpressed EGLN prolyl hydroxylase demonstrated that HIF-1αPP→AG was active and relatively resistant to EGLN-mediated inactivation (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200407079/DC1). In addition, sympathetic neurons coinfected with an adenovirus expressing HIF-1αPP→AG fused to EGFP (Ad-HIF-1αPP→AG/EGFP) and Ad-HRE-luciferase had substantially higher luciferase activities compared with cells coinfected with Ad-EGFP and Ad-HRE-luciferase (Fig. S1 B).
To determine whether HIF-1αPP→AG can influence trophic factor deprivation–induced cell death, neurons were infected with Ad-HIF-1αPP→AG/EGFP or Ad-EGFP and the next day deprived of NGF. By 24 h of NGF deprivation, about half of the Ad-EGFP infected and uninfected neurons showed signs of chromatin condensation and nuclear fragmentation (Fig. 7, A and B). In contrast, the nuclei of nearly all HIF-1αPP→AG/EGFP expressing neurons appeared healthy, closely resembling those of NGF-maintained neurons. By 48 h, over 80% of neurons expressing HIF-1αPP→AG/EGFP remained healthy compared with just 25% of the control cells. Survival was also assessed using a MTT metabolic activity assay. NGF-deprived neurons infected with Ad-HIF-1αPP→AG/EGFP retained significantly more MTT reducing activity than control neurons infected with Ad-EGFP (Fig. 7 C). Thus, expression of a stabilized, prolyl hydroxylase-resistant form of HIF-1α protects neurons, at least transiently, from apoptosis caused by trophic factor deprivation.
Figure 7.

HIF-1αPP → AG inhibits NGF deprivation–induced death by acting at a point downstream to cytochrome c release. Sympathetic neurons were infected with Ad-EGFP or Ad-HIF-1αPP→AG/EGFP virus (MOI = 100) or left uninfected. The next day, the cells were refed with fresh NGF-containing medium or with medium lacking NGF and cultured for an additional 24 or 48 h. (A) The images show Ad-EGFP and Ad-HIF-1αPP→AG/EGFP-infected neurons 24 h after NGF withdrawal. Top panels show EGFP or HIF-1αPP→AG/EGFP fluorescence, whereas the bottom panels show the corresponding Hoechst-stained nuclei in the same field of view (marked by arrowheads). Bar, 15 μm. (B) Cell survival (mean ± SEM, n = 3) was quantified in neurons expressing EGFP or HIF-1αPP→AG/EGFP using the criteria described in Materials and methods. (C) MTT assays were performed on infected cultures 24 and 48 h after NGF withdrawal. Data represent mean ± SEM (n = 3) and are reported as a percentage of the activity in uninfected NGF-maintained neurons. (D) Neurons were infected with Ad-HIF-1αPP→AG/EGFP as above. After an additional 20 h in the presence or absence of NGF, the cells were analyzed for cytochrome c immunofluorescence. The images show Ad-HIF-1αPP→AG/EGFP fluorescence (top), Hoechst-stained nuclei (middle), and cytochrome c immunofluorescence (bottom) for a single field of view for each treatment. Bar, 20 μm. (E) Neurons were infected with Ad-HIF-1αPP→AG/EGFP and then treated with or without NGF as outlined in D. A set of uninfected neurons was transferred to an incubator equilibrated to 1% O2, whereas a second set of uninfected cells and the cells infected with Ad-HIF-1αPP→AG/EGFP remained at 20% O2. After 20 h the cells were analyzed for cytochrome c immunofluorescence. Data represent the percentage of cells with punctate cytochrome c and are the mean ± SEM from three experiments. (F) Caspase assays were performed using the substrate DEVD-AFC and lysates prepared from uninfected neurons or neurons infected with Ad-HIF-1αPP→AG/EGFP or Ad-EGFP. NGF deprivation was for 24 h. Caspase activity is reported as relative fluorescence units/microgram protein (mean ± SEM, n =3).
HIF-1αPP→AG does not block the loss of cytochrome c from mitochondria
We next addressed whether the release of cytochrome c from mitochondria is inhibited in neurons expressing HIF-1αPP→AG. Cells infected with Ad-HIF-1αPP→AG/EGFP were maintained with or without NGF and analyzed by immunofluorescence for cytochrome c. In the presence or absence of NGF, cells expressing HIF-1αPP→AG/EGFP had the same pattern of cytochrome c immunofluorescence as control neurons—punctate and intense in the presence of NGF, diffuse and faint in its absence (Fig. 7, D and E). Despite the apparent loss of mitochondrial cytochrome c in neurons expressing HIF-1αPP→AG and deprived of NGF, caspase activity assays done using a DEVD-based fluorescent substrate showed that the activation of caspase-3–like enzymes during NGF deprivation was inhibited in cells infected with Ad-HIF-1αPP→AG/EGFP (Fig. 7 F). Together, these results suggest that HIF-1αPP→AG, in contrast to low O2, inhibits caspase activation and ultimately cell death without substantially blocking the loss of mitochondrial cytochrome c.
Low O2-mediated protection is partially reversed by disrupting HIF-1α expression and completely abrogated when disruption of HIF-1α is combined with ectopic BIMEL
The activation of HIF after exposure to low O2 and the ability of mutant HIF-1αPP→AG to inhibit death after NGF withdrawal led us to test the significance of HIF-1α for the neuroprotective effect of low O2. For these experiments, we isolated sympathetic neurons from transgenic mice homozygous for a loxP flanked (floxed) HIF-1α gene (HIF-1αfl/fl; Ryan et al., 2000). Previous studies have shown that introducing Cre recombinase into HIF-1αfl/fl cells results in efficient deletion of the HIF-1α gene and loss of HIF-1-dependent gene induction (Ryan et al., 2000; Seagroves et al., 2001).
To determine whether expression of Cre recombinase in HIF-1αfl/fl sympathetic neurons results in a substantial loss of HIF-1α, we infected the neurons with an adenovirus that expresses Cre (Ad-Cre) or with the control Ad-EGFP virus. Immunofluorescence confirmed Cre expression in virtually all of the cells by 24 h after infection (unpublished data). RT-PCR analysis revealed greatly reduced HIF-1α expression specifically in Ad-Cre infected cultures (Fig. 8 A), suggesting that efficient deletion of the floxed HIF-1α gene had occurred. Loss of HIF-1α had no effect on the ability of NGF to promote survival for at least 4 d, and initial NGF deprivation experiments suggested that HIF-1α deletion did not significantly affect short-term protection afforded by low O2 as measured at 24 h after NGF withdrawal (unpublished data; see below).
Figure 8.

Targeted deletion of HIF-1α combined with ectopic BIMEL abolishes the neuroprotective effect of low O2. (A) Sympathetic neurons isolated from HIF-1αfl/fl mice were infected with either Ad-EGFP or Ad-Cre or left uninfected. 2 d later, RNA was extracted and analyzed by RT-PCR for expression of HIF-1α and an unrelated control gene (cyclophilin). (B) HIF-1αfl/fl neurons were microinjected with expression plasmids for Cre recombinase and BIMEL/EGFP, either alone or as an equimolar mixture. Uninjected neurons and neurons injected with an EGFP expression plasmid were included as controls. NGF deprivation was initiated the next day at which time the cells were transferred to an incubator equilibrated to 1% O2. After 48 h, the cells were stained with Hoechst dye and the fraction of injected neurons that remained healthy was determined. Results (mean ± range) were derived from two independent experiments.
Finally, we tested whether suppression of BIMEL and activation of HIF-1 might both be important for the survival enhancing effect of low O2. Thus, we attempted to disrupt HIF-1α and overexpress BIMEL in the same neurons. To ensure efficient coexpression of Cre and BIMEL/EGFP, we microinjected HIF-1αfl/fl neurons with an equimolar mixture of Ad-Cre and Ad-BIMEL/EGFP expression plasmids. Additional neurons were injected with each plasmid alone and with an EGFP expression plasmid. The next day the neurons were deprived of NGF and exposed to 1% O2. After 48 h, nearly 75% of EGFP-expressing neurons and a similar fraction of BIMEL/EGFP-expressing neurons remained healthy, as judged by the appearance of their nuclei after staining with Hoechst dye (Fig. 8 B). In contrast, only ∼30% of the Cre-injected neurons appeared healthy after 48 h, suggesting that HIF-1 is important for the longer-term (>24 h) neuroprotective effect of low O2. The most dramatic result was seen in neurons coinjected with Cre and BIMEL/EGFP. Virtually all of these cells had condensed or fragmented chromatin after 48 h of NGF deprivation. We conclude that both suppressing BIMEL induction and activating HIF-1 are important for the survival enhancing effects of low O2 on trophic factor deprived neurons.
Discussion
Most cell culture studies on neuronal apoptosis are performed using neurons exposed to O2 concentrations well above those experienced in vivo. Because changes in O2 concentration can influence a wide range of biochemical processes, we decided to investigate the effects of reducing O2 on apoptotic mechanisms in a well-characterized model of neurotrophic factor deprivation. The results from this study support a model in which exposure to low O2 inhibits cell death caused by NGF withdrawal at two levels—one upstream of the loss of cytochrome c from mitochondria and a second that is downstream or independent of cytochrome c release. The block to cytochrome c release appears to be due at least partially to inadequate BIMEL expression. Overexpressing BIMEL reversed the block to cytochrome c release but still failed to override the protective effect of low O2, thus revealing a second place where low O2 acts to inhibit apoptosis.
Certain events triggered by NGF withdrawal were not affected by lowering O2. For example, dephosphorylation of ERKs and Akt and activation of JNKs (as judged by c-Jun phosphorylation) occurred similarly in neurons exposed to 20% or 1% O2. These findings differ from those obtained in models of ischemic and hypoxic preconditioning in which neuroprotection requires activation of Akt (Ruscher et al., 2002; Wick et al., 2002). In our paradigm, lowering the O2 concentration interrupted the apoptotic program subsequent to JNK activation, resulting in reduced BIMEL expression. A similar situation has been reported in colon cancer cells, where O2 deprivation was found to down-regulate expression of the pro-apoptotic BCL-2 family protein BID (Erler et al., 2004).
Although the nature of the downstream block to cell death remains uncertain, several observations point to a role for HIF. First, HIF was activated in NGF-deprived neurons exposed to low O2. Second, introducing HIF-1αPP→AG mimicked the downstream block by inhibiting caspase-3 activation and cell death without preventing cytochrome c release. Third, whereas the neuroprotective effect of low O2 was partially reduced in neurons lacking HIF-1α, protection was completely lost in neurons expressing ectopic BIMEL (to override the first block) and lacking HIF-1α.
How might low O2 inhibit caspase activation after cytochrome c release? One possibility is that neurons exposed to low oxygen may express or maintain higher levels of proteins capable of blocking caspase activation. For example, Potts et al. (2003) reported that down-regulation of the X-linked inhibitor of apoptosis protein (XIAP) was necessary for caspase activation and cell death after NGF withdrawal, even after cytochrome c was present in the cytoplasm. Thus, an increase in XIAP expression under reduced O2 conditions might be sufficient to inhibit trophic factor deprivation–induced death. A similar mechanism was recently suggested based on the induction of IAP-2 expression in oxygen-deprived kidney cells (Dong et al., 2003). Additional mechanisms for blocking cell death after cytochrome c release may involve down-regulation of Apaf-1 or downstream caspases (Jia et al., 2001; Devarajan et al., 2002). We are currently investigating whether the expression level of any of these proteins is influenced by lowering O2 tension. Although our efforts so far have focused on well-studied pro-survival and pro-apoptotic pathways, it is important to realize that reducing O2 tension will at some point influence numerous other biochemical processes. Cellular effects of low oxygen can include changes in ion channel function, reactive O2 species generation, and energy metabolism, as well as alterations in the activity of a large number of enzymes (Bickler and Donohoe, 2002).
In cells maintained under standard culture conditions of 20% O2, HIF activity is kept at an extremely low level do to the highly efficient degradation of its α subunits. This occurs in large part because the O2-dependent enzymes responsible for targeting HIF-α subunits for ubiquitination (HIF prolyl hydroxylases) have a Km for O2 that is near its concentration in air (Hirsilä et al., 2003). Because HIF prolyl hydroxylase activity is reduced in cells at physiologic O2 tensions (Epstein et al., 2001), basal HIF activity in most cells in vivo should be substantially greater than in cells in culture. Given this scenario together with a neuroprotective role for HIF-1, down-regulating basal HIF activity may be an important part of the overall cell death program. How could inactivation of HIF-1 be ensured in neurons deprived of NGF? In a previous study, we showed that NGF deprivation results in an increase in the expression of the SM-20 gene (Lipscomb et al., 1999). Recently, SM-20 was found to be the rat orthologue of EGLN3, one of three HIF prolyl hydroxylases in mammals (Freeman et al., 2003). Thus, up-regulation of SM-20/EGLN3 could serve to quench an HIF-dependent neuroprotective pathway. At O2 tensions that are too low to support EGLN3 activity, this mechanism might be compromised resulting in sustained HIF activation and inhibition of cell death (Freeman et al., 2004).
In summary, basic cellular mechanisms controlling cell death and survival are generally characterized in cells maintained under supraphysiologic oxygen conditions. As shown here, some of these mechanisms can be influenced by reducing O2 tension. We believe these results highlight the importance of carefully defining the O2-sensitivity of mechanisms that determine cell viability.
Materials and methods
Materials
Cell culture reagents were purchased from Invitrogen Life Technologies. FBS and NGF were purchased from Harlan Bioproducts for Science. Boc-aspartyl(OMe)-fluoromethylketone (BAF) was purchased from Enzyme Systems Products. MG132 was purchased from BIOMOL Research Laboratories, Inc. and Hoechst 33,342 was purchased from Molecular Probes. Additional reagents were purchased from Sigma-Aldrich unless otherwise indicated.
Cell culture
Primary cultures of sympathetic neurons were prepared from the superior cervical ganglia of embryonic day-21 Sprague-Dawley rats (Harlan) or newborn HIF-1αfl/fl mice (Ryan et al., 2000) as described elsewhere (Lipscomb et al., 1999). After preplating to enrich for neurons, cells were plated on collagen- or polyornithine/laminin-coated dishes in NGF-containing media (90% MEM, 10% FBS, 2 mM l-glutamine, 20 μM uridine, 20 μM fluorodeoxyuridine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml NGF). For NGF deprivation, neurons were rinsed twice with PBS before addition of media lacking NGF and containing neutralizing anti-NGF antiserum. All treatments were initiated 5–6 d after plating. The caspase inhibitor BAF (50 μM) was included in the media for immunoblotting and cytochrome c immunofluorescence experiments. BAF treatment prevents terminal phases of cell death without blocking upstream events including JNK activation, BIMEL up-regulation, and cytochrome c release (Deshmukh and Johnson, 1998; Tsui-Pierchala et al., 2000; Putcha et al., 2001). O2 tension was controlled by incubating cells at 37°C in humidified, O2/CO2-regulated incubators (Thermo Forma) adjusted to 5% CO2 and the indicated O2 tension (balanced by N2), or in a standard CO2-controlled incubator maintained at 5% CO2/95% air (equivalent to ∼20% O2). In these experiments, a relative O2 tension of 1% (in the gas phase) was equivalent to a partial pressure of O2 of ∼6.9–7.3 mm Hg. Note that the cells were continuously exposed (uninterrupted) to reduced oxygen for times that greatly exceeded the 1–2 h needed to equilibrate the culture medium with the O2 in the gas phase.
Assessing viability and caspase activity
After removal from the incubator, cells were quickly rinsed with PBS and then fixed in 4% PFA. The cultures were then stained with Hoechst 33,342 (1 μg/ml in PBS) and visualized under phase-contrast and epi-fluorescence using a 40× objective and a microscope (model Diaphot 300; Nikon). Viability was assessed based on the appearance of the neurons and the Hoechst-stained nuclei (Lipscomb et al., 2001). Healthy neurons had round, smooth, and refractile cell bodies with clearly discernible nuclear membranes and nucleoli, and diffuse Hoechst-stained chromatin. In contrast, dying or dead cells were characterized by condensed or undetectable chromatin and fragmented and atrophied cell bodies. For experiments with recombinant adenoviruses, 150–200 neurons expressing EGFP or HIF-1αPP→AG/EGFP were scored for each treatment per experiment. MTT assays (Maggirwar et al., 1998) and caspase-3 activity assays (Straub et al., 2003) were described previously.
Immunofluorescence
Immediately after removal from the incubator, cells were fixed in 4% PFA for 30 min at 4°C. Immunofluorescence was done as described previously (Lipscomb et al., 2001) using a 1:500 dilution of mouse anti–cytochrome c antibody (BD Biosciences) or a 1:1000 dilution of anti-Cre antibody followed by a TRITC-conjugated goat anti–mouse secondary antibody (Jackson ImmunoResearch Laboratories) diluted 1:300. Cells were rinsed with PBS before staining with Hoechst 33,342. In all NGF-maintained neurons, the cytochrome c immunofluorescence pattern is punctate throughout the cytoplasm and in neuronal processes but excludes the nucleus. After 20 h of NGF deprivation done in the presence of BAF, all of the neurons exhibited a faint fluorescence signal that was evenly distributed throughout the cytoplasm and nucleus. None of the cells showed an obvious partial loss of punctate staining. For each experiment, the pattern of cytochrome c immunofluorescence was scored by an observer blinded to the experimental treatments. Digital images were captured using a microscope (model Diaphot 300; Nikon) equipped with a DAGE-MTI CCD camera and Scion Image software (Scion Corp.).
Immunoblotting
Cells were rinsed once with ice-cold PBS and solubilized in SDS-PAGE sample buffer immediately after removal from the incubator. Lysates were boiled for 10 min and centrifuged for 5 min at 12,000 g before separation by SDS-PAGE. Separated proteins were transferred to nitrocellulose membranes, which were blocked in TBS containing 5% nonfat dry milk and 0.1% Tween-20 (TBST) for 1 h at RT. Membranes were incubated overnight at 4°C in the same buffer containing the following primary antibodies at the indicated dilutions: anti–cytochrome c, 1:500 (BD Biosciences); anti-BIM, 1:1,000 (StressGen Biotechnologies); anti-BAX, 1:1,000 (Upstate Biotechnology); anti–BCL-XL, 1:1,000 (Santa Cruz Biotechnology); anti–phospho-c-Jun (Ser-63), 1:1,000 (Cell Signaling); anti–phospho-Akt (Ser-473), 1:1,000 (Cell Signaling); anti-Akt, 1:1,000 (Cell Signaling); anti–phospho-ERK1/2, 1:1,000 (Cell Signaling); anti-ERK1/2, 1:1,000 (Cell Signaling); anti–phospho-FKHRL1 (Thr-32), 1:1,000 (Upstate Biotechnology); anti-actin, 1:300 (Sigma-Aldrich); anti–HIF-1α, 1:1,000 (Novus Biologicals). The blots were then incubated for 1 h with appropriate HRP-conjugated secondary antibodies diluted 1:10,000 in TBST and detected by ECL using Super Signal West Dura substrate (Pierce Chemical Co.). To detect endogenous HIF-1α in sympathetic neurons, cells were lysed in RIPA buffer [50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40, 1% deoxycholate, 0.1% SDS, 1 mM PMSF, 5 μg/ml leupeptin, and 5 μg/ml aprotinin] and the lysates immunoprecipitated as described previously (Lipscomb et al., 2001) using 2 μg of anti–HIF-1α antibody. Immunoprecipitated proteins were then immunoblotted using the same anti–HIF-1α antibody. For all immunoblotting experiments, the data shown are representative of the results from three or more independent experiments.
RT–PCR
Total RNA was extracted from equal numbers of neurons plated on collagen-coated 6-well dishes using an RNeasy Mini kit (QIAGEN). Semi-quantitative RT-PCR to analyze BIMEL and cyclophilin expression was performed as described previously (Estus et al., 1994). PCR products shown in Fig. 4 C were labeled by incorporation of α-[32P]-dCTP over 21 and 17 cycles of amplification, respectively. HIF-1α and cyclophilin PCR products in Fig. 8 A were amplified over 30 and 25 cycles, respectively, and analyzed on agarose gels stained with ethidium bromide.
Plasmids and site-directed mutagenesis
The plasmid containing HRE-luciferase was a gift from G. Semenza (Johns Hopkins University, Baltimore, MD) and R. Ratan (Harvard Medical School, Boston, MA). For expression of HIF-1α and the HIF-1α/EGFP fusion protein, the ORF of human HIF-1α was generated by PCR using Pfu Turbo DNA polymerase (Stratagene) and confirmed by DNA sequencing. The HIF-1α cDNA was subcloned into pcDNA3 and pEGFP-C1 (CLONTECH Laboratories, Inc.). Site-directed mutagenesis of HIF-1α and HIF-1α/EGFP was done using QuickChange (Stratagene) to change Pro-402 and Pro-564 to Ala and Gly. The resulting plasmids, HIF-1αPP→AG and HIF-1αPP→AG/EGFP, were verified by DNA sequencing.
Ectopic gene expression
Replication-deficient adenovirus vectors were produced using the AdEasy system (Qbiogene) following the manufacturer's protocol. EGFP and HIF-1αPP→AG/EGFP were subcloned into pShuttle-CMV; the HRE-luciferase cassette was subcloned into pShuttle. Recombination between the pShuttle-derived plasmids and pAdEasy-1 was performed in BJ5183 E. coli (Qbiogene). Recombinant plasmids were confirmed by restriction digests, amplified in DH5α E. coli, and then linearized with PacI before transfection into HEK293 cells. Recombinant replication-defective adenoviruses were plaque purified and verified by PCR and immunoblotting using antibodies against HIF-1α or EGFP. High titer viral stocks (∼1010 plaque-forming units/ml) were obtained after amplification in HEK293 cells and purification through CsCl gradients. The purified virus stocks were diluted to the indicated multiplicity of infection (MOI) in complete medium and then added to cells using half the normal culture volume. The medium was replaced after 12 h. Microinjections were performed as described previously (Lipscomb et al., 2001). Plasmid DNAs (50–100 μg/ml) were diluted in injection buffer containing 100 mM KCl, 10 mM potassium phosphate, pH 7.4, and 4 mg/ml rhodamine-dextran (to identify injected cells).
Luciferase assays
Luciferase activities were determined using the Dual-Luciferase Reporter Assay system (Promega) and a TD 20/20 luminometer (Turner) following the manufacturer's protocol.
Online supplemental material
COS-7 cells were maintained in 90% DME and 10% FBS. The pcDNA3-based plasmid expressing rat EGLN3 (also called SM-20) was described previously (Lipscomb et al., 2001). Transfections were done using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Adenovirus infections, immunoblotting, and luciferase assays were performed as described above. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200407079/DC1.
Acknowledgments
We thank Rajiv Ratan and Gregg Semenza for providing the HRE-luciferase plasmid, Craig Miller for permitting us to use his O2-regulated cell culture incubators, and Elena Suk for expert technical assistance.
This work was supported by National Institutes of Health grants NS34400 and NS42224.
Abbreviations used in this paper: BAF, boc-aspartyl(OMe)-fluoromethylketone; EGLN, egg-laying nine; ERK, extracellular signal-regulated kinase; HIF, hypoxia-inducible factor; HRE, hypoxia response element; JNK, c-Jun NH2-terminal kinase; MOI, multiplicity of infection.
References
- Bickler, P.E., and P.H. Donohoe. 2002. Adaptive responses of vertebrate neurons to hypoxia. J. Exp. Biol. 205:3579–3586. [DOI] [PubMed] [Google Scholar]
- Biswas, S.C., and L.A. Greene. 2002. Nerve growth factor (NGF) down-regulates the Bcl-2 homology 3 (BH3) domain-only protein Bim and suppresses its proapoptotic activity by phosphorylation. J. Biol. Chem. 277:49511–49516. [DOI] [PubMed] [Google Scholar]
- Bruick, R.K., and S.L. McKnight. 2001. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 294:1337–1340. [DOI] [PubMed] [Google Scholar]
- Brunet, A., A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, K.C. Arden, J. Blenis, and M.E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96:857–868. [DOI] [PubMed] [Google Scholar]
- Burek, M.J., and R.W. Oppenheim. 1996. Programmed cell death in the developing nervous system. Brain Pathol. 6:427–446. [DOI] [PubMed] [Google Scholar]
- Burgering, B.M., and G.J. Kops. 2002. Cell cycle and death control: long live Forkheads. Trends Biochem. Sci. 27:352–360. [DOI] [PubMed] [Google Scholar]
- Deckwerth, T.L., and E.M. Johnson Jr. 1993. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J. Cell Biol. 123:1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth, T.L., R.M. Easton, C.M. Knudson, S.J. Korsmeyer, and E.M. Johnson Jr. 1998. Placement of the BCL2 family member BAX in the death pathway of sympathetic neurons activated by trophic factor deprivation. Exp. Neurol. 152:150–162. [DOI] [PubMed] [Google Scholar]
- Deshmukh, M., and E.M. Johnson Jr. 1998. Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 21:695–705. [DOI] [PubMed] [Google Scholar]
- Devarajan, E., A.A. Sahin, J.S. Chen, R.R. Krishnamurthy, N. Aggarwal, A.M. Brun, A. Sapino, F. Zhang, D. Sharma, X.H. Yang, et al. 2002. Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene. 21:8843–8851. [DOI] [PubMed] [Google Scholar]
- Dong, Z., J.Z. Wang, F. Yu, and M.A. Venkatachalam. 2003. Apoptosis-resistance of hypoxic cells: multiple factors involved and a role for IAP-2. Am. J. Pathol. 163:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, S.N., and A.M. Tolkovsky. 1994. Characterization of apoptosis in cultured rat sympathetic neurons after nerve growth factor withdrawal. J. Cell Biol. 124:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers, A., J. Whitfield, C. Babij, L.L. Rubin, and J. Ham. 1998. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J. Neurosci. 18:1713–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein, A.C., J.M. Gleadle, L.A. McNeill, K.S. Hewitson, J. O'Rourke, D.R. Mole, M. Mukherji, E. Metzen, M.I. Wilson, A. Dhanda, et al. 2001. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 107:43–54. [DOI] [PubMed] [Google Scholar]
- Erecinska, M., and I.A. Silver. 2001. Tissue oxygen tension and brain sensitivity to hypoxia. Respir. Physiol. 128:263–276. [DOI] [PubMed] [Google Scholar]
- Erler, J.T., C.J. Cawthorne, K.J. Williams, M. Koritzinsky, B.G. Wouters, C. Wilson, C. Miller, C. Demonacos, I.J. Stratford, and C. Dive. 2004. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol. Cell. Biol. 24:2875–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estus, S., W.J. Zaks, R.S. Freeman, M. Gruda, R. Bravo, and E.M. Johnson Jr. 1994. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 127:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman, R.S., D.M. Hasbani, E.A. Lipscomb, J.A. Straub, and L. Xie. 2003. SM-20, EGL-9, and the EGLN family of hypoxia-inducible factor prolyl hydroxylases. Mol. Cells. 16:1–12. [PubMed] [Google Scholar]
- Freeman, R.S., R.L. Burch, R.J. Crowder, D.J. Lomb, M.C. Schoell, J.A. Straub, and L. Xie. 2004. NGF deprivation-induced gene expression: After ten years, where do we stand? Prog. Brain Res. 146:111–126. [DOI] [PubMed] [Google Scholar]
- Gilley, J., P.J. Coffer, and J. Ham. 2003. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 162:613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halterman, M.W., C.C. Miller, and H.J. Federoff. 1999. Hypoxia-inducible factor-1alpha mediates hypoxia-induced delayed neuronal death that involves p53. J. Neurosci. 19:6818–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham, J., C. Babij, J. Whitfield, C.M. Pfarr, D. Lallemand, M. Yaniv, and L.L. Rubin. 1995. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 14:927–939. [DOI] [PubMed] [Google Scholar]
- Harris, C.A., and E.M. Johnson Jr. 2001. BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J. Biol. Chem. 276:37754–37760. [DOI] [PubMed] [Google Scholar]
- Hirsilä, M., P. Koivunen, V. Günzler, K.I. Kivirikko, and J. Myllyharju. 2003. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 278:30772–30780. [DOI] [PubMed] [Google Scholar]
- Ivan, M., K. Kondo, H. Yang, W. Kim, J. Valiando, M. Ohh, A. Salic, J.M. Asara, W.S. Lane, and W.G. Kaelin Jr. 2001. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 292:464–468. [DOI] [PubMed] [Google Scholar]
- Jaakkola, P., D.R. Mole, Y.M. Tian, M.I. Wilson, J. Gielbert, S.J. Gaskell, A. Kriegsheim, H.F. Hebestreit, M. Mukherji, C.J. Schofield, et al. 2001. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 292:468–472. [DOI] [PubMed] [Google Scholar]
- Jia, L., S.M. Srinivasula, F.T. Liu, A.C. Newland, T. Fernandes-Alnemri, E.S. Alnemri, and S.M. Kelsey. 2001. Apaf-1 protein deficiency confers resistance to cytochrome c-dependent apoptosis in human leukemic cells. Blood. 98:414–421. [DOI] [PubMed] [Google Scholar]
- Lei, K., and R.J. Davis. 2003. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA. 100:2432–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R., K. Balmanno, K. Hadfield, C. Weston, and S.J. Cook. 2003. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J. Biol. Chem. 278:18811–18816. [DOI] [PubMed] [Google Scholar]
- Lipscomb, E.A., P.D. Sarmiere, R.J. Crowder, and R.S. Freeman. 1999. Expression of the SM-20 gene promotes death in nerve growth factor-dependent sympathetic neurons. J. Neurochem. 73:429–432. [DOI] [PubMed] [Google Scholar]
- Lipscomb, E.A., P.D. Sarmiere, and R.S. Freeman. 2001. SM-20 is a novel mitochondrial protein that causes caspase-dependent cell death in nerve growth factor-dependent neurons. J. Biol. Chem. 276:5085–5092. [DOI] [PubMed] [Google Scholar]
- Maggirwar, S.B., P.D. Sarmiere, S. Dewhurst, and R.S. Freeman. 1998. Nerve growth factor-dependent activation of NF-kappaB contributes to survival of sympathetic neurons. J. Neurosci. 18:10356–10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D.P., R.E. Schmidt, P.S. DiStefano, O.H. Lowry, J.G. Carter, and E.M. Johnson Jr. 1988. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 106:829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neame, S.J., L.L. Rubin, and K.L. Philpott. 1998. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J. Cell Biol. 142:1583–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret, J.P., D. Mottet, M. Raes, and C. Michiels. 2002. Is HIF-1alpha a pro- or an anti-apoptotic protein? Biochem. Pharmacol. 64:889–892. [DOI] [PubMed] [Google Scholar]
- Potts, P.R., S. Singh, M. Knezek, C.B. Thompson, and M. Deshmukh. 2003. Critical function of endogenous XIAP in regulating caspase activation during sympathetic neuronal apoptosis. J. Cell Biol. 163:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha, G.V., M. Deshmukh, and E.M. Johnson Jr. 2000. Inhibition of apoptotic signaling cascades causes loss of trophic factor dependence during neuronal maturation. J. Cell Biol. 149:1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha, G.V., K.L. Moulder, J.P. Golden, P. Bouillet, J.A. Adams, A. Strasser, and E.M. Johnson. 2001. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 29:615–628. [DOI] [PubMed] [Google Scholar]
- Putcha, G.V., S. Le, S. Frank, C.G. Besirli, K. Clark, B. Chu, S. Alix, R.J. Youle, A. LaMarche, A.C. Maroney, and E.M. Johnson. 2003. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 38:899–914. [DOI] [PubMed] [Google Scholar]
- Ruscher, K., D. Freyer, M. Karsch, N. Isaev, D. Megow, B. Sawitzki, J. Priller, U. Dirnagl, and A. Meisel. 2002. Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J. Neurosci. 22:10291–10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, H.E., M. Poloni, W. McNulty, D. Elson, M. Gassmann, J.M. Arbeit, and R.S. Johnson. 2000. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 60:4010–4015. [PubMed] [Google Scholar]
- Safran, M., and W.G. Kaelin. 2003. HIF hydroxylation and the mammalian oxygen-sensing pathway. J. Clin. Invest. 111:779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seagroves, T.N., H.E. Ryan, H. Lu, B.G. Wouters, M. Knapp, P. Thibault, K. Laderoute, and R.S. Johnson. 2001. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol. Cell. Biol. 21:3436–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza, G.L. 1999. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 15:551–578. [DOI] [PubMed] [Google Scholar]
- Soucek, T., R. Cumming, R. Dargusch, P. Maher, and D. Schubert. 2003. The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron. 39:43–56. [DOI] [PubMed] [Google Scholar]
- Straub, J.A., E.A. Lipscomb, E.S. Yoshida, and R.S. Freeman. 2003. Induction of SM-20 in PC12 cells leads to increased cytochrome c levels, accumulation of cytochrome c in the cytosol, and caspase-dependent cell death. J. Neurochem. 85:318–328. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala, B.A., G.V. Putcha, and E.M. Johnson Jr. 2000. Phosphatidylinositol 3-kinase is required for the trophic, but not the survival-promoting, actions of NGF on sympathetic neurons. J. Neurosci. 20:7228–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virdee, K., A.J. Bannister, S.P. Hunt, and A.M. Tolkovsky. 1997. Comparison between the timing of JNK activation, c-Jun phosphorylation, and onset of death commitment in sympathetic neurones. J. Neurochem. 69:550–561. [DOI] [PubMed] [Google Scholar]
- Whitfield, J., S.J. Neame, L. Paquet, O. Bernard, and J. Ham. 2001. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 29:629–643. [DOI] [PubMed] [Google Scholar]
- Wick, A., W. Wick, J. Waltenberger, M. Weller, J. Dichgans, and J.B. Schulz. 2002. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J. Neurosci. 22:6401–6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, F., S.B. White, Q. Zhao, and F.S. Lee. 2001. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. USA. 98:9630–9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun, J.K., T.S. McCormick, R. Judware, and E.G. Lapetina. 1997. Cellular adaptive responses to low oxygen tension: apoptosis and resistance. Neurochem. Res. 22:517–521. [DOI] [PubMed] [Google Scholar]
- Zaman, K., H. Ryu, D. Hall, K. O'Donovan, K.I. Lin, M.P. Miller, J.C. Marquis, J.M. Baraban, G.L. Semenza, and R.R. Ratan. 1999. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J. Neurosci. 19:9821–9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, W.H., S. Kar, and R. Quirion. 2002. FKHRL1 and its homologs are new targets of nerve growth factor Trk receptor signaling. J. Neurochem. 80:1049–1061. [DOI] [PubMed] [Google Scholar]