

Abstract
The conserved oligomeric Golgi (COG) complex is an evolutionarily conserved multi-subunit protein complex that regulates membrane trafficking in eukaryotic cells. In this work we used short interfering RNA strategy to achieve an efficient knockdown (KD) of Cog3p in HeLa cells. For the first time, we have demonstrated that Cog3p depletion is accompanied by reduction in Cog1, 2, and 4 protein levels and by accumulation of COG complex-dependent (CCD) vesicles carrying v-SNAREs GS15 and GS28 and cis-Golgi glycoprotein GPP130. Some of these CCD vesicles appeared to be vesicular coat complex I (COPI) coated. A prolonged block in CCD vesicles tethering is accompanied by extensive fragmentation of the Golgi ribbon. Fragmented Golgi membranes maintained their juxtanuclear localization, cisternal organization and are competent for the anterograde trafficking of vesicular stomatitis virus G protein to the plasma membrane. In a contrast, Cog3p KD resulted in inhibition of retrograde trafficking of the Shiga toxin. Furthermore, the mammalian COG complex physically interacts with GS28 and COPI and specifically binds to isolated CCD vesicles.
Introduction
The Golgi apparatus is a hub for membrane trafficking pathways, organizing both the anterograde exocytic trafficking of newly synthesized proteins that travel from the ER to the plasma membrane and retrograde endocytic trafficking of cell surface molecules that travel back to the ER (for review see Shorter and Warren, 2002). COP I coat proteins function in intra-Golgi trafficking and in maintaining the normal structure of the Golgi complex (Duden, 2003). We and others have previously shown that the Golgi vesicular coat complex I (COPI)-modulated membrane trafficking used conserved oligomeric Golgi (COG) vesicle tethering complex (Ram et al., 2002; Suvorova et al., 2002; Oka et al., 2004).
COG complex consists of eight subunits (COG1-8; Kingsley et al., 1986; Whyte and Munro, 2001; Suvorova et al., 2001, 2002; Ram et al., 2002; Ungar et al., 2002). A COG role in Golgi membrane trafficking was suggested by biochemical and genetic studies in yeast (VanRheenen et al., 1998, 1999; Suvorova et al., 2002). Yeast COG complex interacts genetically and physically with Rab protein Ypt1p, intra-Golgi SNARE molecules, as well as with COPI (Suvorova et al., 2002).
Mutations in COG subunits disturb both structure and function of the Golgi in eukaryotic cells (Podos et al., 1994; Ram et al., 2002; Suvorova et al., 2002; Ungar et al., 2002; Farkas et al., 2003), but the exact cellular function of the COG complex remained elusive. It was recently shown that mammalian cells that carried either deletion of COG1 and 2 or truncated version of Cog7p are defective in Golgi glycosylation (Wu et al., 2004) and maintain slightly dilated Golgi cisternae with reduced levels of resident proteins GS15 and GS28 and GPP130 (Oka et al., 2004). These mutant phenotypes may reflect either the primary COG malfunction or secondary manifestations that occur in cells after a prolonged adaptation period. Deletion of either COG1 or COG2 in yeast cells is virtually incompatible with membrane trafficking and normal cell growth (VanRheenen et al., 1998; Ram et al., 2002), whereas ΔCOG1 and ΔCOG2 CHO cells are not compromised in growth or protein secretion. One possibility is that the primary COG deletion phenotype in these cells is masked and/or suppressed by secondary mutations in cell genome. To better understand the initial defects associated with the COG complex malfunction it is important to acutely interfere with its activity. Transfection with small interfering RNAs (siRNAs) and microinjection with inhibitory antibodies are two efficient methods for acute protein knockdown (KD) in mammalian cells.
We rationalized that the most evolutionary conserved COG subunit would be the best candidate for the KD. Yeast and mammalian Cog1 and Cog2 proteins do not share significant similarities, whereas the protein sequence of yeast Cog3p is 41% similar to mammalian Cog3p (Suvorova et al., 2001). In this work, we used siRNA strategy for the efficient KD Cog3p in HeLa cells. Cog3p depletion is accompanied by reduction in Cog1, 2, and 4 protein levels and rapid accumulation of intracellular vesicles carrying v-SNAREs GS15 and GS28 and cis-Golgi glycoprotein GPP130. A prolonged COG3 KD induced extensive Golgi fragmentation. Fragmented Golgi membranes were deficient in retrograde trafficking of the Shiga toxin. Native immunoprecipitations (IPs) revealed that the COG complex physically interacts with Golgi SNARE molecules and COPI and specifically binds to isolated GPP130-containing vesicles in vitro. For the first time, we have demonstrated that the acute depletion of the mammalian COG complex results in specific inhibition of tethering of retrograde vesicles and that the efficient targeting of these vesicles is essential for the maintenance of the Golgi structure.
Results
Cog3p KD induces Golgi fragmentation
To interfere with the COG complex function in HeLa cells we depleted Cog3p by using RNA interference technique (Elbashir et al., 2001). Three different COG3-specific RNA duplexes were tested initially and one of them (sense, AGACUUGUGCAGUUUAACA) efficiently induced reduction of the Cog3 protein level (Fig. 1 A). The expression of other lobe A COG subunits Cog1p, Cog2p, and Cog4p was also reduced 72 h after COG3 KD, whereas protein level of the lobe B Cog5-8p subunits remained unchanged. Similarly, cellular levels were unchanged for other tested cell proteins: protein disulphide isomerase (PDI), actin, GS28, syntaxin 5, GPP130, and p115 (Fig. 1 and unpublished data). Cell transfection with a nonspecific siRNA did not change the levels of COG subunits expression (unpublished data).
Figure 1.

SiRNA-induced COG3 KD is destabilizing Lobe A COG complex subunits. (A) Expression of COG subunits after COG3 KD. WB of cell lysates from control and COG3 KD cells. Average levels of the COG subunits (±SD, n = 4) after 72 h of COG3 KD were determined by quantitative WB, and normalized to mock-transfected cells. (B) Membrane localization of COG complex subunits. WB of membrane (P100) and cytosol (S100) fractions. (C) Cog6p localization. Control and COG3 KD cells that stably express GalT-GFP were fixed and analyzed by three-color IF microscopy after immunostaining with anti-Cog6p. DNA was stained with DAPI. Arrows indicate Golgi or Golgi fragments. Bars, 10 μm.
Because the level of the lobe B COG subunits was not affected by the Cog3p depletion we have determined their intracellular localization by both immunofluorescence (IF) and Western blot (WB) assays. We have found that significant amounts of both Cog5p and Cog8p were still associated with the membrane fraction (Fig. 1 B). IF analysis of COG3 KD cells revealed that Cog6p (Fig. 1 C) and other lobe B subunits (unpublished data) were localized on large structures in juxtanuclear region that were colocalized with resident Golgi enzyme GFP-tagged β 1,4-galactosyltransferase (GalT-GFP; Fig. 1 C). Detailed analysis of COG3 KD cells revealed that both GalT-GFP and a Golgi tethering factor GM130 were found on fragmented Golgi membranes (Fig. 2).
Figure 2.
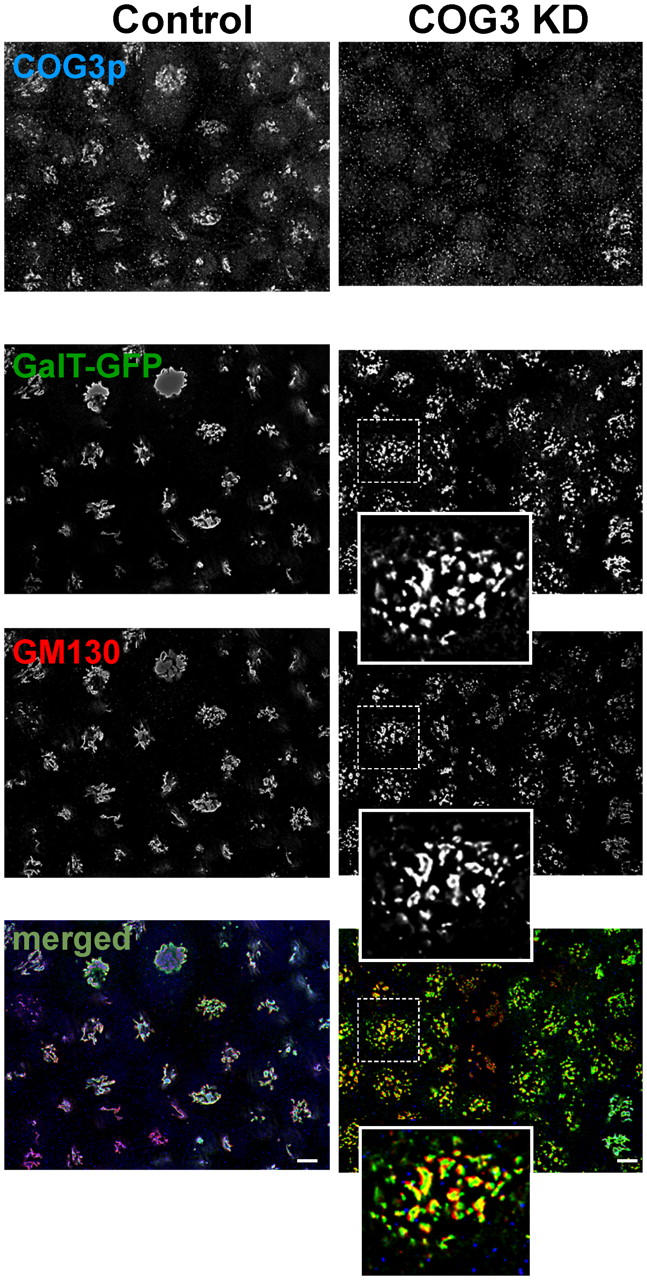
Cog3p depletion induces Golgi fragmentation. GalT-GFP HeLa cells were transfected with COG3 siRNA (right column) or mock transfected (left column). 72 h after transfection, cells were fixed and processed for IF with anti-Cog3p (COG3p row), and anti-GM130 (GM130 row) antibodies. The bottom row represent merged three-color images. Bars, 10 μm.
To test the specificity of COG3 siRNA KD, we took advantage of the fact that human and mouse Cog3 proteins show 95% identity and, therefore, most likely would functionally substitute each other. In the same time, human and mouse COG3 siRNA target region share only 74% of homology with five miss-matched nucleotides (Fig. 3 A) and this difference can be used in gene-replacement siRNA experiments (Puthenveedu and Linstedt, 2004). In good agreement with our prediction, in HeLa cells, that were cotransfected with both hCOG3 siRNA and the plasmid that expressed mouse Cog3p, the Golgi fragmentation was completely or partially prevented (Fig. 3 B). Similar results were obtained when cells were transfected with mCOG3-containing vector 24 h after hCOG3 siRNA transfection (unpublished data). We concluded that the COG3 KD is specific and that the Cog3p depletion directly induces a reversible Golgi fragmentation.
Figure 3.

Mouse COG3p expression partially suppresses hCOG3 KD-induced Golgi fragmentation. (A) DNA alignment of a portion of the hCOG3 and mCOG3 sequences. siRNA target region is highlighted in the box. (B) GalT-GFP HeLa cells were either mock transfected (control), transfected with COG3 siRNA (COG3 KD), or simultaneously transfected with COG3p siRNA and mCOG3-encoding plasmid (COG3 KD +mCOG3) and imaged 72 h after transfection. Bar, 10 μm.
To independently confirm an essential role for the COG complex in Golgi structure maintenance, we used affinity-purified antibodies against Cog3p (Suvorova et al., 2001). These antibodies were previously used for both IF and IP of the endogenous COG complex in mammalian cells (Suvorova et al., 2001; Ungar et al., 2002). We rationalized that upon microinjection into cells anti-Cog3p IgGs would specifically bind to the Cog3p and interfere with the COG complex function. Indeed, we have observed that 4 h after the antibodies microinjection the Golgi ribbon structure was converted into multiple mostly juxtanuclear localized small fragments (Fig. 4). This Golgi phenotype was persistent for up to 20 h after antibody microinjection without any visible signs of cell death. Microinjection with control antibodies did not change the morphology of the Golgi (unpublished data). We have concluded that anti-Cog3p antibodies like the COG3 siRNA act by blocking Cog3p function, which is necessary for the Golgi structure maintenance.
Figure 4.

Microinjection of anti-Cog3 antibodies disrupts Golgi structure. GalT-GFP HeLa cells were microinjected with anti-Cog3p IgGs and imaged immediately (0 min) or 4 h (240 min) after the injection. Texas red was used as an injection marker. Note that in cells injected with anti-Cog3 IgGs Golgi (labeled with asterisks) become fragmented. Microinjections with the preimmune IgGs did not result in Golgi fragmentation (date not depicted).
Loss of Cog3p expression results in accumulation of Golgi v-SNAREs and cis-Golgi resident protein GPP130 in nontethered vesicles
Detailed IF analysis of different Golgi resident proteins in COG3 KD cells revealed that the majority of integral and peripheral membrane Golgi proteins, including cis-Golgi tethering factors p115 (Nelson et al., 1998) and GM130 (Nakamura et al., 1995), cis-Golgi t-SNARE syntaxin 5 (Hay et al., 1998), cis/medial Golgi tethering protein giantin (Linstedt and Hauri, 1993), and trans-Golgi tether p230 (Brown et al., 2001), were present almost exclusively on relatively large (1–3 μm in size) fragmented Golgi membranes (Fig. 5 A; unpublished data). Surprisingly, we have found that a subset of Golgi proteins, intra-Golgi v-SNAREs GS15 (Xu et al., 2002) and GS28 (Hay et al., 1998), and cis-Golgi phosphoprotein GPP130 (Linstedt et al., 1997) were localized preferentially to multiple small vesicle-like structures distributed throughout the cytoplasm of Cog3p KD cells (Fig. 5 B). In control cells all these three proteins were primarily localized to a juxtanuclear Golgi.
Figure 5.
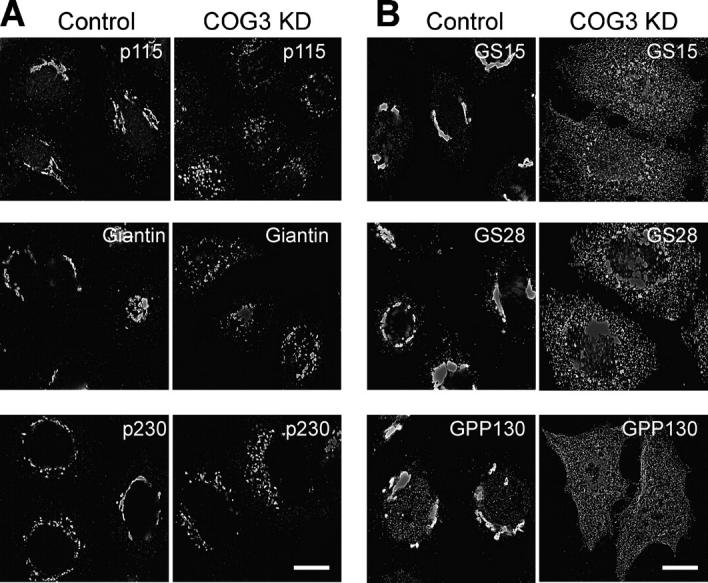
COG3 KD results in accumulation of multiple vesicles that carry v-SNAREs GS15, GS28, and cis-Golgi recycling protein GPP130. Control and COG3 KD cells were fixed 72 h after transfection and analyzed by IF using primary antibodies to indicated proteins and appropriate Alexa 488– and Alexa 595–conjugated secondary antibodies. (A) cis-Golgi tether p115, cis/medial Golgi marker giantin, and trans-Golgi tether p230 are present almost exclusively on Golgi ribbon in control cells and on fragmented juxtanuclear Golgi membranes in COG3 KD cells. (B) v-SNAREs GS15, GS28, and cis-Golgi marker GPP130 are Golgi located in control cells and predominantly localized on multiple small structures distributed throughout the COG3 KD cells. Bars, 10 μm.
Small GPP130-positive structures in COG3 KD cells presumably represent nontethered vesicles, because, in cells permeabilized with the mild detergent digitonin, these structures were efficiently washed away from cells, whereas large Golgi cisternae remained inside the cells (Fig. 6 A). Similar digitonin sensitivity was previously observed for Golgi-derived vesicles that are transiently accumulated during mitosis (Jesch and Linstedt, 1998).
Figure 6.
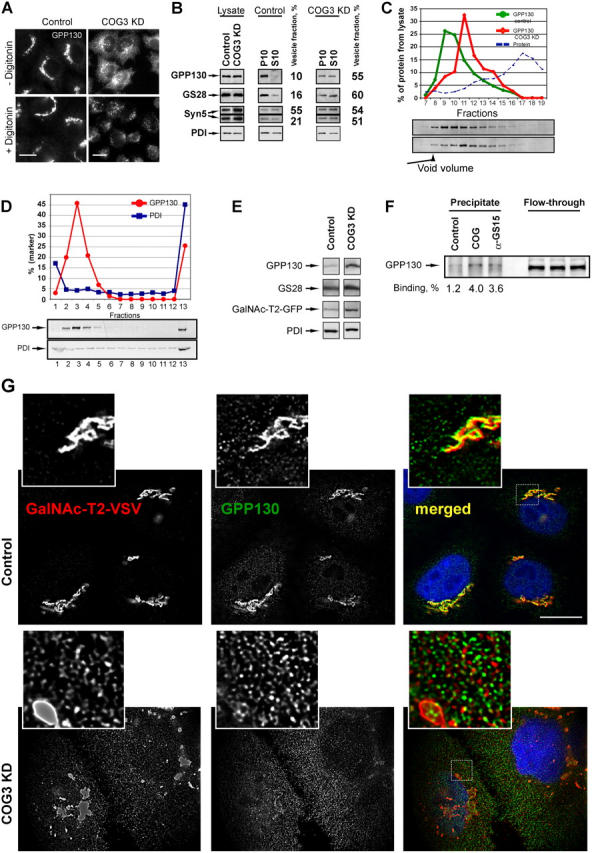
Characterization of CCD vesicles. (A) Apparent release of GPP130-containing vesicles upon digitonin permeabilization. Control or COG3 KD cells were either treated with buffer (−digitonin) or treated with 0.04 mg/ml digitonin, fixed, and analyzed by IF. Note that disperse vesicle-like staining of GPP130 in COG3 KD cells was eliminated after digitonin treatment. Bars, 10 μl. (B) WB analysis of total cell lysates and subcellular fractions prepared from COG3 KD and mock-treated HeLa cells. Cell lysates were fractionated on heavy membranes (P10) and light vesicle-containing fraction (S10). Equal amount of protein (10 μg) was loaded per lane and analyzed by WB with antibodies to GPP130, GS28, syntaxin 5, and PDI. A portion of marker proteins that was found in a vesicular fraction (n = 2) was determined by semi-quantitative WB. (C) Separation of GPP130-containing membranes by gel filtration. PNS from both control and COG3 KD cells was loaded on a Sephacryl S-1,000 column; 0.5-ml fractions were collected and analyzed by semi-quantitative WB. (D) Distribution of GPP130 and PDI on a velocity gradient. PNS from the COG3 KD cells was loaded on a 10–30% glycerol gradient. GPP130 and PDI were analyzed in fractions by semi-quantitative WB. Fraction 1 corresponds to the top of the gradient. (E) Analysis of CCD vesicle fraction. Fractions 3 and 4 from the glycerol gradient were concentrated by ultracentrifugation. Relative concentrations of Golgi and ER proteins were analyzed by WB as described in Materials and methods. (F) CCD vesicles specifically bind to the COG complex in vitro. PNS from COG3 KD cells was incubated with control beads, with beads loaded with the COG complex (COG), or with anti-GS15 IgGs (α-GS15). Precipitates were analyzed by WB with anti-GPP130 IgGs. (G) Accumulation of GalNAcT2 in CCD vesicles. Control or COG3 KD cells that stably express GalNAcT2-VSV were fixed and stained with mAbs to GPP130 or polyclonal anti–VSV-tag and secondary antibodies conjugated with Alexa 594 (GalNAc-T2-VSV) or Alexa 488 (GPP130) as described in Materials and methods. DNA was stained with DAPI. Images were acquired with 63× objective and deconvolved. Double IF labeling revealed that the vesicular pool of GalNAc-T2-VSV increased significantly in COG3 KD cells. Note that some vesicular profiles were double-labeled with both Golgi markers (insets). Bar, 10 μm.
To verify our IF findings we performed a subcellular fractionation of both COG3 KD and mock-treated HeLa cells (Fig. 6 B). We have found that in COG3 KD cells >50% of both GPP130 and GS28 proteins are present in a 10K supernatant, whereas in lysates obtained from control cells these proteins are almost exclusively cofractionated with large membranes. Noticeably, the long syntaxin 5 isoform and the ER resident protein PDI did not significantly change their subcellular distribution in a COG3 KD cells.
To characterize membranes in 10K supernatant we have first used gel-filtration analysis on a Sephacryl S-1000 column (Fig. 6 C). This analysis revealed that in COG3 KD cells the peak of GPP130 was eluted in fractions 11 and 12, whereas in control cells GPP130 was mostly found in fractions 9 and 10. We have concluded that in COG3 KD cells the GPP130 was mostly associated with small vesicles.
To test GPP130 localization by another separation technique we used a glycerol velocity gradient. Post-nuclear supernatant (PNS) from COG3 KD cells was loaded on 10–30% glycerol gradient and membranes were separated by size (Fig. 6 D). WB analysis of collected fractions revealed that majority of GPP130 signal was peaked in a vesicular fraction 3, whereas large ER membranes (PDI lane) were concentrated at the bottom of the gradient. A small fraction (∼25%) of GPP130 was also found at the bottom of the gradient and may represent a fraction of the protein that was associated with large Golgi fragments or vesicle aggregates. Because these vesicles were accumulated as a result of Cog3p depletion, we have named them a COG complex-dependent (CCD) vesicles. GPP130-containing Golgi membranes from identically fractionated control cells were rapidly pelleted to the bottom of the glycerol gradient (Jesch and Linstedt, 1998; unpublished data).
GPP130 is a heavily glycosylated cis-Golgi protein (Linstedt et al., 1997) that cycles through trans-Golgi and endosomal compartments (Puri et al., 2002) and rapidly degrades in both ΔCOG1 and ΔCOG2 CHO cell lines (Oka et al., 2004). We have found that both the protein level and the electrophoretic mobility of GPP130 were identical in both control and COG3-KD cells. Therefore, it can be concluded that in HeLa cells acute Cog3p depletion did not, at least initially, result in GPP130 down-regulation and did not alter its glycosylation.
To further analyze the content of CCD vesicles we prepared lysates from COG3 KD and control cells that stably express GFP-tagged N-acetylgalactosaminyltransferase-2 (GalNAc-T2). Both lysates were subjected to glycerol velocity gradient centrifugation, membranes from vesicle peak fractions 3 and 4 were concentrated and relative concentrations of Golgi and ER proteins were assessed by WB (Fig. 6 E). In an agreement with our IF data, we have found that in COG3 KD cells the amount of GPP130 in the CCD vesicle pool was increased by approximately fourfold. The concentrations of two other Golgi proteins, GS28 and GalNAc-T2-GFP were also increased by approximately twofold, whereas the amount of ER marker PDI remained unchanged. Small but detectable amounts of GPP130, GS28, and GalNAc-T2 in vesicular fractions prepared from control cells may represent a constitutive pool of recycling vesicles.
We also tested whether accumulated CCD vesicles could be recognized by the COG complex in vitro. The COG complex was purified from HeLa cells that transiently express YFP-Cog3p (Suvorova et al., 2001). Protein G–Agarose beads loaded with the COG complex were incubated with PNS fraction obtained from the COG3 KD cells. As a positive control for vesicle binding we used beads loaded with anti-GS15 antibodies and as a negative control for nonspecific binding we used protein G–Agarose beads. As expected, anti-GS15 beads efficiently pulled down GPP130-containing (Fig. 6 F, GS15 lane) and GS28-containing (unpublished data) vesicles. Importantly, the COG beads were also capable to precipitate GPP130-containing CCD vesicles (Fig. 6 F, compare lanes COG and control). We have concluded that CCD vesicles may directly bind to the COG complex and this interaction may be a fist step in vesicle tethering to the cis-Golgi.
Results of the subcellular fractionation experiments confirmed the IF data that showed accumulation of CCD vesicles in COG3 KD cells. It also raised an intriguing possibility that not only recycling SNARE proteins and GPP130, but also Golgi resident proteins like GalNAc-T2 may recycle in the COG complex-dependent manner. To test this hypothesis, we used HeLa cells that stably express VSV-tagged GalNAc-T2. It was shown previously that unlike the GFP-tag the small VSV-tag does not interfere with the GalNAc-T2 folding, function and localization (Storrie et al., 1998). Double IF labeling experiments revealed that the vesicular pool of GalNAc-T2-VSV increased significantly in COG3 KD cells (Fig. 6 G). In the same time, vesicularization of GalNAc-T2–containing Golgi region was not as dramatic as for GPP130-containing membranes and only a few vesicular profiles were double labeled with both Golgi markers. This may reflect different kinetics of GPP130 and GalNAc-T2 recycling as well as a possibility that different Golgi proteins may cycle in different membrane carries.
Does accumulation of CCD vesicles occur before or after COG3 KD-induced Golgi fragmentation? To answer this question we analyzed COG3 KD cells every 12 h after the initial COG3 siRNA transfection. We have found that 48 h after transfection both GS15 (Fig. 7) and GPP130 (unpublished data) were mostly found in CCD vesicles, whereas overall Golgi structure labeled with GalT-GFP has not yet been disturbed in a subpopulation of the COG3 KD cells. These data support the idea that accumulation of CCD vesicles may occur independently and before the Golgi fragmentation.
Figure 7.

Accumulation of CCD vesicles precedes COG3 KD-induced Golgi fragmentation. Control or COG3 KD cells that stably express GalT-GFP were fixed 48 h after siRNA transfection and stained with anti-GS15 and secondary antibodies conjugated with Alexa 594. Images were acquired with 63× objective and deconvolved. Note that the Golgi (arrows) has not yet been fragmented in a subpopulation of the COG3 KD cells, whereas the majority of GS15 was associated with multiple CCD vesicles. Bar, 10 μm.
Fragmented Golgi membranes are capable in supporting anterograde but not retrograde protein trafficking
Although siRNA-induced COG3 KD induced both accumulation of CCD vesicles and Golgi fragmentation, cells were able to multiply and their growth rate was not severely affected (unpublished data). Moreover, fragmented Golgi membranes maintained their juxtanuclear localization (Fig. 5 A). Detailed IF analysis revealed that cis-Golgi p115 (Nelson et al., 1998), and the medial Golgi GalNAc-T2 (Storrie et al., 1998), maintained their overlapping but distinct distribution on both control Golgi ribbon-like structure and Golgi fragments in COG3 KD cells (Fig. 8 A, GalNacT2/p115 frames). Similarly, localization of two medial Golgi proteins GalNAcT2 and giantin (Linstedt and Hauri, 1993) almost completely overlapped in both control and COG3 KD-treated cells (Fig. 8 A, GalNacT2/Giantin frames). And finally trans-Golgi localized p230 (Brown et al., 2001) maintained its relative localization in COG3 KD cells (Fig. 8 A, GalNacT2/p230 frames). We concluded that Cog3p KD resulted in Golgi fragmentation into multiple mini-Golgi stacks.
Figure 8.
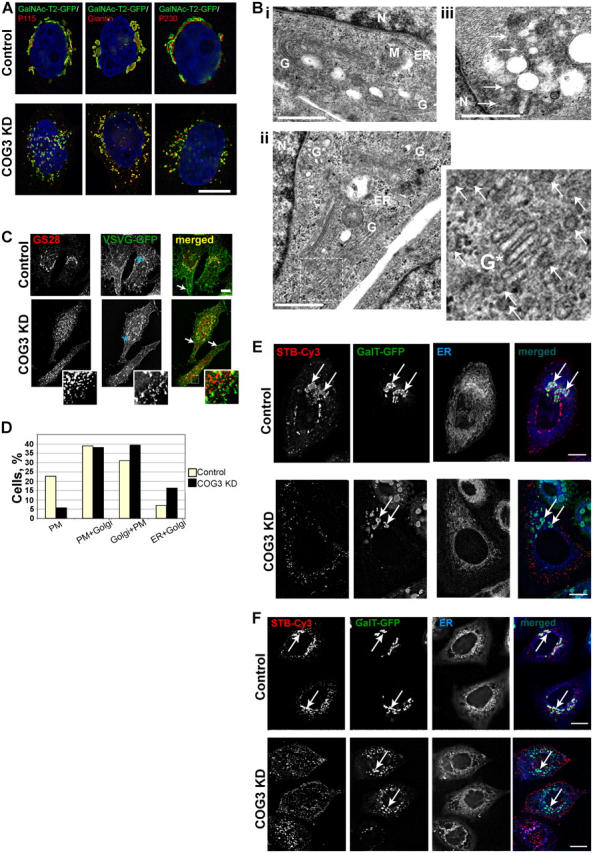
Golgi in COG3 KD cells is disrupted to mini-stacks, which are proficient in anterograde VSVG delivery to plasma membrane and defective in retrograde trafficking of Shiga toxin B subunit. (A) Control and COG3 KD cells that stably express GalNAcT2-GFP were fixed and stained with anti-p115, anti-giantin, or anti-p230 antibodies and secondary antibodies conjugated with Alexa 594. DNA was stained with DAPI. Images were acquired with 100× objective and deconvolved. Bar, 10 μm. (B) Ultrastructural analysis of Golgi in COG3 KD cells. Electron micrographs of the juxtanuclear region in control (i) and COG3 KD (ii and iii) cells. Note the Golgi mini-stack in ii and multiple 60-nm vesicles in COG3 KD cells (ii and iii, arrows). G, Golgi; M, mitochondria; N, nucleus; ER, endoplasmic reticulum. Bars, 1 μm. (C) Control and COG3 KD cells were transfected with the VSVG-GFP-ts045 vector. VSVG was accumulated in the ER for 16 h at 39.5°C. After that cells were transferred to 32°C, incubated for 2 h, fixed, and processed for IF with anti-GS28 antibodies. Both control and COG3 KD cells accumulated VSVG-GFP on the cell surface (merged images, arrows). Some VSVG was also found on GS28-positive Golgi membranes in control cells and on juxtanuclear Golgi fragments in COG3 KD cells. The major pool of GS28 was localized on a VSVG-GFP-negative CCD vesicles in COG3 KD cells (inset). Bar, 10 μm. (D) ∼100 cells in both control and COG3 KD samples were analyzed and each cell was assigned in specific group bases on VSVG localization profile. PM, VSVG localized only on the plasma membrane; PM+Golgi, VSVG localized mostly on the plasma membrane, but partially (<30%) on the Golgi; Golgi+PM, VSVG localized on the plasma membrane, but mostly on the Golgi; and ER+Golgi, accumulation of the VSVG in the ER. All images were acquired with 63× objective and deconvolved. (E) Retrograde trafficking of STB-Cy3. Control and COG3 KD cells that stably express GalT-GFP were pulse incubated with the STB-Cy3 as described in Materials and methods and STB was allowed to internalize for 2 h. Cells were fixed and ER was visualized with ER-Tracker. Note that majority of STB in control cells reached the Golgi (arrows), whereas in COG3 KD cells the STB-Cy3 signal was detected only on cell periphery. Bars, 10 μm. (F) Same as in E, except GalNAc-T2-GFP HeLa cells were used and STB-Cy3 was internalized for 12 h. Bars, 10 μm.
To confirm this conclusion the EM analysis of control and COG3 KD cells (Fig. 8) was performed. We found that in COG3 KD cells Golgi ribbon was disrupted onto multiple fragments, comprising three to four stacked cisternae (Fig. 8 B, ii). In addition to the Golgi fragments, a large number of ∼60-nm vesicles were observed in COG3 KD cells (Fig. 8 B, iii). Thus, Cog3p depletion leads to both vesiculation of Golgi and break up of the ribbon to multiple mini-stacks.
To test whether these Golgi mini-stacks can support two basic Golgi functions, anterograde and retrograde proteins flow, we used GFP-tagged vesicular stomatitis virus G protein (VSVG) as a cargo marker for anterograde protein trafficking (Suvorova et al., 2001) and Cy3-labeled subunit B of the Shiga toxin (STB-Cy3) as a marker for the retrograde trafficking (Johannes et al., 1997).
2 d after siRNA treatment both control and COG3 KD cells were transfected with the vector that directs synthesis of the temperature-sensitive VSVG-GFP protein (Beckers et al., 1987). VSVG was accumulated in the ER for 16 h at the restrictive temperature (39.5°C). After that cells were transferred to the permissive temperature (32°C) to allow VSVG to travel toward plasma membrane and fixed 2 h later. This time frame is sufficient for VSVG delivery from the ER to PM in HeLa cells (Suvorova et al., 2001). Indeed, we have observed that the majority of both control and COG3 KD cells accumulated significant amounts of VSVG on the cell surface (Fig. 8 C, merged images, arrowheads). Some VSVG was also found on GS28-positive Golgi membranes in control cells and on Golgi fragments in COG3 KD cells. In COG3 KD cells the major pool of GS28 was localized on VSVG-negative CCD vesicles (Fig. 8 C, inset).
Detailed analysis of >200 VSVG-GFP-positive cells revealed some minor trafficking defects and/or kinetic delays in the ER-PM delivery of VSVG in COG3 KD cells (Fig. 8 D). Significantly, more cells in a control group demonstrated complete delivery of VSVG to the plasma membrane (22% vs. 6% in COG3 KD cells) and twice as many cells in the COG3 KD group accumulated VSVG in the ER (16% vs. 7% in COG3 KD cells). Nevertheless, because the absolute majority (84%) of the COG3 KD cells were able to deliver at least some VSVG molecules to the cell surface, we concluded that fragmented Golgi is, at least partially, competent to support anterograde protein trafficking. This result is in agreement with previous findings (Ram et al., 2002; Suvorova et al., 2002; Bruinsma et al., 2004) that demonstrate the COG complex defects in yeast cells do not interfere directly with the anterograde protein trafficking.
To investigate if Cog3p-depleted Golgi mini-stacks can function in retrograde plasma membrane to ER protein trafficking we have analyzed STB-Cy3 trafficking by using fluorescent live cell microscopy. In agreement with the data obtained in many different labs (Sandvig et al., 1994; Johannes et al., 1997) STB was rapidly internalized. In the majority of control cells STB signal was detected in juxtanuclear region 2 h after beginning of initial internalization (Fig. 8 E). In control cells 12 h later STB was entirely distributed between the ER and the Golgi cisternae, marked by GalNAc-T2-GFP, (Fig. 8 F, control row). In the COG3 KD cells, even at the 12 h time point, the majority of internalized STB was localized in punctuate structures on cell periphery (Fig. 8 F, COG3 KD row). Some STB was detected in a perinuclear region of transfected cells but these STB-labeled membrane structures were clearly distinct from the GalNAc-T2-GFP–containing Golgi fragments. None of the STB was detected in the ER of COG3 KD cells (Fig. 8, E and F, ER frames) indicating that retrograde trafficking through the Cog3-depleted Golgi was blocked.
COG complex physically interacts with the Golgi SNARE molecules and the COP I vesicular coat
We have previously demonstrated that the yeast COG complex regulates intra-Golgi retrograde trafficking and interacts with the essential components of core machinery of vesicular Golgi traffic, i.e., intra-Golgi SNARE proteins and COPI vesicular coat (Suvorova et al., 2002). To test if the mammalian COG complex may also interact with both SNARE and COPI components, we isolated Golgi from the rat liver and performed an IP of the COG complex, v-SNARE GS28, and PDI from detergent-solubilized membranes. WB analysis of coIP proteins revealed that both GS28 (∼5% from total) and small amounts of t-SNARE Syntaxin5 were coprecipitated with the Cog3p (Fig. 9 A, lane 1; unpublished data). It was previously shown that Syntaxin5 and GS28 form a Golgi-localized SNARE complex (Hay et al., 1997) and our findings suggest that the COG complex can interact with either individual SNARE proteins or with the entire SNARE core complex. In the reciprocal precipitation, Cog3p was coIP by anti-GS28 antibodies (Fig. 9 A, lane 4). Neither Cog3p nor the Golgi SNARE molecules were precipitated with control beads (Fig. 9 A, lane 3). We have also determined that the β subunit of the COPI coat was specifically precipitated with anti-Cog3p antibodies (Fig. 9 B, lane 2). These results indicated that the mammalian COG complex like its yeast homologue could physically interact with both vesicular and Golgi-localized components of membrane transport machinery.
Figure 9.
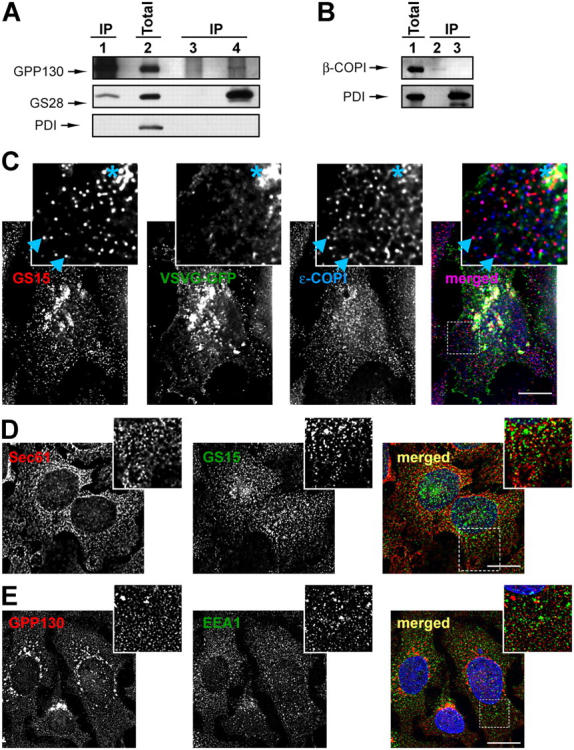
The COG complex interacts with retrograde Golgi SNARE GS28 and β-COPI. (A and B) Protein complexes from detergent-solubilized rat liver Golgi were IP using anti-Cog3p (A, lane 1; B, lane 2), anti-GS28 (A, lane 4), preimmune IgGs (A, lane 3), or anti-PDI (B, lane 3). 10% of the Golgi lysates were loaded as a control (A, lane 2; B, lane 1). Note that Cog3p, GS28, and β-COPI were not recovered with control beads or beads loaded with anti-PDI antibodies (A, lane 3; B, lane 3). (C) CCD vesicles are partially COPI coated and do not carry VSVG. COG3 KD cells that express VSVG-GFP were fixed and processed for IF with mouse anti-GS15 antibodies and rabbit anti–ɛ-COPI antibodies as described in Materials and methods. All images were acquired with 63× objective and deconvolved. Note that a number of GS15-labeled CCD vesicles were colabeled with antibodies to ɛ-COPI coat (inset, arrowheads). VSVG was partially colocalized with the GS15 only on a fragmented Golgi membrane (inset, membranes labeled with asterisk), but not on CCD vesicles. Bar, 10 μm. (D) CCD vesicles do not significantly colocalize with the ER. COG3 KD cells were fixed and stained with rabbit anti-Sec61p (red) and mouse anti-GS15 IgGs (green). Bar, 10 μm. (E) CCD vesicles are distinct from early endosomes. COG3 KD cells were fixed and stained with rabbit IgGs to GPP130 (red) and mouse anti-EEA1 IgGs (green). Bar, 10 μm.
IF experiments indicated that CCD vesicles are distinct from both ER (Fig. 9 D) and early endosomes (Fig. 9 E). To test whether CCD vesicles are COPI-coated we performed double-IF in COG3 KD VSVG-GFP expressing cells (Fig. 9 C). A number of vesicle profiles were labeled with CCD vesicle marker GS15 and some were colabeled with antibodies against the epsilon subunit of COPI coat (Fig. 9 C, inset, arrowheads). In contrast, VSVG was colocalized with the GS15 only on a fragmented Golgi membrane (Fig. 9 C, inset, membranes marked with an asterisk). These data indicated that CCD vesicles do not carry anterograde cargo molecules and most likely represent COPI-coated retrograde intra-Golgi vesicles that bud from distal Golgi compartments and subsequently tether to the cis-Golgi in a COG complex-dependent manner.
Discussion
Although both genetic and biochemical analysis indicated that the COG complex primarily regulates retrograde intra-Golgi membrane trafficking in budding yeast (Ram et al., 2002; Suvorova et al., 2002), the cellular role of the mammalian COG complex is less clear. COG has been implicated in the anterograde intra-Golgi trafficking (Walter et al., 1998), ER to Golgi protein delivery (Loh and Hong, 2002), glycoconjugate synthesis, intracellular protein sorting, and protein secretion (Kingsley et al., 1986; Chatterton et al., 1999; Wu et al., 2004). The most evolutionary conserved COG subunits bear 21–23% identity between yeasts and humans (Whyte and Munro, 2002). Therefore, it is hard to make any functional predictions that are simply based on protein similarities.
In this study, we report that the acute depletion of the Cog3p subunit of the human COG complex results in accumulation of nontethered transport vesicles, dramatic changes in overall Golgi structure and block of Shiga toxin retrograde trafficking. We have also obtained microscopic evidences that in COG3 KD cells the fragmented Golgi maintains its cisternal organization and that these Golgi mini-stacks are capable, at least partially, to support anterograde protein delivery from the ER to the plasma membrane. Finally, our data suggest that the COG complex can directly interact with the CCD retrograde vesicles via binding to both vesicular COPI coat and integral components of vesicular SNARE machinery.
The COG3 KD differentially influences the protein level of other COG subunits. Although the level of Cog1p, Cog2p, and Cog4p is reduced more than twofold, the expression of the other four subunits is not affected. Moreover Cog5-8p continue to localize properly to membranes in a juxtanuclear region. These data support our original model proposing two-lobed COG complex structure (Ungar et al., 2002; Loh and Hong, 2004) and suggests that Lobe B (COGs 5–8) may be attached to the Golgi membrane by its own receptor.
COG3 KD-induced Golgi fragmentation is specific and reversible because either simultaneous or subsequent introduction of the mouse siRNA-insensitive version of the Cog3p is able to restore a wild-type Golgi appearance. We have also demonstrated that the disruption of the Golgi structure is observed in cells microinjected with anti-Cog3p IgGs. The most likely explanation of this phenomenon is that the antibody microinjection induces the proteolytic degradation of the Cog3p similarly to the recently described antibody-induced degradation of the Golgi tethering factor p115 (Puthenveedu and Linstedt, 2001).
Observed Golgi fragmentation phenotype induced by the COG3 KD is distinct from the Golgi appearance in ΔCOG1 and ΔCOG2 CHO cells (Ungar et al., 2002) and indicate that the acute COG3 KD, at least transiently, disrupts normal structural organization of the Golgi complex. In the majority of COG3 KD cells fragmented Golgi membranes retain their juxtanuclear localization and their cisternal organization appear to be normal both at the light microscopy and the EM levels. The Golgi mini-stacks are capable to carry out a plasma membrane delivery of VSVG, though with reduced efficiency compared with a normal Golgi apparatus. This observation sharply differs from the effect of p115 KD on VSVG trafficking (Puthenveedu and Linstedt, 2004); it may reflect different roles of two cis-Golgi localized tethering factors in membrane trafficking. Moreover, in COG3 KD cells p115 is properly expressed (unpublished data) and its localization on the Golgi membranes is virtually undisturbed. Only a small fraction of the COG3 KD cells accumulates VSVG in the ER. These results agree with our previous data that yeast COG mutants do not show any primary anterograde trafficking defects (Suvorova et al., 2002) and that protein secretion in both ΔCOG1 and ΔCOG2 CHO cells is not compromised (Kingsley et al., 1986).
Specific vesicle accumulation is a most striking phenotype observed after the COG3 KD. These CCD vesicles seem to carry at least two different Golgi SNARE molecules, GS15 and GS28, and GPP130, a 130-kD, cis-Golgi protein that continuously cycle between the early Golgi and distal compartments (Linstedt et al., 1997). Significantly, it has been shown that these proteins are rapidly degraded in both ΔCOG1 and ΔCOG2 CHO cells (Oka et al., 2004). The most likely explanation is that after the acute COG3 KD, all three markers are transiently accumulated in nontethered recycling vesicles that are rapidly degraded. We have found that 90 h after COG3 KD the protein level of both GPP130 and GS28 was decreased (unpublished data). A number of Golgi resident proteins, including MG160 (Johnston et al., 1994), GP73 (Puri et al., 2002), GlcNAc T-1, and α-1,2 mannosidase II (Opat et al., 2001a,b) are shown to cycle through the trans-Golgi region. The prediction is that in COG3 KD cells all these proteins will, at least transiently, be accumulated in CCD vesicles.
What is the origin of CCD vesicles? IF data indicated that the major pool of CCD vesicle is clearly distinct from both ER and early endosomes. Some of CCD vesicles are likely to originate from the trans-Golgi because both GS28 and GS15 are SNARE molecules that function in the intra-Golgi trafficking (Xu et al., 2002; Volchuk et al., 2004). Alternatively, some CCD vesicles could originate from TGN/sorting endosomal/recycling compartments. GPP130 was shown to cycle through the trans-Golgi/early endosomal membranes (Linstedt et al., 1997) and both GS28 and GS15 could participate in the early endosomal SNARE complex (Tai et al., 2004). Current models of Golgi trafficking involving cisternal maturation (Pelham, 2001) predict that all Golgi residents move down the stack to the late Golgi. Indeed, a number of cis-Golgi residents in both yeast (Harris and Waters, 1996) and mammalian cells (Johnston et al., 1994; Bachert et al., 2001; Opat et al., 2001a) have been shown to rapidly acquire specific modifications of the late Golgi. The rapid rate of trans-Golgi modifications (Opat et al., 2001a) and relatively slow rate of the recycling through the ER (Miles et al., 2001) suggest that cis- and medial-Golgi resident proteins are recycled directly from the trans-Golgi and/or TGN to the corresponding early Golgi compartment. The COG complex localizes on cis/medial Golgi membranes (Suvorova et al., 2001; Ungar et al., 2002) and most likely regulates recycling of different resident Golgi proteins. We and others have shown that mutations in yCOG3 resulted in abnormal Golgi recycling of yeast Golgi proteins Sec22p and Och1p (Suvorova et al., 2002; Bruinsma et al., 2004).
CCD carriers are most likely to be formed in a COPI-dependent reaction. COPI vesicles bud from all Golgi cisternae and COPI is required for intra-Golgi transport in vitro (Orci et al., 1997). In the in vitro assay, Golgi resident proteins mannosidase II and GS28, were found to be concentrated in COPI vesicles (Lanoix et al., 1999, 2001), and these transport intermediates were able to fuse with cis-Golgi compartment (Love et al., 1998). The COG complex is a good candidate to orchestrate COPI vesicle tethering. Indeed, genetic and biochemical connections between yeast COG and COPI complexes (Ram et al., 2002; Suvorova et al., 2002) have been shown. Oka et al. (2004) reported recently that synthetic phenotypes arose in mutants deficient in both epsilon-COPI and either COG1 or COG2. During the investigation, we have found significant accumulation of COPI-positive vesicular profiles in COG3 KD cells (unpublished data) and demonstrated that some of these vesicles are double-labeled with the CCD vesicle cargo GS15. Finally we have shown that the COG complex binds to CCD vesicles in vitro and could be coIP with the β-COPI.
In conclusion we propose a model (Fig. 10) in which the cis-Golgi localized COG complex acts as a tether for retrograde COPI coated CCD vesicles that originate from distal trans-Golgi/endosomal compartments. The acute COG3 KD and corresponding defects in the Lobe A of the COG complex discontinue normal vesicle recycling. Non-tethered vesicles are transiently accumulated in cell cytoplasm as membrane-depleted Golgi ribbon is fragmented in multiple Golgi mini-stacks. Detailed biochemical analysis of CCD vesicles and the elucidation of exact roles of both lobes of the COG complex should help in our understanding of mechanisms of Golgi maintenance and function.
Figure 10.

Model for the COG complex function in membrane trafficking. Cis-Golgi localized COG complex acts a tether for retrograde COPI-coated CCD vesicles that originate from the trans-Golgi/endosomal compartment(s). The COG3 KD abolishes vesicle tethering to the cis-Golgi. As a result multiple nontethered vesicles are transiently accumulated in cell cytoplasm and the membrane-depleted Golgi ribbon is fragmented into multiple Golgi mini-stacks.
Materials and methods
Reagents and antibodies
Most laboratory reagents were purchased from Sigma-Aldrich. Antibodies used for WB and IF studies were obtained from standard commercial sources and as gifts from generous individual investigators or generated by us (see below). Antibodies (and their dilutions) were as follows: rabbit pAbs: anti-Cog1 (Ungar et al., 2002), anti-Cog2p (Podos et al., 1994; Ungar et al., 2002), anti-Cog3p (Suvorova et al., 2001), anti-Cog4p (Ungar et al., 2002), anti-Cog5p (Walter et al., 1998), anti-Cog6p, anti-Cog7p, anti-Cog8p (Ungar et al., 2002), anti-GPP130 (Covance), anti-giantin (a gift from A.D. Linstedt, Carnegie Mellon University, Pittsburgh, PA), anti-p115 (a gift from M.G. Waters, Merck Research Laboratories, Rahway, NJ), anti–ɛ-COPI (a gift from R. Duden, Royal Holloway University of London, Egham, Surrey), rabbit pAb and monoclonal (18C8) anti-syntaxin 5 (a gift from J. Hay, University of Michigan, Ann Arbor, MI), anti–β-COPI (Sigma-Aldrich), anti-VSVG tag (E11; Delta Biolabs); murine mAbs: anti-GPP130 (A1-118; a gift from A. Linstedt), anti-GM130 (BD Biosciences), anti–GS-28 (BD Biosciences), anti-GS15 (BD Biosciences), anti-p230 (BD Biosciences), and anti-PDI (Affinity BioReagents).
Mammalian cell culture, plasmids, and transfection
Monolayer HeLa cells were cultured in DME/F-12 media supplemented with 15 mM Hepes, 2.5 mM l-glutamine, 5% FBS, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B. Cells were grown at 37°C and 5% CO2 in a humidified chamber. HeLa cells that stably expressed GalNAc-T2 fused to GFP or to a VSV-tag and GalT fused to GFP were provided by B. Storrie (University of Arkansas for Medical Sciences, Little Rock, AR; Storrie et al., 1998). The plasmid encoding VSVGtsO45-GFP was obtained from M.A. McNiven (Mayo Clinic, Rochester, MN). The plasmid pYFP-hCOG3 was described previously (Suvorova et al., 2001). pCMV-SPORT6 with cDNA of mCOG3 (clone ID 4020725; NCBI Accession BC038030) was obtained from Invitrogen and mCOG3 sequence was verified by sequencing. Transfections were performed using TransIT-HeLaMONSTER Kit (Mirus Corporation).
RNA interference experiment
Human COG3 was targeted with a siRNA duplex (target sense, AGACTTGTGCAGTTTAACA). siRNAs were obtained as purified duplexes obtained from Dharmacon Research. Transfection was performed using Oligofectamine (Invitrogen) following the protocol recommended by Invitrogen. For WB, cells were plated in 24-well dishes, grown to a confluency of ∼70%, transfected with siRNA for 24–72 h, and lysed in the SDS-PAGE sample buffer.
IF microscopy
IF microscopy was performed using an epifluorescence microscope (Axiovert 200; Carl Zeiss Microimaging, Inc.) with a Plan-Apochromat 63× oil immersion lens (NA 1.4) at RT. The secondary antibodies conjugated to Alexa Fluor 488 or 594 were obtained from Molecular Probes, Inc. The images were obtained using a QImaging Retiga Fast-EXi camera that was controlled via IP Lab software. During the processing stage, individual image channels were pseudocolored with RGB values corresponding to each of the fluorophore emission spectral profiles. Images were cropped using Adobe Photoshop software. Where indicated, images were digitally deconvolved (Huygens Professional, Scientific Volume Imaging).
Live cell fluorescence microscopy
For live cell fluorescence microscopy, cells were cultured in Lab-TekII Chambered Coverglass System (Nalge Nunc). Cells were maintained on the microscope stage in a chamber at RT in DME/F12 without phenol red and sodium pyruvate (Invitrogen).
For analysis of anterograde transports both COG3 KD and mock-treated HeLa cells 60 h after siRNA treatment were transfected with a vector that encoded VSVGts045-GFP protein and were kept at the 39.5°C for 16 h (Suvorova et al., 2001). After that the temperature was reduced to permissive one (32°C) to allow the VSVG to travel toward the plasma membrane. After 2 h of chase, cells were fixed and stained for GS28.
For analysis of retrograde transport Cy3-Shiga toxin B subunit (STB-Cy3; Mallard et al., 1998) was used. STB-Cy3 was a gift from B. Storrie. 60 h after the start of COG3 KD HeLa cells that stably express the Golgi markers GalT-GFP or GalNacT2-GFP were incubated with STB-Cy3 for 30 min at 4°C, washed, refueled with fresh DME medium without phenol red, and incubated for 2 or 12 h at 37°C and 5% CO2 in a humidified chamber. ER was visualized with the ER-Tracker blue-white PDX (Molecular Probes). Images were obtained as described above.
EM
Cells were fixed with 3% glutaraldehyde in PBS for 2 h at 4°C. Cells were washed three times with PBS, fixed with 2% OsO4 in PBS, washed again, dehydrated in a graded ethanol series, and embedded in Epon 812. Ultrathin sections were obtained using RMC MT-7000 microtome, double stained with uranyl acetate and lead citrate. Specimens were examined using JEOL JEM-1010 electron microscope at 80 kV with magnification ranging from 15,000 to 75,000×.
Microinjection of anti-COG3 antibodies
HeLa cells that stably expressed GalT-GFP were microinjected with anti-Cog3p IgGs with the Narishigee Micromanipulator and imaged immediately and 4 h after the injection using a 40×, 1.3 NA Fluor objective fitted to a confocal microscope (model LSM410; Carl Zeiss MicroImaging, Inc.). The antibody concentration was 2 mg/ml, Texas red was used as an injection marker. Microinjection of the preimmune IgGs was used as the control.
Cell fractionation and preparation of CCD vesicles
HeLa cells were cultured to ∼80% confluence in 60-mm dishes and transfected with COG3 siRNA. 78 h after transfection (KD efficiency of Cog3p ∼80%), cells were washed three times with PBS and then scraped in 0.3 ml of 20 mM Hepes-KOH buffer, pH 7.4, supplemented with a proteinase inhibitor cocktail (Roche Diagnostics Corporation) on ice. The cells were disrupted using a Potter homogenizer. Subcellular fractions were obtained by standard differential centrifugation. The PNS was obtained at 500 g (5 min, 3°C). Heavy membranes, including ER and the Golgi were pelleted at 10,000 g (10 min, 3°C). Light membranes, including transport vesicle were obtained by centrifugation conducted in a rotor (model TLA-100; Beckman Coulter) at 100,000 g (1 h, 3°C). All membrane pellets were resuspended in 2% SDS in volumes equal to volume of original lysate. Membrane and cytoplasmic proteins were denatured by heating at 95°C for 5 min. To normalize the sample loading for WB analysis, protein content was measured using the BCA reagent (Pierce Chemical Co.).
Glycerol velocity centrifugation and gel filtration
Gradient fractionation was prepared as described previously (Jesch and Linstedt, 1998) with some modification. To prepare the lysate, 72 h COG3 KD HeLa cells from one 10-cm plate were collected by trypsinization, pelleted (500 g for 5 min), then washed once in PBS, and once in STE buffer (250 mM sucrose, 10 mM triethylamine, pH 7.4, 1 mM EDTA, with protease inhibitors), homogenized by 20 passages through a 25-gauge needle in 0.5-ml buffer STE without sucrose, and then centrifuged at 1,000 g for 2 min to obtain PNS. This supernatant was used for both gradient fractionation and gel filtration. PNS (1 ml) was layered on linear 10–30% (wt/vol) glycerol gradients (12 ml in 10 mM triethylamine, pH 7.4, and 1 mM EDTA on a 0.5 ml 80% sucrose cushion) and centrifuged at 280,000 g for 60 min in a SW40 Ti rotor (Beckman Coulter).
1 ml fractions were collected from the top. All steps were performed at 4°C. 50 μl of each fraction was combined with sample buffer, then loaded on SDS-PAGE and analyzed by WB.
For gel filtration analysis, PNS was loaded onto 29 × 0.7 cm Sephacryl S-1000 gel filtration column. Material was eluted in STE buffer at flow rate of 0.3 ml/h. 0.5-ml fractions were collected and analyzed by WB.
Interaction of the COG complex with CCD vesicles
The COG complex was isolated from HeLa cells that express YFP-Cog3p (Suvorova et al., 2001). Cells from two 10-cm plates were homogenized in equal volumes of cold CTN buffer (2% CHAPS, 40 mM Tris-HCl, pH 7.4, 300 mM NaCl, protease inhibitor cocktail) on ice. Cell lysates were clarified by centrifugation at 20,000 g for 10 min. The supernatant was transferred into a new presiliconized tube, diluted twice with TBST (TBS with 0.05% Tween 20), and incubated with 50 μl of protein A Sepharose CL-4B (Amersham Biosciences) on a tube rotator for 30 min, then beads were sedimented at 500 g (1 min, 3°C). The supernatant was transferred in a new tube, 2 μg of anti-GFP antibodies were added, and the tube was incubated on the tube rotator for 4 h in a cold room. After that 30 μl of protein G–Agarose beads were added to the samples and incubated for 1 h. Beads were sedimented by centrifugation at 110 g for 1 min and washed four times with TBST. The COG complex-loaded beads were transferred to a new tube, washed again and finally resuspended in 60 μl of TBST. 20 μl of beads were tested for the presence of YFP-Cog3p and other COG subunits by WB.
40 μl of COG complex beads were incubated for 4 h with the CCD vesicles enriched supernatant from the COG3 KD cells. Beads were washed three times and eluted with the sample buffer. Similar Incubation with untreated protein G–agarose beads or protein G–agarose beads preloaded with anti-GS15 antibodies was used as a control.
IP experiments using rat liver Golgi membranes
Rat liver Golgi membranes were purified as described by Hamilton et al. (1991) and frozen in aliquots in liquid nitrogen. All of the following operations were performed in a cold room. Membranes were thawed on ice in an equal volume of cold CTN buffer and incubated for 30 min. 50 μl of protein A Sepharose in TBS was added and incubated on a tube rotator for 30 min. Insoluble material and beads were pelleted at 20,000 g for 10 min at 3°C. The supernatant was transferred to a new tube and diluted twice with TBST. 2 μg of anti-Cog3p, anti-GS28 or anti-PDI antibodies were added to the diluted samples and incubated on tube rotator for 4 h. 20 μl of protein G agarose were added to the samples and incubated for another hour. After incubation the beads were sedimented by low speed centrifugation at 110 g for 1 min and washed four times with TBST. After that beads were transferred to a new tube, resuspended in 30 μl of 2× sample buffer, heated for 5 min at 95°C, loaded on SDS-PAGE, and analyzed by WB.
SDS-PAGE and Western blotting
SDS-PAGE and WB were performed as described by Suvorova et al. (2002). A signal was detected using a chemiluminescence reagent kit (PerkinElmer Life Sciences) and quantitated using ImageJ software (http://rsb.info.nih.gov/ij/).
Acknowledgments
We thank B. Storrie and R. Kurten for help with IF and microinjections and M. Crocker for great help with EM. We are very grateful to O. Pavliv for excellent technical assistance and to A. Shestakova for valuable comments on this manuscript. We also thank R. Duden, J. Hay, F. Hughson (Princeton University, Princeton, NJ), M. Krieger (Massachusetts Institute of Technology, Cambridge, MA), A. Linstedt, D. Ungar (Princeton University), and others who provided antibodies and other valuable reagents.
This work was supported by grants from the National Science Foundation (MCB-0234822) and the Department of Defense (DAMD17-03-1-0243).
Abbreviations used in this paper: CCD, COG complex-dependent; COG, conserved oligomeric Golgi; COPI, vesicular coat complex I; GalNAc-T2, N-acetylgalactosaminyltransferase-2; GalT-GFP, GFP-tagged β 1,4-galactosyltransferase; IF, immunofluorescence; IP, immunoprecipitation; KD, knockdown; PDI, protein disulphide isomerase; PNS, post-nuclear supernatant; siRNA, short interfering RNA; VSVG, vesicular stomatitis virus G protein; WB, Western blot.
References
- Bachert, C., T.H. Lee, and A.D. Linstedt. 2001. Lumenal endosomal and Golgi-retrieval determinants involved in pH-sensitive targeting of an early Golgi protein. Mol. Biol. Cell. 12:3152–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers, C.J., D.S. Keller, and W.E. Balch. 1987. Semi-intact cells permeable to macromolecules: use in reconstitution of protein transport from the endoplasmic reticulum to the Golgi complex. Cell. 50:523–534. [DOI] [PubMed] [Google Scholar]
- Brown, D.L., K. Heimann, J. Lock, L. Kjer-Nielsen, C. van Vliet, J.L. Stow, and P.A. Gleeson. 2001. The GRIP domain is a specific targeting sequence for a population of trans-Golgi network derived tubulo-vesicular carriers. Traffic. 2:336–344. [DOI] [PubMed] [Google Scholar]
- Bruinsma, P., R.G. Spelbrink, and S.F. Nothwehr. 2004. Retrograde transport of the mannosyltransferase Och1p to the early Golgi requires a component of the COG transport complex. J. Biol. Chem. 279:39814–39823. [DOI] [PubMed] [Google Scholar]
- Chatterton, J.E., D. Hirsch, J.J. Schwartz, P.E. Bickel, R.D. Rosenberg, H.F. Lodish, and M. Krieger. 1999. Expression cloning of LDLB, a gene essential for normal Golgi function and assembly of the ldlCp complex. Proc. Natl. Acad. Sci. USA. 96:915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duden, R. 2003. ER-to-Golgi transport: COP I and COP II function (Review). Mol. Membr. Biol. 20:197–207. [DOI] [PubMed] [Google Scholar]
- Elbashir, S.M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 411:494–498. [DOI] [PubMed] [Google Scholar]
- Farkas, R.M., M.G. Giansanti, M. Gatti, and M.T. Fuller. 2003. The Drosophila Cog5 homologue is required for cytokinesis, cell elongation, and assembly of specialized Golgi architecture during spermatogenesis. Mol. Biol. Cell. 14:190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, R.L., A. Moorehouse, and R.J. Havel. 1991. Isolation and properties of nascent lipoproteins from highly purified rat hepatocytic Golgi fractions. J. Lipid Res. 32:529–543. [PubMed] [Google Scholar]
- Harris, S.L., and M.G. Waters. 1996. Localization of a yeast early Golgi mannosyltransferase, Och1p, involves retrograde transport. J. Cell Biol. 132:985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay, J.C., D.S. Chao, C.S. Kuo, and R.H. Scheller. 1997. Protein interactions regulating vesicle transport between the endoplasmic reticulum and Golgi apparatus in mammalian cells. Cell. 89:149–158. [DOI] [PubMed] [Google Scholar]
- Hay, J.C., J. Klumperman, V. Oorschot, M. Steegmaier, C.S. Kuo, and R.H. Scheller. 1998. Localization, dynamics, and protein interactions reveal distinct roles for ER and Golgi SNAREs. J. Cell Biol. 141:1489–1502. (published erratum appears in J. Cell Biol. 1998. 42:following 881). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesch, S.A., and A.D. Linstedt. 1998. The Golgi and endoplasmic reticulum remain independent during mitosis in HeLa cells. Mol. Biol. Cell. 9:623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes, L., D. Tenza, C. Antony, and B. Goud. 1997. Retrograde transport of KDEL-bearing B-fragment of Shiga toxin. J. Biol. Chem. 272:19554–19561. [DOI] [PubMed] [Google Scholar]
- Johnston, P.A., A. Stieber, and N.K. Gonatas. 1994. A hypothesis on the traffic of MG160, a medial Golgi sialoglycoprotein, from the trans-Golgi network to the Golgi cisternae. J. Cell Sci. 107:529–537. [DOI] [PubMed] [Google Scholar]
- Kingsley, D.M., K.F. Kozarsky, M. Segal, and M. Krieger. 1986. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 102:1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoix, J., J. Ouwendijk, C.C. Lin, A. Stark, H.D. Love, J. Ostermann, and T. Nilsson. 1999. GTP hydrolysis by arf-1 mediates sorting and concentration of Golgi resident enzymes into functional COP I vesicles. EMBO J. 18:4935–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoix, J., J. Ouwendijk, A. Stark, E. Szafer, D. Cassel, K. Dejgaard, M. Weiss, and T. Nilsson. 2001. Sorting of Golgi resident proteins into different subpopulations of COPI vesicles: a role for ArfGAP1. J. Cell Biol. 155:1199–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt, A.D., and H.P. Hauri. 1993. Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell. 4:679–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt, A.D., A. Mehta, J. Suhan, H. Reggio, and H.P. Hauri. 1997. Sequence and overexpression of GPP130/GIMPc: evidence for saturable pH-sensitive targeting of a type II early Golgi membrane protein. Mol. Biol. Cell. 8:1073–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh, E., and W. Hong. 2002. Sec34 is implicated in traffic from the endoplasmic reticulum to the Golgi and exists in a complex with GTC-90 and ldlBp. J. Biol. Chem. 277:21955–21961. [DOI] [PubMed] [Google Scholar]
- Loh, E., and W. Hong. 2004. The binary interacting network of the conserved oligomeric Golgi (COG) tethering complex. J. Biol. Chem. 279:24640–24648. [DOI] [PubMed] [Google Scholar]
- Love, H.D., C.C. Lin, C.S. Short, and J. Ostermann. 1998. Isolation of functional Golgi-derived vesicles with a possible role in retrograde transport. J. Cell Biol. 140:541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallard, F., C. Antony, D. Tenza, J. Salamero, B. Goud, and L. Johannes. 1998. Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of shiga toxin B-fragment transport. J. Cell Biol. 143:973–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles, S., H. McManus, K.E. Forsten, and B. Storrie. 2001. Evidence that the entire Golgi apparatus cycles in interphase HeLa cells: sensitivity of Golgi matrix proteins to an ER exit block. J. Cell Biol. 155:543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, N., C. Rabouille, R. Watson, T. Nilsson, N. Hui, P. Slusarewicz, T.E. Kreis, and G. Warren. 1995. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 131:1715–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, D.S., C. Alvarez, Y.S. Gao, R. Garcia-Mata, E. Fialkowski, and E. Sztul. 1998. The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J. Cell Biol. 143:319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka, T., D. Ungar, F.M. Hughson, and M. Krieger. 2004. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol. Biol. Cell. 15:2423–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opat, A.S., F. Houghton, and P.A. Gleeson. 2001. a. Steady-state localization of a medial-Golgi glycosyltransferase involves transit through the trans-Golgi network. Biochem. J. 358:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opat, A.S., C. van Vliet, and P.A. Gleeson. 2001. b. Trafficking and localisation of resident Golgi glycosylation enzymes. Biochimie. 83:763–773. [DOI] [PubMed] [Google Scholar]
- Orci, L., M. Stamnes, M. Ravazzola, M. Amherdt, A. Perrelet, T.H. Sollner, and J.E. Rothman. 1997. Bidirectional transport by distinct populations of COPI-coated vesicles. Cell. 90:335–349. [DOI] [PubMed] [Google Scholar]
- Pelham, H.R. 2001. Traffic through the Golgi apparatus. J. Cell Biol. 155:1099–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podos, S.D., P. Reddy, J. Ashkenas, and M. Krieger. 1994. LDLC encodes a brefeldin A-sensitive, peripheral Golgi protein required for normal Golgi function. J. Cell Biol. 127:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri, S., C. Bachert, C.J. Fimmel, and A.D. Linstedt. 2002. Cycling of early Golgi proteins via the cell surface and endosomes upon lumenal pH disruption. Traffic. 3:641–653. [DOI] [PubMed] [Google Scholar]
- Puthenveedu, M.A., and A.D. Linstedt. 2001. Evidence that Golgi structure depends on a p115 activity that is independent of the vesicle tether components giantin and GM130. J. Cell Biol. 155:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu, M.A., and A.D. Linstedt. 2004. Gene replacement reveals that p115/SNARE interactions are essential for Golgi biogenesis. Proc. Natl. Acad. Sci. USA. 101:1253–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram, R.J., B. Li, and C.A. Kaiser. 2002. Identification of sec36p, sec37p, and sec38p: components of yeast complex that contains sec34p and sec35p. Mol. Biol. Cell. 13:1484–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig, K., M. Ryd, O. Garred, E. Schweda, P.K. Holm, and B. van Deurs. 1994. Retrograde transport from the Golgi complex to the ER of both shiga toxin and the nontoxic shiga B-fragment is regulated by butyric acid and cAMP. J. Cell Biol. 126:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter, J., and G. Warren. 2002. Golgi architecture and inheritance. Annu. Rev. Cell Dev. Biol. 18:379–420. [DOI] [PubMed] [Google Scholar]
- Storrie, B., J. White, S. Rottger, E.H. Stelzer, T. Suganuma, and T. Nilsson. 1998. Recycling of Golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering. J. Cell Biol. 143:1505–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorova, E.S., R.C. Kurten, and V.V. Lupashin. 2001. Identification of a human orthologue of Sec34p as a component of the cis-Golgi vesicle tethering machinery. J. Biol. Chem. 276:22810–22818. [DOI] [PubMed] [Google Scholar]
- Suvorova, E.S., R. Duden, and V.V. Lupashin. 2002. The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J. Cell Biol. 157:631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai, G., L. Lu, T.L. Wang, B.L. Tang, B. Goud, L. Johannes, and W. Hong. 2004. Participation of the syntaxin 5/Ykt6/GS28/GS15 SNARE complex in transport from the early/recycling endosome to the TGN. Mol. Biol. Cell. 15:4011–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar, D., T. Oka, E.E. Brittle, E. Vasile, V.V. Lupashin, J.E. Chatterton, J.E. Heuser, M. Krieger, and M.G. Waters. 2002. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 157:405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen, S.M., X. Cao, V.V. Lupashin, C. Barlowe, and M.G. Waters. 1998. Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J. Cell Biol. 141:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen, S.M., X. Cao, S.K. Sapperstein, E.C. Chiang, V.V. Lupashin, C. Barlowe, and M.G. Waters. 1999. Sec34p, a protein required for vesicle tethering to the yeast Golgi apparatus, is in a complex with Sec35p. J. Cell Biol. 147:729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volchuk, A., M. Ravazzola, A. Perrelet, W.S. Eng, M. Di Liberto, O. Varlamov, M. Fukasawa, T. Engel, T.H. Sollner, J.E. Rothman, and L. Orci. 2004. Countercurrent distribution of two distinct SNARE complexes mediating transport within the Golgi stack. Mol. Biol. Cell. 15:1506–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, D.M., K.S. Paul, and M.G. Waters. 1998. Purification and characterization of a novel 13 S hetero-oligomeric protein complex that stimulates in vitro Golgi transport. J. Biol. Chem. 273:29565–29576. [DOI] [PubMed] [Google Scholar]
- Whyte, J.R., and S. Munro. 2001. The Sec34/35 Golgi transport complex is related to the Exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev. Cell. 1:527–537. [DOI] [PubMed] [Google Scholar]
- Whyte, J.R., and S. Munro. 2002. Vesicle tethering complexes in membrane traffic. J. Cell Sci. 115:2627–2637. [DOI] [PubMed] [Google Scholar]
- Wu, X., R.A. Steet, O. Bohorov, J. Bakker, J. Newell, M. Krieger, L. Spaapen, S. Kornfeld, and H.H. Freeze. 2004. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 10:518–523. [DOI] [PubMed] [Google Scholar]
- Xu, Y., S. Martin, D.E. James, and W. Hong. 2002. GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Mol. Biol. Cell. 13:3493–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]