

Abstract
We previously demonstrated that integrin-dependent adhesion activates STAT5A, a well known target of IL-3–mediated signaling. Here, we show that in endothelial cells the active β1 integrin constitutively associates with the unphosphorylated IL-3 receptor (IL-3R) β common subunit. This association is not sufficient for activating downstream signals. Indeed, only upon fibronectin adhesion is Janus Kinase 2 (JAK2) recruited to the β1 integrin–IL-3R complex and triggers IL-3R β common phosphorylation, leading to the formation of docking sites for activated STAT5A. These events are IL-3 independent but require the integrity of the IL-3R β common. IL-3 treatment increases JAK2 activation and STAT5A and STAT5B tyrosine and serine phosphorylation and leads to cell cycle progression in adherent cells. Expression of an inactive STAT5A inhibits cell cycle progression upon IL-3 treatment, identifying integrin-dependent STAT5A activation as a priming event for IL-3–mediated S phase entry. Consistently, overexpression of a constitutive active STAT5A leads to anchorage-independent cell cycle progression. Therefore, these data provide strong evidence that integrin-dependent STAT5A activation controls IL-3–mediated proliferation.
Introduction
Adhesion of endothelial cells to extracellular matrix is mediated by the integrin family of cell surface receptors (Hynes, 2002). Integrins are heterodimers of an α and β subunit and their binding to matrix proteins triggers multiple signaling pathways, which regulate basic cell functions (Giancotti and Ruoslahti, 1999; Damsky and Ilic, 2002; Howe et al., 2002; Miranti and Brugge, 2002; Giancotti and Tarone, 2003). Integrin-induced signaling also affects gene expression (de Fougerolles and Koteliansky, 2002). Among the early genes responsive to growth signals, c-fos gene has been proposed as a mediator of cell cycle progression controlled by cell adhesion (Wary et al., 1996). The signaling pathway leading to integrin-dependent c-fos transcription is only partially known, and roles for the activated Erk1/Erk2 MAPK (Wary et al., 1996) and the transcriptional factor STAT5A have been proposed (Brizzi et al., 1999).
STAT5 is a known target of IL-3 (Ihle and Kerr, 1995; O'Shea, 1997; Grimley et al., 1999), a T cell–derived cytokine (Wimperis et al., 1989) that, besides promoting hematopoietic progenitor cell proliferation and differentiation, acts as an inducer of endothelial and smooth muscle cell migration and proliferation and as a promoter of neoangiogenesis (Brizzi et al., 1993, 2001; Korpelainen et al., 1995, 1996). Moreover, we reported that CD4/CD25/CD5 + T cells infiltrating breast cancer tissues also express IL-3, which by stimulating endothelial cells can affect vessel assembly (Dentelli et al., 2004). IL-3 has also been found to act as survival factor for tumor-derived endothelial cells (Deregibus et al., 2002), suggesting a pleiotropic role for this cytokine in endothelial cell biology. IL-3 binds to a heterodimeric receptor consisting of a ligand binding α subunit and a β subunit that is shared with GM-CSF and IL-5 receptors and is denoted as β common (Reddy et al., 2000; Geijsen et al., 2001). The type I cytokine receptor β common has a large cytoplasmic domain that plays a pivotal role in downstream signal transduction (Kitamura et al., 1991). IL-3R, which lacks intrinsic kinase activity, interacts with and activates Janus Kinase 2 (JAK2) in response to ligand binding. As a consequence, the β common subunit undergoes tyrosine phosphorylation and cues signaling molecules such as MAPK, the phosphatidylinositol 3-kinase, and the STATs (Reddy et al., 2000; Geijsen et al., 2001). STAT5 proteins consisting of STAT5A and STAT5B are the main targets of IL-3 signaling (Mui et al., 1995a; Ihle, 2001). Upon cytokine stimulation, JAK2 phosphorylates STAT5 (Reddy et al., 2000) and the phosphorylated STAT5 proteins dimerize and translocate into the nucleus, where, by binding DNA, they activate target genes including c-fos (Mui et al., 1996). In addition to this JAK-catalyzed tyrosine phosphorylation, STAT5 may undergo serine phosphorylation in the carboxy-terminal P(M)SP site (Yamashita et al., 1998) in response to prolactin (Decker and Kovarik, 2000). However, functional studies of the effects of serine phosphorylation on STAT5's transcriptional activity have not provided a consistent picture.
In addition to the role played by STAT5 in cytokine receptor signaling, we reported that STAT5A becomes activated in endothelial cells upon cell matrix adhesion (Brizzi et al., 1999). Also STAT1 has been implicated in regulation of cell adhesion, spreading, and migration (Xie et al., 2001), suggesting a pleiotropic role for the STAT pathway in adhesion-dependent signaling.
In nonadherent hematopoietic cells, STAT5 is required for IL-3–mediated cell proliferation (Mui et al., 1995b, 1996). In addition, IL-3 triggers endothelial cell proliferation (Brizzi et al., 1993; Mui et al., 1995b, 1996) and activates STAT5 to induce in vivo neoangiogenesis (Dentelli et al., 1999, 2004), suggesting that STAT5 could also regulate IL-3–dependent cell cycle progression in vascular adherent cells. Here, we show that integrin-dependent JAK2 and STAT5A activation are prerequisites for IL-3–mediated endothelial cell proliferation.
Results
The β common subunit of IL-3R stably interacts with the active β1 integrin
It is well established that integrins trigger specific signaling pathways by directly trans-activating growth factor receptors (Moro et al., 1998; Miranti and Brugge, 2002; Schwartz and Ginsberg, 2002; Giancotti and Tarone, 2003). We showed that in endothelial cells integrins induce ligand-independent STAT5A activation and c-fos gene expression (Brizzi et al., 1999). As shown in Table I, adhesion of endothelial cells to fibronectin (FN) in the absence of IL-3 is not sufficient to promote entry in the S phase of the cell cycle. Consistently, upon adhesion, unlike c-fos mRNA expression, which is strongly up-regulated (Fig. 1 A), cyclin D1 protein is not modified within 15 h of adhesion (Fig. 1 B, gray bars). Its expression is induced by serum in adherent cells (Fig. 1 B, black bars), confirming that in primary cells expression of G1 phase cyclins requires concomitant signals emanated from integrins and soluble ligands (Assoian and Schwartz, 2001). Endothelial cells plated on FN undergo both proliferation and migration in response to IL-3 treatment (Table I), suggesting that IL-3 and its receptor play a crucial role in regulating endothelial cell functions. The fact that STAT5 is a well defined target of IL-3R signaling in endothelial cells (Dentelli et al., 1999) prompted us to evaluate the involvement of the IL-3R in mediating integrin-dependent STAT5 activation. A likely mechanism through which integrins can activate STAT5A may depend on cross talk between β1 integrin and the IL-3R β common. To investigate this possibility, β1 integrin and IL-3R β common were immunoprecipitated (IP) from endothelial cell extracts and the immunoprecipitates were reciprocally immunoblotted (IB) as shown in Fig. 2 A (top panels). Cell extract from MO7-E cells was IP with an antiserum to IL-3R β common and used as positive control (+). These experiments demonstrate that β1 integrin associates with the IL-3R β common, both in cells kept in suspension or adherent to FN, indicating that these two molecules stably interact in endothelial cells independently from the adhesive state. The specificity of the association was also demonstrated by the loss of signal when the same filter was IB with the IL-3 β common antiserum preadsorbed with the immunizing peptide (Fig. 2 A, bottom left). Moreover, no coimmunoprecipitation of IL-3R β common was observed with a control antibody against β2 microglobulin (Fig. 2 A, bottom right).
Table I. IL-3 triggers endothelial cell proliferation and migration.
| Control | IL-3 | bFGF | |
|---|---|---|---|
| Percentage of cells in S phase |
6 ± 1 | 15 ± 2 | 18 ± 2 |
| Number of cells counted upon 6 d of culturea |
3 ± 2 | 22 ± 2 | 39 ± 3 |
| Number of cells migrated across the membrane |
12 ± 2 | 48 ± 3 | 54 ± 4 |
Endothelial cells resuspended at 5 × 104/ml were seeded on FN-coated dishes. For the determination of the percentage of cells in S phase, triplicate cultures (five individual experiments) were pulse labeled with [3H]TdR (4 μCi/ml) for 1 h the second day after the addition of 20 ng/ml IL-3 or 10 ng/ml bFGF, which was used as positive control, and processed by autoradiography (Brizzi et al., 1993). Cell proliferation was assessed 6 d after the addition of the indicated growth factors by direct cell count on triplicate wells. The numbers are the mean ± SD of labeled or counted cells. The migration assay was performed by the modified chambers technique (8-μm pore size filters FN coated) on endothelial cells untreated or treated with 20 ng/ml IL-3 or 10 ng/ml bFGF, which was used as positive control (Dentelli et al., 1999). The numbers represent the mean ± SD of cells counted per 10 fields (×200).
Cells × 104.
Figure 1.
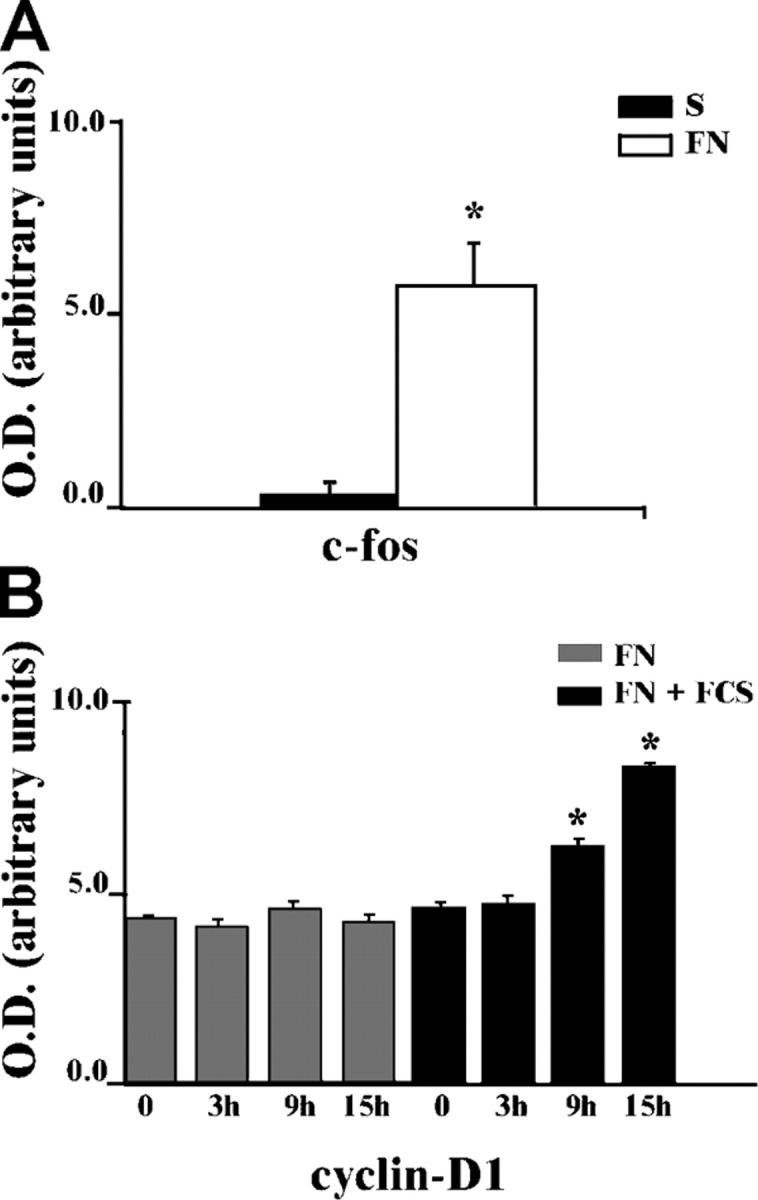
Adhesion-mediated c-fos gene mRNA and cyclin D1 protein expression. (A) RNA from endothelial cells kept in suspension (S) or adherent to FN for 30 min was subjected to Northern blot; c-fos mRNA level was quantified by densitometric analysis. (B) Cell extracts from endothelial cells kept in suspension (0) or let to adhere to FN in the presence or in the absence of FCS for the indicated times were subjected to 12% SDS-PAGE and IB with cyclin D1 antibodies. Cyclin D1 expression was quantified by densitometric analysis and reported as the mean ± SD of four independent experiments. *, P < 0.05 FN versus FN + serum.
Figure 2.

FN-induced association of activated STAT5A with the constitutive β1 integrin–IL-3R β common complex. (A) Cell extracts from starved endothelial cells kept in suspension (S) or plated on FN-coated dishes (20 min) were immunoprecipitated (IP) with mAb TS2/16 to the β1 integrin subunit, and the immunoprecipitates were divided in two aliquots. Western blot was performed with IL-3R β common antibodies (top left) or with the same antibodies preadsorbed on the recombinant IL-3R β common protein (bottom left). In the absence of the positive signal, to reveal IgG, the latter filter was overexposed. The same amount of cell extracts was IP with IL-3R β common antibodies, the immunoprecipitate run on a 6% SDS-PAGE in non-reducing conditions, and IB with β1 integrin mAb (top right). Parallel experiments were IP with anti-β2 microglobulin antibodies. Filters were IB with IL-3R β common antibodies (bottom right). (B) Cell extracts from endothelial cells plated on FN-coated dishes for different times in the presence of 50 μM pervanadate were IP with IL-3R β common antibodies and IB with antiphosphotyrosine antibodies (α PY; top) and reprobed with IL-3R β common antibodies (bottom). (C) Cell extracts from endothelial cells plated as in A were IP with IL-3R β common antibodies, divided in two aliquots, and IB with anti–phospho-STAT5, anti-STAT5A protein, and reimmunoblotted with IL-3R β common antibody or β1 integrin mAb. (D) Cells extracts treated as in A were IP with uncoupled or with STAT5A-coupled Sepharose protein A beads and IB with STAT5A antibody. M07-E cell extracts were used as positive control (+). The results are representative of four independent experiments.
To investigate further the molecular mechanisms leading to adhesion-independent constitutive association of IL-3R β common with β1 integrin, the hematopoietic MO7-E cells, which express both IL-3R (Brizzi et al., 1994) and β1 integrin, was used. Consistent with the fact that in these cells β1 integrin is present as an inactive form that can be activated by bivalent cations (Bazzoni et al., 1995; Mould et al., 1995; Humphries et al., 2003), we found that the β1 integrin was able to coimmunoprecipitate the IL-3R β common only in cells treated with Mn2+ (Fig. 3 A). These data suggest that activation of β1 integrin is a prerequisite for association with the IL-3R β common. Flow cytometry analysis using mAb 12G10, recognizing one epitope expressed only in the FN-competent β1 integrin (Mould et al., 1995), or mAb BV7, recognizing a constitutive β1 integrin epitope, was performed on MO7-E cells and endothelial cells. As shown in Fig. 3 B, mAb 12G10 binds to MO7-E cells only in the presence of the activating bivalent cation Mn2+ (Fig. 3 B, compare c and d). In contrast, in endothelial cells the 12G10-recognizing epitope was already exposed in the absence of any treatment (Fig. 3 B, e), indicating that a large majority of the β1 integrin expressed on the surface of endothelial cells was in the active state. No staining was observed by incubating cells with anti–IL-1β (Fig. 3 B, a) as well as with preimmune IgG (Fig. 3 B, a–e, left curve). Thus, IL-3R β common interacts with β1 integrin only in its active form, indicating that β1 integrin activation allows its association with the IL-3R.
Figure 3.

The IL-3R β common subunit associates with the active form of β1 integrin. (A) Starved MO7-E cells incubated or not with 10 mM of bivalent Mn2+ cations (Mn) with (+) or without (−) IL-3 were detergent extracted. Cell extracts were IP with β1 integrin mAb or IL-3R β common antibodies and IB with IL-3R β common antibodies. (B) MO7-E cells untreated (a–c) or treated with 10 mM Mn2+ (d) were subjected to flow cytometry analysis with IL-1β mAb (a) or preimmune IgG (a–e, left curve) as negative controls, mAb BV7 (b) or mAb 12G10 (c and d). Endothelial cells were subjected to flow cytometry analysis with mAb 12G10 (e). The data expressed as relative cell number (y axis) plotted as a function of fluorescence intensity (x axis) are representative of three experiments. (C) Extracts from MO7-E cells left untreated (−) or treated with Mn2+ (+) were subjected to SDS-PAGE and IB with phospho-STAT5 antibody and STAT5A antibody. Similar results were obtained in four individual experiments.
Upon adhesion, phosphorylated JAK2 and STAT5A associate with β1 integrin–IL-3R β common complex
To evaluate the role of the IL-3R β common in integrin-mediated STAT5A activation, endothelial cells were kept in suspension or plated on FN for different time intervals. Cell extracts were IP with antibodies to the IL-3R β common subunit and IB with antiphosphotyrosine antibodies. The 140-kD IL-3R β common became phosphorylated within 20 min of adhesion and coprecipitated with a phosphorylated band of ∼90 kD in the same conditions (Fig. 2 B), indicating that integrin-dependent adhesion triggers IL-3R β common activation. To identify the 90-kD phosphorylated band, the anti–IL-3R β common immunoprecipitates were divided in two aliquots, run on the same gel, and separately IB with anti–phospho-STAT5 and anti-STAT5A antibodies. As depicted in Fig. 2 C, the 90-kD protein coprecipitating with the IL-3R β common only in cells adherent to FN was most likely the phosphorylated STAT5A. Moreover, the observation that β1 integrin coprecipitated with IL-3R β common and STAT5A indicates that adhesion to FN led to the formation of a macromolecular β1 integrin–IL-3R β common–STAT5A complex (Fig. 2 C). The specificity of this interaction was confirmed by the inability of the antibody to STAT5A to recognize IP bands when beads bound only to Sepharose protein A and not antibody to IL-3R were used (Fig. 2 D). Thus, these data show that integrin-mediated adhesion by phosphorylating the IL-3R β common creates a docking site for the phosphorylated STAT5A.
As shown in Fig. 3 A, in M07-E cells β1 integrin–IL-3R β common complex formation depends on integrin activation. Therefore, we use these cells as a suitable model to answer the question of whether or not β1 integrin–IL-3R β common interaction represents a prerequisite for the activation of STAT5A. The results reported in Fig. 3 C clearly show that STAT5A was phosphorylated only in cells pretreated with Mn2+, a condition required for activating β1 integrin and inducing its association with the IL-3R β common (Fig. 3 A). Thus, these data strongly support the possibility that at least in M07-E cells, IL-3R–β1 integrin complex is essential for STAT5A activation. On the contrary, the results obtained in endothelial cells shown in Fig. 2 indicate that IL-3R–β1 integrin complex is required but not sufficient for downstream signaling events.
We previously demonstrated that integrin-mediated adhesion triggered JAK2 activation (Brizzi et al., 1999). Here, we investigated the role of JAK2 in the formation of the β1 integrin–IL-3R β common–STAT5A complex (Fig. 2 C). As positive control for the inhibitory effect of the JAK2 inhibitor AG-490, M07-E cells were used (Fig. 4 A). Subsequently, endothelial cells pretreated with 100 nM were kept in suspension or plated on FN and cell extracts were IP with the antiserum to the IL-3R β common. The anti–phospho-STAT5 immunoblot in Fig. 4 B shows that the phosphorylated STAT5A did not coimmunoprecipitate with the IL-3R β common in adherent cells pretreated with AG-490. In addition, the finding that STAT5A cannot be detected in the IL-3R β common immunoprecipitate from cells pretreated with AG-490 sustains the possibility that JAK2 kinase activity regulates both STAT5 recruitment and activation (Fig. 4 B). Similarly, in the immunoprecipitates of the IL-3R β common, the phosphorylated bands corresponding to STAT5A and the IL-3R β common were not present when the cells were pretreated with AG490 (Fig. 4 C). Moreover, by immunoprecipitating with antibodies to the β1 integrin, JAK2 was found associated with the β1 integrin–IL-3R β common complex only in cells adherent to FN (Fig. 4 D). Adhesion to FN also triggers JAK2 tyrosine phosphorylation (Fig. 4 E). These data support a potential role of JAK2 in regulating both adhesion-mediated IL-3R β common phosphorylation and STAT5A recruitment and activation.
Figure 4.

FN-dependent STAT5A phosphorylation and recruitment to the IL-3R β common depends on integrin-mediated JAK2 kinase activity. (A) Extracts from M07-E cells, untreated or treated with IL-3 with (+) or without (−) 100 nM of JAK2 kinase inhibitor AG490, were IP with JAK2 antibody, IB with anti-PY, and reimmunoblotted with JAK2 antibodies. (B and C) Extracts from endothelial cells kept in suspension or plated on FN-coated dishes with (+) or without (−) 100 nM AG490 were IP with IL-3R β common antibodies, divided in two aliquots, and run on 8% SDS-PAGE. (B) Filter was IB with phospho-STAT5 antibody or STAT5A antibody (top) and reimmunoblotted with IL-3R β common antibodies (bottom). (C) Immunoblotting was performed with anti-PY antibodies (top) or IL-3R β common antibodies. (D) Cell extracts from endothelial cells were IP with β1 integrin mAb TS2/16 and IB with JAK2 antibodies (top) or IL-3R β common antibody (bottom). (E) Cell extracts from endothelial cells with (+) or without (−) IL-3 were IP with JAK2 antibodies and subjected to SDS-PAGE. Filters were IB with anti-PY antibodies or JAK2 antibodies. The results are representative of three independent experiments.
The intracellular domain of the IL-3R β common is required for STAT5A recruitment by cell adhesion
To evaluate whether or not the intracellular region of the IL-3R β common may act as a STAT5A docking site, HEK293 cells were stably transfected with the full-length IL-3R β common subunit or one of two Myc-tagged deletion mutants, the first lacking the entire intracellular domain including the juxtamembrane region corresponding to the docking sites for JAK and STAT proteins (Δ455) and the second the intracellular region corresponding to the amino acids 545–881 (Δ544), still able to activate receptor signaling (Sakamaki et al., 1992). The basal adhesion-dependent activation of STAT5A was first evaluated in Neo vector-transfected cells. As shown in Fig. 5 A, the anti–phospho-STAT5 IB of total cell extracts revealed a faint band corresponding to the activated STAT5, indicating that a minimal STAT5A activation is detectable in these cells upon adhesion. IL-3–treated M07-E cells were used as positive control (Fig. 5 A, +). Subsequently, coimmunoprecipitation experiments were performed on HEK293 cells expressing the full-length IL-3R β common or one of the two mutants. As shown in Fig. 5 B, in agreement with the data obtained in primary endothelial cells, in response to adhesion tyrosine-phosphorylated STAT5A coimmunoprecipitated with the IL-3R β common in HEK293 cells expressing the full-length receptor. In contrast, phosphorylated STAT5A was undetectable in the immunoprecipitates of the Δ455 truncated form of the IL-3R β common (Fig. 5 C). Also in these experiments IL-3–treated M07-E cells were used as positive control. Consistent with the ability of Δ544 mutant receptor to trigger intracellular signaling (Sakamaki et al., 1992), it was still able to recruit STAT5A (Fig. 5 C).
Figure 5.

The juxtamembrane domain of the IL-3R β common is required for STAT5A recruitment by cell adhesion. HEK293 cells expressing the Neo vector, the full-length IL-3R β common subunit (FL), and the myc-tagged Δ455 or Δ544 mutants of the IL-3R β common subunit were replated on FN-coated dishes for 20 min. (A) Cell extracts from Neo vector–transfected cells were IB with antibodies to phosphorylated STAT5A. (B and C) Cell extracts were IP with IL-3R β common antibodies (B) or mAb 9E10 to the Myc epitope tag (C), IB with phospho-STAT5 antibody or STAT5A antibody (top), and reimmunoblotted with IL-3R β common antibodies (B) or mAb 9E10 (C). (D) Nuclear cell extracts from HEK293 FL or Δ455 mutant, kept in suspension (S) or plated on FN, were subjected to EMSA analysis (left) and supershift assay (right). Arrows indicate the SIE complex and supershift species. (E) Extracts from FL or Δ455 mutant were IP with β1 integrin mAb and IB with mAb 9E10 (left) or IL-3R β common antibodies (right). As positive control (+), IL-3–treated M07-E cells were used. The results are representative of three independent experiments.
The transcriptional role of adhesion-induced STAT5A activation and recruitment to the β1 integrin–IL-3R β common complex was assayed by the electrophoretic mobility shift assay (EMSA) using the Sis-inducible element of c-fos (SIE) sequence as probe. The 32P-labeled SIE sequence was incubated with nuclear extracts prepared from HEK293 cells expressing the wild-type or the Δ455 truncated mutant plated on FN or kept in suspension. The results reported in Fig. 5 D demonstrate that only adhesion to FN induces the formation of a SIE-binding complex in nuclear extracts from HEK293 cells expressing the full-length IL-3R β common. The residual adhesion-dependent SIE-binding complex observed in nuclear extracts from cells expressing the Δ455 truncated form of the IL-3R β common is likely due to the basal level of STAT5 activation reported in Fig. 5 A.
Although the Δ455 truncated form of the IL-3R β common lacked the ability to recruit STAT5A in response to adhesion, it was still physically associated with the β1 integrin subunit (Fig. 5 E), suggesting that the interaction between these two molecules occurs through the extracellular or the trans-membrane regions, as reported for the PDGF and VEGF receptors (Borges et al., 2000). Therefore, these data define the IL-3R β common subunit cytoplasmic domain as a docking site for the activated STAT5A in response to adhesion.
IL-3–dependent STAT5A activation requires cell adhesion
Our experiments, performed in the absence of IL-3, show that in response to adhesion, integrins by activating JAK2 cooperate with the IL-3R β common for the activation of STAT5A. However, when IL-3 was added to endothelial cells plated on FN, not only the level of JAK2 phosphorylation (Fig. 4 E) but also that of STAT5 increased (Fig. 6 A). Kinetics analysis shows that, although FN induced a transient STAT5A phosphorylation, IL-3 addition led to a persistent STAT5A phosphorylation, still detectable within 120 min of treatment (Fig. 6 B). Because it has been previously reported that in endothelial cells, upon IL-3 treatment, either STAT5A or STAT5B undergo activation (Dentelli et al., 1999), endothelial cells kept in suspension or adherent to FN were evaluated for STAT5A and STAT5B activation in response to IL-3. The results presented in Fig. 6 C show that the addition of IL-3 to adherent cells induced an increase in STAT5A phosphorylation, quantified by densitometric analysis (Fig. 6 C, top). Moreover, the amount of STAT5A coimmunoprecipitated with IL-3R β common was also increased in IL-3–treated cells (unpublished data), suggesting that the activation of STAT5A in the IL-3–mediated signaling is the result of a cooperative effect between adhesion and soluble ligand.
Figure 6.
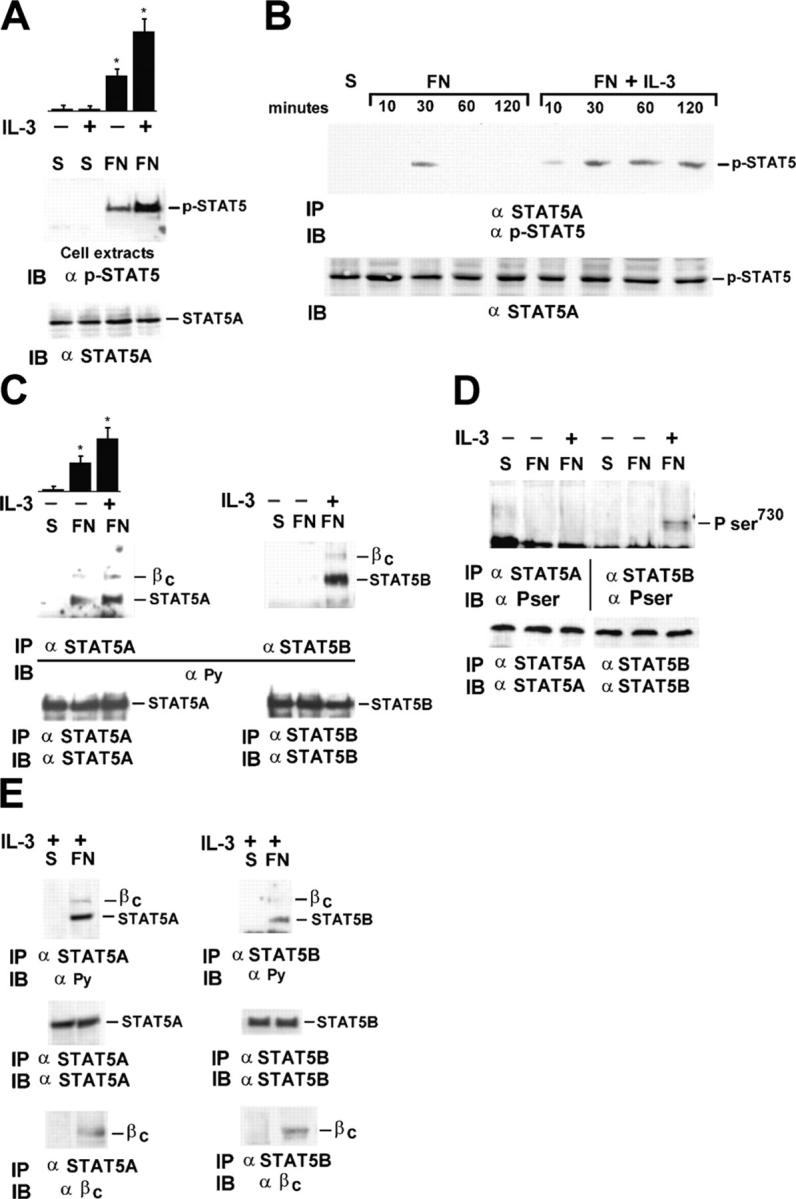
IL-3–dependent STAT5 activation requires cell adhesion. (A) Extracts from endothelial cells kept in suspension or plated on FN-coated dishes with (+) or without (−) IL-3 were IB with phospho-STAT5 antibody or STAT5A antibody. Densitometric analysis of phospho-STAT5 is shown in the top panel. The results are the mean ± SD of four independent experiments; *, P < 0.05 control versus experimental groups. (B) Extracts were IP with STAT5A antibodies and IB with phospho-STAT5 antibody or STAT5A antibody. (C) Cell extracts were divided into two aliquots and IP with specific antibodies to STAT5A or STAT5B. The filters were IB with anti-PY and reimmunoblotted with the anti-STAT5A or the anti-STAT5B antibodies. Densitometric analysis of STAT5A phosphorylation, performed as in A, is reported in the top panel. The results are the mean ± SD of four independent experiments done on separate days; *, P < 0.05 control versus experimental groups. (D) Cell extracts were prepared and IP as in B. The filters were IB with a phospho-serine STAT5 antibody (top) that recognizes P-Ser725 on STAT5A or P-Ser730 on STAT5B (Pser) and reimmunoblotted with STAT5A or STAT5B antibodies (bottom). (E) Cell extracts were separately IP with specific antibodies to STAT5A or STAT5B and IB with the anti-PY antibodies (top), STAT5A/STAT5B antibodies (middle), or IL-3R β common antibodies (bottom). The results are representative of four independent experiments.
Unlike STAT5A, as we already showed (Brizzi et al., 1999), STAT5B was not phosphorylated by integrin-mediated adhesion, but underwent tyrosine phosphorylation when endothelial cells adherent to FN were further stimulated with IL-3 (Fig. 6 C, right). These results indicate that in contrast with STAT5A, whose activation depends on cell adhesion, STAT5B activation strictly depends on IL-3, suggesting that adhesion and IL-3 recruit different signaling targets to achieve IL-3–dependent full biological response. Consistent with this possibility, we found that IL-3, besides its ability to induce STAT5B tyrosine phosphorylation, also triggered serine phosphorylation of STAT5B. By contrast, neither adhesion alone nor adhesion plus IL-3 treatment were able to induce serine phosphorylation of STAT5A (Fig. 6 D).
To analyze further the role played by adhesion in IL-3–mediated activation of STAT5A and STAT5B, endothelial cells were treated with IL-3 both in suspension and in adherent conditions. As depicted in Fig. 6 E, we found that IL-3–dependent tyrosine phosphorylation of STAT5A and STAT5B as well as their interaction with the IL-3R β common only occurred in cells adherent to FN.
Integrin-dependent STAT5A activation is essential for IL-3–mediated cell cycle progression in adherent cells
Cell adhesion to extracellular matrix is a prerequisite for cell cycle progression in non-transformed cells (Assoian, 1997). Although STAT5A has been reported to be involved in IL-3–dependent proliferation of hematopoietic cells (Mui et al., 1996), its role in proliferation of adherent cells has not yet been defined. To evaluate the role of STAT5A in regulating IL-3–dependent cell cycle progression in adherent cells, we reconstituted the IL-3R by transfecting the IL-3R α subunit cDNA into the HEK293 cells expressing the full-length IL-3R β common subunit (HEK293 IL-3R). As reported in the previous section for endothelial cells, also in these cells we were not able to detect STAT5A activation in response to IL-3 in cells kept in suspension (unpublished data). To assess whether or not HEK293 IL-3R cells stimulated with IL-3 progressed through the cell cycle, cells were detached from the plates and either kept in suspension or let to adhere to FN-coated dishes, and cell cycle progression was evaluated upon IL-3 stimulation. As shown in Table II, IL-3 treatment induced G1-S progression in FN-adherent cells (percentage of cells in S phase: 30.5% suspended cells vs. 40.4% adherent cells). Similar results were obtained when cell proliferation was assessed by direct cell count 3 d after the addition of IL-3 (unpublished data). In contrast, IL-3 treatment was unable to induce the G1-S progression in cells kept in suspension (percentage of cells in S phase: 27% untreated vs. 30.5% IL-3–treated), indicating that, consistent with adhesion-dependent STAT5A activation, IL-3–mediated cell cycle progression strictly depends on cell matrix adhesion.
Table II. IL-3–mediated cell cycle progression of HEK293 IL-3R–expressing cells.
| G0/G1 | S | G2/M | |
|---|---|---|---|
| % | % | % | |
| (HEK293 IL-3R) S | 65.5 ± 8 | 27 ± 4 | 6.5 ± 1.5 |
| (HEK293 IL-3R) S + IL-3 | 64.7 ± 7 | 30.5 ± 5 | 4.8 ± 2 |
| (HEK293 IL-3R) FN | 61 ± 3.5 | 30.7 ± 5 | 8.3 ± 4 |
| (HEK293 IL-3R) FN + IL-3 | 51.6 ± 6 | 40.4 ± 4.5 | 8 ± 3 |
HEK293 IL-3R cells serum starved for 24 h were kept in suspension or let to adhere to FN-coated plates in the presence or absence of 20 ng/ml IL-3. After 18 h of IL-3 stimulation, the cells were fixed with ethanol and DNA was stained with propidium iodide. The fluorescence related to DNA content was evaluated by flow cytometry. The percentage of cells in each phase of the cell cycle was determined by ModFit LT Software (Verity Software House Inc.). The numbers are the mean ± SD of three different experiments done on separate days, each performed in triplicate.
To discriminate between STAT5A and STAT5B in mediating IL-3–dependent cell cycle progression, plasmids containing dominant-negative constructs of STAT5 proteins (Mui et al., 1996) or the Neo vector alone were transiently transfected into HEK293 IL-3R cells. As shown in Table III, analysis of the DNA content showed that expression of the dominant-negative STAT5B protein did not modify the percentage of cells in the different phases of the cell cycle if compared with the Neo vector–expressing cells. In contrast, in cells expressing the dominant-negative form of STAT5A, IL-3 treatment was unable to increase the percentage of cells in S phase (Table III).
Table III. Effect of dominant-negative STAT5A and STAT5B expression on IL-3–mediated HEK293 IL-3R cell cycle progression.
| G0/G1 | S | G2/M | |
|---|---|---|---|
| % | % | % | |
| (HEK293 IL-3R + Neo vector) FN |
61 ± 2 | 31 ± 3 | 8 ± 2 |
| (HEK293 IL-3R + Neo vector) FN + IL-3 |
53 ± 3 | 38.5 ± 4 | 8.5 ± 2 |
| (HEK293 IL-3R + Δ STAT5A) FN |
62.5 ± 3 | 30 ± 2 | 7.5 ± 1.5 |
| (HEK293 IL-3R + Δ STAT5A) FN + IL-3 |
63.3 ± 1 | 29.2 ± 2 | 8.5 ± 3 |
| (HEK293 IL-3R + Δ STAT5B) FN |
60 ± 2 | 30.5 ± 2.5 | 9.5 ± 2 |
| (HEK293 IL-3R + Δ STAT5B) FN + IL-3 |
51.6 ± 1 | 39 ± 4 | 9.4 ± 2 |
HEK293 IL-3R cells, transiently transfected with the empty vector (Neo vector) or with dominant-negative STAT5A (ΔSTAT5A) or STAT5B (ΔSTAT5B) constructs, were serum starved for 24 h and let to adhere to FN-coated plates in the absence or presence of 20 ng/ml IL-3. After 18 h of stimulation, the cells were fixed with ethanol and DNA was stained with propidium iodide. The fluorescence related to DNA content was evaluated as described in Table II. The numbers are the mean ± SD of three different experiments done on separate days, each performed in triplicate.
Collectively, these data identify STAT5A as a molecular mediator of adhesion-dependent IL-3–mediated cell cycle progression. However, to further confirm a role of STAT5A in this process, we evaluated if the expression of a constitutive active STAT5A could restore the ability of cells to proliferate in suspension. HEK293 IL-3R–expressing cells were transfected with the constitutive activated STAT5A construct or with an empty vector, and a cell cycle progression assay was performed in cells kept in suspension. As shown in Table IV, HEK293 IL-3R cells transfected with the empty vector did not progress into cell cycle. In contrast, cells transfected with the constitutive active form of STAT5A acquired the ability to enter into the cell cycle in suspension (percentage of cells in S phase: 29.2% empty vector vs. 42% STAT5A-transfected), indicating that the presence of the active form of STAT5A is sufficient to trigger cell cycle progression to S phase in the absence of cell matrix adhesion and of IL-3. Indeed, when cells expressing the constitutive active form of STAT5A were kept in suspension or plated on FN, the addition of IL-3 was unable to further increase the percentage of cells in S phase (Table IV). Together our data show that adhesion-dependent STAT5A activation is a crucial event in the control of IL-3–mediated proliferation.
Table IV. The constitutive active 1*6 STAT5A protein induces adhesion-independent cell cycle progression.
| G0/G1 | S | G2/M | |
|---|---|---|---|
| % | % | % | |
| (HEK293 IL-3R + Neo vector) S |
63.3 ± 8 | 29.2 ± 4 | 7.5 ± 1.5 |
| (HEK293 IL-3R + 1*6 STAT5A) S |
53.2 ± 7 | 42 ± 5 | 4.8 ± 2 |
| (HEK293 IL-3R + 1*6 STAT5A) S + IL-3 |
52.3 ± 5 | 44.9 ± 4 | 2.9 ± 3 |
| (HEK293 IL-3R + 1*6 STAT5A) FN |
57.5 ± 6 | 40.2 ± 3 | 2.3 ± 2 |
| (HEK293 IL-3R + 1*6 STAT5A) FN + IL-3 |
58 ± 5 | 39.7 ± 4 | 2.3 ± 2 |
HEK293 IL-3R cells, transfected with the empty vector (Neo vector) or with the constitutive activated STAT5A construct (1*6 STAT5A), were serum starved for 24 h and kept in suspension or let to adhere to FN-coated plates in the presence or absence of 20 ng/ml IL-3. Cell cycle progression was assayed on ethanol-fixed cells. DNA was stained with propidium iodide. The fluorescence related to DNA content was evaluated as described in Table II. The numbers are the mean ± SD of three different experiments done on separate days, each performed in triplicate.
Discussion
IL-3 is a well known activator of STAT5 signaling pathway in hematopoietic cells (Mui et al., 1995b). Our previous data indicate that, in endothelial cells, STAT5A is a target of integrin-mediated cell matrix interaction (Brizzi et al., 1999), as well as of IL-3 (Dentelli et al., 1999). In the present study, we dissect the molecular mechanisms leading to integrin-dependent STAT5A activation and we demonstrate that, in endothelial cells, β1 integrin cooperates with the IL-3R β common to integrate adhesion- and cytokine-mediated signals. In particular, the IL-3R β common associates with the activated β1 integrin and, only upon adhesion, acquires the ability to recruit STAT5A. Cell matrix adhesion is also required to induce JAK2 tyrosine phosphorylation and recruitment to the β1 integrin–IL-3R β common complex. Moreover, we also identify STAT5A as a crucial molecular target of adhesion-dependent IL-3–mediated signaling leading to cell cycle entry.
Integrins and tyrosine kinase receptors, such as PDGF, insulin, and EGF receptors, physically interact on the cell membrane, as indicated by formation of macromolecular complexes (Vuori and Ruoslahti, 1994; Miyamoto et al., 1996; Sundberg and Rubin, 1996; Schneller et al., 1997; Moro et al., 2002). Our data first report the association of β1 integrin and the IL-3 cytokine receptor, indicating that also receptors devoid of tyrosine kinase activity can cross talk with integrins. β1 integrin specifically associates with the IL-3R β common in an adhesion-independent stable complex in the absence of IL-3. Although we cannot exclude that additional membrane molecules may interplay with this complex, our experimental conditions (i.e., serum deprivation, absence of soluble growth factors) ruled out the possibility that exogenous soluble factors can trigger this association. Moreover, the finding that the β1 integrin–IL-3R β common complex is found in primary cells indicates that this event represents a physiological condition. The truncated form of the IL-3R β common, which lacks the cytoplasmic domain (Δ455), is still able to interact with the β1 integrin subunit, suggesting that this interaction might occur either through the transmembrane or the extracellular domains, as described by the model proposed for the constitutive interaction of β3 integrin subunit with the PDGF or VEGF receptors (Borges et al., 2000). However, comparing the expression of the activated epitope of the β1 integrin in MO7-E cells with endothelial cells, we can conclude that the activation state of β1 integrin is an essential step for its association with the IL-3R β common. These data provide the first evidence of the relevance of β1 integrin activation in mediating the interaction with other membrane-associated receptors.
Integrin growth factor receptor cooperation has been extensively demonstrated, showing that integrins can regulate receptor functions including transactivation, receptor coordination and compartmentalization, and downstream signaling (Miranti and Brugge, 2002; Schwartz and Ginsberg, 2002; Yamada and Even-Ram, 2002). In fact, strong evidence indicates that integrin- and tyrosine kinase receptor–dependent signals need to be integrated at various levels to induce cell proliferation. However, few data are available on integrin cross talk with cytokine receptors on murine hematopoietic cells (Kanda et al., 2003). Our experiments demonstrate that in endothelial cells the constitutive interaction between β1 integrin and the IL-3R β common is not sufficient for inducing downstream signaling events. However, binding and phosphorylation of JAK2, phosphorylation of IL-3R β common, as well as activation of STAT5A are strictly dependent on matrix ligand occupancy of integrins, which in turn impart a stringent control to the action of the IL-3R determining if cells proliferate rather then undergo growth arrest. Moreover, the results obtained by pharmacological inhibition of JAK2 kinase activity are consistent with an essential role of JAK2 kinase for adhesion-dependent IL-3R β common phosphorylation, recruitment, and activation of STAT5A.
Deletion of the membrane proximal region of the IL-3R β common results in loss of activation of JAK2 (Quelle et al., 1994). Upon JAK2 activation, the IL-3R β common becomes phosphorylated and acquires the ability to bind several signal transducing proteins, including STAT5 (Chin et al., 1996; Durstin et al., 1996; Woodcock et al., 1996). Indeed, we report here that the juxtamembrane deletion mutant Δ455 of the IL-3R β common failed to recruit STAT5A upon adhesion, demonstrating that this region, possibly because of the lack of JAK2 kinase binding and activation (Reddy et al., 2000), is not only required for the IL-3 biological response in hematopoietic cells but also for integrin-mediated signaling.
Integrin signaling by itself regulates cell functions, such as cytoskeletal organization, whereas in cooperation with growth factor receptors controls cell migration and cell cycle progression. In fact, signals from integrins intimately coordinate with pathways activated by growth factors at multiple steps during cell proliferation (Assoian, 1997). The molecular mechanisms involved in such events include integrin-dependent activation of growth factor receptors (Moro et al., 2002), enhancement of growth factor signals (Miyamoto et al., 1996; Short et al., 1998), recruitment of crucial transducing proteins to membrane cytoskeletal complexes (Del Pozo et al., 2002), and enhancement of nuclear translocation of transcriptional regulators (Aplin et al., 2001). Our data show that JAK2, the IL-3R β common subunit, and STAT5A are phosphorylated by integrin-dependent adhesion in the absence of ligands. However, these signaling events are further increased by the addition of soluble IL-3. These results provide evidence for a strong cooperation between integrins and IL-3R and suggest that integrin-mediated adhesion, by inducing partial activation of JAK2 and STAT5A, may “prime” the cells to a full activation induced by the ligand.
IL-3 promotes endothelial cell proliferation (Brizzi et al., 1993). Our data show that cell adhesion to FN in the absence of IL-3 leads to STAT5A activation but does not allow cell cycle progression, indicating that, as previously reported (Moro et al., 1998), integrin-dependent activation of JAK2/STAT5A is not sufficient, per se, to sustain cell proliferation. Consistently, the addition of IL-3 confers ability to undergo cell cycle progression. Interestingly, this occurred only in adherent conditions because IL-3 was not able to stimulate proliferation of cells kept in suspension, indicating that cell cycle progression is controlled by multiple consequential steps regulated by both matrix attachment and cytokine receptor ligands.
So far, very few data are available on signaling pathways that are specifically controlled by integrin-mediated anchorage. Herein, we demonstrate that JAK2 and STAT5A are phosphorylated by adhesion and that their phosphorylation increases when IL-3 is added to endothelial cells. Interestingly, IL-3 activates JAK2 or STAT5A only in adherent but not in suspended cells, indicating that these molecules represent specific molecular targets of adhesion-mediated signaling.
We previously reported that IL-3 activates STAT5B in endothelial cells (Dentelli et al., 1999). Here, we show that adhesion does not trigger STAT5B activation. However, when IL-3 is added to adherent cells, STAT5B is phosphorylated both on tyrosine and serine 730. The functional role of STAT5 serine phosphorylation is still unclear (Decker and Kovarik, 2000). Our data showing that STAT5A is not phosphorylated on serine 725 either upon adhesion or in response to IL-3 and that cells expressing dominant-negative STAT5A fail to progress in S phase upon IL-3 suggest that phosphorylation on tyrosine rather than on serine is a crucial event for integrating the signals induced by integrin-mediated adhesion and IL-3. Moreover, expression of the constitutive active form of STAT5A rescues cell cycle progression in cells kept in suspension, indicating that STAT5A renders the cells independent from the adhesive signaling required for ligand-induced biological response. Thus, STAT5A activation is a crucial step in the cooperative pathways leading to anchorage-dependent IL-3–mediated cell growth.
The present work, focused on the cooperative role of integrins in cytokine receptor signals in adherent cells, provides a molecular mechanism to explain how integrins control cell cycle progression in response to the ligand by cross talking with the IL-3R. In addition, we demonstrate that activation of STAT5A overcomes the requirement of cell adhesion and IL-3 stimulation to induce proliferative signals in adherent cells, providing evidence that dysregulation of STAT5A activation, by overcoming anchorage dependence, may induce inappropriate proliferative signals and favor tumorigenesis.
Materials and methods
Reagents and antibodies
FN was purified from human plasma as previously described (Defilippi et al., 1994). M199 medium (endotoxin tested), BSA, and protein A–Sepharose were all obtained from Sigma-Aldrich. Bovine calf serum (endotoxin tested) was obtained from Hyclone. RPMI medium, G418, and lipofectin reagents were purchased from Invitrogen. Trypsin was obtained from Difco Laboratories. α-[32P]dCTP, [3H]TdR, nitrocellulose, HRP protein A, molecular weight markers, poly(dIdC):poly(dIdC), HRP-conjugated secondary antibodies, fluorescein-labeled anti–mouse IgG, and the ECL reagent were obtained from Amersham Biosciences. hIL-3 was a gift from Sandoz Pharma LTD. bFGF was obtained from Euro Clone. The JAK2 kinase inhibitor AG490 was purchased from Calbiochem.
Polyclonal IL-3R β common antibody has been described previously (Dentelli et al., 1999). The mAbs TS2/16 (American Type Culture Collection), BV7 (Immunological Science), and 12G10 (gift of M. Humphries, University of Manchester, Manchester, UK) to the human β1 integrin subunit and mAb 9E10 to the Myc epitope tag (American Type Culture Collection) were affinity purified on protein A–Sepharose and their purity was higher than 95%. mAb PY20 to phosphotyrosine was purchased from Transduction Laboratories (Becton Dickinson). The anti–phospho-STAT5 (tyrosine 694/699) antibody was obtained from Cell Signaling and the anti–phospho-STAT5 (serine725/730) antibody was obtained from Upstate Biotechnology. The antibody to cyclin D1, JAK2, IL-1β, and β2 microglobulin were purchased from Santa Cruz Biotechnology, Inc.
Cell culture and transfection
Endothelial cells were isolated from human umbilical cord vein as described previously (Brizzi et al., 1993). MO7-E were grown as reported in Avanzi et al. (1990) and Brizzi et al. (1994). HEK293 human epithelial cells (American Type Culture Collection) were stably transfected with Neo vector, the full-length IL-3R β common, or one of the IL-3R β common deleted mutants (Δ544 and Δ455; gifts of A. Miyajima, Teikyo University Biotechnology Research Center, Kanagawa, Japan; Sakamaki et al., 1992) by the lipofectin methods and selected with G418. Full-length IL-3R β common–expressing cells were also transfected with the cDNA for the IL-3R α subunit (Kitamura et al., 1991; provided by T. Kitamura, Tokyo University of Science, Tokyo, Japan) and selected with puromycin (HEK293 IL-3R). Activated form of STAT5 (1*6 STAT5A; Onishi et al., 1998) or dominant-negative STAT5A or STAT5B constructs (Mui et al., 1996) were transiently transfected.
Evaluation of cell surface expression of active β1 integrin subunit
MO7-E and confluent endothelial cells were starved for 18 h, put in suspension by 10 mM EDTA treatment, labeled with 10 μg/ml mAb 12G10, mAb BV7, mAb to IL-1β (negative control), or with a preimmune mAb for 30 min at 4°C, washed twice in PBS, and incubated with 10 μg/ml of fluorescein-labeled anti–mouse IgG for the same time. Cell surface expression of β1 integrin subunit was evaluated by flow cytometry (FACScan; Becton Dickinson).
Adhesion experiments
Tissue culture plates were coated with 20 μg/ml FN by overnight incubation as described previously (Moro et al., 1998). Starved HEK293 and endothelial cells were detached by 10 mM EDTA treatment and immediately plated on the tissue culture plates for the indicated times (Moro et al., 2002). When indicated, 50 μM pervanadate was added. Cells were detergent extracted in lysis buffer (1% Triton X-100, 50 mM Pipes, pH 6.8, 100 mM NaCl, 5 mM MgCl2, 300 mM sucrose, 5 mM EGTA, 2 mM sodium orthovanadate, 1 mM PMSF, 10 μg/ml leupeptin, 0.15 U/ml trypsin inhibitory U/ml aprotinin, and 1 μg/ml pepstatin).
Immunoprecipitation, SDS-PAGE, and immunoblotting
Equal amounts of cell extracts were subjected to SDS-PAGE or IP with the indicated antibodies and processed as previously described (Brizzi et al., 1999). The blots were incubated overnight with the indicated antibodies and revealed by HRP-conjugate/chemiluminescent detection method ECL.
Preparation of nuclear extracts and gel retardation assay
Nuclear extracts from transfected HEK293 cells were prepared as described by Sadowski et al. (1993). EMSA analysis was performed as described in Brizzi et al. (1999). The oligonucleotides used were as follows: G GGG CAT TTC CCG TAA ATC and G GGG GAT TTA CGG GAA ATG corresponding to the SIE (Zhong et al., 1994).
Northern blot analysis of c-fos mRNA expression
Cytoplasmic RNA was isolated according to Chomczynski and Sacchi (1987) and Northern blot analysis was performed according to standard methods (Brizzi et al., 1993). c-fos hybridization was quantified by densitometric analysis using a molecular imager (model GS-250; Bio-Rad Laboratories).
Cell proliferation and migration assays
Cell proliferation and migration assays reported in Table I were performed as described previously (Brizzi et al., 1993; Dentelli et al., 1999).
Cell cycle analysis
Endothelial and HEK293 IL-3R cells were kept in suspension or let to adhere to FN-coated dishes with or without IL-3 or bFGF (positive control). After 18 h, cells were fixed with 70% ethanol, processed for propidium iodide fluorescence, and analyzed with FACScan.
Statistical analysis
The results are representative of at least three independent experiments performed in triplicate. Densitometric analysis using a molecular imager was used to calculate differences in the fold induction of protein activation or expression. Significance of differences between experimental and control values was calculated using ANOVA with Newman-Keuls multi-comparison test.
Acknowledgments
We thank Dr. M. Humphries for the kind gift of mAb 12G10, Sara Cabodi for critically reading the manuscript, and Laura Damiano for help in the experiments.
This work was supported by grants of Associazione Italiana per la Ricerca sul Cancro to P. Defilippi and M.F. Brizzi; Ministero dell'Istruzione, dell'Università e della Ricerca to P. Defilippi, G. Tarone, and L. Pegoraro; and Special Project Oncology, Compagnia San Paolo/Fondazione Internazionale in Medicina Sperimentale, to P. Defilippi and G. Tarone.
Abbreviations used in this paper: EMSA, electrophoretic mobility shift assay; FN, fibronectin; IB, immunoblotted; IP, immunoprecipitated; JAK2, Janus Kinase 2; SIE, Sis-inducible element of c-fos.
References
- Aplin, A.E., S.A. Stewart, R.K. Assoian, and R.L. Juliano. 2001. Integrin-mediated adhesion regulates ERK nuclear translocation and phosphorylation of Elk-1. J. Cell Biol. 153:273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assoian, R.K. 1997. Anchorage-dependent cell cycle progression. J. Cell Biol. 136:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assoian, R.K., and M.A. Schwartz. 2001. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Curr. Opin. Genet. Dev. 11:48–53. [DOI] [PubMed] [Google Scholar]
- Avanzi, G.C., M.F. Brizzi, J. Giannotti, A. Ciarletta, Y.C. Yang, L. Pegoraro, and S.C. Clark. 1990. M-07e human leukemic factor-dependent cell line provides a rapid and sensitive bioassay for the human cytokines GM-CSF and IL-3. J. Cell. Physiol. 145:458–464. [DOI] [PubMed] [Google Scholar]
- Bazzoni, G., D.T. Shih, C.A. Buck, and M.E. Hemler. 1995. Monoclonal antibody 9EG7 defines a novel beta 1 integrin epitope induced by soluble ligand and manganese, but inhibited by calcium. J. Biol. Chem. 270:25570–25577. [DOI] [PubMed] [Google Scholar]
- Borges, E., Y. Jan, and E. Ruoslahti. 2000. Platelet-derived growth factor receptor beta and vascular endothelial growth factor receptor 2 bind to the beta 3 integrin through its extracellular domain. J. Biol. Chem. 275:39867–39873. [DOI] [PubMed] [Google Scholar]
- Brizzi, M.F., G. Garbarino, P.R. Rossi, G.L. Pagliardi, C. Arduino, G.C. Avanzi, and L. Pegoraro. 1993. Interleukin 3 stimulates proliferation and triggers endothelial-leukocyte adhesion molecule 1 gene activation of human endothelial cells. J. Clin. Invest. 91:2887–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzi, M.F., M.G. Zini, M.G. Aronica, J.M. Blechman, Y. Yarden, and L. Pegoraro. 1994. Convergence of signaling by interleukin-3, granulocyte-macrophage colony-stimulating factor, and mast cell growth factor on JAK2 tyrosine kinase. J. Biol. Chem. 269:31680–31684. [PubMed] [Google Scholar]
- Brizzi, M.F., P. Defilippi, A. Rosso, M. Venturino, G. Garbarino, A. Miyajima, L. Silengo, G. Tarone, and L. Pegoraro. 1999. Integrin-mediated adhesion of endothelial cells induces JAK2 and STAT5A activation: role in the control of c-fos gene expression. Mol. Biol. Cell. 10:3463–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzi, M.F., L. Formato, P. Dentelli, A. Rosso, M. Pavan, G. Garbarino, M. Pegoraro, G. Camussi, and L. Pegoraro. 2001. Interleukin-3 stimulates migration and proliferation of vascular smooth muscle cells: a potential role in atherogenesis. Circulation. 103:549–554. [DOI] [PubMed] [Google Scholar]
- Chin, H., N. Nakamura, R. Kamiyama, N. Miyasaka, J.N. Ihle, and O. Miura. 1996. Physical and functional interactions between Stat5 and the tyrosine-phosphorylated receptors for erythropoietin and interleukin-3. Blood. 88:4415–4425. [PubMed] [Google Scholar]
- Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162:156–159. [DOI] [PubMed] [Google Scholar]
- Damsky, C.H., and D. Ilic. 2002. Integrin signaling: it's where the action is. Curr. Opin. Cell Biol. 14:594–602. [DOI] [PubMed] [Google Scholar]
- de Fougerolles, A.R., and V.E. Koteliansky. 2002. Regulation of monocyte gene expression by the extracellular matrix and its functional implications. Immunol. Rev. 186:208–220. [DOI] [PubMed] [Google Scholar]
- Decker, T., and P. Kovarik. 2000. Serine phosphorylation of STATs. Oncogene. 19:2628–2637. [DOI] [PubMed] [Google Scholar]
- Defilippi, P., C. Bozzo, G. Volpe, G. Romano, M. Venturino, L. Silengo, and G. Tarone. 1994. Integrin-mediated signal transduction in human endothelial cells: analysis of tyrosine phosphorylation events. Cell Adhes. Commun. 2:75–86. [DOI] [PubMed] [Google Scholar]
- Del Pozo, M.A., W.B. Kiosses, N.B. Alderson, N. Meller, K.M. Hahn, and M.A. Schwartz. 2002. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat. Cell Biol. 4:232–239. [DOI] [PubMed] [Google Scholar]
- Dentelli, P., L. Del Sorbo, A. Rosso, A. Molinar, G. Garbarino, G. Camussi, L. Pegoraro, and M.F. Brizzi. 1999. Human IL-3 stimulates endothelial cell motility and promotes in vivo new vessel formation. J. Immunol. 163:2151–2159. [PubMed] [Google Scholar]
- Dentelli, P., A. Rosso, C. Calvi, B. Ghiringhello, G. Garbarino, G. Camussi, L. Pegoraro, and M.F. Brizzi. 2004. IL-3 affects endothelial cell-mediated smooth muscle cell recruitment by increasing TGF beta activity: potential role in tumor vessel stabilization. Oncogene. 23:1681–1692. [DOI] [PubMed] [Google Scholar]
- Deregibus, M.C., V. Cantaluppi, S. Doublier, M.F. Brizzi, I. Deambrosis, A. Albini, and G. Camussi. 2002. HIV-1-Tat protein activates phosphatidylinositol 3-kinase/ AKT-dependent survival pathways in Kaposi's sarcoma cells. J. Biol. Chem. 277:25195–25202. [DOI] [PubMed] [Google Scholar]
- Durstin, M., R.C. Inhorn, and J.D. Griffin. 1996. Tyrosine phosphorylation of Shc is not required for proliferation or viability signaling by granulocyte-macrophage colony-stimulating factor in hematopoietic cell lines. J. Immunol. 157:534–540. [PubMed] [Google Scholar]
- Geijsen, N., L. Koenderman, and P.J. Coffer. 2001. Specificity in cytokine signal transduction: lessons learned from the IL-3/IL-5/GM-CSF receptor family. Cytokine Growth Factor Rev. 12:19–25. [DOI] [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1999. Integrin signaling. Science. 285:1028–1032. [DOI] [PubMed] [Google Scholar]
- Giancotti, F.G., and G. Tarone. 2003. Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annu. Rev. Cell Dev. Biol. 19:173–206. [DOI] [PubMed] [Google Scholar]
- Grimley, P.M., F. Dong, and H. Rui. 1999. Stat5a and Stat5b: fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 10:131–157. [DOI] [PubMed] [Google Scholar]
- Howe, A.K., A.E. Aplin, and R.L. Juliano. 2002. Anchorage-dependent ERK signaling—mechanisms and consequences. Curr. Opin. Genet. Dev. 12:30–35. [DOI] [PubMed] [Google Scholar]
- Humphries, M.J., P.A. McEwan, S.J. Barton, P.A. Buckley, J. Bella, and A. Paul Mould. 2003. Integrin structure: heady advances in ligand binding, but activation still makes the knees wobble. Trends Biochem. Sci. 28:313–320. [DOI] [PubMed] [Google Scholar]
- Hynes, R.O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell. 110:673–687. [DOI] [PubMed] [Google Scholar]
- Ihle, J.N. 2001. The Stat family in cytokine signaling. Curr. Opin. Cell Biol. 13:211–217. [DOI] [PubMed] [Google Scholar]
- Ihle, J.N., and I.M. Kerr. 1995. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 11:69–74. [DOI] [PubMed] [Google Scholar]
- Kanda, E., Z.H. Jin, D. Mizuchi, A. Arai, and O. Miura. 2003. Activation of Rac and tyrosine phosphorylation of cytokine receptors induced by cross-linking of integrin alpha4beta1 and cell adhesion in hematopoietic cells. Biochem. Biophys. Res. Commun. 301:934–940. [DOI] [PubMed] [Google Scholar]
- Kitamura, T., K. Hayashida, K. Sakamaki, T. Yokota, K. Arai, and A. Miyajima. 1991. Reconstitution of functional receptors for human granulocyte/macrophage colony-stimulating factor (GM-CSF): evidence that the protein encoded by the AIC2B cDNA is a subunit of the murine GM-CSF receptor. Proc. Natl. Acad. Sci. USA. 88:5082–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpelainen, E.I., J.R. Gamble, W.B. Smith, M. Dottore, M.A. Vadas, and A.F. Lopez. 1995. Interferon-gamma upregulates interleukin-3 (IL-3) receptor expression in human endothelial cells and synergizes with IL-3 in stimulating major histocompatibility complex class II expression and cytokine production. Blood. 86:176–182. [PubMed] [Google Scholar]
- Korpelainen, E.I., J.R. Gamble, M.A. Vadas, and A.F. Lopez. 1996. IL-3R expression, regulation and function in cells of the vasculature. Immunol. Cell Biol. 74:1–7. [DOI] [PubMed] [Google Scholar]
- Miranti, C.K., and J.S. Brugge. 2002. Sensing the environment: a historical perspective on integrin signal transduction. Nat. Cell Biol. 4:E83–E90. [DOI] [PubMed] [Google Scholar]
- Miyamoto, S., H. Teramoto, J.S. Gutkind, and K.M. Yamada. 1996. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J. Cell Biol. 135:1633–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro, L., M. Venturino, C. Bozzo, L. Silengo, F. Altruda, L. Beguinot, G. Tarone, and P. Defilippi. 1998. Integrins induce activation of EGF receptor: role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 17:6622–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro, L., L. Dolce, S. Cabodi, E. Bergatto, E.B. Erba, M. Smeriglio, E. Turco, S.F. Retta, M.G. Giuffrida, M. Venturino, et al. 2002. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J. Biol. Chem. 277:9405–9414. [DOI] [PubMed] [Google Scholar]
- Mould, A.P., A.N. Garratt, J.A. Askari, S.K. Akiyama, and M.J. Humphries. 1995. Identification of a novel anti-integrin monoclonal antibody that recognises a ligand-induced binding site epitope on the beta 1 subunit. FEBS Lett. 363:118–122. [DOI] [PubMed] [Google Scholar]
- Mui, A.L., H. Wakao, N. Harada, A.M. O'Farrell, and A. Miyajima. 1995. a. Interleukin-3, granulocyte-macrophage colony-stimulating factor, and interleukin-5 transduce signals through two forms of STAT5. J. Leukoc. Biol. 57:799–803. [DOI] [PubMed] [Google Scholar]
- Mui, A.L., H. Wakao, A.M. O'Farrell, N. Harada, and A. Miyajima. 1995. b. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J. 14:1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mui, A.L., H. Wakao, T. Kinoshita, T. Kitamura, and A. Miyajima. 1996. Suppression of interleukin-3-induced gene expression by a C-terminal truncated Stat5: role of Stat5 in proliferation. EMBO J. 15:2425–2433. [PMC free article] [PubMed] [Google Scholar]
- Onishi, M., T. Nosaka, K. Misawa, A.L. Mui, D. Gorman, M. McMahon, A. Miyajima, and T. Kitamura. 1998. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol. Cell. Biol. 18:3871–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea, J.J. 1997. Jaks, STATs, cytokine signal transduction, and immunoregulation: are we there yet? Immunity. 7:1–11. [DOI] [PubMed] [Google Scholar]
- Quelle, F.W., N. Sato, B.A. Witthuhn, R.C. Inhorn, M. Eder, A. Miyajima, J.D. Griffin, and J.N. Ihle. 1994. JAK2 associates with the beta c chain of the receptor for granulocyte-macrophage colony-stimulating factor, and its activation requires the membrane-proximal region. Mol. Cell. Biol. 14:4335–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, E.P., A. Korapati, P. Chaturvedi, and S. Rane. 2000. IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene. 19:2532–2547. [DOI] [PubMed] [Google Scholar]
- Sadowski, H.B., K. Shuai, J.E. Darnell Jr., and M.Z. Gilman. 1993. A common nuclear signal transduction pathway activated by growth factor and cytokine receptors. Science. 261:1739–1744. [DOI] [PubMed] [Google Scholar]
- Sakamaki, K., I. Miyajima, T. Kitamura, and A. Miyajima. 1992. Critical cytoplasmic domains of the common beta subunit of the human GM-CSF, IL-3 and IL-5 receptors for growth signal transduction and tyrosine phosphorylation. EMBO J. 11:3541–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneller, M., K. Vuori, and E. Ruoslahti. 1997. Alphavbeta3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. EMBO J. 16:5600–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, M.A., and M.H. Ginsberg. 2002. Networks and crosstalk: integrin signalling spreads. Nat. Cell Biol. 4:E65–E68. [DOI] [PubMed] [Google Scholar]
- Short, S.M., G.A. Talbott, and R.L. Juliano. 1998. Integrin-mediated signaling events in human endothelial cells. Mol. Biol. Cell. 9:1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg, C., and K. Rubin. 1996. Stimulation of β1 integrins on fibroblasts induces PDGF independent tyrosine phosphorylation of PDGF β-receptors. J. Cell Biol. 132:741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuori, K., and E. Ruoslahti. 1994. Association of insulin receptor substrate-1 with integrins. Science. 266:1576–1578. [DOI] [PubMed] [Google Scholar]
- Wary, K.K., F. Mainiero, S.J. Isakoff, E.E. Marcantonio, and F.G. Giancotti. 1996. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 87:733–743. [DOI] [PubMed] [Google Scholar]
- Wimperis, J.Z., C.M. Niemeyer, C.A. Sieff, B. Mathey-Prevot, D.G. Nathan, and R.J. Arceci. 1989. Granulocyte-macrophage colony-stimulating factor and interleukin-3 mRNAs are produced by a small fraction of blood mononuclear cells. Blood. 74:1525–1530. [PubMed] [Google Scholar]
- Woodcock, J.M., C.J. Bagley, B. Zacharakis, and A.F. Lopez. 1996. A single tyrosine residue in the membrane-proximal domain of the granulocyte-macrophage colony-stimulating factor, interleukin (IL)-3, and IL-5 receptor common beta-chain is necessary and sufficient for high affinity binding and signaling by all three ligands. J. Biol. Chem. 271:25999–26006. [DOI] [PubMed] [Google Scholar]
- Xie, B., J. Zhao, M. Kitagawa, J. Durbin, J.A. Madri, J.L. Guan, and X.Y. Fu. 2001. Focal adhesion kinase activates Stat1 in integrin-mediated cell migration and adhesion. J. Biol. Chem. 276:19512–19523. [DOI] [PubMed] [Google Scholar]
- Yamada, K.M., and S. Even-Ram. 2002. Integrin regulation of growth factor receptors. Nat. Cell Biol. 4:E75–E76. [DOI] [PubMed] [Google Scholar]
- Yamashita, H., J. Xu, R.A. Erwin, W.L. Farrar, R.A. Kirken, and H. Rui. 1998. Differential control of the phosphorylation state of proline-juxtaposed serine residues Ser725 of Stat5a and Ser730 of Stat5b in prolactin-sensitive cells. J. Biol. Chem. 273:30218–30224. [DOI] [PubMed] [Google Scholar]
- Zhong, Z., Z. Wen, and J.E. Darnell Jr. 1994. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 264:95–98. [DOI] [PubMed] [Google Scholar]