

Abstract
Secretory protein trafficking relies on the COPI coat, which by assembling into a lattice on Golgi membranes concentrates cargo at specific sites and deforms the membranes at these sites into coated buds and carriers. The GTPase-activating protein (GAP) responsible for catalyzing Arf1 GTP hydrolysis is an important part of this system, but the mechanism whereby ArfGAP is recruited to the coat, its stability within the coat, and its role in maintenance of the coat are unclear. Here, we use FRAP to monitor the membrane turnover of GFP-tagged versions of ArfGAP1, Arf1, and coatomer in living cells. ArfGAP1 underwent fast cytosol/Golgi exchange with ∼40% of the exchange dependent on engagement of ArfGAP1 with coatomer and Arf1, and affected by secretory cargo load. Permanent activation of Arf1 resulted in ArfGAP1 being trapped on the Golgi in a coatomer-dependent manner. These data suggest that ArfGAP1, coatomer and Arf1 play interdependent roles in the assembly–disassembly cycle of the COPI coat in vivo.
Introduction
Cytosolic coat proteins that bind reversibly to membranes carry out a central role in membrane trafficking by concentrating macromolecules into specialized membrane patches that deform into coated buds to produce coated carriers (for reviews see Schekman and Orci, 1996; Bonifacino and Lippincott-Schwartz, 2003);. The coatomer (COPI)-type coat helps mediate protein sorting and transport within the Golgi membrane system (Duden et al., 1991; Waters et al., 1991). Comprised of the seven-subunit complex of coatomer (Waters et al., 1991), it is recruited onto membranes by the ras-related, small GTPase-ADP-ribosylation factor (Arf1; Donaldson et al., 1992). Coatomer–Arf1-GTP complexes then undergo low affinity interactions to generate a membrane-deforming lattice capable of budding-coated carriers. The lattice disassembles through the activity of an Arf1-directed GTPase activating protein (GAP), which by hydrolyzing GTP on Arf1 leads to the release of Arf1-GDP into the cytoplasm and the destabilization of coatomer on membranes.
Proper growth and disassembly of the coat lattice is largely dependent on the mechanisms controlling ArfGAP activity (for reviews see Springer et al., 1999; Spang, 2002). These mechanisms guarantee that disassembly of the lattice does not occur before the lattice has grown large enough to deform the membrane and to pinch off a carrier. They further ensure that the lattice disassembles quickly enough so a carrier can fuse with its target membrane.
ArfGAPs comprise a family of proteins with divergent noncatalytic domains and a conserved catalytic domain of ∼70 aa that includes a zinc finger motif (for reviews see Donaldson and Jackson, 2000; Randazzo et al., 2000). Within this family, ArfGAP1 catalyzes GTP hydrolysis on Arf1 within the COPI coat lattice localized on Golgi membranes (Cukierman et al., 1995). Binding of ArfGAP1 to Golgi membranes is mediated through interactions between its noncatalytic domain and the KDEL receptor, a transmembrane protein that constitutively cycles between the ER and Golgi apparatus (Aoe et al., 1997, 1999). Interactions of ArfGAP with the cytoplasmic tails of p23/24 family members (Lanoix et al., 2001; Majoul et al., 2001), with coatomer (Eugster et al., 2000; Lewis et al., 2004), or with SNAREs (Rein et al., 2002) might further stabilize ArfGAP1 on membranes or help recruit it into the coat lattice. Stimulation of ArfGAP1 activity is believed to result from interactions of ArfGAP1 with coatomer (Goldberg, 1999; Szafer et al., 2000) and/or by ArfGAP1 localization to membranes of high curvature, such as a coated bud or vesicle (Bigay et al., 2003).
At first glance, the role of ArfGAP1 in coat lattice dynamics is simple: complexes of Arf1 and coatomer formed on membranes polymerize into a lattice that buds off the membrane as a coated vesicle. ArfGAP1 is then recruited to the lattice and stimulates lattice disassembly by catalyzing GTP hydrolysis on Arf1. Supporting this view are studies showing that COPI-coated vesicles can be assembled in vitro in the absence of ArfGAPs and that these vesicles undergo rapid uncoating upon addition of purified GAP (Bremser et al., 1999; Reinhard et al., 2003). However, other in vitro studies have shown that cargo recruitment into coated vesicles, a process that occurs before vesicle budding, depends on GTP hydrolysis by Arf1 (Nickel et al., 1998; Lanoix et al., 1999). Furthermore, in coated vesicle reconstitution assays using ArfGAP1, coatomer, and Arf1, the purified coated vesicles are enriched in ArfGAP1 and coatomer and relatively depleted of Arf1 (Yang et al., 2002). This has given rise to a different model for ArfGAP1's role in coat lattice dynamics in which ArfGAP1 is a basic component of the coat on membranes (forming a ternary complex with Arf1 and coatomer) that undergoes changes in catalytic activity (from inactive to active) as the coat assembles into a lattice (Yang et al., 2002; Bigay et al., 2003).
To determine which of these models best applies to the living cell what is needed is a way to visualize the temporal and spatial operation of ArfGAP1 and the molecules with which it interacts. Previous studies have successfully visualized Arf1 and coatomer in living cells, but the dynamic behavior of ArfGAP1 has not yet been observed. Here, we use GFP-tagged ArfGAP1, as well as GFP-tagged versions of Arf1, coatomer, and ArfGAP1 lacking its zinc finger–containing GAP domain, to study the dynamics of ArfGAP1 and its role in COPI lattice assembly and disassembly in vivo. Our findings favor a model in which ArfGAP1 is a bona fide component of the coat lattice that regulates the dynamics of the lattice independently of coated vesicle production. Furthermore, we find that levels of ArfGAP1 and coatomer bound to Golgi membranes can be modulated by transiting secretory cargo, implying a role of cargo in the regulation of coat lattice dynamics.
Results
ArfGAP1-GFP localizes to the Golgi and functions as a GAP for Arf1
To study ArfGAP dynamics in living cells, we fused CFP or YFP to the carboxy terminus of ArfGAP1 (ArfGAP1-C/YFP) and generated a stable NRK cell line expressing the chimera at low levels. In these cells, ArfGAP1-YFP could be seen distributed on Golgi membranes (colocalizing with the Golgi marker, galactosyltransferase tagged with CFP) in the cytoplasm (Fig. 1 A) with approximately four times more ArfGAP1-YFP in the cytoplasm than on Golgi membranes. In addition to being present in the cytoplasm and on Golgi membranes, a small amount of ArfGAP1-YFP could be seen associated with peripheral structures containing ɛCOPI-YFP (i.e., ERGIC; Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200410142/DC1). A similar ArfGAP1 distribution pattern has been reported by antibody labeling (Cukierman et al., 1995), indicating that ArfGAP1-YFP distributes in a similar manner to endogenous untagged ArfGAP1.
Figure 1.

In vivo GAP activity of ArfGAP1-YFP. (A) Golgi and cytoplasmic distribution of ArfGAP1-YFP in a stable NRK cell line expressing ArfGAP1-YFP at low levels. (B) Golgi-associated pool of Arf1-CFP (expressed as a percentage of total cellular fluorescence) measured in NRK cells stably expressing Arf1-CFP alone (control) (n = 16) or after cotransfection with ArfGAP1-YFP (n = 16). Note that the steady-state Golgi pool of Arf1-CFP is reduced in cells coexpressing ArfGAP1-YFP. (C) Kinetics of dissociation of Arf1-CFP during BFA treatment (5 μg/ml−1) measured in NRK cells stably expressing Arf1-CFP alone (black Δ), with cotransfected ArfGAP1-YFP (red ○), or with cotransfected ArfGAP1Δ64N-YFP (green □). (D) NRK cells transiently expressing ArfGAP1-CFP and GalT-YFP were imaged 20 h after transfection. In cells with high levels of ArfGAP1-CFP expression (arrows), GalT-YFP was redistributed into the ER while ArfGAP1–YFP was found mainly in the cytoplasm. Bar, 5 μm.
To determine whether ArfGAP1-C/YFP functions as a GAP for Arf1 in vivo, we looked for evidence of increased Arf1 GTP hydrolysis in cells expressing ArfGAP1-C/YFP at moderate-to-high expression levels by transient transfection. Three lines of evidence were found. First, in cells coexpressing ArfGAP1-YFP and Arf1-CFP, the Golgi pool of Arf1-CFP was reduced relative to cells expressing Arf1-CFP alone (Fig. 1 B), which is consistent with increased ArfGAP1 activity from expression of ArfGAP1-YFP shifting the steady-state distribution of Arf1 from membranes to cytoplasm due to increased Arf1 GTP hydrolysis and release from membranes. Second, in cells treated with brefeldin A (BFA), a drug that blocks Arf1 recruitment to membranes (permitting Arf1 dissociation to be observed in the absence of rebinding; Peyroche et al., 1999), Arf1-CFP's dissociation rate was increased in the presence of ArfGAP1-YFP (Fig. 1 C) as expected for conditions in which Arf1 GTP hydrolysis on membranes is increased. No increased Arf1-CFP release was observed in BFA-treated cells overexpressing YFP-tagged ArfGAP1 mutant that lacks the zinc finger motif and has negligible GAP activity (ArfGAP1Δ64N-YFP; Huber et al., 1998). Indeed, in these cells, the Arf1-CFP dissociation rate during BFA treatment was reduced (Fig. 1 C) possibly due to competition between endogenous and mutated ArfGAP1 for binding sites on Golgi membranes (see below). Third, in cells coexpressing ArfGAP1-CFP and the Golgi enzyme marker galactosyltransferase tagged with YFP (GalT-YFP), GalT-YFP redistributed into the ER while ArfGAP1-CFP became cytoplasmic at high ArfGAP1-CFP expression levels (10–50 times greater than in low expressing cells; Fig. 1 D, see arrows). Because this type of redistribution is characteristic of cells with reduced levels of Arf1 GTP on membranes (Dascher and Balch, 1994), the results suggested there was increased ArfGAP1 activity in these cells due to ArfGAP1-YFP expression. Based on these findings, we concluded that ArfGAP1-YFP correctly functions as a GAP for Arf1 when expressed in cells.
ArfGAP's membrane association–dissociation cycle and its modulation by Arf1
Given that ArfGAP1-YFP appropriately targets and functions within cells, we used the chimera in experiments aimed at investigating the kinetic properties of ArfGAP1's association with Golgi membranes. These and subsequent experiments were performed in the stable NRK cell line that expresses ArfGAP1-YFP at low levels (2.5-fold above endogenous ArfGAP1), unless indicated otherwise. We first addressed whether ArfGAP1 resides stably or only transiently after being recruited to Golgi membranes. This was accomplished using FRAP. Upon photobleaching the Golgi pool of fluorescence, we observed rapid recovery from the nonbleached cytoplasmic pool, with the original, prebleach fraction of Golgi fluorescence (i.e., 20% of the total cellular pool) completely restored in just under 1 min (Fig. 2, A and D). As ArfGAP1-YFP in the cytoplasm recovered into a photobleached box with a half-time of 1 s (not depicted), the recovery rate of ArfGAP1-YFP observed after photobleaching the Golgi was determined primarily by membrane association–dissociation processes (and was not limited by ArfGAP1-YFP diffusion through the cytoplasm). We concluded, therefore, that Golgi-associated ArfGAP1-YFP molecules undergo rapid exchange with freely diffusing ArfGAP1-YFP molecules in the cytoplasm, and that an individual ArfGAP1-YFP molecule spends only a short period bound to Golgi membranes. Evidence that all Golgi-associated ArfGAP1-YFP molecules are dynamically associated with the Golgi in this manner was suggested by the fact that when the cytoplasm was repeatedly photobleached using the technique of fluorescence loss in photobleaching (FLIP; for review see Lippincott-Schwartz et al., 2001), virtually all Golgi-associated ArfGAP1-YFP fluorescence was lost over time (Fig. 2 B, FLIP). Hence, ArfGAP1 resides only transiently after being recruited to Golgi membranes.
Figure 2.
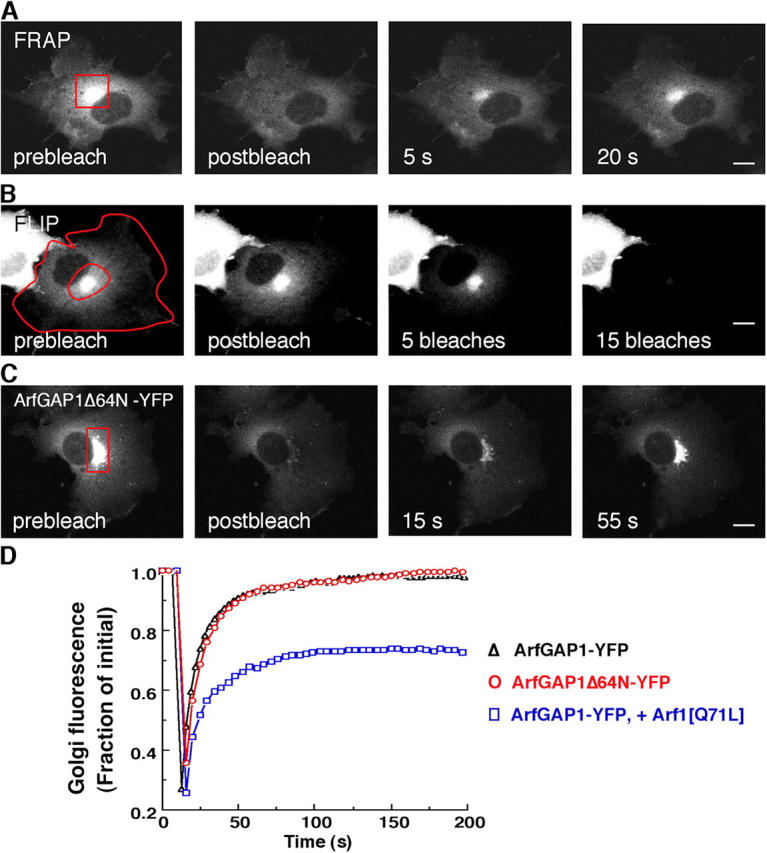
Kinetics of ArfGAP1 binding to and dissociation from Golgi membranes. (A and C) NRK cells stably expressing ArfGAP1-YFP in A or transiently expressing ArfGAP1Δ64N-YFP in C were imaged before (prebleach) and after photobleaching the Golgi region (outlined in red) with high intensity laser light. Note the rapid fluorescence recovery into the Golgi. (B) Repeated photobleaching (FLIP) of the cytoplasm defined by the area between the two red lines caused all Golgi fluorescence within an NRK cell expressing ArfGAP1-YFP to disappear over time. (D) Quantification of the FRAP experiment from A and C, as well as from a similar FRAP experiment in NRK cells stably expressing ArfGAP1-YFP in which an Arf1[Q71L] plasmid was microinjected 9 h before microscopy. Golgi fluorescence in this as well as all other FRAP and BFA experiments was represented as the ratio of Golgi-to-total cell fluorescence divided by the initial ratio. Bars, 5 μm.
To determine whether cycling of ArfGAP1 on and off Golgi membranes depends on ArfGAP1's catalytic activity, we used ArfGAP1Δ64N-YFP. Cells expressing ArfGAP1Δ64N-YFP showed a similar Golgi and cytoplasmic pattern of fluorescence as observed in ArfGAP1-YFP–expressing cells (Fig. 2 C). Moreover, when the Golgi pool of ArfGAP1Δ64N-YFP–expressing cells was photobleached, rapid and complete recovery occurred from nonbleached molecules in the cytoplasm (Fig. 2, C and D). These results indicated that the noncatalytic domain within ArfGAP1 is sufficient both for Golgi targeting of ArfGAP1 and for mediating ArfGAP1 cycling on and off Golgi membranes. Hence, ArfGAP1's capacity to dynamically associate with and dissociate from the Golgi is not dependent on its ability to catalyze the GTP hydrolysis by Arf1.
We next examined whether interactions with Arf1 can affect ArfGAP1's dynamics on the Golgi. ArfGAP1-YFP expressing cells were microinjected with Arf1[Q71L], a GTP-locked form of Arf1 that is unable to hydrolyze GTP and dissociate from membranes. The Golgi pool of ArfGAP1-YFP was then photobleached and the extent of recovery of fluorescence in the Golgi region was monitored over time. Notably, ∼30–40% of the initial ArfGAP1-YFP fluorescence on the Golgi did not recover (Fig. 2 D), suggesting this proportion of ArfGAP1 was irreversibly bound to Golgi membranes (i.e., in a complex with Arf1[Q71L]), whereas the remaining proportion continued to cycle between Golgi membranes and the cytoplasm. Given that there was ∼100% recovery of the fluorescent Golgi pool in cells coexpressing ArfGAP1Δ64N-YFP and Arf1[Q71L] (not depicted), as well as in cells expressing ArfGAP1-YFP or ArfGAP1Δ64N-YFP alone (Fig. 2 D), the results indicated that the membrane dissociation properties of ArfGAP1 are sensitive to the GTP state of Arf1.
ArfGAP1 follows both Arf1-dependent and -independent pathways on Golgi membranes
The above results suggested that ArfGAP1 is recruited to membranes in an Arf1-independent fashion, but once on membranes, its fate differs depending on whether or not it interacts with Arf1 (Fig. 3 A, scheme). With no such interaction, ArfGAP1 releases from membranes via an Arf1-independent pathway, whereas with such an interaction, ArfGAP1 releases via an Arf1-dependent pathway that requires GTP hydrolysis on Arf1. Within either pathway, ArfGAP1 resides on membranes for only short periods (∼30 s) before releasing into the cytoplasm, whereupon it mixes with other ArfGAP1 molecules and then rebinds to Golgi membranes.
Figure 3.

ArfGAP1 follows both Arf1-dependent and –independent pathways on Golgi membrane. (A) Scheme for ArfGAP1 membrane binding and dissociation pathways. (B) Release of Arf1-CFP, ArfGAP1-YFP, and ArfGAP1Δ64N-YFP from Golgi membranes after BFA addition (5 μg/ml−1) in NRK cells stably expressing Arf1-CFP and cotransfected with ArfGAP1-YFP or ARFGAP1Δ64N-YFP. Notice the differences in the rates and extents of release of the three proteins from the Golgi during the BFA treatment. Bars, 5 μm. (C) Quantification of the release kinetics shown in B. (D) After 300 s of BFA treatment, the persisting Golgi pool of ArfGAP1-YFP in an NRK cell such as that shown in B, was photobleached and recovery into the Golgi region was quantified. The results indicated that the BFA-resistant Golgi pool of ArfGAP1-YFP continues to cycle on and off Golgi membranes before Golgi disassembly (which occurs sometime after 600 s in BFA).
To address what proportion of ArfGAP1 follows such Arf1-dependent and -independent pathways on Golgi membranes we used BFA. By preventing Arf1 from binding to Golgi membranes without affecting Arf1's dissociation rate, BFA treatment leads to rapid depletion of Arf1 from membranes (Presley et al., 2002), which later causes the Golgi to disassemble (Sciaky et al., 1997). A consequence of removing Arf1 from Golgi membranes in this manner should be that ArfGAP1 no longer enters the Arf1-dependent pathway. Given that the ArfGAP1 molecules within this pathway still dissociate from membranes, the net effect of BFA treatment should be to redistribute ArfGAP1 molecules within the Arf1-dependent pathway into the cytoplasm.
Upon addition of BFA to ArfGAP1-YFP–expressing cells, ∼40% of the Golgi pool of ArfGAP1-YFP redistributed into the cytoplasm during the first 300 s, with the remaining Golgi pool persisting until Golgi disassembly (Fig. 3, B and C). Four lines of evidence supported the interpretation that the released Golgi fraction of ArfGAP1-YFP corresponded to ArfGAP1 molecules within the Arf1-dependent pathway, whereas the persisting Golgi fraction represented ArfGAP1 molecules that did not interact with Arf1. First, Arf1 levels on the Golgi, assessed in BFA-treated cells expressing Arf1-CFP, were significantly depleted by 300 s of BFA treatment (Fig. 3, B and C), indicating the Arf1-dependent pathway was not operating at this time point. Second, BFA treatment did not cause cytoplasmic redistribution of ARFGAP1Δ64N-YFP, which does not interact with Arf1 and so associates with Golgi membranes only in an Arf1-independent manner (Fig. 3, B and C). Third, when the Golgi pool of ArfGAP1-YFP persisting on the Golgi at 300 s of BFA treatment was photobleached almost complete recovery occurred (Fig. 3 D). This is predicted for ArfGAP1 molecules that cycle on and off Golgi membranes in the absence of interactions with Arf1. Finally, the percentage of the Golgi pool of ArfGAP1 that was released into the cytoplasm during BFA treatment (i.e., ∼40%) was similar to the percentage of the Golgi pool of ArfGAP1 that became immobilized on Golgi membranes during expression of Arf1[Q71L], which stabilizes ArfGAP1 within the Arf1-dependent pathway. These findings led us to conclude that at a steady state, ∼40% of the Golgi pool of ArfGAP1 binds to and dissociates from Golgi membranes in an Arf1-dependent manner, whereas ∼60% does so in an Arf1-independent manner. The rate of Arf1-dependent dissociation of ArfGAP1-YFP from Golgi membranes during BFA treatment was slower than that of Arf1-CFP and resembled that of coatomer as reported in a previous study (Presley et al., 2002).
AlF treatment stabilizes ArfGAP1 within the Arf1-dependent pathway
Addition of AlF to cells leads to the formation of ionic complexes of aluminum and fluoride that can mimic the γ-phosphate of GTP on G proteins, causing them to switch into a stable, GTP-like conformation (Bigay et al., 1985). In small G proteins, the switch to a GTP-like conformation only occurs when AlF binds to them in the presence of their corresponding GAP (Mittal et al., 1996). Given these properties, we used AlF as a tool to investigate the mechanism(s) whereby ArfGAP1 is recruited into the Arf1-dependent pathway and the role of this pathway in coat lattice production.
Addition of AlF to cells expressing ArfGAP1-YFP resulted in ∼40% of ArfGAP1-YFP on the Golgi becoming irreversibly bound, as assessed by photobleaching (Fig. 4 A). Because no stable Golgi pool of ArfGAP1-YFP was observed in untreated cells, we concluded that the irreversibly bound pool represented complexes of fluoride–Arf1–ArfGAP1. The size of the stabilized pool was similar to the ArfGAP1 pool populating the Arf1-dependent pathway, assessed by either BFA treatment or Arf1[Q71L] expression (Fig. 3 C and Fig. 2 D). This suggested that the complexes of fluoride–Arf1–ArfGAP1 were formed within the Arf1-dependent pathway.
Figure 4.

Golgi association–dissociation kinetics of ArfGAP1-YFP in AlF-treated cells. (A, C, E, and F) FRAP of Golgi fluorescence in NRK cells expressing respectively ArfGAP1-YFP (as a stable cell line) in A, ArfGAP1Δ64N-YFP (as a transient transfectant) in C, ɛCOP-YFP (as a stable cell line) in E, or GGA-YFP (as a transient transfectant) in F with or without AlF (50 μM AlCl3, 20 mM NaF) treatment for 10 min. (B) Release kinetics of ArfGAP1-YFP from Golgi membranes after addition of BFA (5 μg/ml−1) with or without AlF pretreatment for 10 min in NRK cells stably expressing ArfGAP1-YFP. (D) ldlF cells were transfected with ArfGAP1-YFP and incubated at 32°C or at 40°C for 3 h to deplete COPI from Golgi membranes. AlF was added for 10 min or not. ArfGAP1-YFP in the Golgi region was then photo-bleached and recovery of Golgi fluorescence was measured over time.
Consistent with this possibility, when BFA was added to AlF-treated cells expressing ArfGAP1-YFP, no drop in ArfGAP1-YFP levels on the Golgi occurred (Fig. 4 B). This can be explained if Golgi-associated ArfGAP1-YFP molecules in these cells consist of a fraction that is stably bound up as fluoride–Arf1–ArfGAP1 complexes within the Arf1-dependent pathway and a fraction that is still cycling on and off Golgi membranes within the Arf1-independent pathway (which is BFA insensitive). In support of this interpretation, AlF treatment was found to have no affect on the dynamics of ArfGAP1 molecules incapable of interacting with Arf1, with no stabilized fluorescent Golgi pool formed in AlF-treated cells expressing ArfGAP1Δ64N-YFP (Fig. 4 C). Hence, AlF treatment only affects ArfGAP1 molecules moving through the Arf1-dependent pathway, causing ArfGAP1 to be trapped as stable fluoride–Arf1–ArfGAP1 complexes within this pathway.
Role of coatomer in the Arf1-dependent pathway
Coatomer is known to be recruited to membranes by Arf1-GTP where it can assemble into a coat lattice. Given this, we asked whether ArfGAP1 molecules moving through the Arf1 dependent pathway serve to catalyze GTP hydrolysis on Arf1 molecules associated specifically with coatomer. To address this, we tested whether stabilized fluoride–Arf1–ArfGAP1 complexes could form in AlF-treated cells depleted of coatomer. We expressed ArfGAP1-YFP in ldlF cells (Guo et al., 1996), which contain a temperature-sensitive form of ɛCOP that is degraded at 40°C. The cells were incubated either at the permissive temperature of 32°C or for 3 h at the restrictive temperature of 40°C (which causes depletion of >75% of all cellular coatomer; Presley et al., 2002). AlF was then added for 10 min. Golgi fluorescence was subsequently photobleached and the extent of fluorescence recovery onto the Golgi was measured.
The effect of AlF in ldlF cells incubated at 32°C was similar to what we observed for other cell types treated with AlF, with ∼40% of the Golgi pool of ArfGAP1-YFP failing to recover after photobleaching (Fig. 4 D). By contrast, after AlF treatment of ldlF cells incubated at 40°C, close to 100% of the Golgi pool of ArfGAP1-YFP recovered after photobleaching. Recovery in these cells was slightly slower than in cells incubated at 32°C in the absence of AlF, potentially due to coatomer not being completely depleted during the 3 h of incubation at 40°C. Together, these results suggested that depletion of coatomer prevents stabilized fluoride–Arf1–ArfGAP1 complexes from forming during AlF treatment. This implies that interactions between coatomer and ArfGAP1 occur within the Arf1-dependent pathway and are necessary for entry of ArfGAP1 into this pathway.
This interpretation would predict that coatomer itself becomes irreversibly bound on Golgi membranes during AlF treatment, which previous studies have shown (Presley et al., 2002) and we confirmed in photobleaching experiments in cells expressing ɛCOP-YFP (Fig. 4 E). Interestingly, AlF treatment did not lead to the stabilization of a different Golgi-associated, Arf1-dependent coat protein, GGA, when YFP-tagged version of this protein was expressed within cells and Golgi fluorescence was photobleached (Fig. 4 F). This suggested that during AlF treatment, fluoride–Arf1–ArfGAP1 complexes associate with coatomer and not other Golgi coat proteins. This property could serve to ensure that ArfGAP1 only interacts with Arf1 molecules that have coatomer associated with them, thereby allowing the Arf1-dependent pathway to function specifically in COPI lattice formation.
To obtain biochemical evidence that ArfGAP1 interacts with coatomer, we examined whether ArfGAP1 and β-COP underwent coimmunoprecipitation. In both COS-7 cells and NRK cells, endogenous β-COP coimmunoprecipitated with endogenous ArfGAP1 (Fig. 5 A). And, in cells expressing ArfGAP1-GFP, immunoprecipitation using GFP antibodies resulted in coimmunoprecipitation of β-COP. The complex between ArfGAP and β-COP was dependent on Arf1-GTP on membranes as BFA treatment, which inactivates Arf1, prevented ArfGAP1 and β-COP from interacting (Fig. 5 B). AlF treatment increased the association between ArfGAP and β-COP, as predicted if AlF stabilizes complexes of fluoride–Arf1–ArfGAP1–coatomer on membranes. This stabilization was dependent on Arf1 being present on membranes because treating cells with BFA before AlF treatment prevented ArfGAP1 and β-COP from associating, whereas treating cells with AlF before BFA treatment did not (Fig. 5 C). The data thus supported our morphological findings suggesting that ArfGAP1–coatomer–Arf1 ternary complexes form within cells and are affected by treatments with BFA or AlF.
Figure 5.

Biochemical evidence for membrane coatomer–ArfGAP1 complex formation. (A) COS-7 and NRK cell lysates were immunoprecipitated with polyclonal anti-ArfGAP1 antibody, and the immunocomplexes were subjected to immunoblot assay with the monoclonal anti–β-COP antibody. (B and C) NRK cells expressing ArfGAP1-YFP were treated without or with BFA (5 μg/ml−1) for 30 min, AlF for 10 min, BFA 30 min then AlF 10 min, or AlF 10 min then BFA 30 min. The cell lysates were immunoprecipitated with monoclonal anti-GFP antibody, and were followed by Western blotting with polyclonal anti–β-COP (top) antibody or anti-GFP antibody (bottom).
Arf1-dependent ArfGAP1 cycling on/off membranes in the absence of vesicle budding
The two opposing models for ArfGAP1's role in COPI lattice formation (Introduction) differ in their respective predictions for when and where ArfGAP1 is recruited to and released from membrane. In the first model, ArfGAP1 is not a coat component but is selectively recruited to the lattice after the lattice has budded off as a coated vesicle (where it then catalyzes vesicle uncoating; Bremser et al., 1999; Reinhard et al., 2003). In the second model, ArfGAP1 is a stoichiometric component of the coat (forming a ternary complex with Arf1 and coatomer) that is recruited to membranes before coat lattice assembly and undergoes changes in its catalytic activity as the coat assembles into a lattice (Yang et al., 2002; Bigay et al., 2003). To test these predictions we examined by photobleaching whether or not Arf1-dependent, ArfGAP1 cycling on and off membranes occurred in cells expressing ArfGAP1-YFP incubated at 4°C, a temperature in which vesicle budding is inhibited (Bremser et al., 1999). Under the first model, cycling should be inhibited because such recruitment and release relies on vesicle budding. Under the second model, cycling should continue because ArfGAP1 recruitment to and release from the lattice does not depend on coated vesicle budding.
Cells expressing ArfGAP1-YFP were incubated at 4°C and their Golgi pool of fluorescence was photobleached. Significantly, complete recovery into the Golgi region (with kinetics similar to that in cells incubated at 37°C) was observed, indicating ArfGAP1 still cycled on and off Golgi membranes at 4°C (Fig. 6 A). To determine whether such cycling involved movement of ArfGAP1 through the Arf1-dependent pathway (e.g., requiring Arf1-dependent GTP hydrolysis catalyzed by ArfGAP1), we tested by photobleaching whether AlF treatment for 10 min could induce an irreversibly bound pool of ArfGAP1 (corresponding to stabilized complexes of fluoride–Arf1–ArfGAP1–coatomer) at this temperature. Notably, ∼40% of the fluorescent Golgi pool showed no recovery after photobleaching (Fig. 6 B), indicating this percentage of ArfGAP1 on the Golgi had become immobilized during the AlF treatment. These data, combined with previous data showing GTP hydrolysis–dependent release of Arf1 and coatomer from membrane at 4°C (Presley et al., 2002), indicated that ArfGAP1-YFP molecules could enter and pass through the Arf1-dependent pathway at 4°C. Hence, ArfGAP1 catalytic activity within the coat lattice is not coupled to or dependent on vesicle budding. The findings thus favored a model in which ArfGAP1 is a stoichiometric component of the coat whose catalytic activity is necessary both for coat lattice assembly and disassembly (Yang et al., 2002; Bigay et al., 2003).
Figure 6.

Membrane association–dissociation kinetics of ArfGAP1-YFP at 4°C. (A) Prebleach and recovery images of NRK cell stably expressing ArfGAP1-YFP whose Golgi pool was photobleached after incubation at 4°C for 1 h or more. Bar, 5 μm. (B) Quantification of FRAP experiment shown in A and from a parallel experiment in which cells at 4°C expressing ArfGAP1-YFP were treated with AlF for 10 min before photobleaching the Golgi. Note the appearance of an immobile pool in the cells treated with AlF, which suggested that both Arf1-independent and Arf1-dependent pathways operate at 4°C.
Modulation of ArfGAP1 association with Golgi membranes in response to increased secretory traffic
ArfGAP1's short presence on Golgi membranes (irrespective of whether it passes through Arf1-dependent or -independent pathways) could serve to allow cells to rapidly modulate membrane-bound pools of ArfGAP1 so that coat lattice assembly can be regulated in response to changes in secretory cargo levels, thereby facilitating the membrane sorting events necessary for secretory transport. If so, then a change in the amount of Golgi-associated ArfGAP1 should occur under changing conditions of cargo transport through the Golgi.
To investigate this possibility, we visualized the behavior of ArfGAP1-YFP during release of a bolus of VSVG-CFP into the secretory pathway by temperature shift from 40°C to 32°C (Bergmann, 1989; Presley et al., 1997; Scales et al., 1997). Notably, a dramatic increase in the amount of Golgi-associated ArfGAP1-YFP was observed at the time when VSVG-CFP molecules passed through the Golgi apparatus, with Golgi-associated ArfGAP1-YFP levels more than doubling (Fig. 6, A and B). When VSVG-CFP molecules reached the plasma membrane, Golgi-associated ArfGAP1-YFP levels dropped back down to the levels observed before release of VSVG cargo from the ER (not depicted). The increased size of the Golgi pool of ArfGAP1-YFP was not a general effect of increased cargo transport through the Golgi, but was specific and dependent on ArfGAP1's ability to interact with Arf1 and coatomer. This was demonstrated in cells expressing ArfGAP1Δ64N-YFP, which showed no change in the size of their Golgi pool during VSVG-CFP transport through the Golgi (Fig. 7, A and B). This was further confirmed by a similar increase in Golgi membrane-bound β-COP in response to VSVG flowing into the Golgi (Fig. 7, A and B). When the Golgi pool of ArfGAP1-YFP was photobleached at the time when VSVG-CFP passed through the Golgi, rapid and complete recovery of Golgi fluorescence occurred (Fig. 7 C). This indicated that the additional ArfGAP1-YFP molecules recruited onto the Golgi during this time period were not stably associated but underwent continuous binding and release, as observed for ArfGAP1-YFP molecules normally associated with the Golgi (Fig. 2 A). Together, these results indicated that ArfGAP1 and coatomer levels on the Golgi are modulated in response to changes in secretory cargo transport through the Golgi. The modulation occurs as ArfGAP1 continuously cycles on and off Golgi membranes and is dependent on ArfGAP1's ability to interact with Arf1.
Figure 7.

Recruitment of coat proteins onto Golgi membranes during cargo transport through the Golgi. (A) COS-7 cells transiently expressing VSVG-CFP only, VSVG-CFP and ArfGAP1-YFP, or VSVG-CFP and ArfGAP1Δ64N-YFP were incubated at 40°C shortly after transfection to retain VSVG-CFP in the ER. Cells were imaged over time upon shift from 40°C to 32°C. Shown are the distributions at 0 and 30 min. To observe the change on Golgi-bound coatomer, COS-7 cells were infected with ts045 VSV and were cultured at 40°C for 4 h. Then the cells were shifted to 32°C for 30 min, fixed, and followed by immunofluorescence staining with anti–β-COP antibody and Alexa Fluor 594–tagged second antibody. (B) To assess changes in ArfGAP1 and coatomer association with Golgi membranes during VSVG transport, the total ArfGAP1-YFP (n = 6), ArfGAP1Δ64N-YFP (n = 6), or β-COP (n = 6) fluorescence associated with the Golgi was measured under nonsaturating conditions, expressed as a fraction of the total cellular fluorescence, and normalized to the initial zero minute value. (C) After shifting from 40°C to 32°C for 30 min, ArfGAP1-YFP in the Golgi region in cells expressing both VSVG-CFP and ArfGAP1-YFP were photobleached, and the fluorescence recovery into the bleached area was monitored over time. The observed rapid recovery of ArfGAP1-YFP fluorescence into the Golgi region indicated that ArfGAP1-YFP molecules continued to cycle between Golgi membranes and the cytoplasm during VSVG-CFP transport through this organelle. Bars, 5 μm.
Discussion
In this study, we have used fluorescence imaging and FRAP techniques to characterize the membrane association–dissociation kinetics of ArfGAP1 in single living cells. Building on models from in vitro reconstitution systems, we have then asked what role these kinetics play in the assembly and disassembly of the COPI coat lattice on Golgi membranes. We found that ArfGAP1 underwent fast exchange between Golgi membranes and cytosol. Once bound to membranes, ArfGAP1 followed either an Arf1-dependent pathway (productive for coat lattice assembly) or was released directly into the cytoplasm in an Arf1-independent manner (nonproductive for coat lattice assembly; Fig. 8, model). The extent of ArfGAP1's cytosol/Golgi exchange could be modulated by secretory cargo load, was independent of vesicle budding, and could be blocked in a coatomer-dependent fashion when Arf1 was permanently activated. These findings lead us to a model, discussed below, in which ArfGAP1, coatomer, and Arf1 play interdependent roles in the assembly–disassembly cycle of the COPI coat.
Figure 8.

Proposed model for ArfGAP1's behavior on Golgi membranes and its role in coat lattice assembly. Upon binding to membranes, ArfGAP1 follows either an Arf1-independent pathway that is nonproductive for coat assembly or an Arf1-dependent pathway that is productive for coat assembly. ArfGAP1 molecules following either pathway persist on Golgi membranes for only short periods. Recruitment of ArfGAP1 into the Arf1-dependent pathway is mediated by coatomer, which by forming a ternary complex with ArfGAP1 and Arf1 generates the basic coat unit. These units diffuse in the membrane and nucleate into small aggregates. Upon assembly of the aggregates into basket-like lattices, the underlying membrane begins to bend, which stimulates ArfGAP1 activity (Bigay et al., 2003). This leads to the release of Arf1 from the lattice, with ArfGAP1 and coatomer becoming destabilized and releasing soon thereafter. Hence, there is a constant flux of Arf1, coatomer, and ArfGAP1 through the lattice during the assembly of the lattice whether or not the lattice pinches off as a coated vesicle.
We found that recruitment of ArfGAP1 to Golgi membranes was not dependent on interactions with Arf1 or coatomer, as ArfGAP1Δ64N-YFP showed the same Golgi membrane binding properties as ArfGAP1-YFP in photobleaching assays. Moreover, such recruitment occurred even at 4°C, in which vesicle production is inhibited. This argued against a model in which ArfGAP1 targets directly from the cytoplasm on to coated vesicles and then releases (Bremser et al., 1999; Reinhard et al., 2003). Rather, the data supports the idea that the initial membrane recruitment of ArfGAP1 can occur at widely dispersed sites on the Golgi, potentially through interactions of ArfGAP1 with the KDEL receptor or with the cytoplasmic tails of p23/24 proteins (Aoe et al., 1997, 1999; Lanoix et al., 2001; Majoul et al., 2001).
Our data showed that ArfGAP1's fate once bound to Golgi membranes differs depending on whether or not it interacts with Arf1 and coatomer. In the absence of such interactions, ArfGAP1 dissociates from membranes by an Arf1-independent manner that is nonproductive for coat lattice assembly as shown by the fact that it was insensitive to BFA or AlF treatment and did not respond to Arf1[Q71L] expression. In the presence of interactions with Arf1 on Golgi membranes, ArfGAP1 follows an Arf1-dependent pathway that is productive for coat lattice assembly. Significantly, release of ArfGAP1 into the cytoplasm from this pathway is dependent on GTP hydrolysis on Arf1. This is supported by our finding that when GTP hydrolysis on Arf1 is inhibited by Arf1[Q71L] expression or by AlF treatment, a proportion of the Golgi pool of ArfGAP1 no longer cycles between Golgi and cytoplasm.
ArfGAP1 entry into the Arf1-dependent pathway was found to be dependent on coatomer. This was supported by our findings that (a) COPI and ArfGAP1 interact in biochemical assays, and (b) in ldlF cells depleted of coatomer and treated with AlF, no formation of stable complexes of fluoride–Arf1–ArfGAP1 occurs. The formation of immobilized pools of ArfGAP1 and coatomer on the Golgi observed normally in cells treated with AlF likely corresponds to complexes of fluoride–Arf1–ArfGAP1–coatomer. This would be consistent with previous studies in yeast showing interactions between coatomer subunits and ArfGAP (Eugster et al., 2000; Lewis et al., 2004), and a recent in vitro system reporting direct binding of coatomer to ArfGAP1 (Lee et al., 2005), as well as with studies examining the effect of AlF on liposomes incubated with Arf1, ArfGAP1, and coatomer (Bigay et al., 2003). Because virtually all coatomer became stabilized on membranes during AlF treatment, compared with ∼40% of ArfGAP1 and <10% of Arf1 (unpublished data), coatomer would appear to be the rate-limiting component of the complex of Arf1–ArfGAP1–coatomer in the cells being examined.
The formation of Arf1–ArfGAP1–coatomer complexes suggests that coatomer serves to spatially localize ArfGAP1 on membranes so that ArfGAP1 catalytic activity is confined to Arf1 molecules in association with the COPI coat. This would imply that different Arf1-dependent coat/effector proteins are used for recruiting GAP proteins to different locations to hydrolyze Arf1 GTP, which recent data showing an interaction between AGAP1 and AP3 on endosomes supports (Nie et al., 2003). A different function of these complexes could be to allow coatomer to exert a stimulatory effect on GTPase activity; for example, by increasing the affinity of ArfGAP1 for Arf1, by assisting ArfGAP1 in orienting the Arf1 catalytic machinery, or by supplying an arginine finger residue to the Arf1 active site (Goldberg, 1999; Szafer et al., 2000; Bigay et al., 2003). Such stimulatory effects, in turn, may depend on changes in membrane curvature caused by assembly of the Arf1–ArfGAP1–coatomer complexes into a coat lattice (Bigay et al., 2003).
Based on these and previous data (Yang et al., 2002; Bigay et al., 2003; Lee et al., 2005), a role for ArfGAP1 in coat lattice dynamics can be proposed (Fig. 8). In this scheme, membrane-bound ArfGAP1 is recruited by coatomer into a ternary unit comprised of ArfGAP1–coatomer–Arf1. This unit serves as a basic coat unit, which upon diffusing in the membrane and encountering other units begins to nucleate into small aggregates. Once the aggregates have grown large enough they begin to bend the membrane by forming basket-like lattices. In this scenario, ArfGAP1's GTPase activity is inactive during the formation/diffusion of individual ternary units, and stimulated once the aggregates have begun to bend the membrane. Once Arf1-GTP is hydrolyzed and released from membranes, coatomer and ArfGAP1 within the lattice become destabilized.
A consequence of this model is that ArfGAP1 would be expected to move into the lattice from the rims (bound to Arf1 and coatomer) and then to be released from the interior (after Arf1 has hydrolyzed its GTP and been released from membranes), resulting in a continuous flux of ArfGAP1 through the lattice whether or not detachment of a coated vesicle occurs (Ehrlich et al., 2004). This would explain why ArfGAP1 is able to cycle on and off Golgi membranes at 4°C and become immobilized on membranes as fluoride–Arf1–ArfGAP1–coatomer complexes upon AlF treatment at this temperature.
The short duration of ArfGAP1 on Golgi membranes would allow Arf1-mediated GTP hydrolysis to be an ongoing process during the formation of the COPI-coated vesicle. This is analogous to the behavior of microtubules, which show similar dynamics (e.g., high energy GTP-bound units entering the polymer are transformed to a lower energy GDP-bound form that dissociates from the structure over time). Just as microtubules maintain their length for certain periods without significant growth or shrinkage (through treadmilling), the coat lattice could be metastable, in which it remains associated with membranes for considerable periods without budding off as a vesicle. An attractive feature of this model is that it allows the coat lattice to be constantly modulated: the lattice can dissipate, go on to pinch off as a coated carrier, or persist as a coated bud. In the latter case, the coated bud could act to stabilize microdomains on Golgi membranes or create membrane tension. The constant flux of coat proteins through the lattice would also allow rapid modulation of the lattice in response to changing levels of cargo moving through the secretory pathway, as our data have demonstrated. The ensuing changes in coat lattice growth or dissipation could thereby serve to facilitate the membrane sorting events involved in secretory transport.
Materials and methods
DNA constructs, reagents, and antibodies
Arf1-GFP, Arf1-CFP, Arf1[Q71L], and GalT-YFP plasmids were described previously (Ward et al., 2001; Presley et al., 2002). ArfGAP1-YFP, ArfGAP1-CFP, and ArfGAP1Δ64N-YFP were made by cloning the rat ArfGAP1 ORF into pECFP-N1 or pEYFP-N1 vector using the Kpn I (5′) and Age I (3′) restriction sites. GGA-YFP was a gift from J.S. Bonifacino (National Institutes of Health, Bethesda, MD 20892). BFA was purchased from Epicentre Technologies and used at 5 μg/ml. In all experiments with AlF, 50 μM AlCl3, and 20 mM NaF were used and were added to medium separately for 10 min.
The following antibodies were used: rabbit polyclonal antiserum to ArfGAP1 (a gift from D. Casso, Technion-Israel Institute of Technology, Haifa, Israel); rabbit polyclonal antibody to β-COP (Affinity BioReagents, Inc.); mouse monoclonal antibody to β-COP (Abcam Inc.); and mouse monoclonal antibody to GFP (BD Biosciences). Alexa Fluor 594–tagged second antibody was purchased from Molecular Probes.
Cell transfection and DNA microinjection
NRK and COS-7 cells were grown in DME supplemented with 10% FCS, 2 mM glutamine, 100 U/ml of penicillin, and 100 U/ml of streptomycin. ldlF cells were maintained in RPMI with full supplement at 32°C. Transient transfections were performed using FuGENE 6 Transfection Reagent according to the manufacturer's instructions (Roche Molecular Biochemicals). Cells were analyzed 15–24 h after transfection. Cell line expressing ArfGAP1-YFP, Arf1-CFP, or ɛCOP-YFP was created by transiently transfection followed by selection with G418.
Microinjections were performed using the Eppendorf Transjector 5242 and micromanipulator 5171 system mounted on an inverted microscope (Carl Zeiss MicroImaging, Inc.). Arf1[Q71L] cDNA were microinjected 9 h before the microscopic observation in NRK cells that have been transfected with ArfGAP1-YFP or ArfGAP1Δ64N-YFP and were growing on etched glass coverslip. Texas red dextrins were included in the injectate to identify the injected cells.
VSV infection and immunofluorescence staining
COS-7 cells growing on sterile glass coverslips were infected with ts045 VSV (50–100 plague-forming units/cell) for 1 h at 32°C. Cells were washed twice in serum-free DME to remove unabsorbed virus, and then shifted in serum-containing DME to 40°C for 4 h to maintain VSVG protein in the ER. Cells were either kept at 40°C or incubated at 32°C for another 30 min, then were fixed by 2% formaldehyde. After two times PBS washing, cells were incubated in PBS/FBS (PBS, pH 7.4, containing 10% FBS) to block nonspecific sites of antibody adsorption. Cells were incubated with polyclonal anti–β-COP plus monoclonal anti-VSVG (P5D4) antibody followed with Alexa Fluor 594–labeled goat anti–rabbit and FITC-labeled goat anti–mouse secondary antibodies, respectively.
Immunoprecipitation and Western blotting
For each immunoprecipitation, 2 μl of polyclonal anti-ArfGAP1 or monoclonal anti-GFP (JL-8) were preincubated with Sepharose 4B beads for 1 h at RT. The antibody-bound beads were washed with PBS and incubated with cell lysates at 4°C for 2 h. The complexes then were washed three times with washing buffer (0.1% Triton X-100 and 5 mM EDTA in 150 mM NaCl, 50 mM Tris-HCl, pH 7.5) and used for Western blotting.
Western blotting was performed as described previously (Liu et al., 1999). In brief, proteins obtained from lysed cells or immunoprecipitates were denatured and loaded on 4–20% Tris-glycine gel (Invitrogen). They were transferred to polyvinylidene difluoride membranes, incubated in 5% nonfat milk, and were stained with specific anti–β-COP or anti-GFP antibody followed by HRP-conjugated goat-anti–rabbit or goat-anti–mouse antibody. The specific bands were detected by ECL plus Western blotting Detection System (Amersham Biosciences).
Fluorescence microscope, imaging and photobleaching analysis
Cells in LabTek chambers (Nalge Nunc International) were imaged in buffered medium with a confocal microscope (model 510; Carl Zeiss MicroImaging, Inc.) with a stage heated to 37°C using a 488-nm laser excitation for GFP, 514 nm for YFP and 413 nm for CFP. For quantification of fluorescence intensities, nonsaturated images were taken with 40×1.3 NA objective and a full-open pinhole, whereas a 63×1.4 NA objective and a pinhole diameter equivalent to 1–3 Airey units were used for nonquantitative imaging. Selective photobleaching was performed using appropriate laser line at full power and the recovery was then monitored by time lapse imaging at 1.5–10-s interval at low intensity illumination. Fluorescence quantification was done as described previously (Hirschberg et al., 1998; Presley et al., 2002). Basically, intensities for total cellular fluorescence or Golgi-associated fluorescence were measured using NIH Image software after subtraction of background outside the cell or cytoplasm overlap. The Golgi-associated fluorescence in BFA or FRAP experiments was represented as the ratio of Golgi-to-total cellular fluorescence at each time point divided by the initial ratio (before BFA treatment or FRAP). Cells were imaged with no saturated pixels. The experiments that were quantified and represented graphically were each done on 16–20 cells under identical conditions. Each of the data points in the graphs, therefore, represent the average value of the 16–20 similarly treated cells at that time point.
Online supplemental material
Fig. S1 shows colocalization of ArfGAP1-GFP with coatomer in Golgi and peripheral ERGIC structures. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200410142/DC1.
Acknowledgments
We are grateful to Nihal Altan-Bonnet and George Patterson for expert technical assistance. We thank Eric Snapp, Julie Donaldson, and Catherine Jackson for critical reading of the manuscript.
Abbreviations used in this paper: BFA, brefeldin A; FLIP, fluorescence loss in photobleaching; GAP, GTPase-activating protein.
References
- Aoe, T., E. Cukierman, A. Lee, D. Cassel, P.J. Peters, and V.W. Hsu. 1997. The KDEL receptor, ERD2, regulates intracellular traffic by recruiting a GTPase-activating protein for ARF1. EMBO J. 16:7305–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoe, T., I. Huber, C. Vasudevan, S.C. Watkins, G. Romero, D. Cassel, and V.W. Hsu. 1999. The KDEL receptor regulates a GTPase-activating protein for ADP-ribosylation factor 1 by interacting with its non-catalytic domain. J. Biol. Chem. 274:20545–20549. [DOI] [PubMed] [Google Scholar]
- Bergmann, J.E. 1989. Using temperature sensitive-mutants of VSV to study membrane protein biogenesis. Methods Cell Biol. 32:85–110. [DOI] [PubMed] [Google Scholar]
- Bigay, J., P. Deterre, C. Pfister, and M. Chabre. 1985. Fluoroaluminates activate transducin-GDP by mimicking the gamma-phosphate of GTP in its binding site. FEBS Lett. 191:181–185. [DOI] [PubMed] [Google Scholar]
- Bigay, J., P. Gounon, S. Robineau, and B. Antonny. 2003. Lipid packing sensed by ArfGAP1 couples COPI coat disassembly to membrane bilayer curvature. Nature. 426:563–566. [DOI] [PubMed] [Google Scholar]
- Bonifacino, J.S., and J. Lippincott-Schwartz. 2003. Coat proteins: shaping membrane transport. Nat. Rev. Mol. Cell Biol. 4:409–414. [DOI] [PubMed] [Google Scholar]
- Bremser, M., W. Nickel, M. Schweikert, M. Ravazzola, M. Amherdt, C.A. Hughes, T.H. Sollner, J.E. Rothman, and F.T. Wieland. 1999. Coupling of coat assembly and vesicle budding to packaging of putative cargo receptors. Cell. 96:495–506. [DOI] [PubMed] [Google Scholar]
- Cukierman, E., I. Huber, M. Rotman, and D. Cassel. 1995. The ARF1 GTPase-activating protein: zinc finger motif and Golgi complex localization. Science. 270:1999–2002. [DOI] [PubMed] [Google Scholar]
- Dascher, C., and W.E. Balch. 1994. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J. Biol. Chem. 269:1437–1448. [PubMed] [Google Scholar]
- Donaldson, J.G., and C.L. Jackson. 2000. Regulators and effectors of the ARF GTPases. Curr. Opin. Cell Biol. 12:475–482. [DOI] [PubMed] [Google Scholar]
- Donaldson, J.G., D. Cassel, R.A. Kahn, and R.D. Klausner. 1992. ADP-ribosylation factor, a small GTP-binding protein, is required for binding of the coatomer protein beta-COP to Golgi membranes. Proc. Natl. Acad. Sci. USA. 89:6408–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duden, R., G. Griffiths, R. Frank, P. Argos, and T.E. Kreis. 1991. Beta-COP, a 110 kd protein associated with non-clathrin-coated vesicles and the Golgi complex, shows homology to beta-adaptin. Cell. 64:649–665. [DOI] [PubMed] [Google Scholar]
- Ehrlich, M., W. Boll, A. Van Oijen, R. Hariharan, K. Chandran, M.L. Nibert, and T. Kirchhausen. 2004. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 118:591–605. [DOI] [PubMed] [Google Scholar]
- Eugster, A., G. Frigerio, M. Dale, and R. Duden. 2000. COP I domains required for coatomer integrity, and novel interactions with ARF and ARF-GAP. EMBO J. 19:3905–3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, J. 1999. Structural and functional analysis of the ARF1-ARFGAP complex reveals a role for coatomer in GTP hydrolysis. Cell. 96:893–902. [DOI] [PubMed] [Google Scholar]
- Guo, Q., M. Penman, B.L. Trigatti, and M. Krieger. 1996. A single point mutation in epsilon-COP results in temperature-sensitive, lethal defects in membrane transport in a Chinese hamster ovary cell mutant. J. Biol. Chem. 271:11191–11196. [DOI] [PubMed] [Google Scholar]
- Hirschberg, K., C.M. Miller, J. Ellenberg, J.F. Presley, E.D. Siggia, R.D. Phair, and J. Lippincott-Schwartz. 1998. Kinetic analysis of secretory protein traffic and characterization of Golgi to plasma membrane transport intermediates in living cells. J. Cell Biol. 143:1485–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber, I., E. Cukierman, M. Rotman, T. Aoe, V.W. Hsu, and D. Cassel. 1998. Requirement for both the amino-terminal catalytic domain and a noncatalytic domain for in vivo activity of ADP-ribosylation factor GTPase-activating protein. J. Biol. Chem. 273:24786–24791. [DOI] [PubMed] [Google Scholar]
- Lanoix, J., J. Ouwendijk, C.C. Lin, A. Stark, H.D. Love, J. Ostermann, and T. Nilsson. 1999. GTP hydrolysis by arf-1 mediates sorting and concentration of Golgi resident enzymes into functional COP I vesicles. EMBO J. 18:4935–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoix, J., J. Ouwendijk, A. Stark, E. Szafer, D. Cassel, K. Dejgaard, M. Weiss, and T. Nilsson. 2001. Sorting of Golgi resident proteins into different subpopulations of COPI vesicles: a role for ArfGAP1. J. Cell Biol. 155:1199–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S.Y., J.S. Yang, W. Hong, R.T. Premont, and V.W. Hsu. 2005. ARFGAP1 plays a central role in coupling COPI cargo sorting with vesicle formation. J. Cell Biol. 168:281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, S.M., P.P. Poon, R.A. Singer, G.C. Johnston, and A. Spang. 2004. The ArfGAP Glo3 is required for the generation of COPI vesicles. Mol. Biol. Cell. 15:4064–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., E. Snapp, and A. Kenworthy. 2001. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2:444–456. [DOI] [PubMed] [Google Scholar]
- Liu, W., A.A. Akhand, M. Kato, I. Yokoyama, T. Miyata, K. Kurokawa, K. Uchida, and I. Nakashima. 1999. 4-Hydroxynonenal triggers an epidermal growth factor receptor-linked signal pathway for growth inhibition. J. Cell Sci. 112:2409–2417. [DOI] [PubMed] [Google Scholar]
- Majoul, I., M. Straub, S.W. Hell, R. Duden, and H.D. Soling. 2001. KDEL-cargo regulates interactions between proteins involved in COPI vesicle traffic: measurements in living cells using FRET. Dev. Cell. 1:139–153. [DOI] [PubMed] [Google Scholar]
- Mittal, R., M.R. Ahmadian, R.S. Goody, and A. Wittinghofer. 1996. Formation of a transition-state analog of the RAS GTPase reaction by Ras•GDP, tetrafluoroaluminate, and GTPase-activating protein. Science. 273:115–117. [DOI] [PubMed] [Google Scholar]
- Nickel, W., J. Malsam, K. Gorgas, M. Ravazzola, N. Jenne, J.B. Helms, and F.T. Wieland. 1998. Uptake by COPI-coated vesicles of both anterograde and retrograde cargo is inhibited by GTPgammaS in vitro. J. Cell Sci. 111:3081–3090. [DOI] [PubMed] [Google Scholar]
- Nie, Z., M. Boehm, E.S. Boja, W.C. Vass, J.S. Bonifacino, H.M. Fales, and P.A. Randazzo. 2003. Specific regulation of the adaptor protein complex AP-3 by the Arf GAP AGAP1. Dev. Cell. 5:513–521. [DOI] [PubMed] [Google Scholar]
- Peyroche, A., B. Antonny, S. Robineau, J. Acker, J. Cherfils, and C.L. Jackson. 1999. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: involvement of specific residues of the Sec7 domain. Mol. Cell. 3:275–285. [DOI] [PubMed] [Google Scholar]
- Presley, J.F., N.B. Cole, T.A. Schroer, K. Hirschberg, K.J. Zaal, and J. Lippincott-Schwartz. 1997. ER-to-Golgi transport visualized in living cells. Nature. 389:81–85. [DOI] [PubMed] [Google Scholar]
- Presley, J.F., T.H. Ward, A.C. Pfeifer, E.D. Siggia, R.D. Phair, and J. Lippincott-Schwartz. 2002. Dissection of COPI and Arf1 dynamics in vivo and role in Golgi membrane transport. Nature. 417:187–193. [DOI] [PubMed] [Google Scholar]
- Randazzo, P.A., Z. Nie, K. Miura, and V.W. Hsu. 2000. Molecular aspects of the cellular activities of ADP-ribosylation factors. Sci. STKE. 2000:RE1. [DOI] [PubMed]
- Rein, U., U. Andag, R. Duden, H.D. Schmitt, and A. Spang. 2002. ARF-GAP-mediated interaction between the ER-Golgi v-SNAREs and the COPI coat. J. Cell Biol. 157:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard, C., M. Schweikert, F.T. Wieland, and W. Nickel. 2003. Functional reconstitution of COPI coat assembly and disassembly using chemically defined components. Proc. Natl. Acad. Sci. USA. 100:8253–8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales, S.J., R. Pepperkok, and T.E. Kreis. 1997. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 90:1137–1148. [DOI] [PubMed] [Google Scholar]
- Schekman, R., and L. Orci. 1996. Coat proteins and vesicle budding. Science. 271:1526–1533. [DOI] [PubMed] [Google Scholar]
- Sciaky, N., J. Presley, C. Smith, K.J. Zaal, N. Cole, J.E. Moreira, M. Terasaki, E. Siggia, and J. Lippincott-Schwartz. 1997. Golgi tubule traffic and the effects of brefeldin A visualized in living cells. J. Cell Biol. 139:1137–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang, A. 2002. ARF1 regulatory factors and COPI vesicle formation. Curr. Opin. Cell Biol. 14:423–427. [DOI] [PubMed] [Google Scholar]
- Springer, S., A. Spang, and R. Schekman. 1999. A primer on vesicle budding. Cell. 97:145–148. [DOI] [PubMed] [Google Scholar]
- Szafer, E., E. Pick, M. Rotman, S. Zuck, I. Huber, and D. Cassel. 2000. Role of coatomer and phospholipids in GTPase-activating protein-dependent hydrolysis of GTP by ADP-ribosylation factor-1. J. Biol. Chem. 275:23615–23619. [DOI] [PubMed] [Google Scholar]
- Ward, T.H., R.S. Polishchuk, S. Caplan, K. Hirschberg, and J. Lippincott-Schwartz. 2001. Maintenance of Golgi structure and function depends on the integrity of ER export. J. Cell Biol. 155:557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters, M.G., T. Serafini, and J.E. Rothman. 1991. ‘Coatomer’: a cytosolic protein complex containing subunits of non-clathrin-coated Golgi transport vesicles. Nature. 349:248–251. [DOI] [PubMed] [Google Scholar]
- Yang, J.S., S.Y. Lee, M. Gao, S. Bourgoin, P.A. Randazzo, R.T. Premont, and V.W. Hsu. 2002. ARFGAP1 promotes the formation of COPI vesicles, suggesting function as a component of the coat. J. Cell Biol. 159:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]