

Abstract
Excessive accumulation of amyloid β-peptide (Aβ) plays an early and critical role in synapse and neuronal loss in Alzheimer's Disease (AD). Increased oxidative stress is one of the mechanisms whereby Aβ induces neuronal death. Given the lessened susceptibility to oxidative stress exhibited by mice lacking p66Shc, we investigated the role of p66Shc in Aβ toxicity. Treatment of cells and primary neuronal cultures with Aβ caused apoptotic death and induced p66Shc phosphorylation at Ser36. Ectopic expression of a dominant-negative SEK1 mutant or chemical JNK inhibition reduced Aβ-induced JNK activation and p66Shc phosphorylation (Ser36), suggesting that JNK phosphorylates p66Shc. Aβ induced the phosphorylation and hence inactivation of forkhead transcription factors in a p66Shc-dependent manner. Ectopic expression of p66ShcS36A or antioxidant treatment protected cells against Aβ-induced death and reduced forkhead phosphorylation, suggesting that p66Shc phosphorylation critically influences the redox regulation of forkhead proteins and underlies Aβ toxicity. These findings underscore the potential usefulness of JNK, p66Shc, and forkhead proteins as therapeutic targets for AD.
Introduction
Alzheimer's Disease (AD) is a neurodegenerative disease that causes progressive cognitive and behavioral deterioration. Although research on AD is extensive, the etiology of the disease is not completely understood. A central hypothesis in AD research is that the accumulation of amyloid β-peptide (Aβ), which is derived from proteolytic processing of amyloid precursor protein, is an early and critical event leading to synapse and neuronal cell loss (Iversen et al., 1995; Selkoe, 2001). Increased oxidative stress has been proposed to play an important role in Aβ-induced neuronal death in AD because reactive oxygen species (ROS) and oxidized lipids and proteins increase and accumulate in AD brain (Behl et al., 1994; Selkoe, 2001; Butterfield et al., 2002; Butterfield and Bush, 2004; Huang et al., 2004). In vitro studies have shown that complexes resulting from the association of Aβ with metals (Fe, Cu, and Zn) exhibit metalloenzyme-like activities leading to the production of neurotoxic H2O2 (Huang et al., 1999a; Opazo et al., 2002). However, the signaling mechanisms whereby Aβ induces oxidative cell death remain unclear.
A heightened resistance to oxidative stress has been shown to increase longevity in Caenorhabditis elegans and mice (Lin et al., 1997; Migliaccio et al., 1999). Mice lacking the adaptor protein p66Shc live 30% longer than control animals, and cells derived from these mice are resistant to ROS-induced apoptosis (Migliaccio et al., 1999). p66Shc plays an important role in signal transduction from tyrosine kinases to Ras protooncogenes. The three mammalian ShcA isoforms (p46, p52, and p66) share structural features including a COOH-terminal Src homology 2 domain, a proline- and glycine-rich region, a collagen-homologous region 1, and an NH2-terminal phosphotyrosine-binding domain (Pelicci et al., 1992; Luzi et al., 2000). In addition, there is a unique collagen-homologous region 2 domain at the NH2 terminus of the p66Shc isoform containing a serine phosphorylation site (Ser36). It is well established that Shc proteins are phosphorylated at tyrosine residues in response to stimulation by a variety of growth factors and cytokines (Pelicci et al., 1992; Rozakis-Adcock et al., 1992; Cutler et al., 1993; Pronk et al., 1993; Ruff-Jamison et al., 1993). The p46 and p52 isoforms transmit signals from receptor tyrosine kinases to the Ras–MAPK pathway by forming a stable complex involving Grb2 and a Ras exchange factor, SOS (Son of Sevenless; Pelicci et al., 1992; Egan et al., 1993; Skolnik et al., 1993; Aronheim et al., 1994; Kavanaugh and Williams, 1994; Ohmichi et al., 1994). However, p66Shc appears to be functionally different from the p46 and p52 isoforms. Unlike the other isoforms, p66Shc undergoes phosphorylation mainly at Ser36 after exposure to oxidative stress such as UV light or H2O2 (Kao et al., 1997; Migliaccio et al., 1999; Yang and Horwitz, 2000; Le et al., 2001; Nemoto and Finkel, 2002), although p66Shc is also transiently phosphorylated at tyrosine residues in response to growth factor stimulation (Bonfini et al., 1996; Migliaccio et al., 1997). Phosphorylation at Ser36 is required for conferring increased susceptibility to oxidative stress and is critical for the cell death response elicited by oxidative damage (Skulachev, 2000). Therefore, prevention of this phosphorylation may have a therapeutic impact on diseases that are associated with oxidative damage.
Forkhead transcription factors (FKHs) are a large family of gene products expressed in species ranging from yeast to human, all of which have a domain with sequence similarly to Drosophila forkhead protein. A forkhead subfamily member, DAF-16 in C. elegans, functions in insulin signaling pathways and influences longevity (Lin et al., 1997, 2001; Ogg et al., 1997; Accili and Arden, 2004; Giannakou et al., 2004; Hwangbo et al., 2004). Previous studies have demonstrated that for each of the three DAF-16 vertebrate homologues, FKHR (FOXO1), FKHR-L1 (FOXO3a), and AFX (FOXO4), phosphorylation results in the retention of the protein in the cytosol and, hence, a reduction in forkhead-dependent transcriptional activity (Biggs et al., 1999; Brunet et al., 1999; Kops et al., 1999; Takaishi et al., 1999; Tang et al., 1999; Burgering and Kops, 2002). Subsequent studies have demonstrated the roles of FKHRL1, FKHR, and AFX in the insulin response, the oxidative stress response, and the growth arrest/apoptosis pathways (S. Guo et al., 1999; Medema et al., 2000; Zheng et al., 2002; Brunet et al., 2004). Strikingly, recent papers show that p66Shc regulates intracellular oxidant levels in mammalian cells and that ROS can negatively regulate forkhead activity (Nemoto and Finkel, 2002; Trinei et al., 2002). In contrast, activation of FKHs leads to transcriptional activation of the manganese superoxide dismutase (MnSOD) gene and increases MnSOD mRNA and protein levels. Elevated MnSOD levels cause a reduction of ROS and protection against cell death (Nemoto and Finkel, 2002; Trinei et al., 2002). The role of p66Shc and FKHs in Aβ-induced oxidative damage in AD is unknown.
In this study, we test the hypothesis that Aβ increases intracellular ROS level, activates the p66Shc signaling pathway, and negatively regulates FKH activity. We demonstrate that treatment with Aβ causes cell death and p66Shc phosphorylation at Ser36, partially in a JNK-dependent manner. Aβ induced the phosphorylation (inactivation) of forkhead FKHRL1 and FKHR transcription factors in established cultures and in mouse primary neuronal cultures. Cells that ectopically expressed p66ShcS36A or were treated with antioxidants were protected against Aβ-induced death and exhibited reduced forkhead phosphorylation. These findings lend strong support to the notion that phosphorylation of Shc66 and forkhead protein play important roles in Aβ toxicity in AD.
Results
Aβ induces phosphorylation of p66Shc at serine 36
p66Shc is phosphorylated on serine/threonine residues in response to cellular stress induced by treatment with UV, H2O2, or Taxol (Kao et al., 1997; Migliaccio et al., 1999; Yang and Horwitz, 2000; Le et al., 2001; Nemoto and Finkel, 2002), although p66Shc also is transiently phosphorylated at tyrosine residues in response to growth factor stimulation (Bonfini et al., 1996; Migliaccio et al., 1997). Here, we set out to investigate the role of p66Shc in Aβ toxicity. Treatment of human neuroblastoma SH-SY5Y and neuronal-like PC12 cells with 30 μM Aβ 25-35 (hereafter Aβ) for 24 h caused cell death in both cell lines, whereas 30 μM of the control peptide Aβ 35-25 had no effect (Fig. 1 A). To determine whether or not p66Shc was phosphorylated on Ser36 in response to Aβ, lysates of Aβ-treated SH-SY5Y cells were analyzed by Western blotting using an antibody that specifically recognized phospho-p66Shc at Ser36. Aβ treatment of cells rapidly induced p66Shc phosphorylation at Ser36, which remained elevated for 15 h (Fig. 1 B, top), whereas Aβ 35-25 treatment of cells was without effect (not depicted). In contrast, protein levels of p66, p52, and p46 isoforms were not affected (Fig. 1 B, bottom). We confirmed these results by immunoprecipitating total Shc protein and performing Western blot analysis using the Ser36 phospho-p66Shc antibody (unpublished data). We also assessed the phosphorylation of p66Shc by using mouse cortical neuronal cultures at 7 d in vitro (DIV). Aβ treatment of cultures caused dose-dependent cell death (Fig. 1, C and D) and induced p66Shc phosphorylation at Ser36, which remained elevated for 24 h (Fig. 1 E), whereas the response of Aβ 35-25–treated cultures was comparable to that of untreated cultures. The phosphorylation of p66Shc was abolished by serine/threonine protein phosphatase 2A1 (PP2A1; Fig. 1 F), further confirming that p66Shc is phosphorylated at Ser36 by Aβ.
Figure 1.

Treatment of Aβ induces the phosphorylation of p66Shc at Ser36 residues in SH-SY5Y cells. (A) SH-SY5Y and PC12 cells were treated with 30 μM Aβ25-35 or Aβ35-25 for 24 h in serum-free media containing N2 supplements. Trypan blue exclusion was used to determine cell death. Data are means ± SEM for three separate experiments performed in duplicate. *, P < 0.05 versus untreated cells. (B) Western blot analysis showing phosphorylated and total p66Shc in SH-SY5Y cells treated with 30 μM Aβ for the indicated times after a 16-h serum starvation period by using a specific anti–phospho-p66Shc (Ser36) antibody (top) and an anti-Shc polyclonal antibody (bottom). The blots are representative of three separate experiments. (C) At 7 DIV, cultures were treated with Aβ for 24 h, whereupon representative photographs were taken to illustrate the changes in cell morphology. (D) At 7 DIV, cells treated with Aβ25-35 or Aβ35-25 for 24 h and Hoechst 33342 labeling were used to determine cell death. Data are the means ± SEM of three separate experiments performed in duplicate. (E) Western blot analysis to detect p66Shc phosphorylation in 7 DIV cultures treated with 25 μM Aβ for the indicated times was performed by using a specific anti–phospho-p66Shc (Ser36) antibody (top) and an anti-Shc polyclonal antibody (bottom). The blots are representative of three separate experiments. (F) SH-SY5Y cells treated with 30 μM Aβ for 30 min after a 16-h serum starvation period. The cells were harvested and the lysates were either treated with PP2A1 or left untreated, and then sequentially blotted with a specific anti–phospho-p66Shc (Ser36) antibody (top) and an anti-Shc polyclonal antibody (bottom). The blots are representative of three separate experiments.
Cells expressing dominant-negative p66ShcS36A are protected against Aβ toxicity
To investigate the functional consequences of the phosphorylation of p66Shc at Ser36 by Aβ, PC12 cells overexpressing wild-type p66Shc (p66Shc) and a serine 36-to-alanine (and therefore nonphosphorylatable) mutant of p66Shc (p66ShcS36A; Fig. 2 A) were treated with Aβ for 24 h. PC12 cells expressing p66Shc (clone A) were sensitive to Aβ, with ∼40% of cells dying as measured by Trypan blue exclusion (Fig. 2 B). A Hoechst labeling assay revealed a fourfold increase in apoptosis in Aβ-treated cells compared with untreated control cells (Fig. 2 C), whereas PC12 cells expressing p66ShcS36A (clone a) were almost completely resistant to Aβ-induced cell death (Fig. 2, B and C). Caspase-3 activity in PC12 cells expressing p66Shc (clone A) increased significantly after Aβ treatment and remained unchanged in PC12 cells expressing p66ShcS36A (clone a) (Fig. 2 D). Two other clones of p66Shc (clones B and C) exhibited sensitivities to Aβ treatment comparable to those of clone A, whereas two additional clones of p66ShcS36A (clones b and c) displayed sensitivities to Aβ treatment comparable to those of clone a (Fig. 2 E). The growth rates of all of the clones were the same as those of the vector populations, supporting the notion that the differences in sensitivity to Aβ were indeed linked to the expression of p66Shc, either wild type or mutant.
Figure 2.
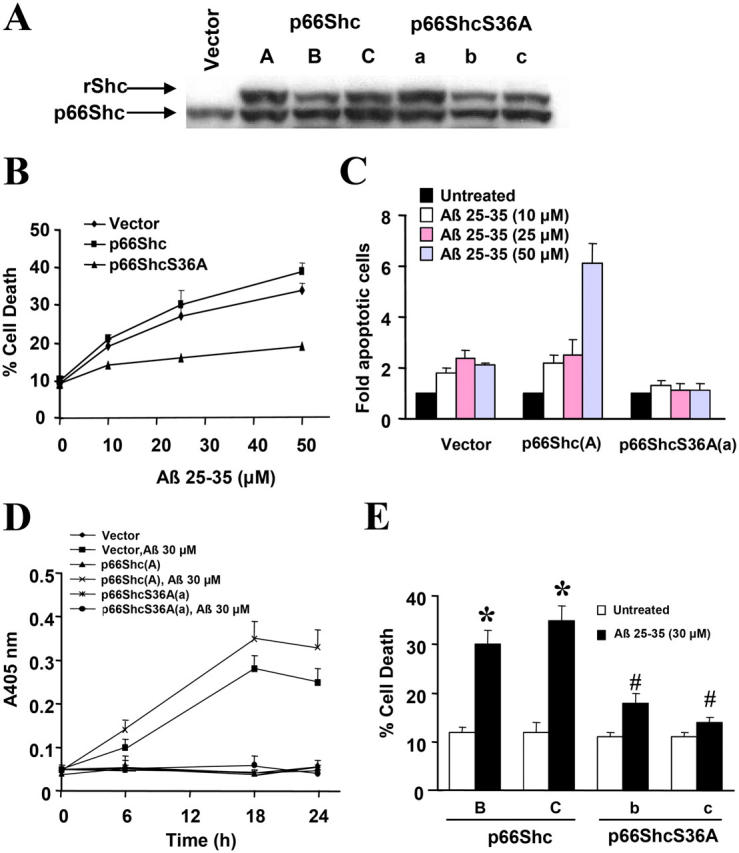
Ectopic expression of dominant-negative p66ShcS36A protects against Aβ-induced PC12 cell death. (A) Western blot analysis of Shc expression in PC12 cells stably transfected with vector, p66Shc (clones A, B, and C), or p66ShcS36A (clones a, b, and c) was performed using an anti-Shc antibody. rShc, recombinant Shc carrying the xpress-tag. (B and C) PC12 populations ectopically expressing either p66Shc (clone A) or p66ShcS36A (clone a) were treated with Aβ for 24 h in serum-free media containing N2 supplements. Cell death was measured by Trypan blue exclusion (panel B) and Hoechst nuclear labeling (panel C). (D) PC12 populations ectopically expressing either p66Shc (clone A) or p66ShcS36A (clone a) were treated with Aβ for the indicated times in serum-free media containing N2 supplements. The activity of caspase-3 in cell extracts was measured colorimetrically by cleavage of DEVD-p-nitroanilide. The results are means ± SEM from five independent experiments. (E) PC12 populations ectopically expressing p66Shc (clones B and C) or p66ShcS36A (clones b and c) were treated with 30 μM Aβ for 24 h in serum-free media containing N2 supplements, whereupon cell death was measured by Trypan blue exclusion. Data are means ± SEM for three separate experiments performed in duplicate.
We further confirmed these results by cotransfecting a GFP expression plasmid with the second plasmid, pcDNA3.1-xpress, expressing either p66Shc or p66ShcS36A or lacking an insert. 24 h after transfection, cells were treated for an additional 24 h with Aβ, and GFP-positive cells were counted using fluorescence microscopy. The transfection efficiencies were similar in all transfection groups (unpublished data). There were significantly more GFP-positive cells in the p66ShcS36A-transfected group than in groups transfected with either vector or p66Shc (Fig. 3). These results support the view that p66Shc is a crucial component in Aβ-induced cell death.
Figure 3.

Transient expression of dominant-negative p66ShcS36A protects against Aβ-induced SH-SY5Y cell death. (A) SH-SY5Y cells were cotransfected with pcDNA3.1-GFP along with either vector pcDNA3.1-xpress, pcDNA3.1-xpress-p66Shc, or pcDNA3.1-xpress-p66ShcS36A at 1:10 ratios for 24 h in 10% FBS DME media, and then incubated with 2% FBS-containing DME and treated with 30 μM Aβ for 24 h. GFP-positive cells were counted by using fluorescence microscopy. Representative photomicrographs for each treatment group are shown: top panels, GFP signals; bottom panels, cell morphology under light microscopy. (B) Quantitation of data in A, representing the cell viability of each treatment group. A minimum of 1,000 cells were counted for each treatment group and the percentage of GFP-positive cells relative to the total number of cells in six random fields was calculated. Data are means ± SEM for three separate experiments performed in duplicate. *, P < 0.05 versus untreated cells; #, P < 0.05 versus Aβ-treated vector cells.
To further understand the protection engendered by p66ShcS36A, intracellular ROS levels were measured by the redox-sensitive fluorophore 2′,7′-dichlorofluorescein (DCF) diacetate (DCFDA). Nonfluorescent DCFDA is converted by oxidation to the fluorescent molecular DCF. Under basal conditions, the level of DCF was very low in vector control-transfected cells as well as in p66Shc- and p66ShcS36A-expressing cell lines (unpublished data). However, when these cells were treated with 30 μM Aβ 25-35 for 4 h, the level of DCF was three- to fourfold lower in p66ShcS36A-expressing cells compared with p66Shc-expressing cells (Fig. 4). Previous papers had shown that Aβ increased the levels of mitochondrial superoxide and peroxynitrite in PC12 cells (Q. Guo et al., 1999), so we also measured cellular superoxide production with hydroethidine (HE), which is converted to ethidium when oxidized by superoxide (ethidium then bends the DNA and is thereby trapped in the cell, accumulating over time). The level of ethidium was two- to threefold lower in p66ShcS36A-expressing cells compared with p66Shc-expressing cells after Aβ treatment for 12 h (Fig. 4 E). The dye dihydrorhodamine (DHR) was used to quantify the relative levels of mitochondrial peroxynitrite. As shown in Fig. 4 F, 12 h after treatment with 30 μM Aβ 25-35, the levels of DHR fluorescent was about twofold lower in p66ShcS36A-expressing cells compared with p66Shc-expressing cells. These results suggested that p66Shc influenced Aβ-induced intracellular ROS, superoxide level, and mitochondrial ROS levels.
Figure 4.

Overexpression of p66ShcS36A lowers intracellular ROS, superoxide level, and mitochondrial ROS levels. (A) Quantification of DCF fluorescence in PC12 cells that were either left untreated or were treated with 30 μM Aβ using confocal microscopy and the METAMORPH image analysis software. Representative DCF fluorescence after treatment with 30 μM Aβ for a 4-h time period in PC12 cells stably transfected with plasmids ectopically expressing p66Shc (clone A; panel B) or p66ShcS36A (clone a; panel C) under confocal photomicrography of DCFDA fluorescence. These experiments were repeated three times with similar results. (D) Quantification of DCF fluorescence in PC12 cells that were either left untreated or treated with 30 μM Aβ by using a CytoFluor Multi-well Plate Reader. (E) Quantification of HE fluorescence in PC12 cells that were either left untreated or treated with 30 μM Aβ for 12 h. (F) Quantification of DHR fluorescence in PC12 cells that were either left untreated or treated with 30 μM Aβ for 12 h. Data are means ± SEM for three separate experiments.
Aβ-induced phosphorylation of p66Shc at Ser36 is partly dependent on JNK activation
JNK is a stress-activated protein kinase that plays an important role in response to oxidative damage. Recently, we demonstrated that activation of the JNK pathway by Aβ causes cell death (Wei et al., 2002b). To investigate whether or not Aβ-induced JNK activation affects p66Shc phosphorylation at serine 36, SH-SY5Y cells expressing either an insertless vector (SYV) or a dominant-negative mutant of SEK1 (an upstream kinase of JNK) SEK-AL were treated with Aβ for the indicated times. Lysates of Aβ-treated cells were analyzed by Western blotting with a specific antibody recognizing phospho-p66Shc (Ser36). SYV-transfected cells showed p66Shc phosphorylation at Ser36 between 15 to 90 min after stimulation with 30 μM Aβ. In contrast, cells expressing SEK1-AL exhibited 65% lower p66Shc phosphorylation at 15 min after Aβ treatment (Fig. 5 A). To determine if this decreased p66Shc phosphorylation correlated with suppression of Aβ-induced neuronal death, cell death was assessed by Trypan blue exclusion (Fig. 5 B) after Aβ treatment of mutant SEK1-AL and control cells. Cells expressing SEK1-AL were significantly protected against Aβ-induced cell death compared with cells transfected with the vector alone. To further confirm these results, SH-SY5Y cells treated with SP600125, an inhibitor of JNK phosphorylation, displayed partially reduced p66Shc phosphorylation and were protected against Aβ toxicity (Fig. 5, C and D). These results suggest that JNK is one of the upstream kinases that phosphorylates p66Shc at Ser36 in response to Aβ treatment.
Figure 5.

Reduction of JNK activation partially blocks Aβ-induced p66Shc phosphorylation. (A) p66Shc phosphorylation at Ser36 in SH-SY5Y cells overexpressing either SYV or SEK1-AL after addition of 30 μM Aβ at various times after a 16-h serum starvation period was detected by Western blot analysis, sequentially using anti–phospho-p66Shc (Ser36) and anti-Shc antibodies. The blots are representative of three separate experiments. (B) Cells were treated with 30 μM Aβ for 24 h in serum-free media containing N2 supplements. Trypan blue exclusion was used to determine cell death. Data are means ± SEM for three separate experiments performed in duplicate. *, P < 0.05 versus untreated cells; #, P < 0.05 versus Aβ-treated SYV cells. (C) SH-SY5Y cells were serum-starved for 16 h and were either left untreated or pretreated with 20 μM SP600125 for 1 h followed by 30-μM Aβ treatment for 10 and 20 min. Cells were harvested and cell lysates were subjected to Western blot analysis using antibodies that recognized phospho-FKHR, phospho-Shc (Ser36), and Shc. The blots are representative of three separate experiments. (D) SH-SY5Y cells were either left untreated or pretreated with 20 μM SP600125 for 1 h, and then treated with 30 μM Aβ for 24 h. Cell death was measured by Trypan blue exclusion. *, P < 0.05 versus untreated cells; #, P < 0.05 versus Aβ-treated cells.
Aβ induces forkhead phosphorylation
Previous results suggest that p66Shc regulates intracellular oxidant levels and that H2O2 can negatively regulate forkhead activity (Nemoto and Finkel, 2002; Trinei et al., 2002). Phosphorylation of forkhead proteins results in the inactivation of forkhead transcriptional activity and the down-regulation of its target genes such as those encoding ROS-scavenging enzymes (MnSOD, catalase, etc.; Nemoto and Finkel, 2002). To investigate whether or not Aβ regulates forkhead phosphorylation, lysates from SH-SY5Y cells and 7 DIV cultures treated with Aβ for various times were subjected to Western blot analysis using antibodies that detect phosphorylated FKHRL1 and FKHR (Fig. 6). Duplicate blots were probed with antibodies recognizing FKHR to verify equal protein loading of samples. As shown in Fig. 6 (A and B), treatment with Aβ led to increases in the phosphorylation of FKHRL1 and FKHR in SH-SY5Y cells and 7 DIV cultures, respectively. Interestingly, FKHR was also phosphorylated in rat PC12 cells expressing p66Shc with patterns similar to those of SH-SY5Y cells (Fig. 6 C). However, Aβ treatment did not increase the phosphorylation of forkhead in p66ShcS36A-expressing PC12 cells (Fig. 6 C), suggesting that Aβ regulated forkhead phosphorylation in a p66Shc-dependent manner. We did not detect the phosphorylation of other FKHs in these cell cultures after Aβ treatment (unpublished data). The different phosphorylated isoforms of FKHs after Aβ treatment in these cell populations may result from species differences.
Figure 6.

Aβ induces forkhead phosphorylation. (A) In SH-SY5Y cells treated with 30 μM Aβ at various times after a 16-h serum starvation period, endogenous FKHRL1 phosphorylation was detected by Western blot analysis using antibodies that recognized phospho-FKHRL1 or FKHR. (B) In 7 DIV cultures treated with 25 μM Aβ for various lengths of time, endogenous FKHR and phosphorylated FKHR were detected by Western blot analysis using antibodies that recognized phospho-FKHR or FKHR, respectively. The blots are representative of three separate experiments. (C) Endogenous FKHR phosphorylation in transfected PC12 cells that ectopically expressed either p66Shc or p66ShcS36A after treatment with Aβ was assessed by Western blotting using antibodies that recognized either phospho-FKHR or FKHR. These experiments were performed at least three times, yielding the same results.
Ebselen blocks Aβ-induced p66Shc and forkhead phosphorylation and cell death
We further investigated the findings that Aβ induced the phosphorylation of forkhead in a p66Shc-dependent manner by examining the effects of ROS scavengers after exposure to Aβ. Treatment of SH-SY5Y cells with the glutathione peroxidase mimetic ebselen reduced Aβ-induced intracellular and mitochondrial ROS production (Fig. 7, C–E), prevented the phosphorylation of p66Shc and forkhead FKHRL1 (Fig. 7 B), and protected against cell death (Fig. 7 A). Similar results were obtained in PC12 cells expressing p66Shc and in 7 DIV cultures pretreated with the glutathione precursor N-acetylcysteine (NAC; unpublished data). These findings suggest that ROS scavengers block Aβ-induced cell death by blocking the phosphorylation of p66Shc and FKHs.
Figure 7.

Effect of ebselen on p66Shc phosphorylation and Aβ toxicity. (A) SH-SY5Y cells were either left untreated or pretreated with 10 μM ebselen for 1.5 h, and then treated with 30 μM Aβ for 24 h, whereupon cell death was measured by Trypan blue exclusion. *, P < 0.05 versus untreated cells; #, P < 0.05 versus Aβ-treated cells. (B) Cells were serum-starved for 16 h, and then were either left untreated or pretreated with 10 μM ebselen for 1.5 h followed by 30-μM Aβ treatment for the time periods shown. Cells were harvested and cell lysates were subjected to Western blot analysis using antibodies that recognized phospho-KHRL1, phospho-Shc (Ser36), or Shc. The blots are representative of three separate experiments. (C) SH-SY5Y cells were either left untreated or pretreated with 10 μM ebselen for 1.5 h, and then treated with 30 μM Aβ for 4 h. DCF fluorescence was quantified in SHSY5Y cells by using a CytoFluor Multi-well Plate Reader. Data are the means ± SEM for three separate experiments performed in duplicate. *, P < 0.05 versus untreated cells; #, P < 0.05 versus Aβ-treated vector cells. (D and E) SH-SY5Y cells were either left untreated or pretreated with 10 μM ebselen for 1.5 h, and then treated with 30 μM Aβ for 12 h. HE and DHR fluorescence were quantified, respectively. Data are means ± SEM for three separate experiments performed in duplicate. #, P < 0.05 versus Aβ-treated vector cells.
Discussion
In this paper, we demonstrate that Aβ caused cell death and induced p66Shc phosphorylation at serine 36, partially in a JNK-dependent manner. Cells overexpressing dominant-negative p66ShcS36A decreased the Aβ-induced levels of intracellular ROS and were more resistant to Aβ-induced cell death. Furthermore, we demonstrate that Aβ induced the phosphorylation (and hence inactivation) of forkhead FKHRL1 and FKHR transcription factors in cells and primary cultures. Cells expressing p66ShcS36A as well as cells treated with ebselen or NAC exhibited more resistance to Aβ-induced death and reduced forkhead phosphorylation. These results suggest that phosphorylation of Ser36 p66Shc and forkhead proteins plays an important role in Aβ toxicity.
The accumulation of oxidative cellular damage has been reported in AD and some other age-related and neurodegenerative diseases, such as Parkinson's, Huntington's, and amyotrophic lateral sclerosis (Sharp and Ross, 1996; Smith et al., 1997; Albers and Beal, 2000; Jellinger, 2001). Previous papers show that Aβ can directly generate ROS including H2O2 through inherent redox activity, such as that derived from the complexing of Aβ with metals (Cu2+ and Zn2+; Huang et al., 1999a,b; Opazo et al., 2002). ROS are freely permeable across tissue boundaries and accumulate intracellularly. However, the molecular mechanisms of Aβ leading to oxidative cell death have not been fully elucidated. Recently, several groups have shown that phosphorylation of the p66Shc adaptor protein plays an important role in signaling events leading to cell death in response to oxidative damage (Migliaccio et al., 1999; Le et al., 2001; Nemoto and Finkel, 2002). Here, we show that Aβ can induce p66Shc phosphorylation at Ser36 (Fig. 1). Ectopic expression of p66ShcS36A decreased Aβ-induced intracellular ROS level and protected against Aβ-induced cell death. Moreover, treatment with antioxidants (ebselen or NAC) decreased intracellular ROS level, reduced p66Shc phosphorylation, and protected against Aβ-induced cell death (Fig. 7). These findings support the notion that Aβ causes neuronal death by mechanisms involving oxidative damage and requiring p66Shc phosphorylation. It has recently been demonstrated that p66Shc-null mouse fibroblasts have enhanced cellular resistance to treatment with oxidants, UV, and H2O2 (Migliaccio et al., 1999). Mice lacking p66Shc are less susceptible to oxidative damage induced by paraquat and have an extended life span (Migliaccio et al., 1999). These findings suggest that phosphorylation of p66Shc at Ser36 is critical for eliciting cell death in response to oxidative stress. Therefore, the prevention of p66Shc phosphorylation may have a therapeutic impact on AD and other age-related diseases that are associated with oxidative damage.
JNK is a stress-activated protein kinase whose function has been associated with the induction of apoptosis by several types of environmental stress and cellular stress (including H2O2) in neuronal cells (Verheij et al., 1996; Le Niculescu et al., 1999; Namgung and Xia, 2000). Recently, we demonstrated that JNK activation was critical for Aβ-induced neuronal death (Wei et al., 2002b). In that paper, overexpression of a dominant-interfering form of SEK1 (MKK4), the upstream kinase of JNK, prevented the cell death induced by Aβ and inhibited JNK activation (Wei et al., 2002b). Here, we show that cells expressing SEK1-AL also reduced p66Shc phosphorylation at Ser36, suggesting that JNK activation plays a role in regulating this phosphorylation. We confirmed this possibility by using a chemical inhibitor of JNK, SP600125, which yielded similar results (Fig. 5). Our findings are also in agreement with previous papers showing that JNK is the kinase that phosphorylates p66Shc at Ser36 in response to UV irradiation (Le et al., 2001), although we cannot exclude the possibility that additional kinases may also phosphorylate p66Shc. Several groups have shown that JNK is activated in degenerating or apoptotic neurons in AD brains (Anderson et al., 1995, 1996; Marcus et al., 1998; Shoji et al., 2000; Zhu et al., 2001). Together, these findings suggest that p66Shc, like c-Jun, is a substrate of JNK during the induction neuronal loss in AD by oxidative damage. However, further studies using AD transgenic mice or AD brain tissue are needed to definitively link the JNK–p66Shc pathway with AD pathogenesis in vivo.
Genetic determinants of longevity include the forkhead-related transcription factor DAF-16 in C. elegans and the p66SHC locus in mice (Lin et al., 1997; Migliaccio et al., 1999). Previous papers have demonstrated that p66Shc regulates intracellular oxidant levels in mammalian cells and that H2O2 can negatively regulate forkhead activity (Nemoto and Finkel, 2002; Trinei et al., 2002). Expression of FKHRL1 results in an increase in both ROS-scavenging activity and oxidative stress resistance (Nemoto and Finkel, 2002). In agreement with this previous literature, we show here that Aβ treatment was capable of inducing the phosphorylation of forkhead FKHRL1 and FKHR transcription factors in cells and primary cultures. Cells expressing p66ShcS36A as well as cells treated with ebselen or NAC exhibited more resistance to Aβ-induced death and reduced forkhead phosphorylation. Previous studies have shown that phosphorylation of FKHs results in their cytoplasmic retention and inactivation, thereby inhibiting the expression of FKH-regulated genes, which control the cell cycle, cell death, cell metabolism, and oxidative stress (Brunet et al., 2001; Burgering and Kops, 2002). This pathway appears to be well conserved throughout evolution from C. elegans to higher eukaryotes (Biggs et al., 1999; Brunet et al., 1999; Kops et al., 1999; Tran et al., 2002). Whereas nonphosphorylated FKHs are localized in the nucleus and activate gene transcription, phosphorylation of FKHs induces their relocalization from the nucleus to the cytoplasm (and hence away from the promoters of target genes) and consequently inhibits their ability to induce the expression of target genes such as MnSOD (Biggs et al., 1999; Brunet et al., 1999; Nakamura et al., 2000; Dijkers et al., 2000a,b; Kops et al., 2002; Tran et al., 2002). These FKH regulatory events likely represent one of the mechanisms underlying Aβ toxicity. Interestingly, in cells expressing p66ShcS36A, phosphorylation of FKHR was diminished, suggesting that p66Shc phosphorylation may be required for Aβ-induced FKH phosphorylation leading to a subsequent down-regulation of target genes such as MnSOD and ensuing cell death. These findings further suggest that p66Shc may function by regulating an intracellular ROS/redox system, which, in turn, modulates forkhead activity. Together with previous papers, we propose that Aβ toxicity is elicited through the generation of ROS leading to the activation of the JNK pathway, which in turn activates p66Shc by phosphorylation at Ser36. Activated p66Shc then triggers the phosphorylation of FKH, thereby down-regulating target genes such as MnSOD and leading to an even greater accumulation of cellular ROS. This biochemical cycle causes cell death. In summary, our findings not only advance our understanding of the intricacies of neuronal loss in AD but also suggest that JNK, p66Shc, and FKHs may be useful as potential therapeutic targets in the treatment of AD and other age-related disorders.
Materials and methods
Materials
Media, N2, and N27 supplements for cell culture were obtained from Invitrogen. Aβ 25-35 and Aβ 35-25 (Biosource International) were dissolved in water and incubated at 37°C for 24 h before use. Hoechst 33342, DCFDA, HE, and DHR were obtained from Molecular Probes. Anti-Shc antibody was purchased from BD Biosciences; anti-FKHR, anti–phospho-FKHR, and anti–phospho-FKHRL1 were purchased from Cell Signaling Technology. Anti–phospho-p66Shc (Ser36) antibody was purchased from Calbiochem. PP2A1, NAC, and ebselen were obtained from Sigma-Aldrich. SP600125 was obtained from BIOMOL Research Laboratories, Inc.
Neuronal cultures
Mouse primary cortical neuronal cultures were derived from a CD-1 outbreed mouse (The Jackson Laboratory) at embryonic 15 to 16. Cortices were dissociated as described previously (Movsesyan et al., 2004); after dissociation, neurons were plated on laminin- and poly-d-lysine–coated plates (BD Biosciences) and cultured in neurobasal medium with the addition of glutamax, B-27 supplement, and penicillin/streptomycin. Under these culture conditions, 95% of cells were neurons. At 7 DIV the neuronal cultures were treated with Aβ.
Cell culture and transfection
SH-SY5Y human neuroblastoma cells were grown in DME with 10% FBS, 1× nonessential amino acid, and 1× antibiotic-antimycotic (100 U/ml penicillin, 100 μg/ml streptomycin, and 2.5 μg/ml fungizone) solutions at 37°C in 5% CO2/95% air. Transfections were performed with LipofectAmine (Invitrogen) according to the manufacturer's protocol. SH-SY5Y cells were transfected with either pcDNA3.zeo or pcDNA.zeo-SEK1-AL constructs (provided by J.R. Woodgett, The Princess Margaret Hospital, Toronto, Canada). SEK1-AL encodes a dominant-negative form of SEK1 containing a double mutation (S220A and T224L). Pooled cells stably expressing pcDNA3.zeo or pcDNA.zeo-SEK1-AL were selected in media containing 200 μg/ml Zeocin (Invitrogen) for 2 mo (Wei et al., 2002b). PC12 cells were grown in DME with 10% FCS. Stable PC12 cell lines expressing human wild-type p66Shc and dominant-negative p66Shc were obtained by transfection and subsequent isolation by limiting dilutions in G418 (Nemoto and Finkel, 2002).
Measurement of cell death and viability
Trypan blue exclusion was used to measure cell death by counting the number of dead (blue) and live cells in the cultures after Aβ peptide treatment. Hoechst labeling of cells to detect apoptotic nuclei was performed as described previously (Dive et al., 1992; Wei et al., 2002a). In brief, cells were fixed with 4% PFA for 30 min at RT and incubated in a solution containing Hoechst 33342 and propidium iodine for 30 min, after which they were examined by fluorescence microscopy. Apoptotic cells were identified as those exhibiting condensed and fragmented nuclei. The percentage of apoptotic cells was calculated as the ratio of apoptotic cells to total cells. A minimum of 400 cells was counted for each treatment.
SH-SY5Y cell viability assay
Cells were cotransfected with pcDNA3.1-GFP along with vector pcDNA3.1-xpress, pcDNA3.1-xpress-p66Shc, or pcDNA3.1-xpress-p66ShcS36A at a 1:10 ratio for 24 h in 10% FBS DME media, and then changed to 2% FBS DME media and treated with Aβ for 24 h. GFP-positive cells were counted by using fluorescence microscopy. A minimum of 1,000 cells were counted in each treatment group. The percentage of GFP-positive cells relative to the total number of cells in six random fields was calculated.
Measurement of cellular caspase-3 activity
Cells were harvested in cell lysis buffer (50 μM Hepes, pH 7.4, 1 mM EDTA, 0.1% CHAPS, and 0.1% Triton X-100). DEVD-p-nitroanilide was the substrate for caspase-3. The experiments were performed according to the manufacturer's protocol (Biosource International).
Measurements of intracellular ROS, superoxide level, and mitochondrial ROS
The levels of cytosolic ROS were measured by DCFDA (Molecular Probes) as previously described (Nemoto and Finkel, 2002; Sponne et al., 2003). In brief, cells were treated with 30 μM Aβ 25-35 at 37°C for 4 h, washed with PBS, and incubated for 45 min with DCFDA, which is initially nonfluorescent and is converted by oxidation to the fluorescent molecular DCF. DCF was quantified using a CytoFluor Multi-well Plate Reader (Series 400; PerSeptive Biosystems) with 485-nm excitation and 538-nm emission filters; fluorescence intensity of cells was quantified using confocal microscopy and the METAMORPH image analysis software. The levels of intracellular superoxide anion radical were measured with HE, which is oxidized to fluorescent ethidium cation by superoxide, by using methods similar to those described previously (Q. Guo et al., 1999). In brief, cells were incubated for 30 min in the presence of 5 μM HE (Molecular Probes) and washed twice with Locke's solution, whereupon confocal images of cell-associated ethidium fluorescence were acquired (488-nm excitation and 590-nm emission). The average pixel intensity in individual cell bodies was determined with METAMORPH image analysis software. DHR was used to quantify relative levels of mitochondrial peroxynitrite by using methods similar to those described previously (Q. Guo et al., 1999). DHR localizes to mitochondria and fluoresces when oxidized to the positively charged rhodamine 123 derivative. In brief, cells were incubated for 30 min in the presence of 5 μM DHR and washed three times with Locke's solution, and confocal images of cellular fluorescence were acquired (488-nm excitation and 510-nm emission) and analyzed as described for DCF fluorescence.
Immunoblot and PP2A1 treatment
Cells were harvested in lysis buffer (20 mM Hepes, pH 7.4, 2 mM EGTA, 50 mM β-glycerophosphate, 1% Triton X-100, 10% glycerol, 1 mM dithiothreitol, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM Na3VO4, and 5 mM NaF). Where indicated, cell lysates (0.1 mg of protein) were treated with PP2A1 for 30 min at 30°C as described previously (Yang and Horwitz, 2000). The reaction was stopped by adding sample buffer and boiling. Lysates were resolved on 4–12% NuPAGE Bis-Tris gels (30 μg/lane) and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were blocked in TBST (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.1% Tween 20) containing 5% nonfat milk and probed with different antibodies. Proteins were detected by using ECL reagents (NEN Life Science Products).
Data analysis
Quantitative data are expressed as arithmetic means ± SEM based on at least three separate experiments performed in duplicate. The difference between two groups was statistically analyzed by t test or an analysis of variance (one-way ANOVA). A P value <0.05 was considered significant.
Acknowledgments
We thank Dr. Siyuan Le, Dr. Ricky Hirschhorn, and Zhong Pei for helpful discussions.
This research was funded by the Intramural Research Program of the National Institute on Aging, National Institutes of Health.
W.W. Smith's present address is Dept. of Psychiatry, Division of Neurobiology, Johns Hopkins University School of Medicine, Baltimore, MD 21205.
Abbreviations used in this paper: Aβ, amyloid β-peptide; AD, Alzheimer's Disease; DCF, 2′,7′-dichlorofluorescein; DCFDA, DCF diacetate; DHR, dihydrorhodamine; DIV, day in vitro; FKH, forkhead transcription factor; HE, hydroethidine; MnSOD, manganese superoxide dismutase; NAC, N-acetylcysteine; PP2A1, serine/threonine protein phosphatase 2A1; ROS, reactive oxygen species.
References
- Accili, D., and K.C. Arden. 2004. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 117:421–426. [DOI] [PubMed] [Google Scholar]
- Albers, D.S., and M.F. Beal. 2000. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J. Neural Transm. Suppl. 59:133–154. [DOI] [PubMed] [Google Scholar]
- Anderson, A.J., C.J. Pike, and C.W. Cotman. 1995. Differential induction of immediate early gene proteins in cultured neurons by beta-amyloid (A beta): association of c-Jun with A beta-induced apoptosis. J. Neurochem. 65:1487–1498. [DOI] [PubMed] [Google Scholar]
- Anderson, A.J., J.H. Su, and C.W. Cotman. 1996. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J. Neurosci. 16:1710–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronheim, A., D. Engelberg, N. Li, N. al Alawi, J. Schlessinger, and M. Karin. 1994. Membrane targeting of the nucleotide exchange factor Sos is sufficient for activating the Ras signaling pathway. Cell. 78:949–961. [DOI] [PubMed] [Google Scholar]
- Behl, C., J.B. Davis, R. Lesley, and D. Schubert. 1994. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 77:817–827. [DOI] [PubMed] [Google Scholar]
- Biggs, W.H., III, J. Meisenhelder, T. Hunter, W.K. Cavenee, and K.C. Arden. 1999. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA. 96:7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfini, L., E. Migliaccio, G. Pelicci, L. Lanfrancone, and P.G. Pelicci. 1996. Not all Shc's roads lead to Ras. Trends Biochem. Sci. 21:257–261. [PubMed] [Google Scholar]
- Brunet, A., A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, K.C. Arden, J. Blenis, and M.E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96:857–868. [DOI] [PubMed] [Google Scholar]
- Brunet, A., S.R. Datta, and M.E. Greenberg. 2001. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 11:297–305. [DOI] [PubMed] [Google Scholar]
- Brunet, A., L.B. Sweeney, J.F. Sturgill, K.F. Chua, P.L. Greer, Y. Lin, H. Tran, S.E. Ross, R. Mostoslavsky, H.Y. Cohen, et al. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 303:2011–2015. [DOI] [PubMed] [Google Scholar]
- Burgering, B.M., and G.J. Kops. 2002. Cell cycle and death control: long live Forkheads. Trends Biochem. Sci. 27:352–360. [DOI] [PubMed] [Google Scholar]
- Butterfield, D.A., and A.I. Bush. 2004. Alzheimer's amyloid beta-peptide (1-42): involvement of methionine residue 35 in the oxidative stress and neurotoxicity properties of this peptide. Neurobiol. Aging. 25:563–568. [DOI] [PubMed] [Google Scholar]
- Butterfield, D.A., A. Castegna, C.M. Lauderback, and J. Drake. 2002. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiol. Aging. 23:655–664. [DOI] [PubMed] [Google Scholar]
- Cutler, R.L., L. Liu, J.E. Damen, and G. Krystal. 1993. Multiple cytokines induce the tyrosine phosphorylation of Shc and its association with Grb2 in hemopoietic cells. J. Biol. Chem. 268:21463–21465. [PubMed] [Google Scholar]
- Dijkers, P.F., R.H. Medema, J.W. Lammers, L. Koenderman, and P.J. Coffer. 2000. a. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 10:1201–1204. [DOI] [PubMed] [Google Scholar]
- Dijkers, P.F., R.H. Medema, C. Pals, L. Banerji, N.S. Thomas, E.W. Lam, B.M. Burgering, J.A. Raaijmakers, J.W. Lammers, L. Koenderman, and P.J. Coffer. 2000. b. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol. Cell. Biol. 20:9138–9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dive, C., C.D. Gregory, D.J. Phipps, D.L. Evans, A.E. Milner, and A.H. Wyllie. 1992. Analysis and discrimination of necrosis and apoptosis (programmed cell death) by multiparameter flow cytometry. Biochim. Biophys. Acta. 1133:275–285. [DOI] [PubMed] [Google Scholar]
- Egan, S.E., B.W. Giddings, M.W. Brooks, L. Buday, A.M. Sizeland, and R.A. Weinberg. 1993. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature. 363:45–51. [DOI] [PubMed] [Google Scholar]
- Giannakou, M.E., M. Goss, M.A. Junger, E. Hafen, S.J. Leevers, and L. Partridge. 2004. Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science. 305:361. [DOI] [PubMed] [Google Scholar]
- Guo, Q., W. Fu, F.W. Holtsberg, S.M. Steiner, and M.P. Mattson. 1999. Superoxide mediates the cell-death-enhancing action of presenilin-1 mutations. J. Neurosci. Res. 56:457–470. [DOI] [PubMed] [Google Scholar]
- Guo, S., G. Rena, S. Cichy, X. He, P. Cohen, and T. Unterman. 1999. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem. 274:17184–17192. [DOI] [PubMed] [Google Scholar]
- Huang, X., C.S. Atwood, M.A. Hartshorn, G. Multhaup, L.E. Goldstein, R.C. Scarpa, M.P. Cuajungco, D.N. Gray, J. Lim, R.D. Moir, et al. 1999. a. The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 38:7609–7616. [DOI] [PubMed] [Google Scholar]
- Huang, X., M.P. Cuajungco, C.S. Atwood, M.A. Hartshorn, J.D. Tyndall, G.R. Hanson, K.C. Stokes, M. Leopold, G. Multhaup, L.E. Goldstein, et al. 1999. b. Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 274:37111–37116. [DOI] [PubMed] [Google Scholar]
- Huang, X., R.D. Moir, R.E. Tanzi, A.I. Bush, and J.T. Rogers. 2004. Redox-active metals, oxidative stress, and Alzheimer's disease pathology. Ann. NY Acad. Sci. 1012:153–163. [DOI] [PubMed] [Google Scholar]
- Hwangbo, D.S., B. Gersham, M.P. Tu, M. Palmer, and M. Tatar. 2004. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 429:562–566. [DOI] [PubMed] [Google Scholar]
- Iversen, L.L., R.J. Mortishire-Smith, S.J. Pollack, and M.S. Shearman. 1995. The toxicity in vitro of beta-amyloid protein. Biochem. J. 311:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger, K.A. 2001. Cell death mechanisms in neurodegeneration. J. Cell. Mol. Med. 5:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, A.W., S.B. Waters, S. Okada, and J.E. Pessin. 1997. Insulin stimulates the phosphorylation of the 66- and 52-kilodalton Shc isoforms by distinct pathways. Endocrinology. 138:2474–2480. [DOI] [PubMed] [Google Scholar]
- Kavanaugh, W.M., and L.T. Williams. 1994. An alternative to SH2 domains for binding tyrosine-phosphorylated proteins. Science. 266:1862–1865. [DOI] [PubMed] [Google Scholar]
- Kops, G.J., N.D. de Ruiter, A.M. Vries-Smits, D.R. Powell, J.L. Bos, and B.M. Burgering. 1999. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 398:630–634. [DOI] [PubMed] [Google Scholar]
- Kops, G.J., R.H. Medema, J. Glassford, M.A. Essers, P.F. Dijkers, P.J. Coffer, E.W. Lam, and B.M. Burgering. 2002. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol. Cell. Biol. 22:2025–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Niculescu, H., E. Bonfoco, Y. Kasuya, F.X. Claret, D.R. Green, and M. Karin. 1999. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol. Cell. Biol. 19:751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, S., T.J. Connors, and A.C. Maroney. 2001. c-Jun N-terminal kinase specifically phosphorylates p66ShcA at serine 36 in response to ultraviolet irradiation. J. Biol. Chem. 276:48332–48336. [DOI] [PubMed] [Google Scholar]
- Lin, K., J.B. Dorman, A. Rodan, and C. Kenyon. 1997. daf-16: an HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 278:1319–1322. [DOI] [PubMed] [Google Scholar]
- Lin, K., H. Hsin, N. Libina, and C. Kenyon. 2001. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 28:139–145. [DOI] [PubMed] [Google Scholar]
- Luzi, L., S. Confalonieri, P.P. Di Fiore, and P.G. Pelicci. 2000. Evolution of Shc functions from nematode to human. Curr. Opin. Genet. Dev. 10:668–674. [DOI] [PubMed] [Google Scholar]
- Marcus, D.L., J.A. Strafaci, D.C. Miller, S. Masia, C.G. Thomas, J. Rosman, S. Hussain, and M.L. Freedman. 1998. Quantitative neuronal c-fos and c-jun expression in Alzheimer's disease. Neurobiol. Aging. 19:393–400. [DOI] [PubMed] [Google Scholar]
- Medema, R.H., G.J. Kops, J.L. Bos, and B.M. Burgering. 2000. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 404:782–787. [DOI] [PubMed] [Google Scholar]
- Migliaccio, E., S. Mele, A.E. Salcini, G. Pelicci, K.M. Lai, G. Superti-Furga, T. Pawson, P.P. Di Fiore, L. Lanfrancone, and P.G. Pelicci. 1997. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J. 16:706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio, E., M. Giorgio, S. Mele, G. Pelicci, P. Reboldi, P.P. Pandolfi, L. Lanfrancone, and P.G. Pelicci. 1999. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 402:309–313. [DOI] [PubMed] [Google Scholar]
- Movsesyan, V.A., B.A. Stoica, and A.I. Faden. 2004. MGLuR5 activation reduces beta-amyloid-induced cell death in primary neuronal cultures and attenuates translocation of cytochrome c and apoptosis-inducing factor. J. Neurochem. 89:1528–1536. [DOI] [PubMed] [Google Scholar]
- Nakamura, N., S. Ramaswamy, F. Vazquez, S. Signoretti, M. Loda, and W.R. Sellers. 2000. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol. Cell. Biol. 20:8969–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namgung, U., and Z. Xia. 2000. Arsenite-induced apoptosis in cortical neurons is mediated by c-Jun N-terminal protein kinase 3 and p38 mitogen-activated protein kinase. J. Neurosci. 20:6442–6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto, S., and T. Finkel. 2002. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 295:2450–2452. [DOI] [PubMed] [Google Scholar]
- Ogg, S., S. Paradis, S. Gottlieb, G.I. Patterson, L. Lee, H.A. Tissenbaum, and G. Ruvkun. 1997. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 389:994–999. [DOI] [PubMed] [Google Scholar]
- Ohmichi, M., K. Matuoka, T. Takenawa, and A.R. Saltiel. 1994. Growth factors differentially stimulate the phosphorylation of Shc proteins and their association with Grb2 in PC-12 pheochromocytoma cells. J. Biol. Chem. 269:1143–1148. [PubMed] [Google Scholar]
- Opazo, C., X. Huang, R.A. Cherny, R.D. Moir, A.E. Roher, A.R. White, R. Cappai, C.L. Masters, R.E. Tanzi, N.C. Inestrosa, and A.I. Bush. 2002. Metalloenzyme-like activity of Alzheimer's disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2. J. Biol. Chem. 277:40302–40308. [DOI] [PubMed] [Google Scholar]
- Pelicci, G., L. Lanfrancone, F. Grignani, J. McGlade, F. Cavallo, G. Forni, I. Nicoletti, F. Grignani, T. Pawson, and P.G. Pelicci. 1992. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 70:93–104. [DOI] [PubMed] [Google Scholar]
- Pronk, G.J., J. McGlade, G. Pelicci, T. Pawson, and J.L. Bos. 1993. Insulin-induced phosphorylation of the 46- and 52-kDa Shc proteins. J. Biol. Chem. 268:5748–5753. [PubMed] [Google Scholar]
- Rozakis-Adcock, M., J. McGlade, G. Mbamalu, G. Pelicci, R. Daly, W. Li, A. Batzer, S. Thomas, J. Brugge, and P.G. Pelicci. 1992. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature. 360:689–692. [DOI] [PubMed] [Google Scholar]
- Ruff-Jamison, S., J. McGlade, T. Pawson, K. Chen, and S. Cohen. 1993. Epidermal growth factor stimulates the tyrosine phosphorylation of SHC in the mouse. J. Biol. Chem. 268:7610–7612. [PubMed] [Google Scholar]
- Selkoe, D.J. 2001. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81:741–766. [DOI] [PubMed] [Google Scholar]
- Sharp, A.H., and C.A. Ross. 1996. Neurobiology of Huntington's disease. Neurobiol. Dis. 3:3–15. [DOI] [PubMed] [Google Scholar]
- Shoji, M., N. Iwakami, S. Takeuchi, M. Waragai, M. Suzuki, I. Kanazawa, C.F. Lippa, S. Ono, and H. Okazawa. 2000. JNK activation is associated with intracellular beta-amyloid accumulation. Brain Res. Mol. Brain Res. 85:221–233. [DOI] [PubMed] [Google Scholar]
- Skolnik, E.Y., A. Batzer, N. Li, C.H. Lee, E. Lowenstein, M. Mohammadi, B. Margolis, and J. Schlessinger. 1993. The function of GRB2 in linking the insulin receptor to Ras signaling pathways. Science. 260:1953–1955. [DOI] [PubMed] [Google Scholar]
- Skulachev, V.P. 2000. The p66shc protein: a mediator of the programmed death of an organism? IUBMB Life. 49:177–180. [DOI] [PubMed] [Google Scholar]
- Smith, M.A., P.L. Richey Harris, L.M. Sayre, J.S. Beckman, and G. Perry. 1997. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J. Neurosci. 17:2653–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sponne, I., A. Fifre, B. Drouet, C. Klein, V. Koziel, M. Pincon-Raymond, J.L. Olivier, J. Chambaz, and T. Pillot. 2003. Apoptotic neuronal cell death induced by the non-fibrillar amyloid-beta peptide proceeds through an early reactive oxygen species-dependent cytoskeleton perturbation. J. Biol. Chem. 278:3437–3445. [DOI] [PubMed] [Google Scholar]
- Takaishi, H., H. Konishi, H. Matsuzaki, Y. Ono, Y. Shirai, N. Saito, T. Kitamura, W. Ogawa, M. Kasuga, U. Kikkawa, and Y. Nishizuka. 1999. Regulation of nuclear translocation of forkhead transcription factor AFX by protein kinase B. Proc. Natl. Acad. Sci. USA. 96:11836–11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, E.D., G. Nunez, F.G. Barr, and K.L. Guan. 1999. Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274:16741–16746. [DOI] [PubMed] [Google Scholar]
- Tran, H., A. Brunet, J.M. Grenier, S.R. Datta, A.J. Fornace Jr., P.S. DiStefano, L.W. Chiang, and M.E. Greenberg. 2002. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 296:530–534. [DOI] [PubMed] [Google Scholar]
- Trinei, M., M. Giorgio, A. Cicalese, S. Barozzi, A. Ventura, E. Migliaccio, E. Milia, I.M. Padura, V.A. Raker, M. Maccarana, et al. 2002. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene. 21:3872–3878. [DOI] [PubMed] [Google Scholar]
- Verheij, M., R. Bose, X.H. Lin, B. Yao, W.D. Jarvis, S. Grant, M.J. Birrer, E. Szabo, L.I. Zon, J.M. Kyriakis, et al. 1996. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 380:75–79. [DOI] [PubMed] [Google Scholar]
- Wei, W., D.D. Norton, X. Wang, and J.W. Kusiak. 2002. a. Abeta 17-42 in Alzheimer's disease activates JNK and caspase-8 leading to neuronal apoptosis. Brain. 125:2036–2043. [DOI] [PubMed] [Google Scholar]
- Wei, W., X. Wang, and J.W. Kusiak. 2002. b. Signaling events in amyloid beta-peptide-induced neuronal death and insulin-like growth factor I protection. J. Biol. Chem. 277:17649–17656. [DOI] [PubMed] [Google Scholar]
- Yang, C.P., and S.B. Horwitz. 2000. Taxol mediates serine phosphorylation of the 66-kDa Shc isoform. Cancer Res. 60:5171–5178. [PubMed] [Google Scholar]
- Zheng, W.H., S. Kar, and R. Quirion. 2002. FKHRL1 and its homologs are new targets of nerve growth factor Trk receptor signaling. J. Neurochem. 80:1049–1061. [DOI] [PubMed] [Google Scholar]
- Zhu, X., A.K. Raina, C.A. Rottkamp, G. Aliev, G. Perry, H. Boux, and M.A. Smith. 2001. Activation and redistribution of c-Jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J. Neurochem. 76:435–441. [DOI] [PubMed] [Google Scholar]