

Abstract
Endoneurial laminins (Lms), β1-integrins, and dystroglycan (DG) are important for Schwann cell (SC) ensheathment and myelination of axons. We now show that SC expression of galactosyl-sulfatide, a Lm-binding glycolipid, precedes that of Lms in developing nerves. This glycolipid anchors Lm-1 and -2 to SC surfaces by binding to their LG domains and enables basement membrane (BM) assembly. Revealingly, non–BM-forming fibroblasts become competent for BM assembly when sulfatides are intercalated into their cell surfaces. Assembly is characterized by coalescence of sulfatide, DG, and c-Src into a Lm-associated complex; by DG-dependent recruitment of utrophin and Src activation; and by integrin-dependent focal adhesion kinase phosphorylation. Collectively, our findings suggest that sulfated glycolipids are key Lm anchors that determine which cell surfaces can assemble Lms to initiate BM assembly and DG- and integrin-mediated signaling.
Introduction
Gene-targeting studies have shown that peripheral nerve development, in which Schwann cells (SCs) envelop, sort, and myelinate axons, depends on the expression and interactions of endoneurial γ1-laminins (Lms), β1-integrins, and dystroglycan (DG); the latter two major receptors are ligated by Lm and other basement membrane (BM) components (Chen and Strickland, 2003; Previtali et al., 2003; Saito et al., 2003). Lm-2 (α2β1γ1) and -8 (α4β1γ1) are particularly important, with compensation provided by Lm-1 (α1β1γ1) (Previtali et al., 2003; Yang et al., 2005).
A signature characteristic of Lms is their intimate association with select cell surfaces that accompany their ability to self-assemble into polymers (Yurchenco et al., 2004). Nerve Lms accumulate on the outer endoneurial surface of the SCs but not on axons, whereas mucosa Lms accumulate on epithelial but not fibroblast surfaces. Because Lms are required for BM assembly (Smyth et al., 1999), this selectivity of interaction determines where a BM can form and, therefore, which cells can be signaled by its ligands. A question that arises is whether there are cell-surface molecules that provide Lm anchorage, enabling cell-specific assembly and signaling.
Recent studies have implicated the Lm LG domains, and, in particular, their sulfated carbohydrate-binding loci, as providing cell anchorage (Li et al., 2002; Tsiper and Yurchenco, 2002). Although β1-integrins and DG have been thought to play this important role, several genetic studies propose that neither receptor is necessary for assembly during development of peripheral nerve and other tissues (Feltri et al., 2002; Saito et al., 2003; Yurchenco et al., 2004). Alternatively, sulfated glycolipids such as the sulfatides might provide this function for two reasons. First, sulfatides can strongly bind to Lm LG domains (Roberts et al., 1985, 1986; Ishizuka, 1997). Second, the most common of these, HSO3-3galactosylβ-1ceramide (gal-sulfatide), is highly expressed in developing and adult peripheral nerves (Mirsky et al., 1990).
In this study, we evaluated Lm-1 and -2 and gal-sulfatide in the peripheral nerve and their interactions in cultured SCs and fibroblasts. Sulfatide expression was found to precede that of Lms in the developing sciatic nerve, and SC gal-sulfatide was found to interact with Lm-1 and -2 with formation of a BM. Furthermore, intercalation of sulfatides into fibroblast plasma membranes rendered the cells competent for BM assembly. Lm assembly on both cell types initiated DG-dependent Src/Fyn activation and utrophin recruitment that contributed to their survival, and when cells were maintained in suspension, β1-integrin–dependent FAK phosphorylation was also observed. Collectively, the data provide evidence that sulfatides are Lm anchors that enable BM self-assembly and the engagement and activation of integrin and DG receptors.
Results
Lm assembly in the peripheral nerve
The order of expression of gal-sulfatide and Lms was evaluated in developing rat sciatic nerves (Fig. 1). Gal-sulfatide was detected as early as E14 in an intracellular and linear pattern in the absence of detectable γ1-Lm. By E17, extracellular α2/γ1-Lms colocalized with the gal-sulfatide in linear BM-like patterns that were more prominent by P1. In the adult nerve, most of the sulfatide was located on the outside BM zone of myelinated axons in a polarized fashion. Thus, sulfatide expression occurs before initiation of BM assembly.
Figure 1.
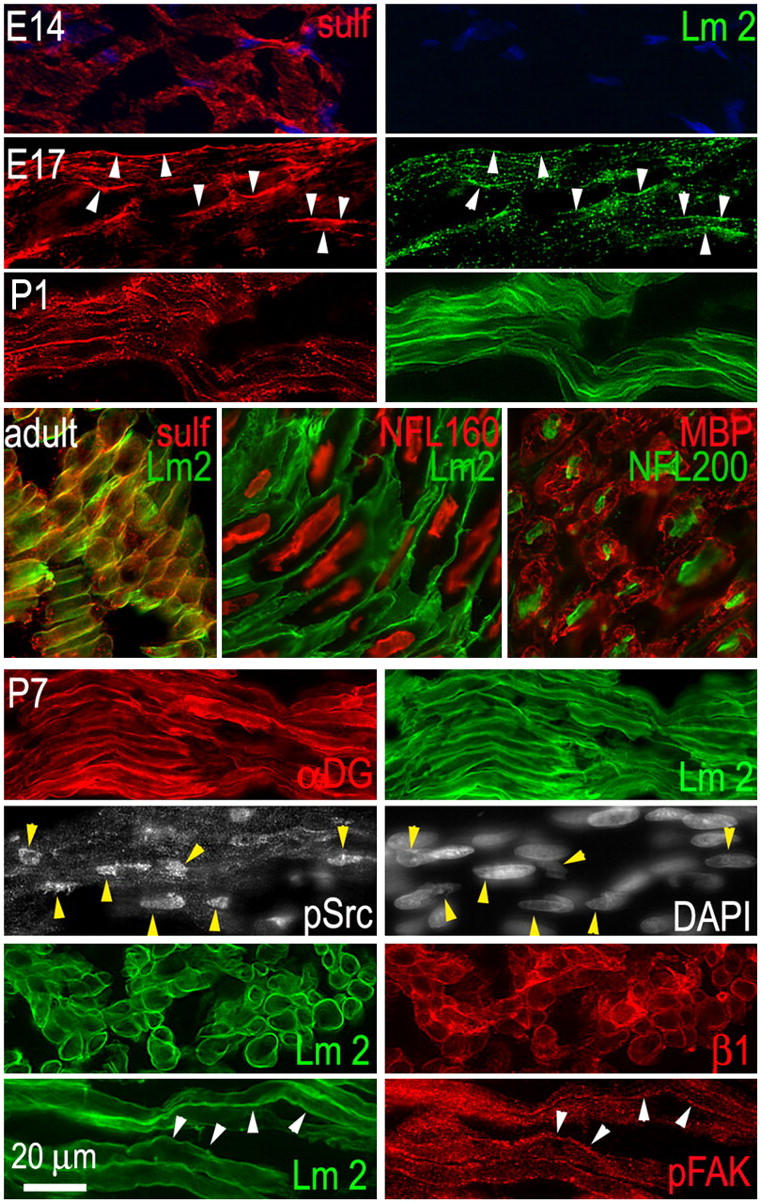
Sciatic nerve. (top three panels) Gal-sulfatide expression precedes that of Lm. Sections of rat embryos containing sciatic nerve (E14 and E17) or isolated sciatic nerve (P1–P7, adult) were immunostained with antibodies for gal-sulfatide (sulf, red) and Lm-2/4 (Lm2, green). Arrowheads indicate colocalizations of antibody immunofluorescence between paired panels, establishing the relationship at various points. (middle) Adult sciatic nerve was immunostained with antibodies for Lm-2/4, gal-sulfatide, neurofilament proteins (NFL160 or NFL200), and SC myelin basic protein (MBP), revealing the colocalization of gal-sulfatide to Lm and the polarized relationship to myelin and axon within nerve bundles. (bottom four panels) Expression of nuclear pSrc and pFAK in P7 nerve. Sections were stained with antibodies to β1-integrin (β1), αDG, c-Src-PY416 (pSrc), tyrosine-phosphorylated focal adhesion kinase (pFAK), and with DAPI.
BM assembly on SCs
Cultured rat sciatic nerve SCs did not possess detectable Lm-γ1 or α2 epitopes on their exposed surfaces or, as determined in immunoblots in cell extracts and conditioned medium (unpublished data), evidence that the cells had lost the ability to synthesize relevant Lms. However, these cells assemble a BM when incubated with exogenous Lm-1 or -2 (Tsiper and Yurchenco, 2002). When subconfluent cells (passage 25–32) were treated with 10 μg/ml Lm-1, Lm epitope, which was initially distributed diffusely (Fig. 2 a), condensed within 30 min into large confluent regions in which the aggregate edges were retracted from one or more of the outer cell borders, allowing for the assessment of colocalization with other components. A similar time course was observed with recombinant Lm-2 (unpublished data). The Lm pattern colocalized with endogenous nidogen-1 and type IV collagen (Fig. 2 b). Accumulation of the latter component was not appreciated previously (Tsiper and Yurchenco, 2002), likely because a short accumulation time in medium (1 h) during culturing was used in that study to permit detection of collagen as now seen with overnight accumulation.
Figure 2.
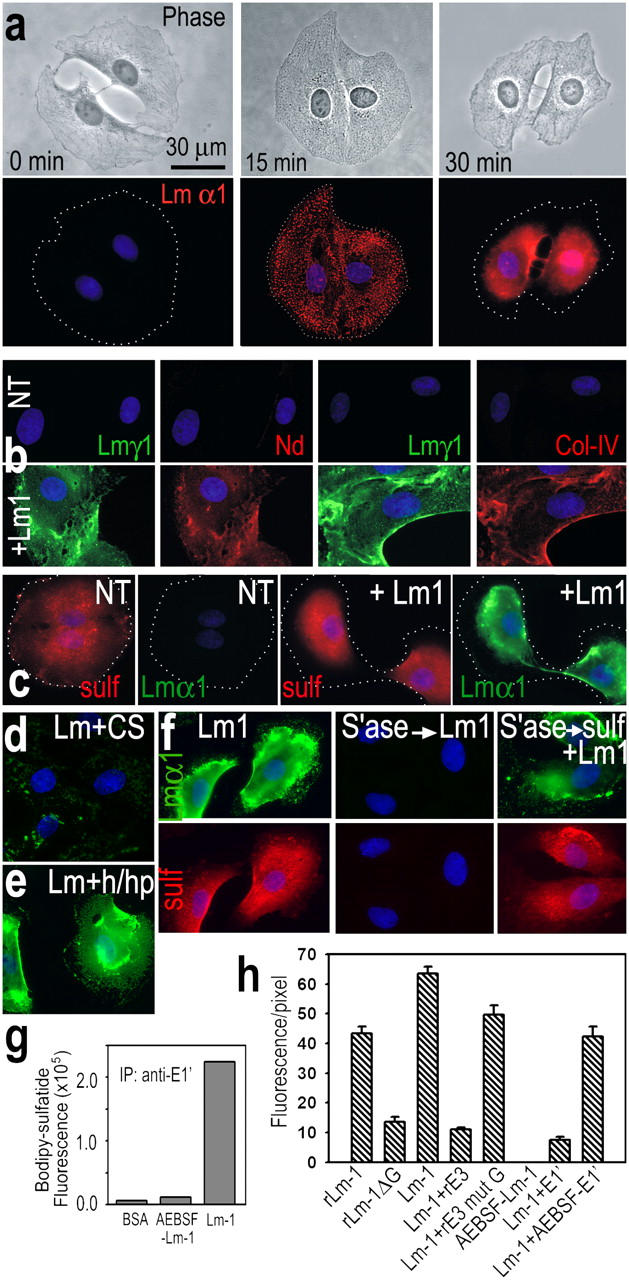
Lm 1–sulfatide associations on cultured SCs. (a) SCs that adhered to plastic were incubated with 10 μg/ml Lm-1 added to medium. Lm-α1 (red; DAPI, blue) initially accumulated in diffusely dispersed aggregates and then condensed into a compact, sheetlike pattern. Dotted lines indicate outer cell borders. (b) Nidogen-1 (red) and type IV collagen (red) were detected in a colocalizing pattern when treated with Lm (green). (c) Gal-sulfatide (nonpermeabilized cells, Cy3, red), which was initially diffuse, condensed and colocalized with Lm-α1 (FITC, green). (d and e) Lm-1 did not accumulate (d) on SCs when incubated for 1 h (10 μg/ml) in the presence of malarial CS protein (150 μg/ml), whereas it accumulated (e) on SCs treated with a mixture (h/hp) of heparitinase (0.1 U/ml) and heparinase (1 U/ml). (f) Sulfatide and Lm-α1 were largely undetectable if 50 U/ml arylsulfatase was added to SC medium. However, if cells were washed and incubated with 10 μM gal-sulfatide/BSA (for 30 min) followed by 10 μg/ml Lm-1, sulfatide and Lm immunofluorescence were restored. (g) Cell gal-sulfatide is bound to Lm-1. SCs were labeled with 5 μM BODIPY-gal-sulfatide; incubated with Lm-1, BSA, or nonpolymerizing AEBSF-Lm-1 (10 μg/ml) for 1 h; lysed with 1% Triton X-100; extracted; centrifuged to remove insoluble material; and were immunoprecipitated with anti-E1′ Lm antibody. The precipitate was solubilized with 1% SDS and BODIPY fluorescence was determined (excitation, 490 nm; emission, 513 nm). (h) Lm–LG domain and polymerization are required for BM. SCs were incubated for 1 h with 10 μg/ml of recombinant Lm-1 (rLm-1) or 10 μg/ml Lm-1 lacking G-domain (rLm-1ΔG) and immunostained for Lm-γ1. SCs were also incubated with either native Lm-1 (Lm-1, 10 μg/ml), alone, or in the presence of competing recombinant E3 (rE3 WT; 100 μg/ml); with E3 mutant G (rE3 Mut G; 100 μg/ml) to block LG-modular interactions; or with fragment E1′ to block Lm polymerization (AEBSF-E1′ is control).
Gal-sulfatide was distributed on the untreated SC surfaces in a diffuse pattern in the absence of Lm treatment (Fig. 2 c). After the addition of 10 μg/ml Lm-1 or -2, sulfatide condensed and colocalized with the cell-surface Lm. When SCs were treated with Helix promatia arylsulfatase (EC 3.1.6.1), an enzyme that hydrolyzes sulfates from sulfatides and seminolipid but not glycosaminoglycans (Friedman and Arsenis, 1972; Waheed and van Etten, 1980), the sulfatide epitope was no longer detected on SC surfaces and the treatment prevented Lm accumulation (Fig. 2 f). SC accumulation of Lm-1 was blocked by treatment with the malarial circumsporozoite (CS) protein (binds sulfatides and cholesterol sulfate; Cerami et al., 1992; Merten and Thiagarajan, 2001), but not by treatment with a mixture of heparitinase and heparanase, chondroitinase ABC, or neuraminidase (Fig. 2, d and e, and not depicted).
Sulfatides can be integrated (i.e., “loaded”) into the outer leaflet of a plasma membrane by treating cells with sulfatide adsorbed onto de-lipidated serum albumin (Monti et al., 1992). Using this technique, intercalation of gal-sulfatide into the previously sulfatase-treated cells restored Lm accumulation (Fig. 2 f). Assembly of fibronectin, a glycoprotein with heparin-binding but little sulfatide-binding activity, was not affected by arylsulfatase (unpublished data).
The sulfatide–Lm interaction was also examined by biochemical means. SCs were loaded with 10 μM BODIPY-tagged gal-sulfatide as labeled and incubated with either Lm-1 or control 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF)–Lm-1 (10 μg/ml) for 1 h (Fig. 2 g). The cells were then extracted with 1% Triton X-100 and, after centrifugation, the lysate was immunoprecipitated with Lm E1′-specific antibody. Fluorescence was high for Lm-1 in contrast to that of the control nonpolymerizing Lm-1 or albumin, which was evidence for Lm–sulfatide binding in the cells. Lm-1 and -2 were also found to accumulate on primary SCs isolated from 2-d-old rat sciatic nerve (not depicted).
The principle sulfatide-binding locus of Lm-1 has been found to reside within LG4 (Andac et al., 1999). Wild-type recombinant Lm-1 (rLm1; 10 μg/ml), but not Lm-1 with an LG1-5 deletion, accumulated and condensed on SCs (Fig. 2 h). Furthermore, when SCs were incubated with native Lm-1 (10 μg/ml) in the presence of either excess recombinant E3 (rα1LG4-5; 100 μg/ml, 1 h) or a mutated recombinant E3 (mutant G; α1LG4-2791KRK2793 to 2791AAA2793) that binds poorly to sulfatide (Li et al., 2002), the wild-type E3 selectively blocked cell-surface accumulation of Lm. The expectation that Lm polymerization is important for BM assembly in SCs (Tsiper and Yurchenco, 2002) was supported by the observation that Lm-1 fragment E1′ (but not AEBSF-inactivated E1′) blocked accumulation of Lm.
BM assembly in fibroblasts
Fibroblasts produce several BM macromolecules but typically do not assemble BM on their cell surfaces, contributing their molecules instead to adjacent BMs (Cornbrooks et al., 1983; Marinkovich et al., 1993). We reasoned that fibroblasts may lack a molecule that anchors Lms to their surface. Mouse embryonic lung fibroblasts (MEFs) did not express detectable γ1-Lm but secreted type IV collagen and nidogen-1 into the culture medium (determined by antibody immunofluorescence of detergent-permeabilized and nonpermeabilized cells with BM-specific antibodies and conditioned medium immunoblots). These cells were intercalated with sulfatides and evaluated for their ability to assemble a BM (Fig. 3). After incubation of untreated MEFs with Lm-1, little Lm or other BM component epitopes were detected on cell surfaces. However, if the MEFs were first loaded with gal-sulfatide, then the added Lm-1 accumulated on their surfaces (Fig. 3 a). Nidogen-1 and type IV collagen epitopes were also now detected on the exposed fibroblast surfaces. If the gal-sulfatide–treated MEFs were subsequently incubated with arylsulfatase and then incubated with Lm-1, Lm, then cell-surface nidogen and type IV collagen were not detected (Fig. 3 b).
Figure 3.
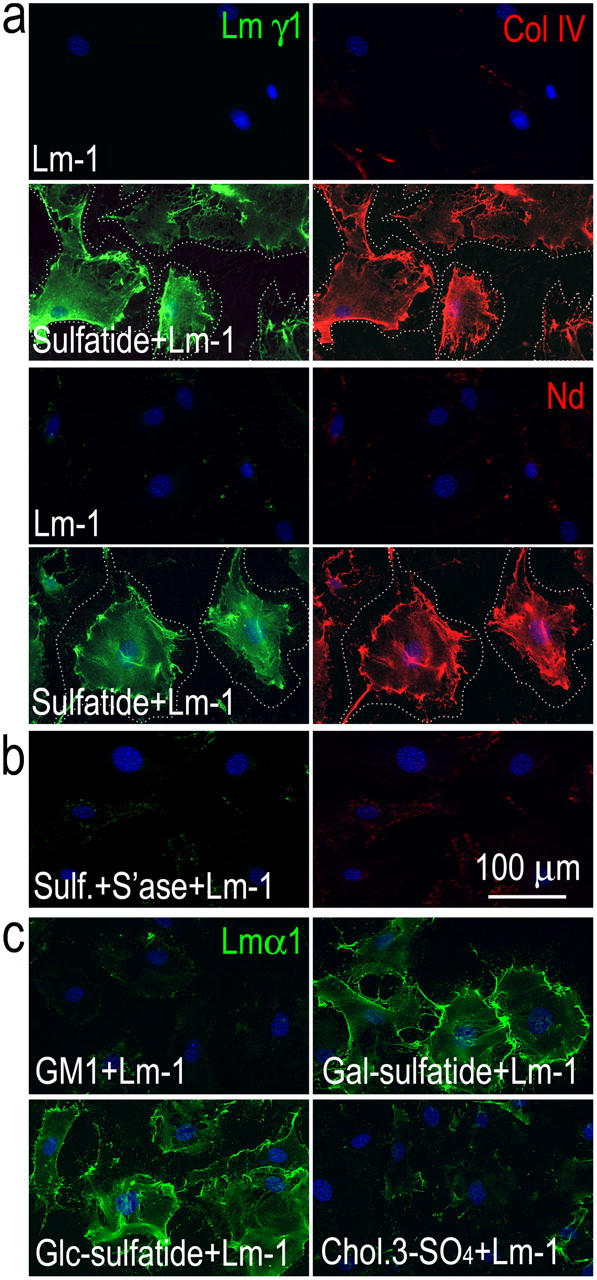
Sulfatide loading renders fibroblasts competent for BM assembly. (a) Mouse embryonic fibroblasts were incubated for 1 h with 10 μg/ml Lm-1, treated with 10 μM sulfatide/BSA (1:1 molar ratio), and incubated with 10 μg/ml Lm-1, or (b) treated with sulfatide/BSA in the presence of 50 U/ml arylsulfatase, followed by incubation with Lm-1. Lm, type IV collagen, and nidogen were detected in a colocalized pattern on fibroblast surfaces only if they were first loaded with sulfatide but not treated with arylsulfatase. The dotted line indicates outer cell borders. (c) Adherent fibroblasts were treated with BSA coupled to GM1-ganglioside, gal-sulfatide, glc-sulfatide, or cholesterol-3-sulfate and treated for 1 h with exogenous Lm-1 (10 μg/ml) followed by Lm-α1 immunostaining. Only the sulfatides enabled Lm accumulation.
Acidic lipids that might also interact with Lms include other sulfatides such as HSO3-3glucosylβ-1ceramide (glc-sulfatide), cholesterol sulfate, gangliosides, and phosphorylated lipids. To examine the specificity of the lipid requirement for cell-surface anchorage, MEFs were loaded separately with 10 μM glc-sulfatide, gal-sulfatide, GM1-ganglioside, GT1b-ganglioside, cholesterol-3-sulfate, and phosphatidylserine (Fig. 3 c and not depicted). Only gal-sulfatide and glc-sulfatide supported the accumulation and condensation of Lm-1. The failure of Lm to accumulate on fibroblasts loaded with other acidic lipids is consistent with the Lm-1 lipid-binding specificity determined by solid phase assay (Roberts et al., 1985).
Effect of sulfatide on cell-surface ultrastructure
SCs and sulfatide-treated MEFs that were maintained in confluent cultures and incubated with Lm-1 were found to achieve maximal Lm surface immunostaining with 20–40 μg/ml of protein, extending over almost the entire surface. The SCs and MEFs treated under these conditions were examined by transmission EM (Fig. 4). After incubation with Lm-1, nearly the entire SC surface was covered by a continuous BM deposit (lamina densa overlying a lamina lucida) that was absent in untreated cells or in cells treated with arylsulfatase. Sulfatide loading of the arylsulfatase-treated SCs restored the linear ECM deposit. The deposit was dependent primarily on Lm deposition rather than type IV collagen and was shown by incubating the SCs in the presence of Lm-1 and bacterial collagenase (which eliminated detectable type IV collagen immunostaining). MEFs had almost no extracellular deposits either without or with Lm-1 incubation. However, if cells were first loaded with sulfatide, and then treated with Lm-1, a continuous BM was noted in all sections examined. Treatment of the MEFs with arylsulfatase after sulfatide loading prevented the appearance of an ECM deposit on the exposed fibroblast plasma membrane.
Figure 4.
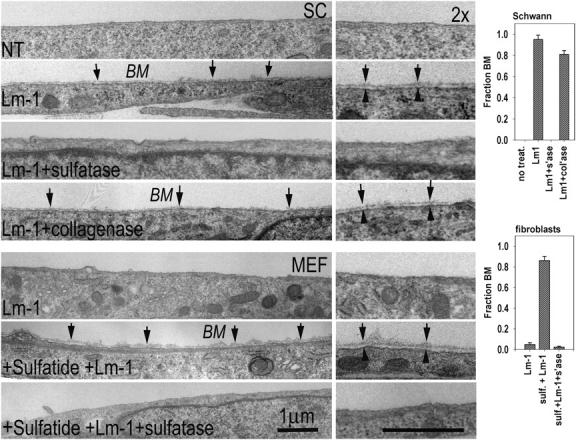
Ultrastructural evidence of cell-surface BM assembly. (top four panels) SCs were prepared for EM untreated (NT), treated with 20 μg/ml Lm-1 for 1 h, treated with Lm-1 in the presence of 50 U/ml arylsulfatase (sulfatase, s'ase), or treated with Lm-1 plus 250 U/ml of bacterial collagenase (col'ase). (bottom three panels) MEFs were treated with 40 μg/ml Lm-1 for 1 h, loaded with 10 μM sulfatide (sulf), and treated with Lm-1 or treated with Lm in the presence of arylsulfatase. Arrows indicate BM and arrowheads delimit plasma membrane. Cross sections through cells that are adhered to plastic with exposed cell surfaces are shown above. (right) Degree of BM coverage is determined from different cross-sectional levels (mean ± SEM; SCs, n = 3–4; fibroblasts, n = 5–7).
Association of Lm, DG, and utrophin
The topographical associations of Lm-1 with DG and utrophin were examined in adherent SCs (Fig. 5). Lm-1 treatment (10 μg/ml for 1 h) induced condensation of previously diffusely distributed α-DG and utrophin, confirming earlier observations (Tsiper and Yurchenco, 2002). However, if the cells were also treated with arylsulfatase, their condensation was not observed (Fig. 5 a). Although α-DG is known to bind to Lms (Ervasti and Campbell, 1993), such complexes have not been shown to occur during BM assembly. To evaluate this, SCs were incubated with Lm-1 under the above conditions, detergent extracted, immunoprecipitated with β-DG antibody, and immunoblotted with Lm-α1 antibody (Fig. 5 b). Lm was detected in the Lm-1–treated cell fraction without changing total amount of DG. β-DG–containing immunoprecipitates of SC detergent lysates were also examined for the presence of utrophin in immunoblots (Fig. 5, c and d). Utrophin was seen in the DG complex only from lysates extracted from cells treated with Lm-1 and was prevented with DG-blocking antibody, whereas the total amount of cellular utrophin and β-DG remained constant; i.e., utrophin was recruited to a sulfatide-associated Lm–DG complex by Lm interaction with DG.
Figure 5.
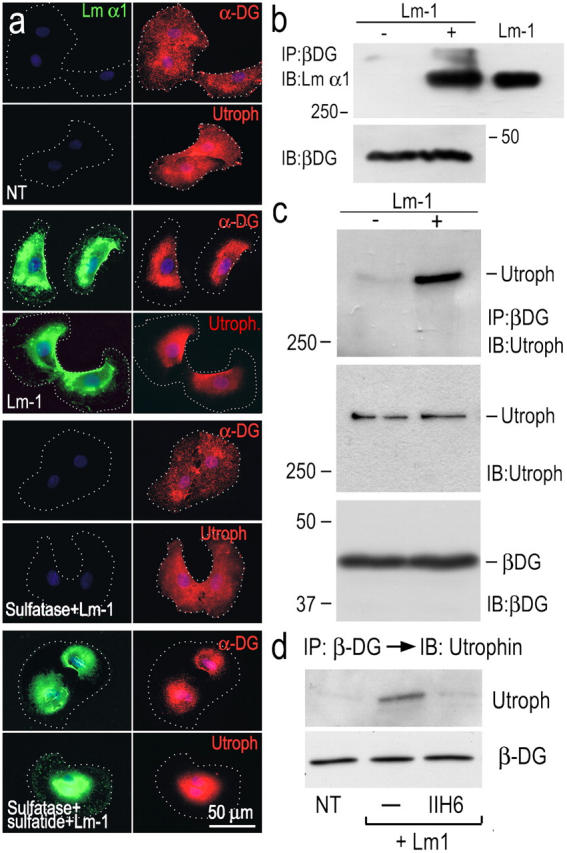
Lm-1 binding to sulfatide recruits DG and utrophin in SCs. (a) SCs, untreated (NT), treated with 10 μg/ml Lm-1 (for 1 h), treated with 50 U/ml arylsulfatase followed by Lm-1, or treated with arylsulfatase and then loaded with sulfatide followed by Lm-1, were immunostained for the indicated components (dots indicate cell borders). α-DG and utrophin condensation (and Lm colocalization) were prevented if the cells were treated with arylsulfatase. (b) Lm-1–DG association. SCs untreated (−) or treated (+) with 10 μg/ml Lm-1 (for 1 h) were washed and extracted with 1% Triton X-100. The cell lysates were immunoprecipitated (IP) with anti–β-DG antibody and probed for Lm-α1 chain. The input cell lysates were also immunoblotted (IB) with anti–β-DG antibody. Exogenous Lm was detected in the precipitates when added to the medium. (c and d) Recruitment of utrophin to DG. The immunoprecipitates formed with anti–β-DG antibody were subjected to immunoblotting with utrophin-specific antibody (c). Utrophin recruitment was blocked by antibody IIH6 (d).
Tyrosine phosphorylation of Src and focal adhesion kinase
The possibility that anchorage-dependent BM assembly enabled SC signaling was investigated (Fig. 6). c-Src became tyrosine phosphorylated at its activating residue Y416 (Fig. 6 a), beginning within 15 min of Lm treatment and peaking by 30–60 min. Fyn, another Src family member present in SCs in which the activation-specific antibody shows cross-reactivity, was also activated by Lm treatment (Fig. 6 b). If the SCs were incubated with arylsulfatase, Lm treatment failed to induce Src activation (Fig. 6 c). Immunoprecipitation of SC detergent lysates with β-DG antibody followed by immunoblotting with c-Src–specific antibody revealed that c-Src was associated with the DG-containing complex regardless of whether or not the cells were Lm treated (Fig. 6 d). c-Src underwent a transition from a dispersed pattern to a condensed one, colocalizing with Lm (Fig. 6 e). pY416-Src, on the other hand, was only weakly detected in untreated SCs and strongly detected in Lm-treated SCs. The epitope, although increased throughout the cells, was seen to be predominantly associated with nuclei, and pSrc was not detected in soluble detergent lysates of the cells (Fig. 6 e and not depicted). The data suggest that Lm-activated Src translocates to the nucleus; however, alternative mechanisms cannot be ruled out. Nuclear pSrc was also detected in a patchy distribution of sciatic nerves between P1 and P7 (Fig. 1).
Figure 6.
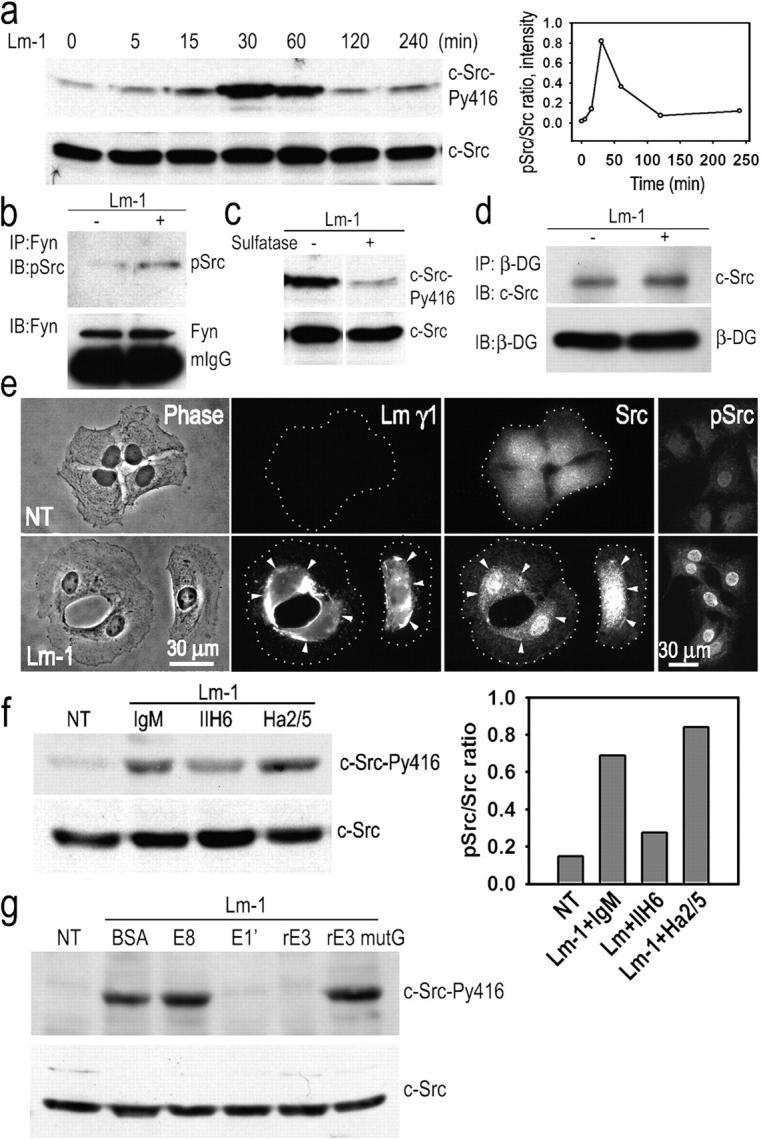
Schwann cell c-Src is activated in response to Lm-1. (a) Transient Src activation in response to Lm: SCs were incubated with 10 μg/ml Lm-1, harvested at the indicated times, lysed, and analyzed for c-Src-PY416 and c-Src. Time course immunoblot and densitometry plot of pSrc/total Src ratio are shown. (b) Fyn activation in response to Lm: lysates from SCs treated as above for 1 h were immunoprecipitated with Fyn-specific antibody followed by immunoblotting with phospho-Src (PY416) antibody that also detects pFyn. (c) Src activation depends on the presence of gal-sulfatide. SCs were treated with Lm-1 as above for 1 h in the presence (+) or absence (−) of 50 U/ml arylsulfatase and analyzed for c-Src phosphorylation. (d) c-Src coimmunoprecipitates with β-DG. SCs untreated or treated with 10 μg/ml Lm-1 for 1 h were extracted with 1% Triton X-100–Tris buffer. Cell lysates were immunoprecipitated with anti–β-DG antibody and the immunoprecipitates were subjected to immunoblot analysis with c-Src-–specific antibody. (e) SCs were untreated or treated with 10 μg/ml Lm-1 for 1 h and immunostained for Lm-γ1, Src, and Src-PY416 (pSrc). Diffusely distributed Src immunofluorescence coalesces into dense plaques that overlap with Lm immunofluorescence after Lm-1 treatment, whereas most of Src-PY416 is associated with the nucleus (arrowheads indicate colocalizations of antibody immunofluorescence between paired panels, establishing the relationship at various points). (f) Anti–α-DG antibody IIH6 inhibits Src phosphorylation, whereas anti–β1-integrin (Ha2/5) does not. The bar graph shows the phospho-Src/total Src ratio based on the immunoblot densitometry. (g) Lm-1 fragments E1′ and E3, but not E8 or E3 mutG (which lack a sulfatide-binding sequence), block c-Src phosphorylation. SCs were incubated for 1 h with Lm-1 in the presence of 100 μg/ml BSA, 250 μg/ml E8, 250 μg/ml E1′, 100 μg/ml rE3, or 100 μg/ml rE3 mutant G. Cells were washed, lysed in 1% SDS-Tris buffer, and immunoblotted.
Treatment of SCs with β1-integrin–blocking antibody Ha2/5 did not block Src phosphorylation, whereas a substantial reduction was seen with αDG-blocking antibody IIH6 (Fig. 6 f). On the other hand, tyrosine phosphorylation of β-DG and caveolin-1 was not detected in response to Lm (unpublished data). Finally, SCs were incubated with Lm-1 in the presence of excess Lm-1 fragments known to inhibit different activities; i.e., E8 (α6 integrin-binding activity), E1′ (polymerization), and E3; and E3 mutated to prevent its sulfatide/DG binding (Li et al., 2002). Of these fragments, only E1′ and E3 blocked Src activation (Fig. 6 g).
Sulfatide-treated fibroblasts were then evaluated for Src tyrosine phosphorylation (Fig. 7) in response to Lm-1. Lm-1 induced a similar transient activation of c-Src in sulfatide-loaded cells that was maximal at 1 h (Fig. 7 a). This was not observed if the fibroblasts were treated with Lm but not loaded with sulfatide (Fig. 7 b). Src phosphorylation was blocked partially by fragment E3 (as seen with SCs) and fully by the DG antibody IIH6, but not with antibody Ha2/5 to β1-integrin or by Lm fragment E8 that possesses the α6β1-integrin–binding locus (Fig. 7 c). To further examine the role of DG and β1-integrin in Src activation, we evaluated cultures of fibroblasts isolated from differentiated mouse embryonic stem (ES) cells that were genetically null for DG or for β1-integrin, and compared these with fibroblasts derived from wild-type ES cells or ones that were transfected with a construct to enable expression of β1-integrin (β1AGD25 cells; Wennerberg et al., 1996). The cells were cultured on plastic, loaded with gal-sulfatide, and incubated in the presence of Lm-1. Src activation was observed in response to Lm-1 in the wild-type, but not the DG-null, fibroblasts (Fig. 7 d). In contrast, Lm-1 stimulated an increased Src activation in both control and β1-integrin–null fibroblasts, although at an approximately twofold higher level in the control cells (Fig. 7 e). Because β1-integrin did not colocalize with Lm under these conditions, and because β1-integrin–blocking antibody Ha2/5 did not inhibit Lm-induced Src phosphorylation, it was thought likely that Src activation was primarily dependent on DG and that the integrin contribution was independent of Lm assembly. Lm-1 accumulated on both DG-null and β1-integrin–null fibroblasts (Fig. 7, f and g). Finally, caveolin-1 became transiently phosphorylated at tyrosine-14 with a similar time course in fibroblasts, unlike SCs (Fig. 7 h). Inhibition of Src kinases with two structurally different inhibitors (PP2 and SU6656) inhibited caveolin-1 phosphorylation (Fig. 7 i), suggesting caveolin-1 was a downstream target of the Lm-activated Src.
Figure 7.
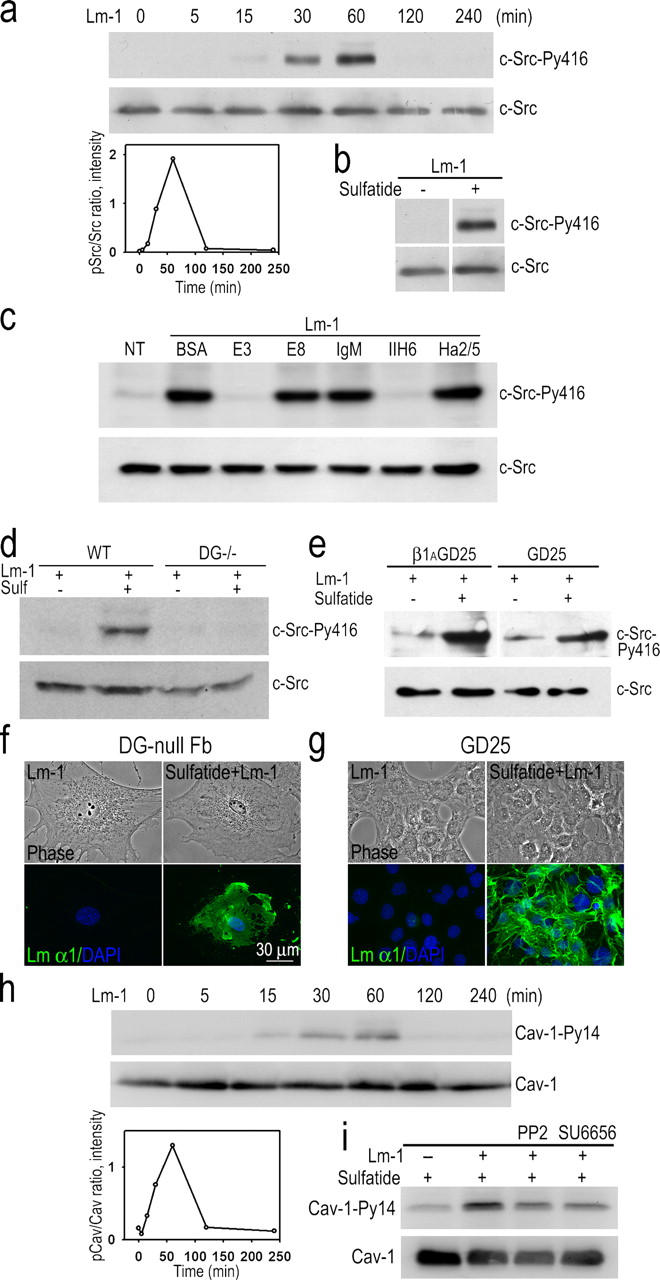
Tyrosine phosphorylation of c-Src and caveolin-1 in sulfatide-loaded fibroblasts. (a) MEFs were loaded with gal-sulfatide and treated with 10 μg/ml Lm-1. Equal protein loads of cell lysates were analyzed in immunoblots. Transient Src activation (PY416) was detected within 30 min after Lm-1 treatment. (bottom left) Ratio of Src-PY416/total Src. (b) Lm-1 does not induce Src phosphorylation in fibroblasts in the absence of sulfatide loading. Fibroblasts with or without sulfatide loading were incubated with 10 μg/ml Lm-1 for 1 h. Cell lysates were immunoblotted with either c-Src-Py416 or c-Src–specific antibodies. (c) αDG antibody and Lm-1 fragment E3 inhibit Src phosphorylation in sulfatide-loaded fibroblasts treated with Lm-1. Gal-sulfatide–loaded fibroblasts were treated with 10 μg/ml Lm-1 for 1 h in the presence of either 100 μg/ml BSA,100 μg/ml E3, 250 μg/ml E8, 10 μg/ml mouse IgM, 10 μg/ml IIH6, or 10 μg/ml of β1-integrin antibody Ha2/5; lysed; and immunoblotted for pSrc and c-Src. (d) DG expression is required for Lm induction of Src activation in sulfatide-loaded fibroblasts. Fibroblasts derived from wild-type or DG-null embryonic stem cells treated with gal-sulfatide were incubated with 10 μg/ml Lm-1 for 1 h and analyzed for pSrc and total Src. (e) Ablation of the β1-integrin gene does not prevent Lm-1–induced Src phosphorylation in sulfatide-loaded fibroblasts. β1-integrin–deficient fibroblasts (GD25) and β1-integrin–transduced GD25 control cells were treated the same as described in d, with lysates analyzed for pSrc and total Src. (f and g) Lm-1 assembly on sulfatide-loaded fibroblast surfaces does not require DG or β1-integrin. DG-null (f) and β1-integrin–null (g) fibroblasts, loaded with gal-sulfatide and incubated with 10 μg/ml Lm-1 for 1 h, were fixed and immunostained for Lm α1. (h) Caveolin-1 phosphorylation is induced by Lm-1 in sulfatide-treated embryonic lung fibroblasts. Fibroblasts were treated the same as described in panel a, and analyzed for Py14-caveolin-1 (Cav-1). The densitometry plot (caveolin-1-Py14/total caveolin-1) is also shown. (i) Src inhibition decreases Lm-induced caveolin-1 phosphorylation. Sulfatide-loaded fibroblasts were treated with Lm-1 plus Src kinase inhibitor PP2 (2 μM) or SU6656 (2 μM) for 1 h. Cell lysates were analyzed in immunoblots for caveolin-1-Py14 (Cav-1-Py14) or total caveolin-1 (Cav-1).
To determine whether cell survival was affected by Lm assembly, SCs and MEFs were incubated in suspension in the absence of serum in order to eliminate the possibility that the plastic substrate and serum component were alternative substrate and soluble inhibitors of apoptosis (Fig. 8). Both cells cultured in this manner formed cell aggregates in spherical clusters with scattered single cells. When the suspended SCs were treated with Lm-1 in the presence or absence of receptor-blocking antibodies, c-Src phosphorylation was induced by Lm and was selectively blocked with the DG-specific reagent (Fig. 8 a), which provided evidence that the Lm-initiated process occurred in suspended SCs as well as in adherent SCs in a DG-dependent, but integrin-independent, fashion (Fig. 8 a). SC clusters and sulfatide-loaded fibroblasts were treated with the same concentration of Lm-1 for 24 h. Sections of cell aggregates were immunostained for Lm and activated caspase-3, which is a marker for apoptosis (Fig. 8, b–d). SC and MEF cell clusters that were not treated with Lm-1 developed irregular borders and were stained with antibody specific for activated caspase-3. In those SC and MEF cell clusters treated with Lm-1, the cell edges were smooth, and prominent pericellular and peri-spherule Lm immunostaining was noted, and was accompanied by almost no detectable activated caspase-3. The SC clusters were also treated with Lm-1 and PP2 or SU6656: apoptosis was observed in the presence of Lm-1 at levels approaching those of cells incubated without Lm, implicating Src family kinases as mediators of Lm-dependent cell survival.
Figure 8.
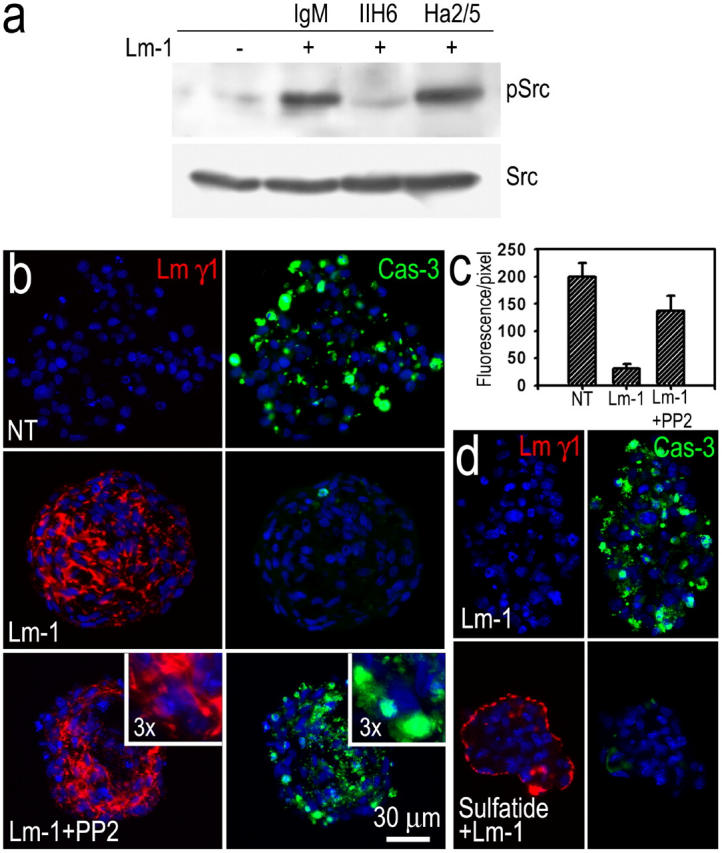
Inhibition of Src kinase reduces the Lm-dependent protection from apoptosis. SCs and MEFs grew as spherical clusters in suspension. (a) SCs were cultured in suspension in the absence (−) or presence (+) of Lm-1 (10 μg/ml) plus DG-blocking antibody IIH6 (10 μg/ml), β1-integrin–blocking antibody Ha2/5, or control IgM for 6 h. Cell lysates were subjected to immunoblot analysis for c-Src-Py416 (pSrc) and total c-Src (Src). (b) SCs were grown in suspension in the absence (NT), presence of Lm-1 (10 μg/ml), or Lm-1 + Src kinase inhibitor PP2 (2 μM) in serum-free medium for 24 h, cryosectioned, and stained for Lm-γ1, activated caspase-3, and DAPI (blue). The Lm-treated cell clusters developed linear, pericellular deposits of Lm. Caspase-3 fluorescence that was detected in untreated cells was almost completely absent in the Lm-treated cells. Src inhibition eliminated the Lm-dependent survival effect. (c) Relative quantitation of SC apoptosis. The ratio of cleaved caspase-3 immunostain summed intensities, divided by the DAPI-stained nuclear areas, was plotted (mean ± SEM; n = 10 fields). (d) Lm matrix assembly protects MEFs from apoptosis. Cells untreated or treated with gal-sulfatide were cultured in suspension and incubated for 24 h with 20 μg/ml Lm-1 in serum-free DME. Cell clusters were immunostained for Lm and cleaved caspase-3. Lm-1 accumulated in a linear pattern, mostly on the surface of sulfatide-loaded MEF clusters with the cells showing little apoptosis. In untreated MEFs, Lm-1 did not accumulate in the cell clusters with the cells undergoing substantial apoptosis.
β1-integrin association and FAK phosphorylation in cell suspension
In our analysis of Lm on adherent SCs (Tsiper and Yurchenco, 2002; this study), we did not observe a colocalization of Lms with β1-integrin and found that BM assembly could occur in the absence of β1-integrin. Although this was useful in ruling out a role for integrin in BM assembly, in agreement with observations made in embryoid bodies and supported by genetic studies (Li et al., 2003), the lack of integrin colocalization was nonetheless puzzling, as this was known to occur in developing peripheral nerves (Previtali et al., 2003). Because β1-integrin staining was largely confined to the basal adherent side of the SCs, we reasoned that adhesion to the plastic substrate may have recruited most of the integrin, leaving little to interact with the upper exposed cell surface. We therefore evaluated SCs grown in suspension culture. When Lm-1 was added to the medium, it accumulated between the cells of the cluster (Fig. 9 a) and colocalized with β1-integrin. Furthermore, tyrosine phosphorylation of FAK greatly increased after Lm treatment overnight under suspension conditions (Fig. 9 b). Substrate-adherent cells, on the other hand, revealed constitutively high levels of pFAK, likely obscuring a Lm response (unpublished data). To further examine the relationship between β1-integrin and FAK phosphorylation, fibroblasts isolated from mouse ES cells that were null for β1-integrin (DG25) were compared with ones that had been transfected with a construct that allowed the cells to express β1-integrin (β1AGD25). These cells were first sulfatide loaded, and then grown in suspension. Both in the absence or presence of exogenous 10 μg/ml Lm-1, the fibroblasts formed spherical aggregates. After incubation for 18 h, the cells were harvested, extracted with detergent, precipitated with FAK antibody, and evaluated for tyrosine phosphorylation in immunoblots (Fig. 9 c). Low levels of pFAK were detected in the untreated controls. Treatment of integrin-expressing cells with Lm resulted in about a threefold increase of phosphorylation without a significant change in total FAK. In contrast, treatment of the β1-integrin–null cells with Lm caused no detectable increase in pFAK.
Figure 9.

β1-integrin association with Lm and FAK phosphorylation. SCs or ES cell–derived fibroblasts were cultured as aggregates in poly-HEMA–coated dishes for 24 h in the presence or absence of 10 μg/ml Lm-1. (a) Cryosections of SC aggregates were immunostained for Lm-α1 and β1-integrin. The integrin colocalized with Lm at cell edges. Arrowheads indicate colocalizations of antibody immunofluorescence between paired panels, establishing the relationship at various points. (b) Lm-1 induces FAK phosphorylation in suspended SCs. SC aggregates were maintained overnight in the absence (−) or presence (+) of 10 μg/ml Lm-1. The aggregates were washed, lysed in 1% Triton X-100–Tris buffer, immunoprecipitated with anti-FAK antibody (IP: FAK), and immunoblotted with antiphosphotyrosine (Py) antibody. The immunoblot of total FAK was used as the control. (c) Lm-1 induction of FAK phosphorylation in fibroblasts is dependent on β1-integrin. GD25 ES cell–derived fibroblasts and GD25 cells transduced with a β1-integrin expression vector (control) were loaded with sulfatide and grown in suspension overnight in the absence (−) or presence (+) of 10 μg/ml Lm-1. Cell extracts were analyzed for tyrosine phosphorylation of FAK and total FAK as described above. Lm-1 treatment increased FAK phosphorylation level in the control β1AGD25 cells but not the β1-integrin–deficient GD25 fibroblasts.
Discussion
SC development was characterized by early expression of gal-sulfatide followed by Lm deposition in a polarized fashion, suggesting that sulfated glycolipids are involved in endoneurial BM assembly. This hypothesis was supported by data from the current study on SCs. Furthermore, the observation that intercalation of sulfatides into fibroblast membranes enables a non–BM-forming cell to assemble a BM suggests that expression of sulfatides may regulate competency of different cells to assemble BMs.
Lm anchorage to sulfatides and BM assembly
Gal-sulfatide is a glycosphingolipid found in different tissues and enriched in the peripheral nerve. It, along with other sulfatides, binds to Lms (Roberts et al., 1985, 1986) through lysine- and arginine-rich sequences within the exposed loops of the fourth LG domain of Lm-1 and fourth and fifth domains of Lm-2 (Tisi et al., 2000; Wizemann et al., 2003). A study of Lm-1 interactions with synthetic sulfatide bilayers revealed that Lm polymerization facilitated binding through a cooperative interaction, leading to an aggregation of Lm on the lipid surface (Kalb and Engel, 1991) and providing the first evidence to implicate sulfatides in BM assembly. In this study on living SCs and fibroblasts, we found that the surface assembly of Lm-1 depended on Lm polymerization and anchorage to sulfatide through LG4, enabling the incorporation of nidogen-1 and type IV collagen into the Lm matrix. Analysis of the ultrastructure of confluent cultures revealed that a continuous BM, similar to that observed in the peripheral nerve, had formed in response to Lm–sulfatide interactions.
We also found that rat kidneys possess gal-sulfatide that colocalizes with the Lm epitopes of tubular and other BMs in a polarized fashion (unpublished data), suggesting that a sulfatide-anchoring function is not confined to nerves. Nonetheless, we think it unlikely that gal-sulfatide provides universal Lm anchorage for two reasons. First, glc-sulfatide may similarly anchor Lms, and the finding that glc-sulfatide can replace gal-sulfatide may explain the presence of BMs in the gal-sulfatide–negative but glc-sulfatide–positive peripheral nerves of UDP-galactosyl transferase-null mice, which is a phenomenon that may also occur with ceramide sulfotransferase-null mice (Coetzee et al., 1996; Honke et al., 1996, 2002; Bosio et al., 1998). Second, we have found that the embryonic BM is sulfate dependent and arylsulfatase sensitive in the absence of detectable gal-sulfatide in developing embryoid bodies (unpublished data).
Recently, the LG4-5 domains of Lm-1 were deleted by gene targeting and studied in early embryos and embryoid bodies, where its absence was found to affect epiblast polarization without losing subendodermal BM (Scheele et al., 2005). Although Scheele et al. (2005) proposed that LG4-5 serves a signaling, rather than an assembling role, the concept of LG4 anchorage is supported by evidence that Lm-10 (α5β1γ1) is also expressed in the embryonic BM and that inactivation of the Lamα1 gene does not prevent either BM assembly or failure of epiblast polarization caused by functional Lm redundancy (Miner et al., 2004). Thus, as a general mechanism, it may be that several sulfated glycolipids can provide Lm anchorage through different LG domains, whereas other acidic lipids lack this activity. A separate, unanswered question concerns which molecules anchor α3, α4, and α5-Lms.
DG– and integrin-dependent signaling
The evidence of this and other studies (Smirnov et al., 2002; Yurchenco et al., 2004) demonstrates that the formation of BM on cell surfaces through anchorage, Lm polymerization, and the binding of other BM components to Lms is largely one of self-assembly. Cell signaling, on the other hand, requires the separate binding of receptors to the newly assembled BM. In this study, contributions mediated by DG and β1-integrins of this type were observed. These receptors can transduce signals and lead to the association of the ECM with the actin cytoskeleton. Furthermore, they have shown that they become colocalized with the BM in the developing SC of the peripheral nerve (Previtali et al., 2003). In SCs and sulfatide-loaded fibroblasts, Lm sulfatide-mediated assembly triggered the phosphorylation of c-Src at its activating tyrosine 416, and this was accompanied by Fyn phosphorylation (a known mediator of myelination in the central nervous system; Umemori et al., 1999). Src activation was inhibited by an antibody that blocks the binding of Lm, perlecan, and agrin to α-DG, implicating DG in Src activation. The resulting activated Src was detected in the nucleus, suggesting that Src is translocated to the nucleus from the plasma membrane. A corresponding nuclear distribution of activated Src was seen in the developing sciatic nerve, a phenomenon which was also found after calcium-induced keratinocyte differentiation (Zhao et al., 1992). Lm assembly in sulfatide-treated fibroblasts similarly led to transient Src activation, which was accompanied by downstream phosphorylation of caveolin-1. Antibody binding to α-DG inhibited the Lm-initiated response, and sulfatide-loaded fibroblasts that were null for DG were unable to activate Src in response to Lm-1, suggesting that DG was the primary receptor that mediated Src activation in both SCs and fibroblasts. The finding that Src activation was associated with the promotion of Lm-dependent cell survival leads one to expect that if the DG–Src pathway is a major mediator of cell survival, then reduced Src family activation and increased apoptosis might be seen in DG-null peripheral nerve SCs (Saito et al., 2003).
β1-integrins, like DG, were not required for Lm assembly on sulfatide-containing cell surfaces. On the other hand, their Lm-initiated signaling, a prediction given their role (especially α6β1) in peripheral nerve radial sorting (Feltri et al., 2002), was appreciated under suspension culture conditions. In particular, Lm-1 colocalized with β1-integrins and induced tyrosine phosphorylation of FAK.
A working hypothesis
The evidence from both SCs and sulfatide-treated fibroblasts constitutes the first demonstration of a critical mediator of Lm anchorage (Fig. 10). Interpreting the new cell findings in the context of the biophysical evidence (Kalb and Engel, 1991; Yurchenco and Cheng, 1993), we propose that sulfated glycolipids such as gal-sulfatide facilitate Lm polymerization by specifically binding to the Lm–LG domains and increasing the local Lm concentration at the cell surface. This initial ECM then binds to nidogens, type IV collagens, and other BM components to complete BM assembly. Signaling functions, on the other hand, are reserved for transmembrane protein receptors whose binding to the Lm scaffolding and/or attached BM components is enabled by anchorage and Lm polymerization. Two receptor classes implicated in SCs, and mirrored in the sulfatide-loaded fibroblasts, are DG and β1-integrins. The signals detected in response to DG and integrin were c-Src/Fyn and FAK tyrosine phosphorylation, respectively; the former is implicated in protecting the cells from apoptosis. In addition, utrophin, which can bind to both β-DG and F-actin, was recruited to the BM zone in a step that required DG and its ECM interaction. Thus, BM assembly consists of its self-assembly, initiated by Lm polymerization and anchorage through sulfated glycolipids, and its signal transduction, which is enabled by assembly and is mediated through integrin and DG receptor interactions.
Figure 10.

Working model distinguishing Lm anchorage from signaling. Lm (blue) binds to sulfatides (red lipids in membrane) through its LG domains and polymerizes to create a nascent BM scaffolding that captures and binds to nidogen and type IV collagen. These self-assembly steps are distinguished from those of the binding of the BM to DG (orange) and β1-integrins (β1, blue), the former being directly or indirectly bound to c-Src/Fyn. Src/Fyn becomes activated through tyrosine phosphorylation and may translocate to the nucleus. Lm assembly recruits cytoskeletal utrophin to β-DG, which, in turn, can bind to the actin cytoskeleton. β1-integrin mediates FAK tyrosine phosphorylation.
Materials and methods
Proteins and antibodies
Lm-1 and its fragments.
Lm-1, E1′, E3 (α1LG4-5), AEBSF-treated Lm-1, and AEBSF-E1′ were prepared as previously described (Li et al., 2002). AEBSF strongly binds to the E1′ region of Lm-1 to block self-assembly and E1′ inhibition of polymerization (Colognato et al., 1999).
The 9,564-bp fragment coding for mouse Lm-α1 (Yurchenco et al., 1997) with an NH2-terminal FLAG tag and BM40 signal sequence was excised from ma1-pRCX3 and subcloned into the pcDNA3.1 vector. A construct for mouse Lm-α1 without a G domain was made by replacing a 3,928-bp SacII–AflII fragment of the full-length α1 construct with an analogous 1,091-bp PCR fragment, which introduces a STOP codon after the sequence SIKVAVSADRD. The human β1 chain with an NH2-terminal hemagglutin tag was subcloned into pcDNA3.1/zeo+ vector (Invitrogen) from a pCIS vector containing human Lm-β1 (Yurchenco et al., 1997). The human Lm-γ1 construct was made as described previously (Smirnov et al., 2002). HEK 293 cells were transfected with the expression constructs. Recombinant wild-type E3 and E3 with the sulfatide-DG–binding site KRK in LG4 that was replaced with AAA (mutant G) were also expressed in HEK 293 cells. Recombinant Lm-1 and fragments were purified by FLAG affinity chromatography. FLAG-tagged recombinant human Lm-2 was expressed and purified as described previously (Smirnov et al., 2002). cDNA for malarial CS protein was obtained from the Malaria Research and Reference Reagent Resource Center. Recombinant CS protein with a 6x His-tag was expressed in Escherichia coli M15 and purified to homogeneity by metal chelation affinity chromatography (QIAGEN).
Antibodies were used with the following specificities: rG50 (Lm-α1–LG domains 4–5) rabbit polyclonal pAb, Lm-α2G, Lm2/4 (which reacts with α2 and γ1 subunits), and nidogen-1 pAbs (Cheng et al., 1997; Li et al., 2002), Lm-γ1 rat mAb (Upstate Biotechnology), β1-integrin hamster mAb Ha2/5 (BD PharMingen), α-DG mouse mAb IIH6 (gift of K. Campbell, University of Iowa, Iowa City, IA), purified gal-sulfatide mAb Sulf I (a gift of P. Fredman, Sahlgreuska University Hospital, Mölndal, Sweden; Fredman et al., 1988), utrophin mAb (DRP2; Novocastra), S100 mAb (Chemicon), rabbit collagen type IV pAb (Rockland Immunochemicals), rabbit-cleaved caspase-3 pAb (Cell Signaling Technology), NFL200 pAb and NFL160 mAb (Sigma-Aldrich), myelin basic protein mAb (Sternberger Monoclonals), rabbit c-Src pAb (Santa Cruz Biotechnology, Inc.), rabbit c-Src-Py416 pAb (which cross-reacts with activated Fyn; Cell Signaling Technology), caveolin-1-Py14 pAb (BD Biosciences), caveolin-1 pAb, and Fyn mAb (Sigma-Aldrich).
Cell culturing and sulfatide loading of plasma membranes
SCs isolated from sciatic nerves were maintained as described previously (Tsiper and Yurchenco, 2002), used between passages 25 and 35, and plated onto 22-mm2 glass coverslips in 6-well dishes 1 d before protein treatments. Primary SCs were isolated from neonatal rat sciatic nerves as described previously (Carey and Stahl, 1990), cultured on polylysine-coated glass coverslips in DME containing 10% FBS (D/F), and used 4 d after isolation. MEFs, provided by Margaret Schwarz (Robert W. Johnson Medical School), were cultured in D/F and used between passages 3 and 5. β1-integrin–null fibroblasts (GD25) and control β1AGD25 cells (provided by Reinhardt Fässler, Max Planck Institute, Martinsreid, Germany; Wennerberg et al., 1996) were cultured in D/F. DG-null and control wild-type fibroblasts were derived from 10-d-old embryoid bodies cultured from DG-null and wild-type R1 ES cells (Li et al., 2002). The embryoid bodies were digested with trypsin-EDTA, and dissociated cells were plated in bacteriological petri dishes. Cells with fibroblast morphology were enriched by selective trypsinization and panning on collagen I–coated dishes.
Confluent cells were trypsinized and cultured in 100-mm dishes coated with poly-HEMA (Sigma-Aldrich) for 24 h in D/F. Cell aggregates were allowed to settle by gravity, washed once in PBS, and replated onto poly-HEMA–coated dishes in serum-free or serum-containing medium.
Sulfatide–BSA complex was prepared as previously described (Viani et al.,1989; Monti et al., 1992). Gal-sulfatide (Avanti Polar Lipids, Inc.), glc-sulfatide (provided by Ineo Ishizuka, Teikyo University, Tokyo, Japan), or BSA-BODIPY-gal-sulfatide (Watanabe et al., 1999) dissolved in chloroform/methanol (1:1 [vol/vol]) was evaporated under a stream of argon, reconstituted in DMSO, and heated at 60°C for 10 min. Sulfatides were then mixed with an equal mole of de-lipidated BSA in PBS, pH 7.4, and incubated at 37°C for 20 min. The sulfatide–BSA complex was diluted (mM) with serum-free DME and added to cells for 30 min (final, 10 mM). Cells were then rinsed three times with PBS and used immediately for experiments.
Fluorescence microscopy
Cells that were grown on glass coverslips were rinsed with PBS and fixed in 3% PFA for 30 min. Suspended cell aggregates were collected by sedimentation, washed with PBS, fixed in 3% PFA, embedded in optimal cutting temperature compound (Tissue-Tek), and sectioned on a cryostat. Cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min on ice when staining of intracellular epitopes was desired. For detection of surface-bound antigens, the detergent step was omitted. Slides were blocked with 5% goat serum and stained with primary and appropriate secondary antibodies conjugated with FITC, Cy3, or Cy5 (Jackson ImmunoResearch Laboratories). Control staining was performed using preimmune IgG or IgM. Slides were nuclear counterstained with DAPI. Immunofluorescence and phase microscopy were performed on an inverted microscope (model IX70; Olympus) with IX-FLA fluorescence and CCD camera, and the data were collected and analyzed in IPLab v.3.52 as described previously (Li et al., 2002).
Electron microscopy
Cells were plated in 60-mm Permanox dishes (Nunc) 2 d before the experiment. H. promatia arylsulfatase (Sigma-Aldrich) or bacterial collagenase (CLS; Worthington) were added to cell cultures 30 min before the addition of 20–40 μg/ml Lm-1. Cells adherent to plastic were embedded, sectioned, stained, and imaged with an electron microscope (model JEM-1200EX; JEOL USA) as described previously (Tsiper and Yurchenco, 2002). The degree of BM coverage was determined as the ratio of measured length of continuous ECM present divided by the total measured length of exposed cell surface in random cross sections cut at different depths within the Epon block.
Immunoprecipitation and immunoblotting
Cells were washed with cold PBS and disrupted in lysis buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 0.5 mM EDTA, 1% Triton X-100, 1% SDS, and protease and phosphatase inhibitor cocktails [diluted 1:10 and 1:100, respectively, with lysis buffer; Sigma-Aldrich]). Immunoprecipitation was performed with SDS removed from the lysis buffer (Li et al., 2002). Equal amounts of proteins were separated by SDS-PAGE on 12% (caveolin-1), 8% (c-Src), or 6% (utrophin) gels under reducing conditions. Proteins were transferred to polyvinylidene difluoride membranes, blocked, and incubated with primary antibodies followed by HRP-conjugated secondary antibodies (Pierce Chemical Co.). Blots were developed with ECL reagents. Band intensities were quantified from the membrane or scanned films using Quantity 1 software after data acquisition with a gel documentation system (model ChemiDoc XRS; Bio-Rad Laboratories).
Acknowledgments
We thank Dr. Pam Fredman for the Sulf I antibody, Dr. Reinhard Fässler for GD25 and control ES-derived fibroblasts, and Dr. Ineo Ishizuka for glucosyl-sulfatide.
This study was supported by National Institutes of Health grants R01-NS38469 and DK36425.
Abbreviations used in this paper: AEBSF, 4-(2-aminoethyl)benzenesulfonyl fluoride; BM, basement membrane; CS, circumsporozoite; DG, dystroglycan; ES, embryonic stem; gal-sulfatide, HSO3-3galactosylβ-1ceramide; glc-sulfatide, HSO3-3glucosylβ-1ceramide; Lm, laminin; MEF, mouse embryonic lung fibroblast; SC, Schwann cell.
References
- Andac, Z., T. Sasaki, K. Mann, A. Brancaccio, R. Deutzmann, and R. Timpl. 1999. Analysis of heparin, alpha-dystroglycan and sulfatide binding to the G domain of the laminin alpha1 chain by site-directed mutagenesis. J. Mol. Biol. 287:253–264. [DOI] [PubMed] [Google Scholar]
- Bosio, A., E. Binczek, W.F. Haupt, and W. Stoffel. 1998. Composition and biophysical properties of myelin lipid define the neurological defects in galactocerebroside- and sulfatide-deficient mice. J. Neurochem. 70:308–315. [DOI] [PubMed] [Google Scholar]
- Carey, D.J., and R.C. Stahl. 1990. Identification of a lipid-anchored heparan sulfate proteoglycan in Schwann cells. J. Cell Biol. 111:2053–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami, C., F. Kwakye-Berko, and V. Nussenzweig. 1992. Binding of malarial circumsporozoite protein to sulfatides [Gal(3-SO4)beta 1-Cer] and cholesterol-3-sulfate and its dependence on disulfide bond formation between cysteines in region II. Mol. Biochem. Parasitol. 54:1–12. [DOI] [PubMed] [Google Scholar]
- Chen, Z.L., and S. Strickland. 2003. Laminin γ1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. J. Cell Biol. 163:889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Y.S., M.F. Champliaud, R.E. Burgeson, M.P. Marinkovich, and P.D. Yurchenco. 1997. Self-assembly of laminin isoforms. J. Biol. Chem. 272:31525–31532. [DOI] [PubMed] [Google Scholar]
- Coetzee, T., N. Fujita, J. Dupree, R. Shi, A. Blight, K. Suzuki, and B. Popko. 1996. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 86:209–219. [DOI] [PubMed] [Google Scholar]
- Colognato, H., D.A. Winkelmann, and P.D. Yurchenco. 1999. Laminin polymerization induces a receptor–cytoskeleton network. J. Cell Biol. 145:619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornbrooks, C.J., D.J. Carey, J.A. McDonald, R. Timpl, and R.P. Bunge. 1983. In vivo and in vitro observations on laminin production by Schwann cells. Proc. Natl. Acad. Sci. USA. 80:3850–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti, J.M., and K.P. Campbell. 1993. A role for the dystrophin–glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 122:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri, M.L., D.G. Porta, S.C. Previtali, A. Nodari, B. Migliavacca, A. Cassetti, A. Littlewood-Evans, L.F. Reichardt, A. Messing, A. Quattrini, et al. 2002. Conditional disruption of β1 integrin in Schwann cells impedes interactions with axons. J. Cell Biol. 156:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman, P., L. Mattsson, K. Andersson, P. Davidsson, I. Ishizuka, S. Jeansson, J.E. Mansson, and L. Svennerholm. 1988. Characterization of the binding epitope of a monoclonal antibody to sulphatide. Biochem. J. 251:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, Y., and C. Arsenis. 1972. The resolution of aryl sulfatase and heparin sulfamidase activities from various rat tissues. Biochem. Biophys. Res. Commun. 48:1133–1139. [DOI] [PubMed] [Google Scholar]
- Honke, K., M. Yamane, A. Ishii, T. Kobayashi, and A. Makita. 1996. Purification and characterization of 3′-phosphoadenosine-5′-phosphosulfate:GalCer sulfotransferase from human renal cancer cells. J. Biochem. (Tokyo). 119:421–427. [DOI] [PubMed] [Google Scholar]
- Honke, K., Y. Hirahara, J. Dupree, K. Suzuki, B. Popko, K. Fukushima, J. Fukushima, T. Nagasawa, N. Yoshida, Y. Wada, and N. Taniguchi. 2002. Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc. Natl. Acad. Sci. USA. 99:4227–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka, I. 1997. Chemistry and functional distribution of sulfoglycolipids. Prog. Lipid Res. 36:245–319. [DOI] [PubMed] [Google Scholar]
- Kalb, E., and J. Engel. 1991. Binding and calcium-induced aggregation of laminin onto lipid bilayers. J. Biol. Chem. 266:19047–19052. [PubMed] [Google Scholar]
- Li, S., D. Edgar, R. Fässler, W. Wadsworth, and P.D. Yurchenco. 2003. The role of laminin in embryonic cell polarization and tissue organization. Dev. Cell. 4:613–624. [DOI] [PubMed] [Google Scholar]
- Li, S., D. Harrison, S. Carbonetto, R. Fässler, N. Smyth, D. Edgar, and P.D. Yurchenco. 2002. Matrix assembly, regulation, and survival functions of laminin and its receptors in embryonic stem cell differentiation. J. Cell Biol. 157:1279–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinkovich, M.P., D.R. Keene, C.S. Rimberg, and R.E. Burgeson. 1993. Cellular origin of the dermal-epidermal basement membrane. Dev. Dyn. 197:255–267. [DOI] [PubMed] [Google Scholar]
- Merten, M., and P. Thiagarajan. 2001. Role for sulfatides in platelet aggregation. Circulation. 104:2955–2960. [DOI] [PubMed] [Google Scholar]
- Miner, J.H., C. Li, J.L. Mudd, G. Go, and A.E. Sutherland. 2004. Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation. Development. 131:2247–2256. [DOI] [PubMed] [Google Scholar]
- Mirsky, R., C. Dubois, L. Morgan, and K.R. Jessen. 1990. 04 and A007-sulfatide antibodies bind to embryonic Schwann cells prior to the appearance of galactocerebroside; regulation of the antigen by axon-Schwann cell signals and cyclic AMP. Development. 109:105–116. [DOI] [PubMed] [Google Scholar]
- Monti, E., A. Preti, A. Novati, M.F. Aleo, M.L. Clemente, and S. Marchesini. 1992. Uptake and metabolism of a fluorescent sulfatide analogue in cultured skin fibroblasts. Biochim. Biophys. Acta. 1124:80–87. [DOI] [PubMed] [Google Scholar]
- Previtali, S.C., A. Nodari, C. Taveggia, C. Pardini, G. Dina, A. Villa, L. Wrabetz, A. Quattrini, and M.L. Feltri. 2003. Expression of laminin receptors in Schwann cell differentiation: Evidence for distinct roles. J. Neurosci. 23:5520–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, D.D., C.N. Rao, J.L. Magnani, S.L. Spitalnik, L.A. Liotta, and V. Ginsburg. 1985. Laminin binds specifically to sulfated glycolipids. Proc. Natl. Acad. Sci. USA. 82:1306–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, D.D., C.N. Rao, L.A. Liotta, H.R. Gralnick, and V. Ginsburg. 1986. Comparison of the specificities of laminin, thrombospondin, and von Willebrand factor for binding to sulfated glycolipids. J. Biol. Chem. 261:6872–6877. [PubMed] [Google Scholar]
- Saito, F., S.A. Moore, R. Barresi, M.D. Henry, A. Messing, S.E. Ross-Barta, R.D. Cohn, R.A. Williamson, K.A. Sluka, D.L. Sherman, et al. 2003. Unique role of dystroglycan in peripheral nerve myelination, nodal structure, and sodium channel stabilization. Neuron. 38:747–758. [DOI] [PubMed] [Google Scholar]
- Scheele, S., M. Falk, A. Franzen, F. Ellin, M. Ferletta, P. Lonaio, B. Andersson, R. Timpl, E. Forsberg, and P. Ekblom. 2005. Laminin alpha1 globular domains 4-5 induce fetal development but are not vital for embryonic basement membrane assembly. Proc. Natl. Acad. Sci. USA. 102:1502–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov, S.P., E.L. McDearmon, S. Li, J.M. Ervasti, K. Tryggvason, and P.D. Yurchenco. 2002. Contributions of the LG modules and furin processing to laminin-2 functions. J. Biol. Chem. 277:18928–18937. [DOI] [PubMed] [Google Scholar]
- Smyth, N., H.S. Vatansever, P. Murray, M. Meyer, C. Frie, M. Paulsson, and D. Edgar. 1999. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J. Cell Biol. 144:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisi, D., J.F. Talts, R. Timpl, and E. Hohenester. 2000. Structure of the C-terminal laminin G-like domain pair of the laminin alpha2 chain harbouring binding sites for alpha-dystroglycan and heparin. EMBO J. 19:1432–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiper, M.V., and P.D. Yurchenco. 2002. Laminin assembles into separate basement membrane and fibrillar matrices in Schwann cells. J. Cell Sci. 115:1005–1015. [DOI] [PubMed] [Google Scholar]
- Umemori, H., Y. Kadowaki, K. Hirosawa, Y. Yoshida, K. Hironaka, H. Okano, and T. Yamamoto. 1999. Stimulation of myelin basic protein gene transcription by Fyn tyrosine kinase for myelination. J. Neurosci. 19:1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viani, P., S. Marchesini, B. Cestaro, and S. Gatt. 1989. Correlation of the dispersion state of pyrene cerebroside sulfate and its uptake and degradation by cultured cells. Biochim. Biophys. Acta. 1002:20–27. [DOI] [PubMed] [Google Scholar]
- Waheed, A., and R.L. van Etten. 1980. Chemical characterization and substrate specificity of rabbit liver aryl sulfatase A. Biochim. Biophys. Acta. 614:92–101. [DOI] [PubMed] [Google Scholar]
- Watanabe, R., K. Asakura, M. Rodriguez, and R.E. Pagano. 1999. Internalization and sorting of plasma membrane sphingolipid analogues in differentiating oligodendrocytes. J. Neurochem. 73:1375–1383. [DOI] [PubMed] [Google Scholar]
- Wennerberg, K., L. Lohikangas, D. Gullberg, M. Pfaff, S. Johansson, and R. Fässler. 1996. β 1 integrin-dependent and -independent polymerization of fibronectin. J. Cell Biol. 132:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wizemann, H., J.H. Garbe, M.V. Friedrich, R. Timpl, T. Sasaki, and E. Hohenester. 2003. Distinct requirements for heparin and alpha-dystroglycan binding revealed by structure-based mutagenesis of the laminin alpha2 LG4-LG5 domain pair. J. Mol. Biol. 332:635–642. [DOI] [PubMed] [Google Scholar]
- Yang, D., J. Bierman, Y.S. Tarumi, Y.P. Zhong, R. Rangwala, T.M. Proctor, Y. Miyagoe-Suzuki, S. Takeda, J.H. Miner, L.S. Sherman, et al. 2005. Coordinate control of axon defasciculation and myelination by laminin-2 and -8. J. Cell Biol. 168:655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenco, P.D., and Y.S. Cheng. 1993. Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J. Biol. Chem. 268:17286–17299. [PubMed] [Google Scholar]
- Yurchenco, P.D., Y. Quan, H. Colognato, T. Mathus, D. Harrison, Y. Yamada, and J.J. O'Rear. 1997. The alpha chain of laminin-1 is independently secreted and drives secretion of its beta- and gamma-chain partners. Proc. Natl. Acad. Sci. USA. 94:10189–10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenco, P.D., P.S. Amenta, and B.L. Patton. 2004. Basement membrane assembly, stability and activities observed through a developmental lens. Matrix Biol. 22:521–538. [DOI] [PubMed] [Google Scholar]
- Zhao, Y., M. Sudol, H. Hanafusa, and J. Krueger. 1992. Increased tyrosine kinase activity of c-Src during calcium-induced keratinocyte differentiation. Proc. Natl. Acad. Sci. USA. 89:8298–8302. [DOI] [PMC free article] [PubMed] [Google Scholar]