

Abstract
The Rho GTPases play a critical role in initiating actin polymerization during phagocytosis. In contrast, the factors directing the disassembly of F-actin required for fission of the phagocytic vacuole are ill defined. We used fluorescent chimeric proteins to monitor the dynamics of association of actin and active Cdc42 and Rac1 with the forming phagosome. Although actin was found to disappear from the base of the forming phagosome before sealing was complete, Rac1/Cdc42 activity persisted, suggesting that termination of GTPase activity is not the main determinant of actin disassembly. Furthermore, fully internalized phagosomes engineered to associate constitutively with active Rac1 showed little associated F-actin. The disappearance of phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) from the phagosomal membrane closely paralleled the course of actin disassembly. Furthermore, inhibition of PI(4,5)P2 hydrolysis or increased PI(4,5)P2 generation by overexpression of phosphatidylinositol phosphate kinase I prevented the actin disassembly necessary for the completion of phagocytosis. These observations suggest that hydrolysis of PI(4,5)P2 dictates the remodeling of actin necessary for completion of phagocytosis.
Introduction
The ability of leukocytes to engulf and subsequently eliminate foreign particles is essential for immune function. Soluble immune complexes can be internalized by clathrin-mediated endocytosis, but uptake of particles larger than ≈0.5 μm involves an actin-dependent process termed phagocytosis. Phagocytosis is triggered by the association of ligands on the surface of the target particle with receptors on the leukocyte membrane. A variety of phagocytic receptor types have been described in mammalian neutrophils and macrophages. Perhaps the best characterized of these is the Fcγ family of receptors (FcγR), which recognize the constant domain of IgG. Upon cross-linking by their cognate ligand, FcγR activate signaling pathways that trigger a highly dynamic and coordinated set of cytoskeletal rearrangements that culminate in particle internalization (Aderem and Underhill, 1999; Aderem, 2002; Greenberg and Grinstein, 2002; Underhill and Ozinsky, 2002).
Actin polymerization at the forming phagosome is thought to be controlled by GTPases of the Rho family. Specifically, Rac1 and Cdc42 are known to be stimulated upon engagement of FcγR and are essential for the extension of the pseudopods that surround and engulf the phagocytic particle (Cox et al., 1997; Massol et al., 1998; Hoppe and Swanson, 2004). The tips of the advancing pseudopods eventually meet and fuse, sequestering the target particle in an intracellular vacuole, or phagosome. Detachment of the phagocytic vacuole from the plasma membrane is accompanied by, and likely requires, extensive dissociation of the actin meshwork that drives pseudopodial extension. This is suggested by the inability of phagocytosis to reach completion in cells treated with inhibitors of phosphatidylinositol 3′-kinase (PI3-K). In such cells actin polymerization at the phagocytic cup persists for an extended period, yet particle internalization is frustrated (Araki et al., 1996).
Although much has been learned about the steps leading to actin assembly at the phagosome, considerably less is known about its disassembly. Because dynamic studies of the behavior of the cytoskeleton during phagocytosis are scarce, it is not clear if actin surrounding the phagosome depolymerizes suddenly and symmetrically upon completion of internalization, or whether the depolymerization is gradual and polarized. More importantly, the factors dictating the disassembly of actin during phagocytosis have not been explored. Although recent work has shed light on the activation kinetics of Rho-family proteins during phagosome formation (Hoppe and Swanson, 2004), it has yet to be established if actin disassembly is merely the result of inactivation of Rac1 and Cdc42, or whether other controlling factors are involved. To address these issues, we generated phagocytic cells stably transfected with GFP-actin and monitored the distribution of the fluorescent protein in live cells during the course of phagocytosis. The spatial and temporal changes displayed by actin were compared with the pattern of activation of Rac1 and Cdc42. In addition, we devised a system whereby the persistence of actin around the nascent phagosome could be studied while ensuring a sustained activation of the Rho GTPases. Our results suggest that inactivation of the GTPases is not the main factor controlling the disassembly of polymerized actin from the phagocytic cup and that phosphoinositide metabolism plays an essential role in these events.
Results
Actin dynamics during phagocytosis
To study actin dynamics during phagocytosis, RAW 264.7 macrophages (referred to hereafter as RAW cells) were stably transfected with GFP-actin. Phagocytosis was induced by exposure of the cells to latex beads opsonized with IgG and the distribution of actin was monitored in live cells by laser confocal microscopy. As illustrated in Fig. 1 and reported previously (Allison et al., 1971; Henry et al., 2004), there is a marked accumulation of actin in the region of the forming phagosome. At the earliest stages, extension of actin-rich pseudopods around the latex bead was consistently observed (Fig. 1, B and C). Upon phagosome closure, when the advancing pseudopods meet and fuse, actin transiently surrounds the entire phagosome, appearing as a nearly homogeneous, continuous ring (Fig. 1 D). Importantly, actin disassembly occurs asymmetrically after phagosomal sealing, with loss of fluorescence occurring initially at the base of the phagosome (Fig. 1 E, the innermost half), whereas a “cap” of actin persists for at least 240 s in the space between the phagosome and the plasmalemma. Eventually, this outermost cap also disassembles and actin accumulation is no longer detectable around the internalized phagosome (Fig. 1 F). For beads of 3.1-μm diam, such as those used in Fig. 1, the entire process is completed in 4–5 min at 37°C. Predictably, this time course is slightly more protracted than that reported by Defacque and colleagues (Defacque et al., 2000), who monitored actin polymerization using smaller (1 μm) beads. Slower kinetics of actin association was reported by Henry et al. (2004) who used somewhat larger particles, namely sheep RBCs.
Figure 1.

Association of GFP-actin with forming phagosomes. Phagocytosis was initiated by addition of IgG-opsonized latex beads (3.1-μm diam) to RAW cells stably transfected with GFP-actin. Fluorescence was monitored by confocal microscopy. (A–F) Representative time course. The numbers indicate the time in seconds after a bead makes contact at the site indicated by the open arrow in A. Note that another bead has just been engulfed near the top of the cell and still shows remnant actin at the site of sealing. Insets show enlargements of the phagosome noted by arrow. Open arrows point to forming phagosomes and closed arrows point to formed, sealed phagosomes. Bar, 5 μm. (G) The phagosomal accumulation of GFP-actin above the cytosolic level was quantified and binned into 20-s intervals as detailed in Materials and methods. Abscissa: time in seconds after the bead made contact with cell. Ordinate: relative fluorescence. To allow comparison between experiments, phagosomal fluorescence was normalized to the maximum recorded for each individual phagosome. Data are means ± SEM of seven individual determinations.
Actin accumulation around the phagosome was quantified by image analysis, using the level of cytosolic GFP-actin as a threshold (see Materials and methods). The data of seven cells from four experiments are summarized in Fig. 1 G. Actin association with the particles peaked ∼150 s after the onset of phagocytosis, coinciding with the moment of phagosomal closure (Fig. 1 D). This was followed by rapid dissociation of actin, approaching background levels ∼300 s after the onset of phagocytosis. Depolymerization of actin at the innermost aspect of the phagosome was apparent as early as 100 s after initiation of phagocytosis.
Active Rac/Cdc42 persists after actin depolymerization
The preceding observations indicate that actin depolymerization commences shortly after phagosomal closure and that it occurs asymmetrically. Because activation of Rac1 and Cdc42 is thought to underlie the assembly of actin at the phagosome (Cox et al., 1997; Caron and Hall, 1998; Massol et al., 1998) we considered the possibility that deactivation of the GTPases was responsible for the pattern of actin disassembly. To this end, we measured the kinetics of activation of the GTPases using a fusion protein consisting of the p21-binding domain of PAK fused to YFP (PBD-YFP; Srinivasan et al., 2003). In quiescent RAW cells the PBD-YFP probe was distributed almost exclusively in the cytosol, indicating minimal activation of Rac1 and Cdc42 (Fig. 2 A). Upon addition of opsonized beads, PBD-YFP accumulated in the budding pseudopods (Fig. 2 B) and was ultimately visible all around the phagosome, closely resembling the early stages of actin accumulation (Fig. 2, D and E). Unlike actin, however, the deactivation of Rac and/or Cdc42 occurred homogeneously throughout the phagosomal circumference (Fig. 2, E and F). Moreover, the association of PBD-YFP with the phagosome persisted after actin disassembly was obvious. The differential behavior of actin and of the active GTPases became more apparent when the kinetics of PBD-YFP association with the phagosome was quantified as described above. The results of six experiments, summarized in Fig. 2 G, indicate that Rac/Cdc42 activation is maximal only after ≈200 s and is still detectable after 300 s, clearly lagging behind the kinetics of actin assembly.
Figure 2.
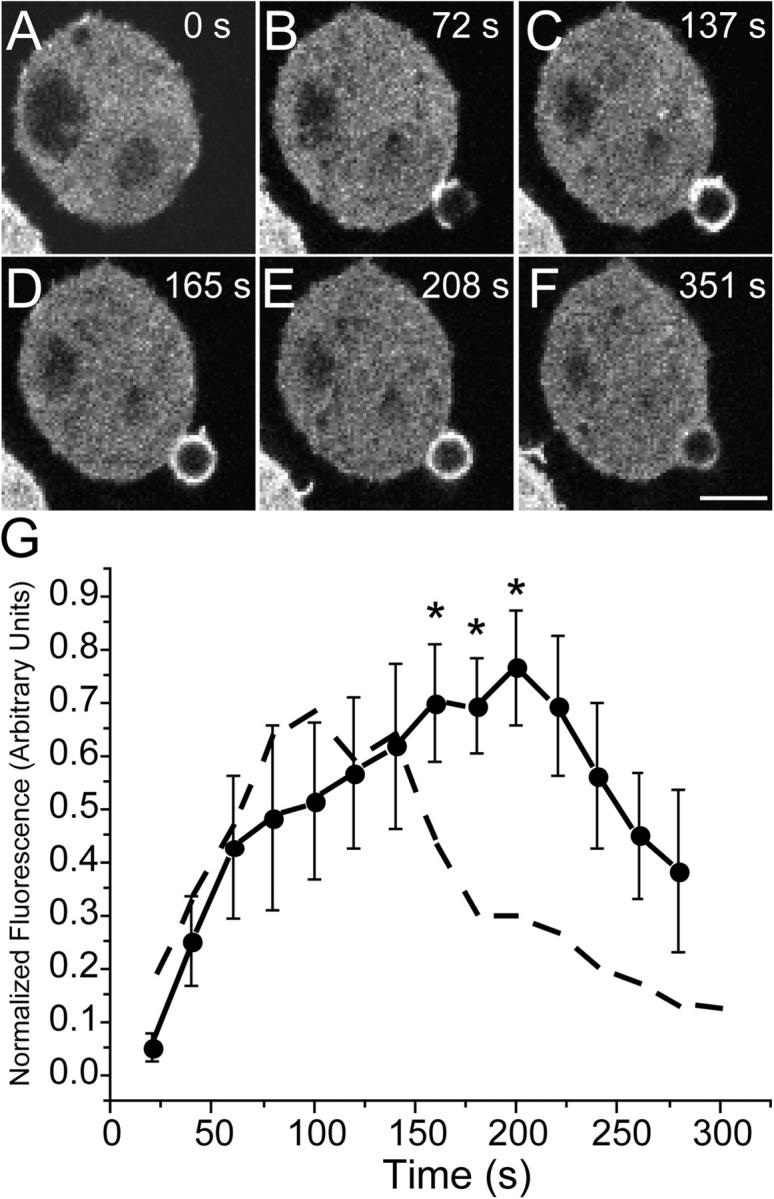
Assessment of Rac/Cdc42 activity in forming phagosomes. Phagocytosis was initiated by addition of IgG-opsonized latex beads to RAW cells transiently transfected with PBD-YFP. Fluorescence was monitored by confocal microscopy. (A–F) Representative time course. The numbers indicate the time in seconds after a bead makes contact with the cell. Bar, 5 μm. (G) The phagosomal accumulation of PBD-YFP above the cytosolic level was quantified, normalized and binned into 20-s intervals as in Fig. 1 (solid line). Abscissa: time in seconds after the bead made contact with cell. Ordinate: relative fluorescence, normalized as in Fig. 1 G. Data are means ± SEM of six individual determinations. The dashed line is a reproduction of the GFP-actin data of Fig. 1, for comparison. Asterisks indicate instances where the difference between the curves is significant (P < 0.05).
Because of the similar spectral properties of GFP and YFP, actin recruitment and the activation of Rac/Cdc42 were studied separately in the experiments of Figs. 1 and 2. It was conceivable that the differences noted were fortuitous, or that the expression of the PBD-YFP had resulted in an artifactual retardation of phagocytosis. To eliminate these possibilities, we analyzed actin distribution in the same cells transfected with the PBD-YFP probe. This was accomplished by fixation of cells at various stages during phagocytosis, followed by permeabilization and staining of F-actin using labeled phalloidin. As shown in Fig. 3 (A–C), there was an exquisite colocalization of F-actin with PBD-YFP on the forming phagosome at the early (1–2 min) stages of phagocytosis. Note that the less abundant cortical actin found in regions of the membrane not involved in formation of the phagocytic cup was not associated with PBD-YFP, implying that it remains polymerized in a manner that is independent of Rac/Cdc42 activation. The asymmetric accumulation of actin observed after phagosomal closure was also detectable using phalloidin (Fig. 3 D), validating the GFP-actin observations. More importantly, the activation of Rac/Cdc42 clearly persisted in recently closed phagosomes at a time when actin had nearly disappeared from their base (Fig. 3, E and F). These findings confirm the asynchrony between actin disassembly and the deactivation of Rac/Cdc42 and suggest that other factors are involved in the dissociation of actin.
Figure 3.

Comparison of Rac/Cdc42 activity and F-actin in forming phagosomes. RAW cells were transiently transfected with the PBD-YFP chimeric probe (B, C, E, and F, green) or with Cdc42Q61L-GFP (H and I, green) and exposed to IgG-opsonized latex beads for the time indicated. The cells were immediately fixed with 4% PFA, permeabilized and stained with rhodamine-phalloidin to visualize F-actin (A, C, D, F, G, and I, red). (A–C) PBD-YFP expressing cells fixed 2 min after addition of beads. (D–F) PBD-YFP expressing cells fixed 4 min after addition of beads. (G–I) Cdc42Q61L-GFP expressing cells fixed after 4 min. Open arrows point to forming phagosomes and closed arrows point to sealed phagosomes. Inset in G–I shows enlargement of phagosome denoted by the box in main panel. Bars, 5 μm. Images are representative of at least three experiments of each type.
As both Cdc42 and Rac1 can signal through PAK1, the PBD-YFP chimera is, in principle, a probe for the activity of both GTPases. However, it has been claimed that PBD-YFP associates preferentially with Rac1, being poorly sensitive to changes in the activation of Cdc42 (Srinivasan et al., 2003). It is therefore conceivable that termination of Cdc42 activity occurred before dissociation of the probe was complete and that the activity of Cdcd42 is essential for actin to remain assembled. To analyze this possibility we transfected cells with a constitutively active allele of Cdc42. As expected, GFP-tagged (Fig. 3 H) and myc-tagged Cdc42Q61L constructs accumulated in the plasma membrane. The PBD-YFP construct was recruited to the membrane when coexpressed with myc-Cdc42Q61L (not depicted), confirming that the latter was active. Under the conditions used, expression of Cdc42Q61L did not prevent phagocytosis of IgG-opsonized particles. Importantly, the active form of Cdc42 could be seen to remain present on the phagosomal membrane after actin had dissociated and was no longer detectable (Fig. 3, G–I). Therefore, termination of Cdc42 activity is not the primary determinant of actin depolymerization and dissociation from the phagosomal membrane. Jointly, these observations suggest factors other than Rho-family proteins contribute to direct actin remodeling during phagocytosis.
Actin dissociates from phagosomes despite the sustained activation of Rac1
To more conclusively dissociate the disassembly of actin from the deactivation of Rac1 we sought to prolong the association of Rac1-GTP with the phagosomal membrane. This was accomplished using the system illustrated in Fig. 4 A, originally described by Castellano et al. (2000), designed to recruit constitutively active Rac1 (myc-Rac1-V12) to the cytoplasmic face of the plasma membrane at the site of bead attachment. The system consists of RBL cells transfected with two separate vectors: one encodes for a soluble form of myc-tagged Rac1-V12 and the other for a transmembrane membrane protein with an extracellular epitope (CD25). Each one of the constructs is in addition fused to different rapamycin-binding moieties. Upon addition of rapamycin, which is membrane permeant, the active Rac1 is recruited to the membrane, where it associates with the transmembrane construct (Fig. 4 A′). The latter can then be clustered on the surface by binding to beads bearing anti-CD25 antibodies (Fig. 4 A′′). The result of these joint maneuvers is the recruitment of constitutively active Rac1 to the cytosolic face of the membrane lining the bead. This was shown earlier to suffice for particle engulfment, recapitulating phagocytosis (Castellano et al., 2000). As shown in Fig. 4 C, the recruitment of Rac1 to the sites of bead attachment can be verified by immunostaining for the myc epitope linked to the GTPase. When the beads have not been fully internalized, as shown by their accessibility to externally added antibodies (Fig. 4 D), polymerization of F-actin can be readily demonstrated at sites where phagosomes are being formed, by staining with phalloidin derivatives (Fig. 4 B). At later times, the beads become fully internalized, because they can no longer be stained by externally added antibodies (Fig. 4 G), and are displaced from the cell periphery toward the center of the cell (Fig. 4 F). Remarkably, F-actin is no longer associated with these fully internalized beads, even though they remain lined by active Rac1, which was confirmed by immunostaining the myc epitope (Fig. 4, E and F). These findings clearly indicate that F-actin can dissociate from the phagosomal membrane despite the continued presence of active Rac1. Together with the preceding findings, these observations imply that factors other than deactivation of the Rho family GTPases are responsible for the rapid and asymmetric disassembly of phagosomal actin.
Figure 4.
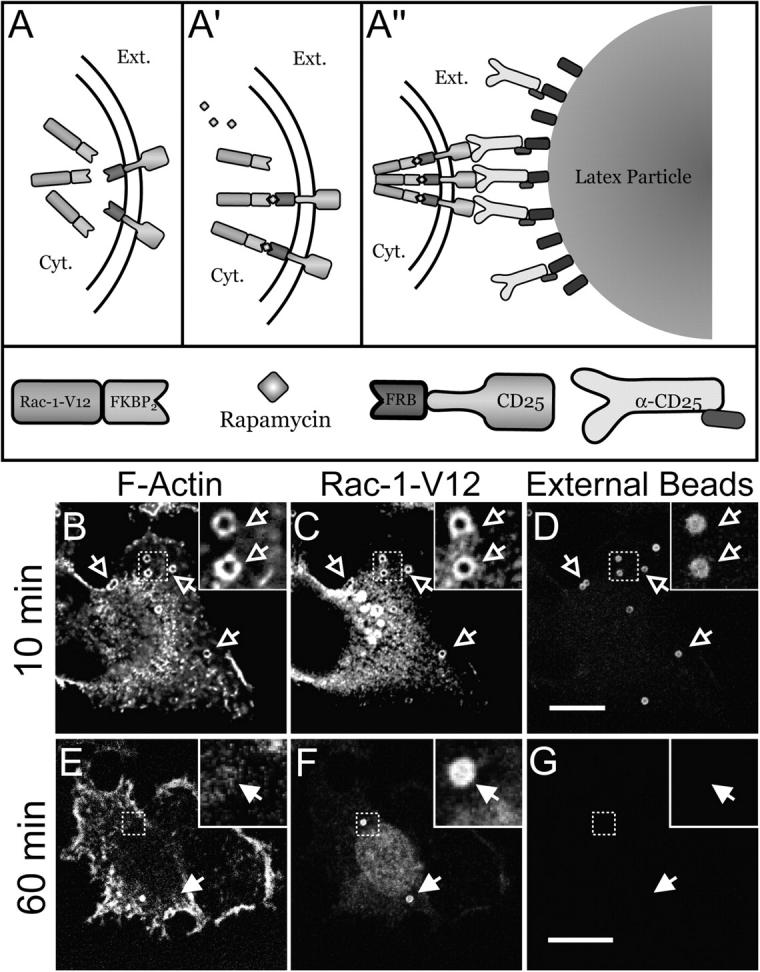
Actin association with myc-Rac1-V12–induced phagosomes. (A) Strategy used to generate phagosomes by recruitment of active Rac1 to the membrane. RBL-2H3 cells were stably transfected with two separate constructs: a soluble, myc-tagged Rac1-V12/FKBP2 chimera (myc-Rac1-V12) and a transmembrane CD25/FRB chimera (A). Association between the two chimeras is induced by addition of rapamycin (A′). Beads coated with anti-CD25 antibodies are then added to cross-link the complexes at defined sites (A′′). (B–D) After binding the beads, the cells were incubated for 10 min at 37°C, then rapidly cooled to 4°C and treated with Cy5-conjugated anti–mouse antibodies to identify beads that were accessible from the medium, i.e., not completely internalized. The cells were next fixed, permeabilized, and stained for F-actin and immunostained with anti-myc antibodies to reveal the location of myc-Rac1-V12. (E–G) After binding the beads, the cells were incubated for 60 min at 37°C and treated as in B–D. Bars, 5 μm. Open arrows point to forming phagosomes and closed arrows point to sealed phagosomes. Insets show a magnification of the area indicated by the square in the main panels. Images are representative of three experiments.
Similarity between the kinetics of phosphatidylinositol-4,5-bisphosphate hydrolysis and the dissociation of phagosomal actin
Phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) plays multiple roles in the nucleation, elongation, and bundling of actin filaments (Hilpela et al., 2004). It is therefore conceivable that this phosphoinositide contributes to the remodeling of F-actin during phagocytosis. We therefore compared the distribution and dynamics of PI(4,5)P2 during phagocytosis with that of actin. A chimeric construct consisting of GFP attached to the PH domain of PLCδ (PHPLCδ-GFP; Stauffer et al., 1998; Varnai and Balla, 1998; Botelho et al., 2000) was used to monitor PI(4,5)P2 in live macrophages during the course of particle ingestion. As illustrated in Fig. 5, PI(4,5)P2 is present throughout the plasmalemma before phagocytosis and is clearly visible in the advancing pseudopods during the early stages of particle ingestion (A–C). However, the density of the inositide drops sharply at the base of the phagosome as the pseudopods meet at the top of the particle. PI(4,5)P2 in fact begins to disappear from the base of the phagocytic cup before sealing (Fig. 5, C and D), which is more evident when larger (e.g., 8–μm diam) particles are used (Botelho et al., 2000). The hydrolysis of PI(4,5)P2 during phagocytosis can be validated by conventional chemical methods such as thin layer chromatography (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200412162/DC1), although such global methods are not optimal to detect localized changes. The dynamics of association of PI(4,5)P2 with the phagocytic particle is summarized in Fig. 5 G, where the course of actin assembly is also shown (dashed line) for comparison. Clearly, the asymmetric and rapid disappearance of PI(4,5)P2 from forming phagosomes closely parallels the spatial and temporal pattern of F-actin disassembly. Because PI(4,5)P2 is an important factor in the control of actin polymerization, it is conceivable that hydrolysis of the inositide dictates the disassembly of actin from sealing phagosomes.
Figure 5.
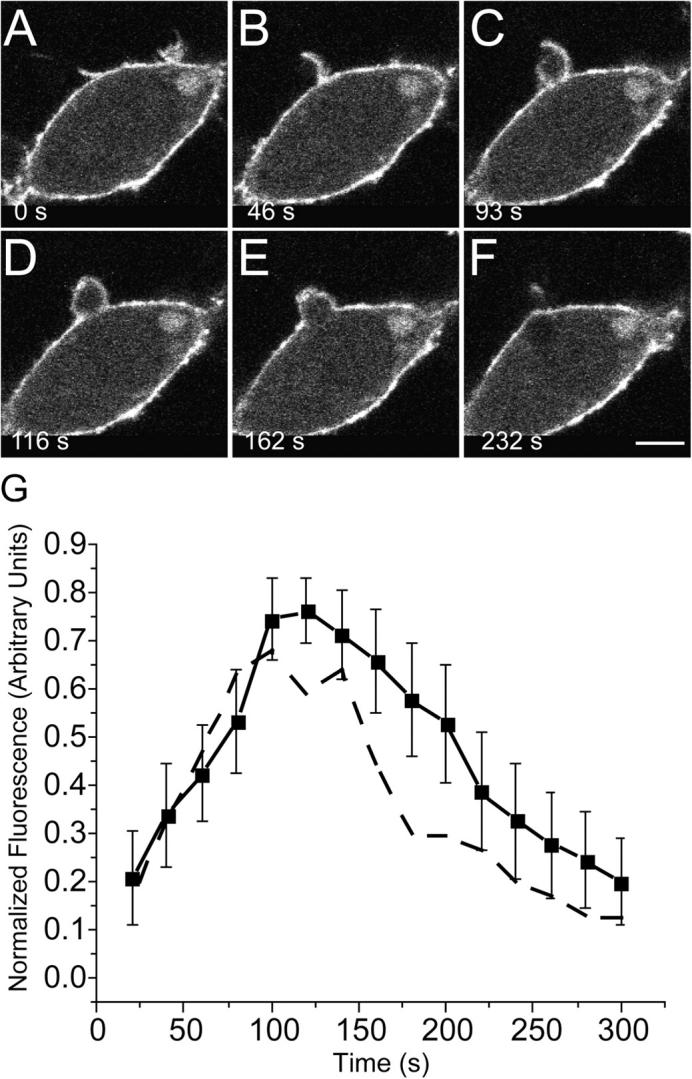
Association of PI(4,5)P2 with forming phagosomes. Phagocytosis was initiated by addition of IgG-opsonized latex beads to RAW cells transiently transfected with PHPLCδ-GFP. Fluorescence was monitored by confocal microscopy. (A–F) Representative time course. The numbers indicate the time in seconds after a bead makes contact with the cell. Bar, 5 μm. (G) The phagosomal accumulation of PHPLCδ-GFP above the cytosolic level was quantified, normalized, and binned into 20-s intervals as in Fig. 1 (solid line). Data are means ± SEM of eight individual determinations. The dashed line is a reproduction of the GFP-actin data of Fig. 1, for comparison. The curves were not significantly different (P > 0.05) at any point.
Loss of PI(4,5)P2 correlates with actin disassembly in myc-Rac1-V12 phagosomes
As described in connection with Fig. 4, actin dissociates from phagosomes despite the sustained presence of active Rac1. We therefore investigated whether this dissociation is similarly accompanied by the hydrolysis of PI(4,5)P2 from myc-Rac1-V12–induced phagosomes. To this end, we transiently transfected the engineered RBL phagocytes with the PH domain-GFP chimera to monitor the fate of PI(4,5)P2. After internalization of the particles the amount of PI(4,5)P2 associated with the phagocytic vacuoles is greatly reduced compared with the originating plasma membrane (Fig. 6 A). It is noteworthy that this PI(4,5)P2 depletion occurred in spite of the continued presence of active Rac1 that can directly associate with and activate phosphatidylinositol phosphate kinase type I (PIPKI), the enzyme responsible for PI(4,5)P2 generation (Tolias and Carpenter, 2000; Weernink et al., 2004). Therefore, stimulation of PI(4,5)P2 catabolism probably accounts for the observed drop in its density during phagocytosis, whereas inhibition of synthesis is less likely.
Figure 6.

Disappearance of PI(4,5)P2 from Rac1-V1–induced phagosomes. RBL-2H3 cells engineered to express stably the two constructs described in Fig. 4 were transiently transfected with PHPLCδ-GFP (A). Beads coated with anti-CD25 antibodies were then added and the cells were incubated for 60 min at 37°C, then rapidly cooled to 4°C and treated with Cy5-conjugated anti–mouse antibodies to identify beads that were accessible from the medium, i.e., not completely internalized (C). A small amount of light refracted by latex is visible in both internal and external beads. This refraction is apparent only at the center of the beads and is clearly distinguishable from the more peripheral antibody labeling. The cells were next fixed, permeabilized and immunostained with anti-myc antibodies to reveal the location of myc-Rac1-V12 (B). Open arrows point to externally accessible beads and closed arrows point to sealed phagosomes. Bar, 5 μm. Insets show a magnification of the area indicated by the square in the main panels. (D) Phagocytosis was induced in the transfected RBL-2H3 cells as described in Fig. 4. The cells were otherwise untreated, or were pretreated with the PLC inhibitors U73122 (1 μM) or ET-18-OCH3 (25 μM) for 10 min before phagocytosis. Extracellular beads were labeled and the number of beads ingested after 20 min was quantified under a fluorescence microscope. The phagocytic index was normalized to allow comparison of multiple experiments. The data are the means ± SEM of three experiments, each scoring at least 50 cells.
In the case of professional phagocytes, disappearance of PI(4,5)P2 from forming phagosomes is due largely to hydrolysis by PLC (Azzoni et al., 1992; Liao et al., 1992). To verify that PI(4,5)P2 hydrolysis plays a role in phagocytosis induced by recruitment of active Rac1, the RBL cells expressing myc-Rac1-V12 were treated with the specific PLC inhibitors ET-18-OCH3 and U73122. As shown earlier for cells that are naturally phagocytic through a receptor-mediated process (Botelho et al., 2000), inhibitors of PLC also prevented particle internalization by the engineered myc-Rac1-V12 phagocytes (Fig. 6 D). Conversion of PI(4,5)P2 to diacylglycerol and inositol-1,4,5-trisphosphate is required for phagocytosis, despite the fact that neither increased cytosolic calcium nor activation of PKC are universally required for particle ingestion (Di Virgilio et al., 1988; Greenberg et al., 1991, 1993; Hishikawa et al., 1991). This suggests that perhaps the disappearance of the substrate, rather than the generation of the products, is the critical event for successful phagocytosis. These findings are consistent with the notion that PI(4,5)P2 hydrolysis is responsible for the disassembly of actin that facilitates phagosomal sealing.
Impairment of PI(4,5)P2 hydrolysis inhibits phagocytosis at an early stage
The preceding findings imply that inhibitors of PLC must block phagocytosis at a step preceding the disassembly of phagosomal actin. We therefore analyzed in greater detail the stage at which phagocytosis was arrested in RAW cells treated with U73122. Compared with untreated cells, which displayed distinct actin recruitment to sites of phagocytosis (Fig. 7, A–C), cells pretreated with the PLC inhibitor showed little accumulation of actin at the sites where opsonized particles adhered (Fig. 7, D–F). Instead, the cells appeared rounder, with a thick layer of submembranous F-actin. The increased association of actin with the membrane could be documented both by measurement of the thickness of the actin layer or by integration of the amount of subcellular F-actin in single cells (Fig. 7 G), and by quantitation of total cellular F-actin in populations (Fig. 7 H). These findings suggest that increased deposition of cortical F-actin before exposure to phagocytic particles increased the rigidity of the membrane, impairing the ability of the cells to extend pseudopods and ingest particles.
Figure 7.

Effect of PLC inhibition on F-actin distribution. IgG-opsonized latex beads were added to RAW cells that were otherwise untreated (A–C) or that had been pretreated with 1 μM U73122 for 10 min (D–F). After 15 min at 37°C, the cells were cooled to 4°C and extracellular beads identified by addition of FITC-conjugated secondary antibody (B and E). The cells were next fixed, permeabilized, and stained with rhodamine-phalloidin (A and D). Corresponding differential interference contrast (DIC) images are shown in C and F. Insets show a magnification of the area indicated by the square in the main panels. Open arrows point to externally accessible beads and closed arrows point to sealed phagosomes. (G) Cortical F-actin thickness, quantified from line scans (white bars) and total cortical actin, calculated by integration (black bars) in control and in U73122-treated cells. Data are the means ± SEM of three experiments, each scoring at least 20 cells. (H) High throughput analysis of F-actin during phagocytosis, determined by averaging the integrated fluorescence of rhodamine-phalloidin in populations of cells. Circles, moderate number of beads. Triangles, high number of beads. Stars, moderate number of beads added after pretreatment with 5 μM U73122 for 10 min. Data show the integrated fluorescence of at least 2,500 cells per time point. Standard error bars were smaller than the size of the symbols.
The validity of these conclusions, which were based on analysis of a limited number of single cells, was confirmed using an automated high-throughput microscopic analysis. This population-wide approach yielded an actin polymerization profile (Fig. 7 H, circles) consistent with that of the single cell technique (compare with Fig. 1 G). The slightly longer association of actin with the phagosomes determined in the automated measurements is likely a consequence of asynchrony of onset of phagocytosis in different cells of the population, despite our efforts to initiate engulfment synchronously. Accordingly, we found that the duration of the actin polymerization transient was increased further by addition of a higher number of beads (Fig. 7 H, triangles). Regardless of this asynchrony, we found that pretreatment of cells with U73122 (Fig. 7 H, stars) not only increased the cellular F-actin content to levels above those of resting cells but, more importantly, prevented any further actin polymerization in response to the addition of opsonized beads.
Inhibition of PI(4,5)P2 hydrolysis during the late stages of phagocytosis prevents actin remodeling and completion of phagocytosis
When added before exposure of the cells to phagocytic targets, U73122 prevented cup formation and failed to provide evidence of the role of PI(4,5)P2 hydrolysis in cytoskeletal disassembly during the course of particle internalization. To circumvent the effects of PLC inhibitors on the early stages of actin remodeling, we attempted to use the inhibitors only after phagocytosis had been initiated. However, their slow permeation rate requires extended incubation periods, precluding this experimental paradigm. Instead, we sought to design experiments where phagocytosis could be arrested after formation of the phagocytic cup, but before completion of phagocytosis, affording us the opportunity to load the cells with PLC antagonists and assess their effect on actin disassembly. This was made possible by the use of LY294002, a reversible PI3-K inhibitor (Vlahos et al., 1994). Treatment with PI3-K inhibitors arrested phagocytosis at an intermediate stage, after actin assembly and partial extension of pseudopodia (Fig. 8, C and D; Araki et al., 1996; Cox et al., 1999; Vieira et al., 2001). The profound inhibition of phagocytosis exerted by LY294002 is illustrated in Fig. 8 G. Unlike wortmannin, which covalently inactivates PI3-K, LY294002 is reversible and most of the phagocytic activity was restored shortly after the inhibitor was removed (Fig. 8 G, second column). The actin accumulated at the aborted phagocytic cup disappeared almost entirely from the periphery of the formed phagosomes when the inhibitor was removed (not depicted). This enabled us to abort phagocytosis after actin cups formed, and subsequently incubate the cells for 10 min with U73122 to allow its entry to the cells. LY294002 was then removed while maintaining U73122 in the incubation medium, and the effects of inhibition of PLC on phagocytosis and on the disassembly of the actin cup were monitored. Fig. 8 G shows that U73122 was as potent a blocker of phagocytosis when added after cup formation, as it was when added before phagocytosis (compare third column with fourth column). More importantly, the PLC antagonist prevented the disassembly of F-actin from the cups, which remained fully formed yet unable to seal (Fig. 8, E and F). These observations support the concept that PI(4,5)P2 hydrolysis is required for the disassembly of actin that is associated with phagosomal closure.
Figure 8.
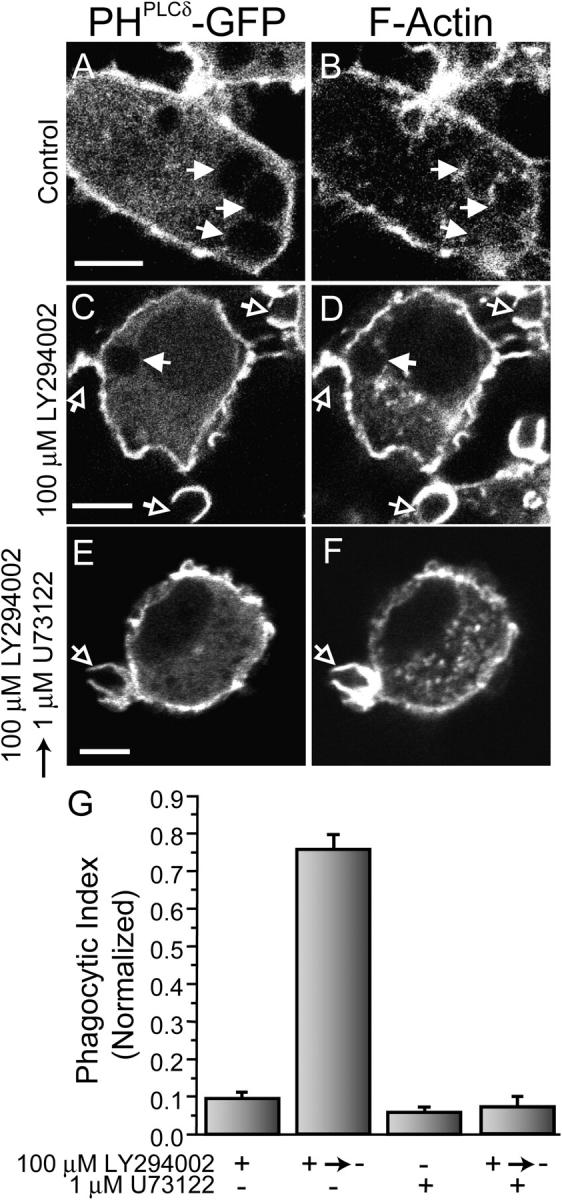
Effect of PLC inhibition on the completion of phagocytosis. RAW cells transiently transfected with PHPLCδ-GFP were either left untreated (A and B), treated with 100 μM LY294002 for 30 min (C and D), treated with 1 μM U73122 for 10 min (not depicted), or treated with 100 μM LY294002 for 30 min, with 1 μM U73122 present for the last 10 min of the incubation, followed by removal of LY294002 while maintaining the U73122 for an additional 10 min (E and F). The cells were then exposed to IgG-opsonized particles for 10 min and finally fixed, permeabilized, and stained with rhodamine-phalloidin (B, D, and F). PHPLCδ-GFP fluorescence is shown in A, C, and E. Bars, 5 μm. (G) The phagocytic index of cells treated as above was determined as described in Materials and methods. Data are the means ± SEM of three experiments, each scoring at least 50 cells.
Overexpression of PIPKI inhibits phagocytosis
We interpret the effects of PLC inhibitors on phagocytosis to mean that removal of PI(4,5)P2 from the cup is necessary for phagosome completion. If this interpretation is correct, a similar impairment of phagocytosis should occur if disappearance of the inositide is prevented not by inhibition of its hydrolysis, but by increasing its synthesis. To test this prediction, we transfected macrophages with PIPKI, the enzyme believed responsible for conversion of phosphatidylinositol-4-phosphate to PI(4,5)P2 at the plasma membrane. Three main isoforms of PIPKI have been described and termed α, β, and γ. We engineered GFP chimeras of the three murine isoforms and overexpressed them transiently in RAW cells before assessment of phagocytic efficiency.
The PIPKI isoforms distributed largely to the plasmalemma of RAW cells, in agreement with earlier observations in various cell types (Stephens et al., 1991; Doughman et al., 2003). Opsonized particles bound normally to the overexpressing cells and phagocytic cups formed in these cells. Actin could be seen to accumulate at the cups, as in normal cells (Fig. 9, D and F). Remarkably, closure of the phagosomes was aborted and engulfment was rarely completed. In multiple experiments, where 150 cells were counted, the phagocytic efficiency was inhibited by 95, 72, and 79% by PIPKI α, β, and γ, respectively. Direct assessment of PI(4,5)P2 under these conditions using CFP-tagged PH domain constructs in cells transfected with YFP-PIPKI confirmed that the phosphoinositide remains present and in fact is enriched at the base of the cup (Fig. 9, A and B). Although other explanations can be envisaged, these findings can be most simply explained by the persistence of PI(4,5)P2 at the base of the phagosome due to excessive synthesis.
Figure 9.

Effect of PIPKI overexpression on phagocytosis. RAW cells transfected with the plasmids indicated below were exposed to IgG-opsonized particles for 8–10 min, then analyzed by laser confocal microscopy either immediately (A and B) or after fixation and staining with Alexa 633-phalloidin (D) or rhodamine-phalloidin (E and F). (A and B) RAW cells transiently transfected with PIPKIβ-YFP and PHPLCδ-CFP. (C and D) Cells transiently transfected with PIPKIβ-YFP. (E and F) Cells transiently transfected with PIPKIγ-GFP. (G) Quantification of the phagocytic efficiency of untransfected cells (control) or cells transiently transfected with PIPKIα-CFP, PIPKIβ-YFP, or PIPKIγ-GFP. Data are the means ± SEM of three experiments, each scoring at least 100 cells.
Discussion
Actin disassembly from nascent phagosomes appears to be required for their proper sealing. In support of this concept, O'Reilly et al. (2003) reported that stabilization of polymerized actin with jasplakinolide inhibits phagocytosis. Although actin polymerization is recognized to be essential for phagosome formation, there is also a suggestion that its removal from newly formed phagosomes is required for their maturation. The capacity of mycobacteria to survive within the host's macrophages has been attributed by some to their ability to arrest phagosomal maturation and this has been proposed to result from retention of a coronin (TACO) and actin meshwork around the early phagosomes, which precludes fusion with late endosomes and lysosomes (Ferrari et al., 1999). However, this view has been disputed by Schüller and colleagues (Schüller et al., 2003). Furthermore, there is growing evidence to suggest that at later times, successive waves of actin polymerization and depolymerization promote maturation by facilitating fusion with the appropriate compartments (Anes et al., 2003; Kjeken et al., 2004; Yam and Theriot, 2004).
Dynamic analysis of actin distribution during the course of particle ingestion revealed that actin disassembly at the base of the cup occurs at a surprisingly early stage, preceding completion of phagosomal sealing, a feature that is more noticeable when larger particles are used. Remarkably, actin depolymerization was clearly apparent within 2–3 min of initiation of phagocytosis, contrasting with earlier measurements of Rho-family GTPase activation, which was reported to persist longer (Niedergang et al., 2003). Rac1 activity was nearly identical between 1–5 min and declined noticeably only after 10 min. Cdc42 was activated even later and remained stimulated for up to 20 min (Niedergang et al., 2003). These estimates were made using biochemical determinations in extracts obtained from populations of macrophages. Even though the onset of phagocytosis was synchronized in these studies, heterogeneity among cells of the population may have artifactually prolonged the measured activation of the GTPases. To circumvent the problem of cellular heterogeneity and asynchrony in a population of cells, we resorted instead to measurements in single cells using the Rac1/Cdc42-binding domain of PAK. Although in these experiments the overall duration of the GTPase activation was shorter than that recorded by biochemical means, we nevertheless confirmed the observation of Niedergang and colleagues that Rac1/Cdc42 activation persists after actin depolymerization is well underway. Furthermore, our observations are consistent with earlier single-cell measurements made by Hoppe and colleagues (Hoppe and Swanson, 2004) who monitored actin and PBD localization during phagocytosis and found that the association of Rac with phagosomes persisted after actin was lost. Moreover, actin disassembly was found to occur also using the engineered system where phagocytosis was induced by recruitment of constitutively active Rac1. The observation that phagocytosis is completed and actin shed from nascent phagosomes despite the continued presence of active Rac1 on the cytoplasmic face of the vacuole is the most compelling evidence that factors other than inactivation of the Rho-family GTPases contribute to the disassembly of F-actin.
Unlike the delayed inactivation of Rac1 and Cdc42, the disappearance of PI(4,5)P2 from the phagocytic cup parallels actin dissociation very closely. The precise contribution of individual metabolic pathways to the disappearance of PI(4,5)P2 from forming phagosomes is not known, but several candidates exist. PLC is one of the principal effectors, because DAG is formed at sites of phagocytosis with a kinetics and spatial distribution that closely parallel the disappearance of PI(4,5)P2 (Botelho et al., 2000). In addition, conversion to PI(3,4,5)P3 by class I PI3K also contributes to the disappearance of PI(4,5)P2. Accumulation of 3′-polyphosphoinositides, a process dependent on wortmannin-sensitive isoforms of PI3K, is one of the earliest events recorded at the phagocytic cup (Marshall et al., 2001). Inhibition of PI3K and of PLC prevent the disappearance of PI(4,5)P2 from the phagocytic cup (Fig. 8). It is also conceivable that termination of synthesis, perhaps due to PIPKI inactivation, contributes to the depletion of PI(4,5)P2. Lastly, the activation of other pathways, e.g., phosphoinositide phosphatases, cannot be ruled out. Of particular interest is the mammalian phosphatase synaptojanin 2, which was shown recently to associate with Rac1 (Malecz et al., 2000) and may contribute to PI(4,5)P2 degradation during phagocytosis.
Several lines of evidence suggest that conversion of PI(4,5)P2 to other chemical species contributes to the termination of actin assembly during the late stages of phagocytosis: (a) PI(4,5)P2 disappears from the forming phagosome with a spatial pattern and temporal course that closely resemble those of actin depolymerization; (b) impairment of PI(4,5)P2 hydrolysis by PLC blocks actin detachment from the forming phagosomes and (c) a similar phenotype is observed when class I PI3K is blocked; and (d) promoting the accelerated synthesis of PI(4,5)P2 by overexpression of PIPKI also inhibited phagocytosis. Accordingly, removal of PI(4,5)P2 from the membrane has been shown to be associated with weakening of the underlying actin skeleton and loss of membrane rigidity in other systems (Niebuhr et al., 2002). This effect can be envisaged to occur at multiple sites, because PI(4,5)P2 is known to play several distinct roles in actin assembly and remodeling. The inositide stimulates de novo actin nucleation (Prehoda et al., 2000; Rohatgi et al., 2000), is capable of uncapping barbed ends of existing filaments (Schafer et al., 1996) and of severing filaments, thereby unmasking additional barbed ends filaments (Janmey and Stossel, 1987). Filament cross-linking is also aided by PI(4,5)P2 (Fukami et al., 1994). Thus, elimination of PI(4,5)P2 from the forming phagosome by PLC and/or PI3K is expected to lead to actin disassembly, even if the Rho GTPases are active. In the engineered Rac1 phagocytes, it is likely that PI(4,5)P2 is hydrolyzed by PLCβ, which is reportedly activated by Rac (Snyder et al., 2003) or by synaptojanin 2 (Malecz et al., 2000).
The proposed role of PI(4,5)P2 in completion of phagocytosis sheds some light on the phenotype of macrophages derived from Syk-deficient animals. These cells were unable to engulf IgG-opsonized particles, yet managed to assemble an actin cup under the targets (Kiefer et al., 1998). Syk is one of the two main kinases proposed to activate PLCγ downstream of immunoreceptors and its deletion likely caused impaired PI(4,5)P2 degradation. The other kinase suggested to stimulate PLCγ is Btk, which in turn requires PI(3,4,5)P3 for its activation. This may explain the profound inhibitory effect of LY294002 on the disappearance of PI(4,5)P2 from the cup (Fig. 8 C). The PI3K antagonist may be exerting dual effects, precluding phosphorylation by PI3K and, as a result, also activation of PLCγ by Btk.
In summary, on the basis of our findings, we propose that phosphoinositide metabolism, and in particular disappearance of PI(4,5)P2 have a critical role in the termination of actin assembly and in its ensuing disassembly from the phagocytic cup. This event liberates elements of the cytoskeletal machinery for assembly elsewhere, including the leading edges of pseudopodia, facilitates curving of the membrane for particle enclosure, and creates access for incoming endomembrane organelles targeted to fuse with the forming phagosome. It is also tempting to speculate that PI(4,5)P2 plays an equivalent role in other processes, such as chemotaxis and macropinocytosis, where rapid actin remodeling is required.
Materials and methods
Materials
Rabbit polyclonal antibodies to c-Myc (A-14) were purchased from Santa Cruz Biotechnology, Inc. Rabbit polyclonal antibodies to sheep RBCs and sheep RBCs were purchased from ICN Biomedicals. FITC, Cy3, and Cy5-conjugated donkey anti–rabbit IgG, Cy5-conjugated donkey anti–mouse IgG, and FITC-conjugated goat anti–human IgG were purchased from Jackson ImmunoResearch Laboratories. Both U73122 and ET-18-OCH3 were purchased from BIOMOL Research Laboratories, Inc. Phalloidin conjugates were acquired from Molecular Probes. Unconjugated latex beads were supplied by Bangs Laboratories Inc. and streptavidin-labeled latex beads were purchased from Sigma-Aldrich. Tissue culture supplies were obtained from CellGro and Wisent. All other reagents were obtained from Sigma-Aldrich, unless otherwise stated.
Cell culture, plasmids, and transfection
RAW 264.7 cells were obtained from the American Tissue Culture Collection. RBL-2H3-15BE22 cells were previously described (Castellano et al., 2000). All cell lines were maintained in DME supplemented with 10% FBS at 37°C under 5% CO2. Cells were seeded on 25-mm glass coverslips the day before transfection.
Human cytoplasmic brain β-actin fused to eGFP (GFP-actin) was expressed from the EF1-a promoter using the vector pEF6-GFP-actin. This vector was constructed by isolating the GFP-actin fusion from pEGFP-actin (CLONTECH Laboratories, Inc.) as a NheI–XbaI fragment and cloning it into the SpeI site of pEF6/Myc-His A (Invitrogen). RAW 264.7 cells were transfected by electroporation using a modification of Cassady et al. (1991). Constitutive expression of the urokinase plasminogen activator gene in murine RAW 264.7 macrophages involves distal and 5′ noncoding sequences that are conserved between mouse and pig (Cassady et al., 1991). Cells were harvested and washed in PBS and resuspended to 5 × 107 cells/ml in PBS. Aliquots of 200 μl of cells were added to 0.4-cm cuvettes containing DNA in 50 μl PBS and the cells pulsed once at 960 μF and 280 V. Cells were washed and plated in complete medium and allowed to recover overnight. Blasticidin was added to 3 μg/ml the next day to select stable cell lines expressing GFP-actin. Highly expressing clones were picked directly from the culture dish under epifluorescence microscopy using a manual pipette.
The plasmid encoding the fusion of the PBD-YFP was a gift of G. Bokoch (The Scripps Research Institute, La Jolla, CA). Plasmids encoding the PH domain of PLCδ fused to the cyan and GFPs (PHPLCδ-CFP and PHPLCδ-GFP respectively) were provided by T. Meyer (Stanford University, Stanford, CA). The myc- and GFP-tagged Cdc42Q61L mutants were provided by A. Kapus (Toronto Hospital, Ontario, Canada) and K. Hahn (University of North Carolina, Chapel Hill, NC), respectively.
The mouse cDNAs encoding PIPKα and β were cloned into pECFP and pEYFP (CLONTECH Laboratories, Inc.) using BamHI and NotI cloning sites and the cDNA for PIPKγ was cloned similarly into pEGFP.
Plasmids for transfection were purified using the QIA-filter Maxi Prep Kit (QIAGEN). For transfection RAW cells were grown to 50–60% confluence and treated with the plasmids plus FuGene 6 (Roche Diagnostics) according to the manufacturer's instructions. The RBL-2H3–derived stable cells were grown to 50–60% confluence and transfected with the indicated plasmids using Superfect (QIAGEN). The cells were maintained in the presence of Superfect plus DNA in serum-free DME for at least 16 h before washing. All cells were used for experiments 16–32 h after initiation of the transfection protocol.
Phagocytosis of RBCs and latex beads
A volume of 500 μl of a 10% suspension of sheep RBCs was opsonized with 10 μl of rabbit anti–sheep RBC antibodies for 1 h while rotating at RT. The cells were gently washed three times with PBS and resuspended in 1 ml of PBS. Approximately 40 μl of this suspension was added to each coverslip to initiate phagocytosis. When required, external RBCs were lysed by hypotonic shock, accomplished by brief (20 s) incubation in deionized water.
To opsonize 3.1-μm latex beads, a 6.67% suspension of beads was incubated with 1.67 mg/ml human IgG for 1 h at RT. The beads were then washed three times with PBS, and resuspended in 1 ml of PBS. Approximately 40 μl of this suspension was added to each coverslip to initiate phagocytosis. When required, extracellular beads were labeled by placing the cells on ice and incubating with fluorophore-coupled anti–human IgG (1:500) antibodies for 10 min at 4°C.
For phagocytosis by RBL-2H3 stably transfected cells, streptavidin-coupled latex beads were incubated with 10 μg/ml murine monoclonal anti-CD25 antibodies conjugated to biotin (Immunotech) for 15 min at 37°C. To recruit the myc-Rac1-V12 fusion protein to the plasma membrane, the cells were treated with 100 μM rapamycin 2 h before phagocytosis and rapamycin was maintained throughout the experiment. The beads were washed twice with PBS, centrifuged onto the cells, and incubated on ice for 20 min. The cells were washed once with cold PBS, and placed in prewarmed DME for the appropriate times. To label extracellular beads, the sample was cooled on ice and incubated with Cy5-conjugated anti–mouse IgG antibodies (1:500) for 10 min at 4°C.
Confocal imaging
For live imaging, cells were seeded on 25-mm glass coverslips one day before use. They were washed once with PBS, and then placed in a thermostatted Leiden chamber holder on the stage of a LSM 510 laser confocal microscope (Carl Zeiss MicroImaging, Inc.) in bicarbonate-free medium RPMI 1640 supplemented with 20 mM Hepes and maintained at 37°C. Opsonized latex beads or red cells were added to the chamber and the cells were visualized with a 100× oil immersion objective. To quantify the recruitment of soluble probes to the phagosome, we defined a region of interest encompassing the bead and integrated the fluorescence above the cytosolic level. This was accomplished by setting a lower threshold equivalent to the mean cytosolic fluorescence plus two SDs, thus ensuring that only fluorescence levels significantly higher than the cytosolic intensity were taken into account. To avoid ambiguity due to the contribution of plasmalemmal-associated probes, only the innermost half of the phagosome, which is clearly separated from the nonphagosomal plasma membrane, was selected for quantification. These manipulations were all performed using the MetaMorph V5.0 software (Universal Imaging Corp.). To allow comparison between experiments, the phagosomal fluorescence was normalized to the maximum recorded within each experiment. For graphic representation and statistical evaluation the data were binned into 20-s intervals.
High-throughput quantification of actin dynamics
Phagocytosis was synchronized by centrifugation (1 min, 300 g) of opsonized latex beads onto cells plated directly on 24-well plates. Cells were fixed with 4% PFA in PBS for 20 min at RT and washed with 100 mM glycine in PBS for 10 min. The fixed cells were then permeabilized and blocked with 0.1% Triton X-100 containing 5% powdered milk overnight at 4°C. The cells were stained with rhodamine-phalloidin as per the manufacturer's instructions, washed and maintained in PBS. The plates were analyzed with a Cellomics KineticScan HCS Reader (Cellomics) and the integrated fluorescence intensity of at least 2,500 valid objects per condition was determined.
Immunostaining and fluorescence microscopy
Cells were fixed with 4% PFA in PBS for 30 min at RT and washed with 100 mM glycine in PBS for 10 min. The fixed cells were then permeabilized and blocked with 0.1% Triton X-100 containing 5% powdered milk for 1 h at RT or overnight at 4°C. The primary antibodies, followed by the secondary antibodies, were added to the coverslips in PBS with for 45 min each at 37°C. Rabbit anti–c-myc as used at a 1:100 dilution. The coverslips were mounted onto glass slides using Fluorescent Mounting Medium (DakoCytomation). Fluorescence images were captured using the LSM 510 confocal microscope under a 100× oil immersion objective. Digital images were prepared using Adobe Photoshop 6.0 and Adobe Illustrator 10 software. To quantify cortical actin thickness, a line scan across individual cells was performed using ImageJ analysis software (NIH) and the number and intensity of consecutive pixels above background fluorescence was determined.
Online supplemental material
Fig. S1 shows a thin layer chromatographic profile of the phosphoinositides extracted from whole macrophages during the course of phagocytosis of 3.1-μm latex beads. Also presented is the summary of kinetic analyses of phosphatidylinositol bisphosphate levels from four such experiments. Methodological details are provided in the supplementary figure legend. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200412162/DC1.
Acknowledgments
The authors would like to thank Ms. Analyn Yu and Mr. Benjamin Steinberg for their help in performing and analyzing some of the experiments.
This work was supported by the Arthritis Society of Canada and the CIHR. C.C. Scott and R.J. Botelho are recipients of graduate studentships from the CIHR. D.A. Knecht is supported by National Institutes of Health grant GM30599. S. Grinstein is the current holder of the Pitblado Chair in Cell Biology.
Abbreviations used in this paper: FcγR, Fcγ family of receptors; PIPKI, phosphatidylinositol phosphate kinase type I; PI(4,5)P2, phosphatidylinositol-4,5-bisphosphate; PBD-YFP, p21-binding domain of PAK fused to YFP; PI3-K, phosphatidylinositol 3′-kinase.
References
- Aderem, A. 2002. How to eat something bigger than your head. Cell. 110:5–8. [DOI] [PubMed] [Google Scholar]
- Aderem, A., and D.M. Underhill. 1999. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17:593–623. [DOI] [PubMed] [Google Scholar]
- Allison, A.C., P. Davies, and S. De Petris. 1971. Role of contractile microfilaments in macrophage movement and endocytosis. Nat. New Biol. 232:153–155. [DOI] [PubMed] [Google Scholar]
- Anes, E., M.P. Kuhnel, E. Bos, J. Moniz-Pereira, A. Habermann, and G. Griffiths. 2003. Selected lipids activate phagosome actin assembly and maturation resulting in killing of pathogenic mycobacteria. Nat. Cell Biol. 5:793–802. [DOI] [PubMed] [Google Scholar]
- Araki, N., M.T. Johnson, and J.A. Swanson. 1996. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 135:1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzoni, L., M. Kamoun, T.W. Salcedo, P. Kanakaraj, and B. Perussia. 1992. Stimulation of FcγRIIIA results in phospholipase C-γ 1 tyrosine phosphorylation and p56lck activation. J. Exp. Med. 176:1745–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho, R.J., M. Teruel, R. Dierckman, R. Anderson, A. Wells, J.D. York, T. Meyer, and S. Grinstein. 2000. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J. Cell Biol. 151:1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron, E., and A. Hall. 1998. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 282:1717–1721. [DOI] [PubMed] [Google Scholar]
- Cassady, A.I., K.J. Stacey, K.A. Nimmo, K.M. Murphy, D. von der Ahe, D. Pearson, F.M. Botteri, Y. Nagamine, and D.A. Hume. 1991. Constitutive expression of the urokinase plasminogen activator gene in murine RAW264 macrophages involves distal and 5′ non-coding sequences that are conserved between mouse and pig. Nucleic Acids Res. 19:6839–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano, F., P. Montcourrier, and P. Chavrier. 2000. Membrane recruitment of Rac1 triggers phagocytosis. J. Cell Sci. 113:2955–2961. [DOI] [PubMed] [Google Scholar]
- Cox, D., P. Chang, Q. Zhang, P.G. Reddy, G.M. Bokoch, and S. Greenberg. 1997. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 186:1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, D., C.C. Tseng, G. Bjekic, and S. Greenberg. 1999. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J. Biol. Chem. 274:1240–1247. [DOI] [PubMed] [Google Scholar]
- Defacque, H., M. Egeberg, A. Habermann, M. Diakonova, C. Roy, P. Mangeat, W. Voelter, G. Marriott, J. Pfannstiel, H. Faulstich, and G. Griffiths. 2000. Involvement of ezrin/moesin in de novo actin assembly on phagosomal membranes. EMBO J. 19:199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio, F., B.C. Meyer, S. Greenberg, and S.C. Silverstein. 1988. Fc receptor-mediated phagocytosis occurs in macrophages at exceedingly low cytosolic Ca2+ levels. J. Cell Biol. 106:657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughman, R.L., A.J. Firestone, and R.A. Anderson. 2003. Phosphatidylinositol phosphate kinases put PI4,5P2 in its place. J. Membr. Biol. 194:77–89. [DOI] [PubMed] [Google Scholar]
- Ferrari, G., H. Langen, M. Naito, and J. Pieters. 1999. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell. 97:435–447. [DOI] [PubMed] [Google Scholar]
- Fukami, K., T. Endo, M. Imamura, and T. Takenawa. 1994. alpha-Actinin and vinculin are PIP2-binding proteins involved in signaling by tyrosine kinase. J. Biol. Chem. 269:1518–1522. [PubMed] [Google Scholar]
- Greenberg, S., P. Chang, and S.C. Silverstein. 1993. Tyrosine phosphorylation is required for Fc receptor-mediated phagocytosis in mouse macrophages. J. Exp. Med. 177:529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg, S., and S. Grinstein. 2002. Phagocytosis and innate immunity. Curr. Opin. Immunol. 14:136–145. [DOI] [PubMed] [Google Scholar]
- Greenberg, S., J. el Khoury, F. di Virgilio, E.M. Kaplan, and S.C. Silverstein. 1991. Ca2+-independent F-actin assembly and disassembly during Fc receptor-mediated phagocytosis in mouse macrophages. J. Cell Biol. 113:757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, R.M., A.D. Hoppe, N. Joshi, and J.A. Swanson. 2004. The uniformity of phagosome maturation in macrophages. J. Cell Biol. 164:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilpela, P., M.K. Vartiainen, and P. Lappalainen. 2004. Regulation of the actin cytoskeleton by PI(4,5)P2 and PI(3,4,5)P3. Curr. Top. Microbiol. Immunol. 282:117–163. [DOI] [PubMed] [Google Scholar]
- Hishikawa, T., J.Y. Cheung, R.V. Yelamarty, and D.W. Knutson. 1991. Calcium transients during Fc receptor-mediated and nonspecific phagocytosis by murine peritoneal macrophages. J. Cell Biol. 115:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe, A.D., and J.A. Swanson. 2004. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol. Biol. Cell. 15:3509–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janmey, P.A., and T.P. Stossel. 1987. Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature. 325:362–364. [DOI] [PubMed] [Google Scholar]
- Kiefer, F., J. Brumell, N. Al-Alawi, S. Latour, A. Cheng, A. Veillette, S. Grinstein, and T. Pawson. 1998. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol. Cell. Biol. 18:4209–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeken, R., M. Egeberg, A. Habermann, M. Kuehnel, P. Peyron, M. Floetenmeyer, P. Walther, A. Jahraus, H. Defacque, S.A. Kuznetsov, and G. Griffiths. 2004. Fusion between phagosomes, early and late endosomes: a role for actin in fusion between late, but not early endocytic organelles. Mol. Biol. Cell. 15:345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, F., H.S. Shin, and S.G. Rhee. 1992. Tyrosine phosphorylation of phospholipase C-gamma 1 induced by cross-linking of the high-affinity or low-affinity Fc receptor for IgG in U937 cells. Proc. Natl. Acad. Sci. USA. 89:3659–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecz, N., P.C. Mc, C. Cabe, R. Spaargaren, Y. Qiu, Y. Chuang, and M. Symons. 2000. Synaptojanin 2, a novel Rac1 effector that regulates clathrin-mediated endocytosis. Curr. Biol. 10:1383–1386. [DOI] [PubMed] [Google Scholar]
- Marshall, J.G., J.W. Booth, V. Stambolic, T. Mak, T. Balla, A.D. Schreiber, T. Meyer, and S. Grinstein. 2001. Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fcγ receptor-mediated phagocytosis. J. Cell Biol. 153:1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massol, P., P. Montcourrier, J.C. Guillemot, and P. Chavrier. 1998. Fc receptor-mediated phagocytosis requires CDC42 and Rac1. EMBO J. 17:6219–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niebuhr, K., S. Giuriato, T. Pedron, D.J. Philpott, F. Gaits, J. Sable, M.P. Sheetz, C. Parsot, P.J. Sansonetti, and B. Payrastre. 2002. Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. EMBO J. 21:5069–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedergang, F., E. Colucci-Guyon, T. Dubois, G. Raposo, and P. Chavrier. 2003. ADP ribosylation factor 6 is activated and controls membrane delivery during phagocytosis in macrophages. J. Cell Biol. 161:1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly, P.J., J.M. Hickman-Davis, I.C. Davis, and S. Matalon. 2003. Hyperoxia impairs antibacterial function of macrophages through effects on actin. Am. J. Respir. Cell Mol. Biol. 28:443–450. [DOI] [PubMed] [Google Scholar]
- Prehoda, K.E., J.A. Scott, R.D. Mullins, and W.A. Lim. 2000. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science. 290:801–806. [DOI] [PubMed] [Google Scholar]
- Rohatgi, R., H.Y. Ho, and M.W. Kirschner. 2000. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4, 5-bisphosphate. J. Cell Biol. 150:1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, D.A., P.B. Jennings, and J.A. Cooper. 1996. Dynamics of capping protein and actin assembly in vitro: uncapping barbed ends by polyphosphoinositides. J. Cell Biol. 135:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüller, S., J. Neefjes, T. Ottenhoff, J. Thole, and D. Young. 2003. Coronin is involved in uptake of Mycobacterium bovis BCG in human macrophages but not in phagosome maintenance. Cell. Microbiol. 3:785–793. [DOI] [PubMed] [Google Scholar]
- Snyder, J.T., A.U. Singer, M.R. Wing, T.K. Harden, and J. Sondek. 2003. The pleckstrin homology domain of phospholipase C-beta2 as an effector site for Rac. J. Biol. Chem. 278:21099–21104. [DOI] [PubMed] [Google Scholar]
- Srinivasan, S., F. Wang, S. Glavas, A. Ott, F. Hofmann, K. Aktories, D. Kalman, and H.R. Bourne. 2003. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 160:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer, T.P., S. Ahn, and T. Meyer. 1998. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 8:343–346. [DOI] [PubMed] [Google Scholar]
- Stephens, L.R., K.T. Hughes, and R.F. Irvine. 1991. Pathway of phosphatidylinositol(3,4,5)-trisphosphate synthesis in activated neutrophils. Nature. 351:33–39. [DOI] [PubMed] [Google Scholar]
- Tolias, K., and C.L. Carpenter. 2000. In vitro interaction of phosphoinositide-4-phosphate 5-kinases with Rac. Methods Enzymol. 325:190–200. [DOI] [PubMed] [Google Scholar]
- Underhill, D.M., and A. Ozinsky. 2002. Phagocytosis of microbes: complexity in action. Annu. Rev. Immunol. 20:825–852. [DOI] [PubMed] [Google Scholar]
- Varnai, P., and T. Balla. 1998. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol–8labeled phosphoinositide pools. J. Cell Biol. 143:501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira, O.V., R.J. Botelho, L. Rameh, S.M. Brachmann, T. Matsuo, H.W. Davidson, A. Schreiber, J.M. Backer, L.C. Cantley, and S. Grinstein. 2001. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J. Cell Biol. 155:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos, C.J., W.F. Matter, K.Y. Hui, and R.F. Brown. 1994. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269:5241–5248. [PubMed] [Google Scholar]
- Weernink, P.A., K. Meletiadis, S. Hommeltenberg, M. Hinz, H. Ishihara, M. Schmidt, and K.H. Jakobs. 2004. Activation of type I phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42. J. Biol. Chem. 279:7840–7849. [DOI] [PubMed] [Google Scholar]
- Yam, P.T., and J.A. Theriot. 2004. Repeated cycles of rapid actin assembly and disassembly on epithelial cell phagosomes. Mol. Biol. Cell. 15:5647–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]