

Abstract
Atherosclerotic plaque forms in regions of the vasculature exposed to disturbed flow. NF-κB activation by fluid flow, leading to expression of target genes such as E-selectin, ICAM-1, and VCAM-1, may regulate early monocyte recruitment and fatty streak formation. Flow-induced NF-κB activation is downstream of conformational activation of integrins, resulting in new integrin binding to the subendothelial extracellular matrix and signaling. Therefore, we examined the involvement of the extracellular matrix in this process. Whereas endothelial cells plated on fibronectin or fibrinogen activate NF-κB in response to flow, cells on collagen or laminin do not. In vivo, fibronectin and fibrinogen are deposited at atherosclerosis-prone sites before other signs of atherosclerosis. Ligation of integrin α2β1 on collagen prevents flow-induced NF-κB activation through a p38-dependent pathway that is activated locally at adhesion sites. Furthermore, altering the extracellular matrix to promote p38 activation in cells on fibronectin suppresses NF-κB activation, suggesting a novel therapeutic strategy for treating atherosclerosis.
Introduction
Atherosclerotic plaque develops in response to a localized inflammatory reaction in the vessel wall (Ross, 1999). Although known risk factors for atherosclerosis, such as hyperlipidemia, hypertension, diabetes, and smoking, play major roles in the incidence and progression of the disease, these factors are uniform throughout the circulation. The observation that atherosclerotic plaque forms preferentially at sites of disturbed blood flow (VanderLaan et al., 2004) suggests that flow patterns can regulate the chronic inflammation associated with atherogenesis (Caro et al., 1969; Ku et al., 1985; Glagov et al., 1988). In support of this idea, application of prolonged laminar flow to endothelial cells in culture is antiinflammatory and atheroprotective, whereas disturbed flow stimulates endothelial cell turnover, expression of prothrombotic and proinflammatory proteins, and altered redox regulation (Topper et al., 1996; Mohan et al., 1997; De Keulenaer et al., 1998; Brooks et al., 2002). These data suggest that disturbed flow patterns initiate the local inflammatory reaction seen during atherosclerosis, with amplification of lesions by humoral risk factors driving progression of the disease.
The NF-κB family of transcription factors are involved in numerous cellular processes, including differentiation, inflammation, proliferation, and apoptosis, and are postulated to contribute to atherogenesis (Collins and Cybulsky, 2001; Kutuk and Basaga, 2003). Current data implicate NF-κB as a key regulator of shear stress–induced inflammatory gene expression. NF-κB dimers, particularly the p50/p65 heterodimer, bind to a shear stress responsive element found in the promoter of several atherogenic genes, including ICAM-1, VCAM-1, and MCP-1 (monocyte chemotactic protein-1), which regulate monocyte recruitment, as well as PDGF, which stimulates smooth muscle growth and migration (Resnick et al., 1993; Khachigian et al., 1995; Huo and Ley, 2001). NF-κB expression and activity, as well as ICAM-1 and VCAM-1 expression, are elevated in atherosclerosis-prone regions before or in the absence of fatty streak formation, indicating that they are very early events in atherosclerotic progression (Brand et al., 1996; Nakashima et al., 1998; Iiyama et al., 1999; Wilson et al., 2000). In vitro, disturbed shear stimulates prolonged NF-κB activation and NF-κB–dependent gene expression; in contrast, acute onset of laminar shear stress activates NF-κB but only transiently (Lan et al., 1994; Khachigian et al., 1995; Mohan et al., 1997). We hypothesize that these differences in signaling pathways induced by laminar versus disturbed flow are due in part to differences in adaptation mechanisms, such that changes in flow velocity and direction associated with disturbed flow prevents down-regulation of the responses so that signals activated transiently by laminar flow are sustained in disturbed flow. Thus, data from both in vivo and in vitro models suggest that NF-κB contributes to the initiation of atherosclerosis by fluid shear stress.
Studies in vitro have shown that acute onset of laminar shear triggers conversion of integrins to a high affinity state, followed by their binding to the subendothelial ECM (Tzima et al., 2001). Resultant integrin signaling mediates activation of NF-κB through the small GTPase Rac (Tzima et al., 2002). Endothelial cells normally reside on a basement membrane comprised mainly of collagen (Coll) IV and laminin (LN). Coll binds primarily integrins α2β1 and α1β1, whereas LN binds mainly α6β1 and α6β4 (Belkin and Stepp, 2000; Heino, 2000). Ligation of these integrins is associated with a quiescent cell phenotype, consistent with the low turnover observed in endothelial cells in vivo (Schwartz and Assoian, 2001). Inflammation or injury can trigger the deposition of transitional ECM proteins such as fibronectin (FN) and fibrinogen (FG) into the subendothelial matrix (Sechler et al., 1998). In endothelial cells, FN primarily ligates α5β1 whereas FG ligates αvβ3, although other integrins may also be involved. Signals from α5β1 and αvβ3 are associated with enhanced endothelial cell proliferation and migration, processes important for injury-induced endothelial remodeling (Schwartz and Assoian, 2001). Many studies have shown that different integrins transduce distinct signals (Schwartz and Assoian, 2001). The involvement of integrins in shear stress–dependent signaling thus suggests that the composition of the subendothelial ECM may modulate cellular responses to fluid flow. Therefore, we investigated the role of the subendothelial ECM in the endothelial response to fluid shear stress.
Results
Subendothelial matrix regulates shear stress–induced NF-κB activation
To test whether or not shear stress–induced NF-κB activation depends on the underlying matrix, bovine aortic endothelial (BAE) cells plated on glass slides coated with Coll I, LN, FN, or FG for 4 h were subjected to laminar shear stress (12 dyne/cm2) in a parallel plate flow chamber. NF-κB is normally held inactive in the cytoplasm through interactions with IκB, but stimulus-induced IκB degradation results in NF-κB nuclear targeting and initiation of transcription (Chen and Greene, 2004; Yamamoto and Gaynor, 2004). As previously reported for endothelial cells under standard conditions (Scatena et al., 1998; Klein et al., 2002; Guo et al., 2004), cells on FN or FG showed enhanced nuclear translocation of NF-κB after flow; in contrast, cells on LN responded poorly and cells on Coll showed a higher baseline under static conditions, which decreased in response to flow (Fig. 1 A).
Figure 1.

Shear stress–induced NF-κB activation is matrix specific. (A) BAE cells plated on Coll I, LN, FN, or FG for 4 h were sheared for 60 min, and p65 localization was assessed by immunocytochemistry. The percentage of cells showing nuclear staining for NF-κB was determined. Values are means ± SD (>100 cells counted per condition per experiment; n = 3). *, P < 0.05; **, P < 0.01. A representative p65 stain for these conditions is shown on the right. (B) Endothelial cells were grown on different matrix proteins and sheared for 0, 5, 15, 30, and 60 min. Phosphorylation of p65 on Ser536 was assessed by immunoblotting total cell lysates with a phosphorylation site-specific antibody. Values are means ± SD following normalization for total p65 (n = 4). A representative blot is shown for each condition. (C) Human umbilical vein endothelial cells were plated on different matrix proteins and sheared for 5 h, and the level of ICAM-1 expression was assessed by Western blotting. Densitometric quantification was normalized to actin. n = 3; *, P < 0.05. Immunocytometric staining for ICAM-1 surface expression is shown on the right.
In addition to its nuclear targeting, numerous posttranslational modifications contribute to NF-κB's transcriptional activity (Wang and Baldwin, 1998; Yang et al., 2003). Phosphorylation of the p65 subunit of NF-κB on Ser536 in the transactivation domain alters NF-κB–dependent transcription, presumably by regulating its interaction with other cis-acting elements. In addition, this site is phosphorylated by the IκB kinase (IKK) complex, and its phosphorylation is inhibited by interaction with IκB, suggesting it is a good measure for pathway activation. Ser536 phosphorylation was stimulated in cells on FN or FG, but showed only small changes on LN; on Coll, NF-κB phosphorylation showed an elevated baseline that decreased after shear stress (Fig. 1 B). Cells on Coll IV behaved identically to Coll I (unpublished data). This result was expected because Coll I and IV bind the same integrins (Defilippi et al., 1991; Tulla et al., 2001). To test if these matrix-specific signals translate to altered gene expression, we assayed expression of ICAM-1, an NF-κB–responsive gene. Both Western blotting and surface staining were performed in human umbilical vein endothelial cells due to poor antibody cross-reactivity with bovine ICAM-1. Both methods showed that, in cells on Coll, ICAM-1 expression diminished after shear stress, whereas expression increased in cells on FN and FG (Fig. 1 C). Thus, NF-κB nuclear translocation, phosphorylation, and target gene expression correlate closely. We conclude that the transitional ECM proteins FN and FG support flow-induced NF-kB activation whereas LN and Coll do not.
ECM remodeling in vivo
The distinct effects of different ECM proteins in flow-induced NF-κB activation in vitro prompted us to ask whether or not these effects were relevant to plaque development in vivo. To test whether FN and FG deposition are associated with focal expression of NF-kB target genes in atherosclerosis-prone regions of the vasculature, these proteins were examined in mice. Serial sections from atherosclerosis-susceptible sites were stained for ECM proteins, for ICAM-1 and VCAM-1 as markers of NF-κB activation, and for the leukocyte integrin Mac-2 as a measure of monocyte recruitment. Atherosclerosis-resistant C57/B6 mice and atherosclerosis-prone apolipoprotein E (ApoE) null mice were fed either a normal chow diet or a high fat, Western diet for 10 wk. Sites associated with atherosclerotic plaque formation, such as the innominate artery and the carotid arteries, were then analyzed by immunohistochemistry (IHC; Nakashima et al., 1994).
In C57/B6 mice fed the low fat, chow diet, sites that are susceptible to atherosclerosis in other models demonstrated focal increases in ICAM-1 and VCAM-1 staining and increased staining for FN, although no Mac-2 staining was apparent (Fig. 2 A). We did not detect any change in FG (Fig. 2 A) or LN (not depicted) staining and no staining for FN in regions outside these zones. ApoE null mice fed a chow diet have elevated cholesterol and develop atherosclerotic lesions over the course of their lifespan (Daugherty, 2002). In these mice, similar colocalization of ICAM-1, VCAM-1, FN, and FG was observed when nearby sections were stained (Fig. 2 B). FN has been reported in well-defined fatty streaks (Stenman et al., 1980; Labat-Robert et al., 1985; Shekhonin et al., 1987; Tanouchi et al., 1992), but these data are the first evidence that FN is deposited before fatty streak development. In contrast, no changes in PECAM staining were evident (unpublished data). VCAM-1 staining in vascular smooth muscle cells is indicative of smooth muscle activation and is associated with atherosclerosis, which is consistent with our interpretation of these regions as early atherogenesis (Li et al., 1993).
Figure 2.
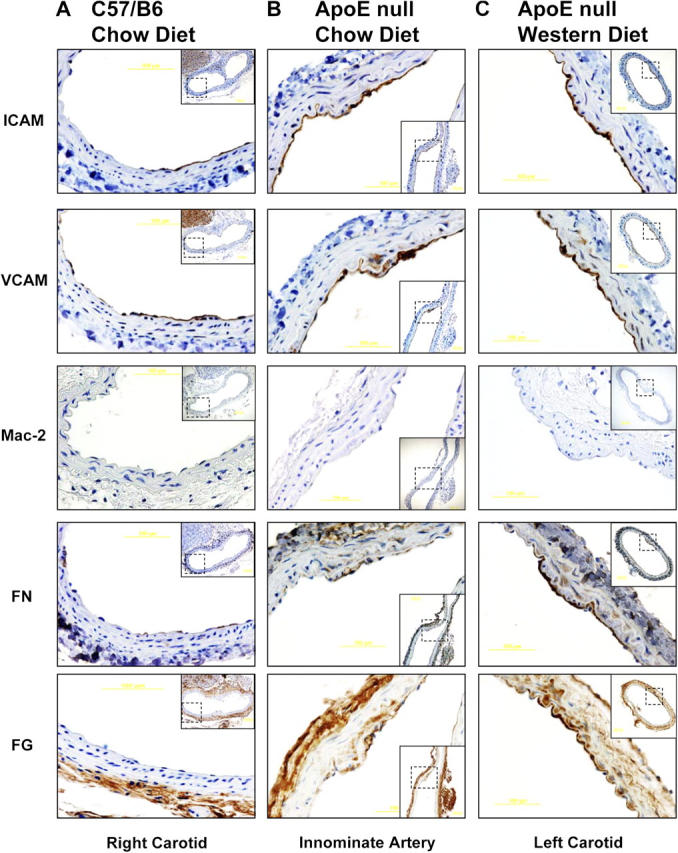
Matrix remodeling and inflammatory gene expression in vivo . C57/B6 mice were fed a chow diet (A), male ApoE null mice were fed a chow diet for 10 wk (B), and ApoE null mice were fed a Western diet for 10 wk (C). Mice were killed and the indicated arteries were removed and embedded in paraffin. Serial sections were stained for FN, FG, ICAM-1, VCAM-1, and Mac-2 and shown at high magnification, with lower magnification views of the entire vessels shown as insets.
ApoE null mice fed a Western diet develop atherosclerosis at greatly accelerated rates (Daugherty, 2002). Under these conditions, FN, FG, VCAM-1, and ICAM-1 staining was strongly enhanced in atherosclerosis-prone sites compared with C57/B6 or ApoE null mice fed a chow diet; however, no Mac-2 staining was apparent (Fig. 2 C). As before, these changes were restricted to atherosclerosis-prone regions near bifurcations. Together, these data show that ECM is remodeled in regions of disturbed flow in vivo before other indicators of atherosclerosis and that NF-κB activation correlates with these changes in the subendothelial ECM. Furthermore, conditions favoring hypercholesterolemia, such as depletion of ApoE or a high fat diet, enhance matrix remodeling specifically at sites of disturbed flow, suggesting a potential synergy between the atherogenic effects of hypercholesterolemia and disturbed flow in vivo.
Coll-specific p38 signaling inhibits shear-induced NF-κB activation
In addition to NF-κB, p38 MAP kinase is also involved in stimulating inflammatory protein expression in a variety of systems. However, p38 shows differential effects on NF-κB, either enhancing or suppressing NF-κB depending on the stimulus (Schwenger et al., 1998; Alpert et al., 1999; Bowie and O'Neill, 2000; Bradbury et al., 2001). To examine p38 in this system, BAE cells were plated on different ECM proteins for 4 h and effects of shear were tested. Western blotting with an antibody against the phosphorylated and activated form of p38 showed that shear stress stimulated a transient increase in p38 phosphorylation in cells on Coll but not on other ECM proteins (Fig. 3 A). This result is consistent with previous reports showing that the Coll-binding integrin α2β1 stimulates p38 activation (Ivaska et al., 1999). To test if this Coll-specific p38 activation mediates inhibition of shear stress–induced NF-κB activation, cells were preincubated with the p38 inhibitor SB202190 (1 μM for 1 h) before fluid flow. Inhibition of p38 with SB202190 restored shear stress–induced NF-κB nuclear translocation and NF-κB phosphorylation in cells on Coll but had no effect on cells on FN (Fig. 3, B and C). To confirm this result, endothelial cells were transiently transfected with a dominant-negative mutant of p38 (p38-AGF). This construct did not alter baseline levels of NF-κB activity but enhanced flow-induced NF-κB nuclear translocation in cells on Coll (Fig. 3 D). Thus, selective activation of p38 mediates suppression of NF-κB in cells on Coll.
Figure 3.

p38 inhibits shear stress–induced NF-κB activation on Coll. (A) BAE cells plated on Coll, LN, FN, or FG were sheared for the indicated times. Phosphorylation of p38 was assessed by Western blotting with phosphorylation site-specific antibodies, quantified by densitometry, and normalized to total p38. Values are means ± SD (n = 3). A representative Western blot is shown for each condition. (B) Cells were treated with the pharmacological p38 inhibitor SB202190 (1 μM for 1 h), and NF-κB nuclear translocation was assessed with or without shear stress for 30 min. Greater than 100 cells were counted per condition per experiment; n = 3; *, P < 0.05; **, P < 0.01. (C) Cells were preincubated with SB202190 and shear stress–induced p65 phosphorylation (Ser536) was assessed as previously described. Densitometric quantification was normalized to total p65. n = 3. (D) Cells were transiently transfected with dominant-negative p38 (p38-AGF) and sheared for 60 min, and p65 nuclear translocation was assessed. Transfected cells were identified by costaining for total p38. Greater than 100 cells were counted per condition per experiment; n = 3; *, P < 0.05. A representative stain is shown.
Although Coll binds both integrins α2β1 and α1β1, expression of α1β1 is restricted to microvascular endothelial cells (Defilippi et al., 1991; Heino, 2000). To test whether integrin α2β1 mediates Coll-specific signaling after shear, endothelial cells were pretreated with the α2β1 integrin–blocking antibody R2-8C8 (20 μg/ml for 30 min) or, as a control, with the nonblocking α2β1 antibody 12F1 (20 μg/ml for 30 min). With this short exposure to blocking antibodies, we can inhibit new integrin binding without significantly affecting existing adhesions or cytoskeletal structure (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200410073/DC1). Blocking α2β1 integrin ligation prevented the shear stress–induced activation of p38 in cells on Coll (Fig. 4 A) and rescued NF-κB phosphorylation (Fig. 4 B). The control antibody had no effect. Preventing new ligation of integrin α6β1 with the blocking antibody GoH3 (20 μg/ml for 30 min) did not enhance flow-induced p65 nuclear translocation or phosphorylation in cells on LN, instead, the already low level of NF-κB was reduced further. These results suggest α6β1 is not suppressive (Fig. S2, A–C, available at http://www.jcb.org/cgi/content/full/jcb.200410073/DC1).
Figure 4.

Integrin α2β1 mediates p38 activation and NF-κB inhibition. (A) BAE cells were incubated for 30 min with either the α2β1 blocking (R2-8C8) or the α2β1 nonblocking antibody (12F1) and sheared for 5 or 30 min. Lysates were analyzed for p38 phosphorylation by immunoblotting. Bands were quantified by densitometry and normalized for total p38 protein. Values are means ± SD (n = 4). *, P < 0.05; **, P < 0.01. (B) Cells on Coll were incubated with 12F1 or R2-8C8 and sheared for 5 or 30 min, and the phosphorylation of p65 was analyzed by immunoblotting. Bands were quantified by densitometry and normalized for total p65. **, P < 0.01 (n = 5). (C) Cells plated on Coll I or FN were pretreated with the p38 inhibitor SB202190 (1 μM for 1 h) and integrin ligation was induced with the β1 integrin–activating antibody TS2/16 (15 μg/ml for 1 h). p65 localization was assessed by immunostaining, and the percentage of cells showing nuclear localization was scored (n = 3; *, P < 0.05). (D) TS2/16-induced p65 phosphorylation of p65 on Ser536 was assessed by Western blotting. Bands were quantified by densitometry and normalized to total p65 (n = 3–6).
To test whether or not activating integrins, as occurs in response to fluid flow, is sufficient to induce these pathways, cells were treated with the β1 integrin–activating antibody TS2/16 (15 μg/ml). Antibody-induced integrin activation resulted in a similar, matrix-specific nuclear translocation (Fig. 4 C) and phosphorylation (Fig. 4 D) of NF-κB on FN but not Coll. Furthermore, TS2/16-induced NF-κB activation on Coll was rescued by the p38 inhibitor SB202190 (Fig. 4, C and D). These data support the conclusion that integrin activation and ECM binding initiate these pathways.
Flow-induced NF-κB activation on mixed matrices
Cells in vivo are usually exposed to multicomponent matrices of varying composition. To test if Coll can show dominant effects in this system, cells were plated on increasing concentrations of FN in either the absence or presence of an underlying Coll matrix. The amount of FN adsorbed to the coverslips was assayed in separate experiments. In the absence of Coll, p65 nuclear translocation and Ser536 phosphorylation displayed a dose-dependent increase as FN concentration increased (Fig. 5, A and B). The dose-dependent effect reached a maximum between 4 and 6 μg of deposited FN, which corresponds to 20 μg/ml in the coating solution. When examined on coverslips first coated with Coll, the amount of FN necessary to elicit NF-κB activation was notably higher, and maximal p65 activation was reduced even at the highest doses of FN. These results support the notion that Coll signaling actively suppresses the FN-dependent activation of NF-κB by flow. However, suppression of signaling may work in the opposite direction because FN deposition inhibited flow-induced p38 phosphorylation in cells on Coll in a dose-dependent manner (Fig. 5 C).
Figure 5.
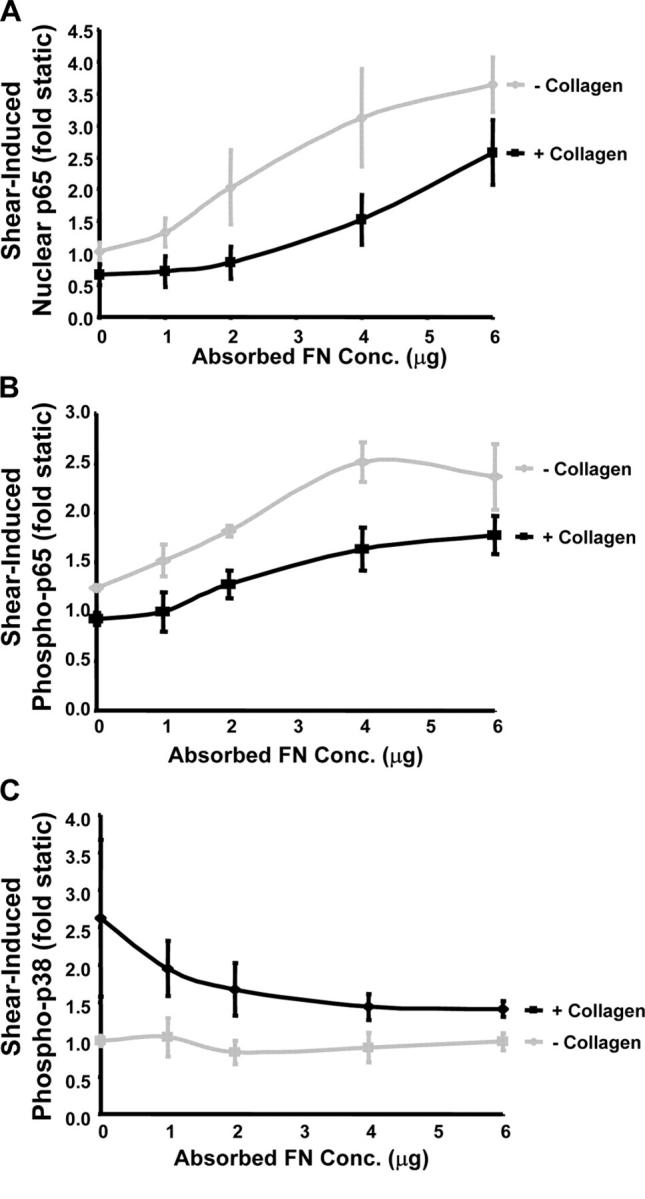
Coll/FN mixed matrices. BAE cells were plated on increasing amounts of FN with or without precoating with 10 μg/ml Coll. (A) Cells were sheared for 30 min and nuclear p65 accumulation was assessed as previously described. The abscissa represents the amount of FN adsorbed to the coverslips under these conditions. Values reflect the increase in p65 nuclear translocation relative to cells under static conditions. n = 4. (B) Cells were treated as in A, and p65 phosphorylation on Ser536 was assessed by Western blotting. Bands were quantified by densitometry and normalized to total p65. Values represent the increase in p65 phosphorylation relative to cells under static conditions. n = 3. (C) Cells were exposed to shear stress for 5 min and p38 phosphorylation was assessed by Western blotting. Bands were quantified by densitometry and normalized to total p38, and data was expressed as the fold increase in p38 phosphorylation compared with cells under static conditions. n = 3.
Signaling is localized to sites of adhesion
To test if elevated p38 activity on Coll suppresses NF-κB activation in a more general way, cells were treated with TNFα, a cytokine that strongly induces this pathway. Interestingly, no suppression of TNFα-induced NF-κB was observed (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200410073/DC1). Furthermore, transfection with a constitutively active MKK3 (MKK3 EE), a kinase that is upstream of p38 and activates p38 globally (not depicted), did not prevent the shear stress– or TNFα-induced activation of NF-κB in cells on FN (Fig. S3 B).
These results appear paradoxical but could be explained if the inhibitory p38 signal from Coll occurs locally within a specific compartment. To test this hypothesis, endothelial cells on Coll or FN were sheared for 5 min and active p38 was localized. Cells on Coll showed colocalization of phospho-p38 with β1 integrins, which was increased by shear stress (Fig. 6 A). Cells on FN showed only low levels of phospho-p38 staining, which is consistent with results from Western blotting.
Figure 6.

Localized activation of p38 and IKK at adhesion sites. (A) Cells were plated on Coll or FN, sheared for 5 min or kept under static conditions, and stained for phosphorylated p38 and β1 integrin. Images are representative of four experiments. (B) BAE cells were plated on Coll, LN, FN, or FG and sheared for the indicated times, and IKK phosphorylation was assessed by Western blotting. Results are representative of four to six experiments. (C) Cells were plated on Coll or FN, sheared for 30 min or kept as static controls, and stained for phosphorylated IKK and β1 integrin. Results are representative of three experiments. (D) Cells on Coll or FN were treated with 10 U/ml TNFα and stained for phosphorylated IKK and β1 integrin. Images are representative of three experiments.
The IKK complex consists of two kinase subunits (IKKα and IKKβ) and a regulatory subunit named NEMO or IKKγ (Israel, 2000). Numerous stimuli activate NF-κB through this complex, resulting in phosphorylation of the kinase subunits (Ser176/Ser180 for IKKα or Ser177/Ser181 for IKKβ; Israel, 2000). Shear stress stimulates NF-κB activation through the integrin- and Rac-dependent activation of IKKα/β, possibly through Nck-interacting kinase (Bhullar et al., 1998; Tzima et al., 2002; Hay et al., 2003). Immunoblotting with phospho-specific antibodies reveals that shear stress stimulates a matrix-specific phosphorylation profile of IKKβ (Fig. 6 B) that correlated well with activation of NF-κB. These results suggest that the divergent matrix-specific signaling occurs upstream of IKK activation. Curiously, shear stress–induced IKK phosphorylation appeared to specifically target IKKβ. This result may be related to recent findings that IKKβ is the primary component involved in NF-κB activation, whereas IKKα has distinct functions (Karin et al., 2004). Staining of fixed cells on Coll with antibody to phospho-IKK showed distinct colocalization of phospho-IKK with β1 integrins in unsheared cells, which was diminished after exposure to shear stress (Fig. 6 C). In cells on FN, phospho-IKK also colocalized with β1 integrins, and this signal increased after shear stress. In contrast, TNFα-induced IKK phosphorylation resulted in diffuse cytoplasmic and nuclear staining (Fig. 6 D). These results suggest that the localized activation of p38 within integrin adhesions specifically inhibits the local, but not global, activation of NF-κB.
Altering the FN matrix to inhibit NF-κB
The selective activation of NF-κB in atherosclerosis-prone regions of the vasculature, together with the importance of NF-κB target genes in atherosclerosis, suggest that inhibition of this transcription factor could have therapeutic value. However, NF-κB has important roles in cell survival and immune protection, thus, global inhibition of the pathway is likely to be deleterious. In contrast, local inactivation of NF-κB within cell–ECM adhesions in atherosclerotic plaque could be beneficial. Although FN does not normally activate p38, a peptide derived from the first type III repeat in FN, termed III-1C, alters the structure of the FN matrix and stimulates p38 activation (Yi and Ruoslahti, 2001; Klein et al., 2003). When cells on FN were treated with this peptide, activated p38 colocalized with integrins at sites of adhesion (Fig. 7, A and B). To test whether or not FNIII-1C might also suppress NF-κB activation, BAE cells on FN were treated for 4 h with FNIII-1C or as controls with heat denatured III-1C or the analogous peptide from the second type III repeat, FNIII-2C. Cells were sheared for 30 min and NF-κB was assayed. FNIII-1C blocked shear stress–induced NF-κB nuclear translocation and phosphorylation, whereas FNIII-2C and heat-denatured FNIII-1C had no effect (Fig. 7, B and D). Furthermore, NF-κB activation was rescued by p38 inhibition in cells treated with FNIII-1C (Fig. 7, C and D). These results show that the proatherosclerotic FN ECM can be modified to locally activate p38 and thereby suppress NF-κB.
Figure 7.
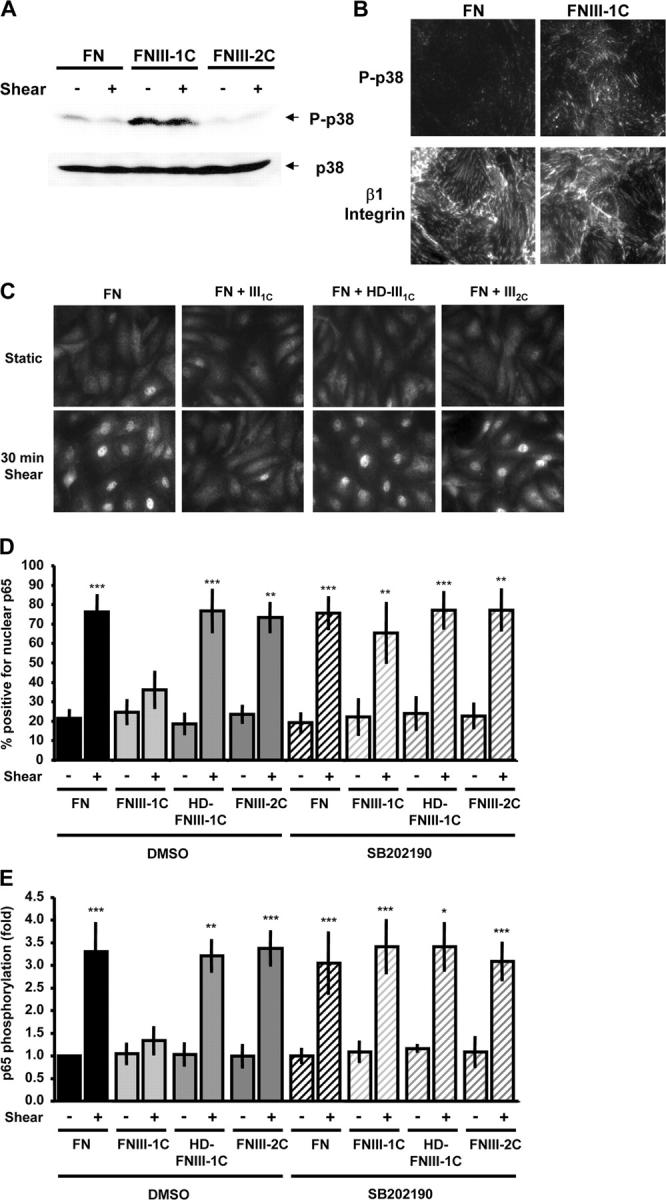
FNIII-1C activates p38 and blocks shear stress–induced NF-κB activation. (A) BAE cells on FN were treated with the FNIII-1C or FNIII-2C peptides for 4 h. Then, cells were either left under static conditions or sheared for 30 min and phosphorylation of p38 was assessed by Western blotting with phosphorylation site-specific antibodies (n = 5). (B) Cells were plated on FN, treated with FNIII-1C for 4 h, and stained for phosphorylated p38 and β1 integrin (n = 3). (C) BAE cells were plated on FN, incubated overnight, and treated with 10 μM FNIII1C, 10 μM of heat-denatured FNIII1C, or 10 μM FNIII2C for 4 h. Cells were sheared for 30 min, and NF-κB nuclear translocation was assessed by immunocytochemistry (n = 3–5). (D) Cells were pretreated with FN peptides as in C, followed by incubation with the p38 inhibitor SB202190 (1 μM) for 1 h before onset of shear stress. Cells were sheared for 30 min and nuclear translocation of p65 was assessed. Values are means ± SD (>100 cells counted per condition per experiment; n = 3–8). **, P < 0.01; ***, P < 0.001. (E) Cells were pretreated with the FN peptides as indicated, incubated with SB202190, and sheared for 30 min. p65 phosphorylation at Ser536 was assessed by Western blotting. Bands were quantified by densitometry and normalized to total p65. Values are means ± SD from four to seven experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
We present evidence that ECM composition leading to specific integrin signals can determine the activation of NF-κB by shear stress. We also show that comparable ECM remodeling occurs in vivo and correlates with activation of NF-κB in regions of disturbed shear stress. Therefore, these results suggest that this interplay might control early atherogenesis in vivo.
The role of the extracellular matrix as a key regulator of the behavior of advanced atherosclerosis is widely accepted, with ECM remodeling thought to affect both the size and stability of the fibrous atherosclerotic plaque (Katsuda and Kaji, 2003). However, a critical role for the subendothelial matrix in development of atherosclerosis has not been suspected. Our data show that deposition of FN and FG into the subendothelial matrix in regions of disturbed flow precedes fatty streak formation in vivo. These proteins in the subendothelial matrix promote activation of NF-κB by shear stress in vitro, whereas the normal basement membrane component Coll suppresses NF-κB through α2β1-dependent p38 activation. Importantly, p38 is activated locally within adhesion sites and prevents the shear stress–induced activation of IKKβ and NF-κB but not the global activation of NF-κB by cytokines. Finally, we demonstrate that FN matrix can be altered to stimulate p38 and suppress shear stress–induced NF-κB activation, suggesting that FN is a possible target for therapeutic intervention.
ECM remodeling in atherogenesis
The normal subendothelial ECM is a basement membrane whose major integrin-binding components are type IV Coll and LNs (Yurchenco and O'Rear, 1994). Injury, inflammation, or angiogenesis result in degradation of the basement membrane and deposition of transitional ECM proteins such as FN and FG. We show here that the presence of transitional matrix proteins in the subendothelial matrix correlates with NF-κB activation and target gene expression in regions of disturbed shear stress. These data are consistent with the idea that transitional matrix deposition regulates inflammatory gene expression in early atherogenesis; however, the causal relationships in vivo between ECM and NF-κB remain to be elucidated. Effects of disturbed flow on FN matrix deposition could involve changes in endothelial permeability to plasma proteins (Friedman and Fry, 1993; Phelps and DePaola, 2000; Himburg et al., 2004), changes in endothelial expression of ECM genes (Brooks et al., 2002), or changes in integrin expression or function (Brooks et al., 2002). Identifying the mechanisms involved in local matrix remodeling at atherosclerosis-prone sites in arteries will be an interesting direction for future work.
We also observed marked increases in both the ECM proteins FN and FG and the inflammatory markers ICAM-1 and VCAM-1 in atherosclerosis-susceptible regions of arteries from ApoE null mice fed a Western diet compared with mice on a chow diet, suggesting that the Western diet may further increase ECM remodeling. This effect would be predicted to further enhance endothelial responses to disturbed flow, thus defining a positive feedback loop that would contribute to disease progression.
Inhibition of NF-κB activation by locally activated p38
Although p38 enhances NF-κB activity in many systems (Carter et al., 1999; Korus et al., 2002), there are several stimuli for which activation of p38 inhibits NF-κB, including vitamin C, salicylate, indomethacin, sorbitol, hydrogen peroxide, and arsenite (Schwenger et al., 1998; Alpert et al., 1999; Bowie and O'Neill, 2000; Bradbury et al., 2001). Drosophila melanogaster p38 isoforms have been shown to limit antimicrobial peptide production, a known target of D. melanogaster NF-κB (Han et al., 1998). In none of these cases has the mechanism by which active p38 represses NF-κB activation been elucidated. Binding of integrin α2β1 to Coll also activates p38 (Ivaska et al., 1999). Consistent with our model in which shear stress stimulates integrin activation and ligand binding (Jalali et al., 2001; Tzima et al., 2001; Katsumi et al., 2004), we found that shear stress stimulated α2β1-dependent p38 activation in cells on Coll. Activation of p38 inhibited both IKKβ and NF-κB activation. Importantly, both active p38 and active IKKβ localized to sites of adhesion. This colocalization correlated with the integrin specificity of the functional interaction because Coll did not inhibit cytokine activation of NF-κB and global activation of 38 did not inhibit shear stress activation of NF-κB. Therefore, the data provide a striking example where signaling specificity is conferred by compartmentalization. Future work will be required to elucidate the pathway by which p38 inhibits IKKβ in this system
Altering the FN matrix
Selective inhibition of NF-κB activation by shear stress offers potential benefits for treatment of atherosclerosis, but it is essential that inhibitors act locally both to be effective and to avoid global inhibition of NF-κB, which would likely have undesirable side effects. A peptide from the first type III repeat of FN that was reported to trigger p38 activation in cells on FN induced local activation of p38 at sites of adhesion on a FN matrix and inhibited shear stress–induced activation of NF-κB. The effects of FNIII-1C are poorly understood. FNIII-1C has been reported to trigger assembly of soluble FN into fibrils and increase matrix FN (Morla and Ruoslahti, 1992), inhibit deposition of matrix FN (Bourdoulous et al., 1998), and alter its conformation without changing the total amount of matrix (Klein et al., 2003). Changes in FN matrix structure could conceivably alter signaling through a previously bound receptor, perhaps by changing its spatial organization, or could expose new cell binding sites or block existing cell binding sites required to prevent p38 activation. FNIII-1C binding to FN masks the EDA domain present in certain forms of FN (Klein et al., 2003), which is consistent with this last hypothesis. Curiously, mice engineered to lack the EDA domain of FN show reduced atherosclerosis, suggesting a potential role for this interaction in atherogenesis (Tan et al., 2004).
FNIII-1C itself is not a likely candidate for therapy, at least as an injected reagent, because it binds to soluble FN in the plasma and has been reported to induce assembly of this FN into fibrils, an event that could trigger thromboses or other deleterious events. Thus, further investigation into the mechanisms of ECM remodeling and p38 activation will be needed to develop more suitable methods for altering subendothelial ECM or cellular responses to it. Nevertheless, these results point toward an entirely novel strategy for treating atherosclerosis. The substrate dependence for proatherosclerotic gene expression may also have important implications in determining suitable signaling-based therapeutic approaches and designing bioengineered vascular grafts.
We conclude that transitional matrix proteins, such as FN and FG, are required for the flow-induced activation of NF-κB and suggest that deposition of FN and FG into the subendothelial matrix at atherosclerosis-prone sites in vivo may regulate the early changes in inflammatory gene expression associated with atherogenesis. Furthermore, this shear stress–induced inflammatory signal appears to be inhibited by the endogenous Coll basement membrane through integrin α2β1–dependent activation of p38. Exploiting this fact, we show that alteration of the FN matrix with the FNIII-1C peptide prevents the shear stress–induced activation of NF-κB through localized p38 signaling. These results suggest a model in which subendothelial matrix remodeling mediates atherosclerotic progression through integrin-dependent mechanotransduction. Further work in which the subendothelial matrix or integrin signals are altered will be required to rigorously test this new model.
Materials and methods
Cell culture, parallel plate flow chamber, and transfection
BAE cells were maintained in DME containing 10% FBS, 1% penicillin/streptomycin, and 2 mM L-glutamine (Invitrogen). Cells were plated on 38 × 75 mm2 glass slides (Corning) precoated with 40 μg/ml Coll I, 20 μg/ml LN, 20 μg/ml FN, or 20 μg/ml FG. For mixed matrix experiments, slides coated with or without Coll I were subsequently coated with increasing concentrations of FN. Western blotting was used to determine the amount of FN adsorbed to the dishes with and without Coll coating. After 4 h, cells were fully attached and spread and formed a confluent monolayer. Slides were loaded onto a parallel plate flow chamber in either 0.1% BSA or 0.5% FBS and 12 dynes/cm2 of shear stress was applied for varying times. Transient transfection of dominant-negative p38 (AGF mutation; 10 μg/ml) and constitutively active MKK3 (EE mutation; 10 μg/ml) was performed by electroporation at 170 V and 960 μF. Cells were replated and incubated for 48 h to allow protein expression before loading onto glass slides for exposure to shear stress.
Immunocytochemistry
Cells were fixed with PBS containing 2% formaldehyde, permeabilized with 0.2% Triton X-100 when applicable, and blocked overnight in PBS containing 1% BSA and 10% goat serum. Primary antibodies were incubated with cells in blocking buffer as follows: rabbit anti-p65 (Santa Cruz Biotechnology, Inc.; 1:200 for 1 h), rabbit anti-ICAM (Santa Cruz Biotechnology, Inc.; 1:100 for 1 h), rabbit anti–phospho-IKK (Cell Signaling Technology; 1:100 overnight), rabbit anti–phospho-p38 (Cell Signaling Technology; 1:100 overnight), and mouse anti-ligated β1 integrins (12G10; 1 μg/ml for 1 h). Cells were incubated in 1 μg/ml Alexa 488–conjugated goat anti–rabbit IgG or Alexa 568–conjugated goat anti–mouse IgG (Molecular Probes). Slides were mounted with Fluoromount G and images were taken using the 60× oil immersion objective on a microscope (model DiaPhot; Nikon) equipped with a CoolSnap video camera (Photometrics) using the Inovision ISEE software program.
Immunoblotting
Cell lysis and immunoblotting were performed as previously described (Orr et al., 2002). Rabbit anti–phospho-p38 (Cell Signaling Technology), mouse anti-p38 (Santa Cruz Biotechnology, Inc.), rabbit anti–phospho-p65 (Ser536; Cell Signaling Technology), rabbit anti-p65 (Santa Cruz Biotechnology, Inc.), rabbit anti–phospho-IKKα/β (Cell Signaling Technologies), and rabbit anti-IKKα/β (Santa Cruz Biotechnology, Inc.) were all used at 1:1000 dilutions.
Animals
Six male ApoE-deficient mice on a C57BL/6 background (Jackson ImmunoResearch Laboratories), 8–12 wk old and weighing 18–20 g, were used in these experiments. Four mice were fed a Western-type atherogenic diet (TD 88137; Harlan-Teklad; containing 21% fat by weight, 0.15% by weight cholesterol, and 19.5% by weight casein without sodium cholate) for 10 wk before killing. Control mice were fed a chow diet during this time. Comparable wild-type C57/B6 mice were fed the chow diet during the same time frame.
Vessel harvest
At 20 wk old (10 wk on diet), mice were perfused with 4% formaldehyde and the innominate, left carotid, right carotid, and short segments of the descending abdominal aorta near the renal arteries and the iliac bifurcation were processed for paraffin embedding.
IHC
5-μm paraffin sections were obtained for IHC. IHC for adhesion molecules VCAM-1, ICAM-1, Mac-2, and PECAM-1 (Santa Cruz Biotechnology, Inc.) was performed as previously described (McPherson et al., 2001). After microwave antigen retrieval with antigen unmasking solution (Vector Laboratories), rabbit anti-FN (1:400; Sigma-Aldrich) and rabbit anti-LN (1:500; Sigma-Aldrich) were applied. Detection of antibodies was done with Vectastain Elite Kit (Vector Laboratories). Visualization was done with DAB (Deko Corp). Goat anti-FG (1 μg/ml; Accurate Chemical) was applied as for the other antigens, except that no antigen retrieval was required. Images were acquired using the 20 or 40× objective on a microscope (model BX51; Olympus) equipped with a digital camera (model DP70; Olympus) using the ImagePro Plus software program in the Academic Computing Health Sciences Center at the University of Virginia.
Online supplemental material
In Fig. 4, we demonstrate that incubation with the α2β1 integrin–blocking antibody R2-8C8 can block shear-induced p38 activation on Coll and rescue NF-κB activation in cells on Coll, presumably by preventing the interaction of newly activated α2β1 with Coll. To demonstrate that this effect was not due to changes in cell adhesiveness, we show that short-term incubation with R2-8C8 does not affect the organization of the actin cytoskeleton (Fig. S1). To show that this effect was specific to Coll, we also prevented the interaction between α6β1 and LN with the α6β1-blocking antibody GoH3, which did not rescue flow-induced NF-κB activation (Fig. S2). Finally, we show that flow-induced p38 activation on Coll prevents flow-dependent activation of NF-κB. To determine if this effect was specific for flow, the ability of TNFα to activate NF-κB in cells on different matrix proteins or overexpressing a constitutively active MKK3 construct (MKK3-EE) was assessed (Fig. S3). Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200410073/DC1.
Acknowledgments
The authors are grateful to Tom Sturgill and Carol Chrestensen (University of Virginia, Charlottesville, VA) for dominant-negative p38 and constitutively active MKK3 constructs, Mark Ginsberg (Scripps Research Institute, La Jolla, CA) for R2-8C8 α2β1 integrin–blocking antibody, Martin Humphries (University of Manchester, Manchester, UK) for 12G10 anti-ligated β1 integrin antibody, Paula J. McKeown-Longo (Albany Medical College, Albany, NY) for FNIII-1C and FNIII-2C peptides, and Andrew Prior and Brett Blackman (University of Virginia, Charlottesville, VA) for human umbilical vein endothelial cells. The authors would also like to acknowledge Daniel Bennet and Elizabeth Thao Phan for their assistance in vessel isolation and image acquisition.
This work was supported by U.S. Public Health Service grant RO1 HL75092 to M.A. Schwartz, American Heart Association Mid-Atlantic Affiliate fellowship 0325654U to A.W. Orr, and National Institutes of Health grant 1RO1HL66264 to I.J. Sarembock.
Abbreviations used in this paper: ApoE, apolipoprotein E; BAE, bovine aortic endothelial; Coll, collagen; FG, fibrinogen; FN, fibronectin; IHC, immunohistochemistry; IKK, IκB kinase; LN, laminin.
References
- Alpert, D., P. Schwenger, J. Han, and J. Vilcek. 1999. Cell stress and MKK6b-mediated p38 MAP kinase activation inhibit tumor necrosis factor-induced IkappaB phosphorylation and NF-kappaB activation. J. Biol. Chem. 274:22176–22183. [DOI] [PubMed] [Google Scholar]
- Belkin, A.M., and M.A. Stepp. 2000. Integrins as receptors for laminins. Microsc. Res. Tech. 51:280–301. [DOI] [PubMed] [Google Scholar]
- Bhullar, I.S., Y.S. Li, H. Miao, E. Zandi, M. Kim, J.Y. Shyy, and S. Chien. 1998. Fluid shear stress activation of IkappaB kinase is integrin-dependent. J. Biol. Chem. 273:30544–30549. [DOI] [PubMed] [Google Scholar]
- Bourdoulous, S., G. Orend, D.A. MacKenna, R. Pasqualini, and E. Ruoslahti. 1998. Fibronectin matrix regulates activation of RHO and CDC42 GTPases and cell cycle progression. J. Cell Biol. 143:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie, A.G., and L.A. O'Neill. 2000. Vitamin C inhibits NF-kappa B activation by TNF via the activation of p38 mitogen-activated protein kinase. J. Immunol. 165:7180–7188. [DOI] [PubMed] [Google Scholar]
- Bradbury, C.M., S. Markovina, S.J. Wei, L.M. Rene, I. Zoberi, N. Horikoshi, and D. Gius. 2001. Indomethacin-induced radiosensitization and inhibition of ionizing radiation-induced NF-kappaB activation in HeLa cells occur via a mechanism involving p38 MAP kinase. Cancer Res. 61:7689–7696. [PubMed] [Google Scholar]
- Brand, K., S. Page, G. Rogler, A. Bartsch, R. Brandl, R. Knuechel, M. Page, C. Kaltschmidt, P.A. Baeuerle, and D. Neumeier. 1996. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J. Clin. Invest. 97:1715–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, A.R., P.I. Lelkes, and G.M. Rubanyi. 2002. Gene expression profiling of human aortic endothelial cells exposed to disturbed flow and steady laminar flow. Physiol. Genomics. 9:27–41. [DOI] [PubMed] [Google Scholar]
- Caro, C.G., J.M. Fitz-Gerald, and R.C. Schroter. 1969. Arterial wall shear and distribution of early atheroma in man. Nature. 223:1159–1160. [DOI] [PubMed] [Google Scholar]
- Carter, A.B., K.L. Knudtson, M.M. Monick, and G.W. Hunninghake. 1999. The p38 mitogen-activated protein kinase is required for NF-kappaB-dependent gene expression. The role of TATA-binding protein (TBP). J. Biol. Chem. 274:30858–30863. [DOI] [PubMed] [Google Scholar]
- Chen, L.F., and W.C. Greene. 2004. Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5:392–401. [DOI] [PubMed] [Google Scholar]
- Collins, T., and M.I. Cybulsky. 2001. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J. Clin. Invest. 107:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty, A. 2002. Mouse models of atherosclerosis. Am. J. Med. Sci. 323:3–10. [DOI] [PubMed] [Google Scholar]
- De Keulenaer, G.W., D.C. Chappell, N. Ishizaka, R.M. Nerem, R.W. Alexander, and K.K. Griendling. 1998. Oscillatory and steady laminar shear stress differentially affect human endothelial redox state: role of a superoxide-producing NADH oxidase. Circ. Res. 82:1094–1101. [DOI] [PubMed] [Google Scholar]
- Defilippi, P., V. van Hinsbergh, A. Bertolotto, P. Rossino, L. Silengo, and G. Tarone. 1991. Differential distribution and modulation of expression of α1/β1 integrin on human endothelial cells. J. Cell Biol. 114:855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, M.H., and D.L. Fry. 1993. Arterial permeability dynamics and vascular disease. Atherosclerosis. 104:189–194. [DOI] [PubMed] [Google Scholar]
- Glagov, S., C. Zarins, D.P. Giddens, and D.N. Ku. 1988. Hemodynamics and atherosclerosis. Insights and perspectives gained from studies of human arteries. Arch. Pathol. Lab. Med. 112:1018–1031. [PubMed] [Google Scholar]
- Guo, M., S.K. Sahni, A. Sahni, and C.W. Francis. 2004. Fibrinogen regulates the expression of inflammatory chemokines through NF-kappaB activation of endothelial cells. Thromb. Haemost. 92:858–866. [DOI] [PubMed] [Google Scholar]
- Han, Z.S., H. Enslen, X. Hu, X. Meng, I.H. Wu, T. Barrett, R.J. Davis, and Y.T. Ip. 1998. A conserved p38 mitogen-activated protein kinase pathway regulates Drosophila immunity gene expression. Mol. Cell. Biol. 18:3527–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay, D.C., C. Beers, V. Cameron, L. Thomson, F.W. Flitney, and R.T. Hay. 2003. Activation of NF-kappaB nuclear transcription factor by flow in human endothelial cells. Biochim. Biophys. Acta. 1642:33–44. [DOI] [PubMed] [Google Scholar]
- Heino, J. 2000. The collagen receptor integrins have distinct ligand recognition and signaling functions. Matrix Biol. 19:319–323. [DOI] [PubMed] [Google Scholar]
- Himburg, H.A., D.M. Grzybowski, A.L. Hazel, J.A. LaMack, X.M. Li, and M.H. Friedman. 2004. Spatial comparison between wall shear stress measures and porcine arterial endothelial permeability. Am. J. Physiol. Heart Circ. Physiol. 286:H1916–H1922. [DOI] [PubMed] [Google Scholar]
- Huo, Y., and K. Ley. 2001. Adhesion molecules and atherogenesis. Acta Physiol. Scand. 173:35–43. [DOI] [PubMed] [Google Scholar]
- Iiyama, K., L. Hajra, M. Iiyama, H. Li, M. DiChiara, B.D. Medoff, and M.I. Cybulsky. 1999. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ. Res. 85:199–207. [DOI] [PubMed] [Google Scholar]
- Israel, A. 2000. The IKK complex: an integrator of all signals that activate NF-kappaB? Trends Cell Biol. 10:129–133. [DOI] [PubMed] [Google Scholar]
- Ivaska, J., H. Reunanen, J. Westermarck, L. Koivisto, V.M. Kahari, and J. Heino. 1999. Integrin α2β1 mediates isoform-specific activation of p38 and upregulation of collagen gene transcription by a mechanism involving the α2 cytoplasmic tail. J. Cell Biol. 147:401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalali, S., M.A. del Pozo, K. Chen, H. Miao, Y. Li, M.A. Schwartz, J.Y. Shyy, and S. Chien. 2001. Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ECM) ligands. Proc. Natl. Acad. Sci. USA. 98:1042–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin, M., Y. Yamamoto, and Q.M. Wang. 2004. The IKK NF-kappa B system: a treasure trove for drug development. Nat. Rev. Drug Discov. 3:17–26. [DOI] [PubMed] [Google Scholar]
- Katsuda, S., and T. Kaji. 2003. Atherosclerosis and extracellular matrix. J. Atheroscler. Thromb. 10:267–274. [DOI] [PubMed] [Google Scholar]
- Katsumi, A., A.W. Orr, E. Tzima, and M.A. Schwartz. 2004. Integrins in mechanotransduction. J. Biol. Chem. 279:12001–12004. [DOI] [PubMed] [Google Scholar]
- Khachigian, L.M., N. Resnick, M.A. Gimbrone Jr., and T. Collins. 1995. Nuclear factor-kappa B interacts functionally with the platelet-derived growth factor B-chain shear-stress response element in vascular endothelial cells exposed to fluid shear stress. J. Clin. Invest. 96:1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, R.M., M. Zheng, A. Ambesi, L. Van De Water, and P.J. McKeown-Longo. 2003. Stimulation of extracellular matrix remodeling by the first type III repeat in fibronectin. J. Cell Sci. 116:4663–4674. [DOI] [PubMed] [Google Scholar]
- Klein, S., A.R. de Fougerolles, P. Blaikie, L. Khan, A. Pepe, C.D. Green, V. Koteliansky, and F.G. Giancotti. 2002. Alpha 5 beta 1 integrin activates an NF-kappa B-dependent program of gene expression important for angiogenesis and inflammation. Mol. Cell. Biol. 22:5912–5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korus, M., G.M. Mahon, L. Cheng, and I.P. Whitehead. 2002. p38 MAPK-mediated activation of NF-kappaB by the RhoGEF domain of Bcr. Oncogene. 21:4601–4612. [DOI] [PubMed] [Google Scholar]
- Ku, D.N., D.P. Giddens, C.K. Zarins, and S. Glagov. 1985. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis. 5:293–302. [DOI] [PubMed] [Google Scholar]
- Kutuk, O., and H. Basaga. 2003. Inflammation meets oxidation: NF-kappaB as a mediator of initial lesion development in atherosclerosis. Trends Mol. Med. 9:549–557. [DOI] [PubMed] [Google Scholar]
- Labat-Robert, J., M. Szendroi, G. Godeau, and L. Robert. 1985. Comparative distribution patterns of type I and III collagens and fibronectin in human arteriosclerotic aorta. Pathol. Biol. (Paris). 33:261–265. [PubMed] [Google Scholar]
- Lan, Q., K.O. Mercurius, and P.F. Davies. 1994. Stimulation of transcription factors NF kappa B and AP1 in endothelial cells subjected to shear stress. Biochem. Biophys. Res. Commun. 201:950–956. [DOI] [PubMed] [Google Scholar]
- Li, H., M.I. Cybulsky, M.A. Gimbrone Jr., and P. Libby. 1993. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler. Thromb. 13:197–204. [DOI] [PubMed] [Google Scholar]
- McPherson, J.A., K.G. Barringhaus, G.G. Bishop, J.M. Sanders, J.M. Rieger, S.E. Hesselbacher, L.W. Gimple, E.R. Powers, T. Macdonald, G. Sullivan, et al. 2001. Adenosine A(2A) receptor stimulation reduces inflammation and neointimal growth in a murine carotid ligation model. Arterioscler. Thromb. Vasc. Biol. 21:791–796. [DOI] [PubMed] [Google Scholar]
- Mohan, S., N. Mohan, and E.A. Sprague. 1997. Differential activation of NF-kappa B in human aortic endothelial cells conditioned to specific flow environments. Am. J. Physiol. 273:C572–C578. [DOI] [PubMed] [Google Scholar]
- Morla, A., and E. Ruoslahti. 1992. A fibronectin self-assembly site involved in fibronectin matrix assembly: reconstruction in a synthetic peptide. J. Cell Biol. 118:421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima, Y., A.S. Plump, E.W. Raines, J.L. Breslow, and R. Ross. 1994. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler. Thromb. 14:133–140. [DOI] [PubMed] [Google Scholar]
- Nakashima, Y., E.W. Raines, A.S. Plump, J.L. Breslow, and R. Ross. 1998. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 18:842–851. [DOI] [PubMed] [Google Scholar]
- Orr, A.W., M.A. Pallero, and J.E. Murphy-Ullrich. 2002. Thrombospondin stimulates focal adhesion disassembly through Gi- and phosphoinositide 3-kinase-dependent ERK activation. J. Biol. Chem. 277:20453–20460. [DOI] [PubMed] [Google Scholar]
- Phelps, J.E., and N. DePaola. 2000. Spatial variations in endothelial barrier function in disturbed flows in vitro. Am. J. Physiol. Heart Circ. Physiol. 278:H469–H476. [DOI] [PubMed] [Google Scholar]
- Resnick, N., T. Collins, W. Atkinson, D.T. Bonthron, C.F. Dewey Jr., and M.A. Gimbrone Jr. 1993. Platelet-derived growth factor B chain promoter contains a cis-acting fluid shear-stress-responsive element. Proc. Natl. Acad. Sci. USA. 90:4591–4595 (published erratum appears in Proc. Natl. Acad. Sci. USA. 1993. 90:7908). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, R. 1999. Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 340:115–126. [DOI] [PubMed] [Google Scholar]
- Scatena, M., M. Almeida, M.L. Chaisson, N. Fausto, R.F. Nicosia, and C.M. Giachelli. 1998. NF-κB mediates αvβ3 integrin-induced endothelial cell survival. J. Cell Biol. 141:1083–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, M.A., and R.K. Assoian. 2001. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J. Cell Sci. 114:2553–2560. [DOI] [PubMed] [Google Scholar]
- Schwenger, P., D. Alpert, E.Y. Skolnik, and J. Vilcek. 1998. Activation of p38 mitogen-activated protein kinase by sodium salicylate leads to inhibition of tumor necrosis factor-induced IkappaB alpha phosphorylation and degradation. Mol. Cell. Biol. 18:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechler, J.L., S.A. Corbett, M.B. Wenk, and J.E. Schwarzbauer. 1998. Modulation of cell-extracellular matrix interactions. Ann. NY Acad. Sci. 857:143–154. [DOI] [PubMed] [Google Scholar]
- Shekhonin, B.V., S.P. Domogatsky, G.L. Idelson, V.E. Koteliansky, and V.S. Rukosuev. 1987. Relative distribution of fibronectin and type I, III, IV, V collagens in normal and atherosclerotic intima of human arteries. Atherosclerosis. 67:9–16. [DOI] [PubMed] [Google Scholar]
- Stenman, S., K. von Smitten, and A. Vaheri. 1980. Fibronectin and atherosclerosis. Acta Med. Scand. Suppl. 642:165–170. [DOI] [PubMed] [Google Scholar]
- Tan, M.H., Z. Sun, S.L. Opitz, T.E. Schmidt, J.H. Peters, and E.L. George. 2004. Deletion of the alternatively spliced fibronectin EIIIA domain in mice reduces atherosclerosis. Blood. 104:11–18. [DOI] [PubMed] [Google Scholar]
- Tanouchi, J., M. Uematsu, A. Kitabatake, T. Masuyama, H. Ito, Y. Doi, M. Inoue, and T. Kamada. 1992. Sequential appearance of fibronectin, collagen and elastin during fatty streak initiation and maturation in hypercholesterolemic fat-fed rabbits. Jpn. Circ. J. 56:649–656. [DOI] [PubMed] [Google Scholar]
- Topper, J.N., J. Cai, D. Falb, and M.A. Gimbrone Jr. 1996. Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc. Natl. Acad. Sci. USA. 93:10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulla, M., O.T. Pentikainen, T. Viitasalo, J. Kapyla, U. Impola, P. Nykvist, L. Nissinen, M.S. Johnson, and J. Heino. 2001. Selective binding of collagen subtypes by integrin alpha 1I, alpha 2I, and alpha 10I domains. J. Biol. Chem. 276:48206–48212. [DOI] [PubMed] [Google Scholar]
- Tzima, E., M.A. del Pozo, S.J. Shattil, S. Chien, and M.A. Schwartz. 2001. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 20:4639–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima, E., M.A. Del Pozo, W.B. Kiosses, S.A. Mohamed, S. Li, S. Chien, and M.A. Schwartz. 2002. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 21:6791–6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderLaan, P.A., C.A. Reardon, and G.S. Getz. 2004. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler. Thromb. Vasc. Biol. 24:12–22. [DOI] [PubMed] [Google Scholar]
- Wang, D., and A.S. Baldwin Jr. 1998. Activation of nuclear factor-kappaB-dependent transcription by tumor necrosis factor-alpha is mediated through phosphorylation of RelA/p65 on serine 529. J. Biol. Chem. 273:29411–29416. [DOI] [PubMed] [Google Scholar]
- Wilson, S.H., N.M. Caplice, R.D. Simari, D.R. Holmes Jr., P.J. Carlson, and A. Lerman. 2000. Activated nuclear factor-kappaB is present in the coronary vasculature in experimental hypercholesterolemia. Atherosclerosis. 148:23–30. [DOI] [PubMed] [Google Scholar]
- Yamamoto, Y., and R.B. Gaynor. 2004. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem. Sci. 29:72–79. [DOI] [PubMed] [Google Scholar]
- Yang, F., E. Tang, K. Guan, and C.Y. Wang. 2003. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J. Immunol. 170:5630–5635. [DOI] [PubMed] [Google Scholar]
- Yi, M., and E. Ruoslahti. 2001. A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc. Natl. Acad. Sci. USA. 98:620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenco, P.D., and J.J. O'Rear. 1994. Basement membrane assembly. Methods Enzymol. 245:489–518. [DOI] [PubMed] [Google Scholar]