

Abstract
Degradation of specific protein substrates by the anaphase-promoting complex/cyclosome (APC) is critical for mitotic exit. We have identified the protein Xenopus nuclear factor 7 (Xnf7) as a novel APC inhibitor able to regulate the timing of exit from mitosis. Immunodepletion of Xnf7 from Xenopus laevis egg extracts accelerated the degradation of APC substrates cyclin B1, cyclin B2, and securin upon release from cytostatic factor arrest, whereas excess Xnf7 inhibited APC activity. Interestingly, Xnf7 exhibited intrinsic ubiquitin ligase activity, and this activity was required for APC inhibition. Unlike other reported APC inhibitors, Xnf7 did not associate with Cdc20, but rather bound directly to core subunits of the APC. Furthermore, Xnf7 was required for spindle assembly checkpoint function in egg extracts. These data suggest that Xnf7 is an APC inhibitor able to link spindle status to the APC through direct association with APC core components.
Introduction
Key events in mitosis such as sister chromatid separation and inactivation of the cyclin-dependent kinase Cdc2 are regulated by the anaphase-promoting complex/cyclosome (APC), a large multisubunit ubiquitin ligase that promotes ubiquitylation and proteasomal degradation of its substrates (Zachariae and Nasmyth, 1999; Harper et al., 2002; Peters, 2002). Although the APC was originally identified as a cyclin B ubiquitin ligase (Irniger et al., 1995; King et al., 1995; Sudakin et al., 1995), it has since been shown to ubiquitylate numerous cell cycle regulatory proteins including cyclin A, securin, Xkid1, Cdc20, Plk1, and Ase1, as well as regulatory proteins involved in other cellular processes such as transcription and development (Hershko and Ciechanover, 1998; Peters, 2002).
The APC is present throughout the cell cycle, but its activity is highly regulated. It is active from prometaphase until late G1, and inactive in S phase and G2 when cyclin B accumulates. Activation of the APC is controlled in part by phosphorylation of at least three of its subunits (Cdc16, Cdc23, and Cdc27) by the mitotic kinases Cdk1/cyclin B and Plk1 (Kotani et al., 1998; Rudner and Murray, 2000; Golan et al., 2002). However, the relative importance of these two kinases in the direct phosphorylation of the APC remains unclear, as data suggesting both kinases (Golan et al., 2002), only Plk1 (Kotani et al., 1998), or only Cdk1 (Rudner and Murray, 2000; Kraft et al., 2003) directly phosphorylate the APC have yet to be reconciled. APC activation is also controlled by the binding of activators, the WD40-repeat–containing proteins Cdc20 and Cdh1. In somatic cells, Cdc20 and Cdh1 binding to the APC is regulated, resulting in a peak in APCCdc20 activity in anaphase and APCCdh1 activity in G1 (Fang et al., 1998b; Zachariae et al., 1998; Kramer et al., 2000). Until recently, it was thought that Cdh1 was not expressed in the early Xenopus laevis embryo, and that only APCCdc20 was active (Lorca et al., 1998). A recent paper suggests, however, that Cdh1 is present in stage VI oocytes, and that it might have a role during the first meiotic cell cycle (Papin et al., 2004). Although the widely accepted model for the role of Cdc20/Cdh1in APC activation defines these proteins as essential for substrate recognition (Burton and Solomon, 2001; Pfleger et al., 2001; Schwab et al., 2001), several recent studies have shown that the core subunits of the APC, rather than Cdc20/Cdh1, are directly responsible for substrate recognition (Meyn et al., 2002; Passmore et al., 2003; Yamano et al., 2004) and that Cdc20/Cdh1 are required for activation of the APC in a still undefined manner.
Tight regulation of the APC is critical to ensure the appropriate sequential destruction of APC substrates during the cell cycle. For example, cyclin A is degraded during prometaphase, and securin, Xkid1, and cyclin B are destroyed at the metaphase–anaphase transition (Funabiki and Murray, 2000; den Elzen and Pines, 2001; Geley et al., 2001). During anaphase and telophase, APCCdh1 is activated and mediates the destruction of Plk1, Ase1, and Cdc20 (Juang et al., 1997; Shirayama et al., 1998). The precise sequence of degradative events that must occur to ensure the proper timing of mitosis suggests that additional factors control the timing of APC-mediated ubiquitylation. One such factor is Emi1 (early mitotic inhibitor 1), an inhibitor of the APC that binds directly to Cdc20 to prevent premature APC activation (Reimann et al., 2001). Another recently reported APC inhibitor, XErp1 (Xenopus Emi1 related protein 1), also prevents premature APCCdc20 activation, although the mechanism of inhibition remains unknown (Schmidt et al., 2005).
APCCdc20 activity is also regulated by the spindle checkpoint, a mechanism that is critical for the maintenance of genomic stability and acts by delaying sister chromatid separation until all chromosomes are properly attached to the mitotic spindle (Amon, 1999; Shah and Cleveland, 2000; Yu, 2002). Upon activation of the checkpoint at unattached kinetochores, checkpoint protein complexes containing BubR1, Bub3, and Mad2 bind to and inhibit the APC activator Cdc20 (Fang et al., 1998a; Hwang et al., 1998; Sudakin et al., 2001; Tang et al., 2001; Fang, 2002). Recent studies show that not only are the spindle checkpoint proteins and Cdc20 spatially regulated but that spindle checkpoint components are required to recruit the APC to kinetochores, suggesting that the APC could be regulated directly by the spindle checkpoint (Acquaviva et al., 2004; Vigneron et al., 2004).
Much of our knowledge of cell cycle progression has emerged from studies in model systems, including yeast and X. laevis. Here, we have used the Xenopus egg extract, in which APC regulation has been extensively analyzed. Unfertilized Xenopus eggs are arrested in metaphase of meiosis II due to the stabilization of cyclin B by an activity known as cytostatic factor (CSF; Tunquist and Maller, 2003). Crushing the eggs by centrifugation releases intracellular Ca2+ stores, activating calcium/calmodulin-dependent protein kinase II and triggering APC activation and degradation of cyclin B (Lorca et al., 1993). However, preparing extracts in the presence of EGTA prevents the activation of calcium/calmodulin-dependent protein kinase II, allowing high levels of cyclin B to remain and arresting extracts in meiosis. These CSF-arrested extracts can then be released from their meiotic state by addition of Ca2+, leading to activation of the APC and progression into interphase.
In performing experiments designed to identify novel cyclin B2 interactors from Xenopus egg extracts that might regulate its function, we have uncovered an additional APC inhibitor, Xenopus nuclear factor 7 (Xnf7). Xnf7 was originally cloned during a screen for proteins that accumulate in embryonic nuclei during the midblastula transition (Reddy et al., 1991). Xnf7 is a maternally expressed protein found in oocyte nuclei. After release into the cytoplasm during maturation, where it remains throughout the early embryonic cell divisions, it reenters nuclei at the midblastula transition. El-Hodiri et al. (1997b) demonstrated that Xnf7 plays a role in dorso-ventral patterning of the Xenopus embryo, but no specific molecular function has been shown to account for this phenotype.
Here, we demonstrate that Xnf7 has the unexpected ability to inhibit APC function. Although we isolated Xnf7 via its affinity for cyclin B2, Xnf7 appears to have universal effects on APC-dependent ubiquitylation. Immunodepletion of Xnf7 from CSF-arrested egg extracts accelerates the degradation of cyclin B1, cyclin B2, and securin upon addition of Ca2+, thereby accelerating the transition to interphase. Moreover, we demonstrate that Xnf7 itself has intrinsic ubiquitin ligase activity, and that this activity is required for inhibition of the APC. Unlike previously identified inhibitors of APCCdc20 (e.g., Mad2 and Emi1), Xnf7 does not inhibit APCCdc20 activity via direct binding to Cdc20, but rather appears to act independently of Cdc20 via association with a core APC subunit. We also find that Xnf7 is required for the spindle assembly checkpoint in egg extracts. Collectively, our data suggest that Xnf7 inhibits APC activation in a manner distinct from known APC regulators, likely via interaction with a core APC subunit.
Results
Xnf7 binds to the cyclin B2 cytoplasmic retention sequence (CRS)
Previous work from our laboratory and others has demonstrated that the subcellular localization of cyclins B1 and B2 is controlled by a region of the cyclin proteins known as the CRS (Pines and Hunter, 1994). However, the CRSs of these two cyclins differ significantly. The cyclin B1 CRS contains a Crm1-binding nuclear export sequence that promotes nuclear export of cyclin B1 throughout interphase and is inactivated by phosphorylation to allow nuclear accumulation at the onset of mitosis (Yang et al., 1998; Hagting et al., 1999). In contrast, the cyclin B2 CRS lacks a classical nuclear export sequence and may serve to anchor cyclin B2 in the cytoplasm (Jackman et al., 1995). The cyclin B2 CRS, when injected into Xenopus oocytes, has also been reported to promote formation of monopolar rather than bipolar spindles, suggesting that this region might interact with proteins important for spindle polarity (Yoshitome et al., 1998). Therefore, we sought to purify novel factors interacting with the cyclin B2 CRS. Upon passage of Xenopus egg extract over a column of immobilized recombinant cyclin B2 CRS protein (amino acids 94 to 204), we observed a prominent band of ∼80 kD bound specifically to the cyclin B2 CRS (Fig. 1 A). Analysis by mass spectrometry identified this band as Xnf7.
Figure 1.
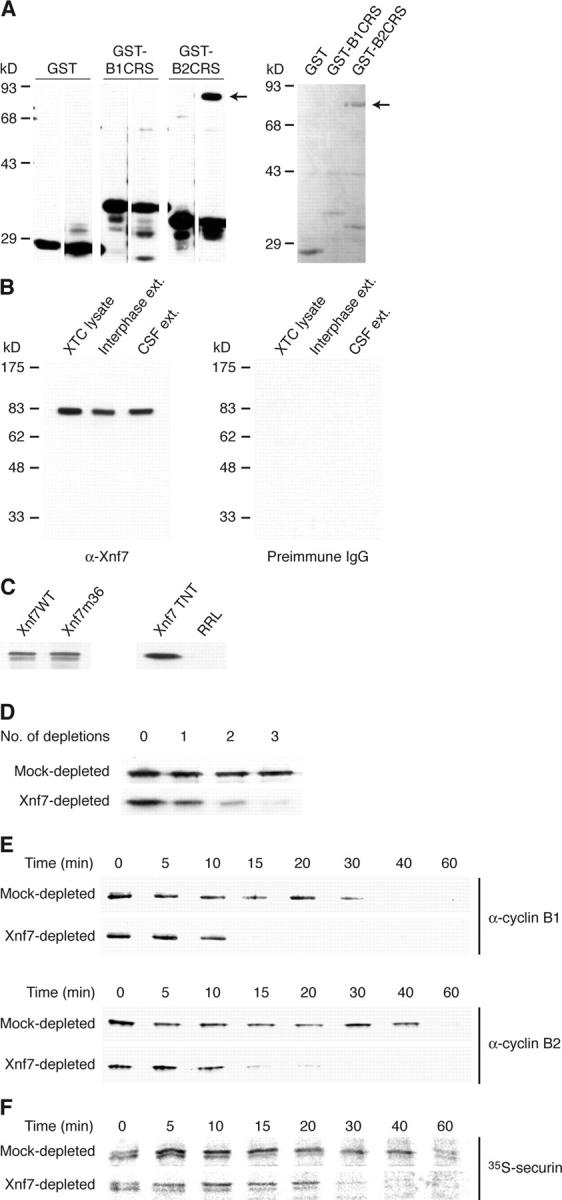
Immunodepletion of Xnf7 accelerates exit from CSF arrest. (A) Xnf7 interacts with the cyclin B2 CRS. Glutathione-Sepharose beads were coupled to GST, GST-cyclin B1 CRS, or GST-cyclin B2 CRS and incubated with egg extracts. (left) Beads were washed, and bound material was conjugated to biotin. Biotinylated material was resolved by SDS-PAGE, transferred to nitrocellulose, and probed with HRP-streptavidin. The left lane of each panel shows proteins present on the beads alone; the right lanes show proteins bound after incubation with extract. (right) Beads were washed and bound proteins were analyzed by SDS-PAGE and Coomassie blue staining. The arrows indicate the ∼80-kD band that was identified by mass spectrometry as Xnf7. (B) Characterization of Xnf7 antibodies. Xenopus XTC cell lysate and interphase and CSF-arrested egg extracts were resolved by SDS-PAGE and immunoblotted with affinity-purified Xnf7 antibodies (left) or purified preimmune IgG (right). (C) Xnf7 wild-type (Xnf7WT) and Xnf7 mutant 36 (Xnf7m36) expressed in Escherichia coli, rabbit reticulocyte lysate (RRL) programmed with Xnf7 (Xnf7 TNT), and unprogrammed RRL were resolved by SDS-PAGE and immunoblotted with affinity-purified Xnf7 antibodies. (D) Immunodepletion of Xnf7 from egg extracts. Purified anti-Xnf7 antibodies or purified preimmune IgG were coupled to protein A–Sepharose and incubated with extracts. Three consecutive depletions were performed and depleted extracts were resolved by SDS-PAGE and immunoblotted with anti-Xnf7 antibodies. (E) Immunodepletion of Xnf7 accelerates the degradation of cyclin B1 and cyclin B2 during exit from CSF arrest. CSF extracts were depleted and incubated at 23°C for 30 min. CaCl2 was added to extracts, and aliquots removed at the indicated times after CaCl2 addition were analyzed by SDS-PAGE and immunoblotting with anti–cyclin B1 or anti–cyclin B2 antibodies. (F) Immunodepletion of Xnf7 accelerates the degradation of securin during exit from CSF arrest. CSF extracts were depleted, supplemented with 35S-securin, and incubated at 23°C for 30 min. CaCl2 was added to extracts, and aliquots removed at the indicated times were analyzed by SDS-PAGE and autoradiography.
Depletion of Xnf7 accelerates the ubiquitylation of APC substrates and mitotic exit
Given the previously reported effects of the cyclin B2 CRS on spindle polarity, we sought to determine if Xnf7 had a discernible role in spindle formation. Toward this end, we produced anti-Xnf7 antibodies for immunodepletion from Xenopus egg extracts. Affinity-purified Xnf7 antibodies recognized a protein of ∼80 kD in Xenopus XTC lysates and Xenopus interphase and CSF-arrested egg extracts, whereas preimmune IgG did not (Fig. 1 B). Antibodies also recognized recombinant Xnf7 and in vitro translated Xnf7, but did not detect a protein in unprogrammed rabbit reticulocyte lysate (RRL; Fig. 1 C).
Immunodepletion of Xnf7 from interphase or CSF extracts was accomplished with three sequential depletions (Fig. 1 D). The absence of Xnf7 had no detectable effect on bipolar spindle formation, cyclin B2 localization, or DNA replication (unpublished data). However, we found that CSF extracts immunodepleted of Xnf7 behaved in an unexpected manner. Specifically, when Xnf7-depleted CSF extracts were released into interphase by Ca2+, we found that they transited from the CSF-arrested state to interphase considerably more rapidly than mock-depleted (control) extracts, reforming interphase nuclei 40–50 min after Ca2+ addition compared with 60–80 min in control extracts (unpublished data). (Note that to start with a high level of Cdc2/cyclin B activity and monitor its decline, extracts were first incubated at room temperature for 30 min before Ca2+ was added.) Consistent with this observation, depletion of Xnf7 accelerated the degradation of cyclin B1, cyclin B2, and securin (Fig. 1, E and F). The extracts shown in Fig. 1 (E and F) are distinct, and the timing of exit from CSF arrest shown here is representative of the timing observed in numerous experiments. Although the absolute timing of exit differed between extracts, the relative rates of exit under different treatment conditions remained constant.
Because CSF extracts are arrested in metaphase of meiosis II, and therefore have characteristics distinct from extracts that mimic the first embryonic M phase (Tunquist and Maller, 2003), we sought to determine whether depletion of Xnf7 accelerated exit from an extract prepared so as to recapitulate the first embryonic mitosis or if this acceleration was unique to the meiotic cell cycle. To this end, we prepared CSF extracts, immediately added Ca2+ (without prior room temperature incubation, thereby promoting a more rapid release from the arrest), and monitored meiotic exit and exit from the following mitosis. As expected, Xnf7-depleted extracts progressed to interphase more rapidly than control extracts, degrading cyclin B by 10 min (compared with 20 min; Fig. 2 A) and reforming nuclei at 20 min (compared with 30 min; unpublished data). Once in the subsequent mitosis, cyclin B degradation (Fig. 2 B, 140 min) and nuclear reformation (Fig. 2 C, 160 min) in depleted extracts proceeded ahead of control extracts, suggesting that Xnf7 affects mitotic exit as well as meiotic exit. We also observed that depletion of Xnf7 from CSF extracts did not cause spontaneous release from CSF arrest without the addition of Ca2+ (Fig. 2 D), nor did the depletion of Xnf7 from interphase extracts induce spontaneous degradation of mitotic substrates (Fig. 2 E). Therefore, Xnf7 is not required for maintenance of the CSF arrest. Moreover, depletion of Xnf7 appeared to alter the rate of cyclin B degradation rather than the lag time until initiation of degradation in that both Xnf7-depleted and mock-depleted extracts appeared to initiate degradation immediately after Ca2+ addition to CSF extracts.
Figure 2.

Xnf7 depletion accelerates exit from mitosis but does not cause spontaneous activation of the APC. (A–C) CSF-arrested Xenopus egg extracts were depleted, supplemented with demembranated sperm chromatin and CaCl2, and incubated at 23°C. (A and B) Aliquots removed at the indicated times after CaCl2 addition were analyzed by SDS-PAGE and immunoblotting with anti–cyclin B2 antibodies. (C) Aliquots of extract were removed at the indicated times and nuclear morphology was visualized by Hoechst 33258 staining and fluorescence microscopy. (D) CSF extracts were depleted, supplemented with demembranated sperm chromatin, and incubated at 23°C. Aliquots removed at the indicated times were analyzed as in A. (E) Interphase extracts were depleted, supplemented with 35S–cyclin B1, and incubated at 23°C. Aliquots removed at the indicated times were analyzed by SDS-PAGE and autoradiography.
After finding that immunodepletion of Xnf7 accelerates the degradation of all APC substrates examined, we wanted to determine whether this effect was specific to APC substrates or if the absence of Xnf7 affected the degradation of other proteins, perhaps causing a global acceleration of proteasome-mediated degradation. To evaluate the effect of Xnf7 immunodepletion on the degradation of a non-APC substrate, we examined an inhibitor of apoptosis (IAP), a protein that does not exhibit distinct cell cycle–regulated turnover. IAPs are E3 ubiquitin ligases that can autoubiquitylate in vivo, marking themselves for destruction by the 26S proteasome (Yang et al., 2000). IAP autoubiquitylation and accelerated proteasomal degradation can be triggered by binding the proapoptotic protein reaper (Holley et al., 2002). Examination of IAP protein levels in control and Xnf7-depleted extracts supplemented with reaper revealed that IAP was degraded at nearly the same rate in the presence or absence of Xnf7 (Fig. 3 A), suggesting that the acceleration of degradation in the absence of Xnf7 was not universal and was likely specific to APC substrates. Together, these data indicated that either Xnf7 or associated codepleted proteins acted to suppress APC activation.
Figure 3.

Xnf7 modulates mitotic exit by altering ubiquitylation of APC substrates. (A) Xenopus egg extracts were depleted, supplemented with 35S-IAP, and treated with reaper and the broad-spectrum caspase inhibitor zVAD-fmk. Extracts were incubated at 23°C, and aliquots removed at the indicated times were analyzed by SDS-PAGE and autoradiography and quantitated by Phosphorimager. (B) Addition of anti-Xnf7 antibodies mimics immunodepletion of Xnf7. CSF extracts were treated with purified preimmune IgG, purified anti-Xnf7 antibodies, or purified anti-Xnf7 antibodies blocked with a COOH-terminal fragment of Xnf7 and incubated at 23°C for 15 min. After CaCl2 addition, aliquots were removed at the indicated times and analyzed by SDS-PAGE and immunoblotting with anti–cyclin B2 antibodies (left) or anti-Xnf7 antibodies (right). (C) Addition of recombinant Xnf7 to Xnf7-depleted extracts restores the normal timing of cyclin degradation. CSF extracts were depleted, treated with buffer or recombinant Xnf7, and incubated at 23°C for 30 min. CaCl2 was added to extracts, and aliquots removed at the indicated times after CaCl2 addition were analyzed as in B. (D) Addition of excess Xnf7 delays cyclin B degradation and mitotic exit. CSF extracts were treated with buffer or Xnf7WT and incubated at 23°C for 20 min. After CaCl2 addition, aliquots were removed at the indicated times and analyzed as in B. (E) Excess Xnf7 delays cyclin B ubiquitylation. Methyl-ubiquitin was added to the experiment described in D. Aliquots were removed at the indicated times, analyzed by SDS-PAGE and autoradiography, and quantitated by Phosphorimager.
To determine if removal of Xnf7 itself, rather than associated proteins, contributed to accelerated APC activation, we examined the effect of anti-Xnf7 antibody addition to extracts. Before Ca2+ addition, we treated CSF extracts with preimmune IgG, anti-Xnf7 antibodies, or anti-Xnf7 antibodies blocked with recombinant Xnf7 protein. Extracts treated with anti-Xnf7 antibodies exited meiosis more rapidly (as observed by microscopy) and degraded cyclin B more rapidly than extracts treated with preimmune IgG. Moreover, the effects of the anti-Xnf7 antibodies were neutralized by treatment with recombinant Xnf7 (Fig. 3 B, left; unpublished data). Note that addition of anti-Xnf7 antibodies did not alter the amount of Xnf7 in the extract (Fig. 3 B, right). Together with the effects of immunodepletion, these data suggested that Xnf7 protein antagonized APC-mediated substrate degradation. To address this issue further, we tested the ability of recombinant wild-type Xnf7 (Xnf7WT) protein to restore the “normal” timing of meiotic exit in extracts from which Xnf7 had been immunodepleted.
After depletion, CSF extracts were treated with buffer or Xnf7WT for 30 min at room temperature before Ca2+ addition. As expected, Xnf7-depleted extracts degraded cyclin B more rapidly than control extracts (Fig. 3 C). Addition of recombinant Xnf7 to depleted extracts restored the normal timing of APC-mediated substrate degradation, slowing cyclin B degradation to a rate similar to that observed in mock-depleted extracts. These data reinforce our conclusion that Xnf7 protein has an inhibitory effect on APC-mediated substrate degradation. Because addition of recombinant Xnf7WT could complement the inhibitory activity of the endogenous protein, we reasoned that the addition of excess recombinant Xnf7WT to egg extracts might delay or even prevent exit from CSF arrest. To examine the effect of increasing the Xnf7 concentration in the extract, we added buffer or Xnf7WT to CSF extracts before Ca2+ addition. Although the control extract reformed interphase nuclei 30 min after Ca2+ addition, Xnf7WT addition caused a meiotic arrest. Accordingly, cyclin B was destroyed by 12 min after Ca2+ addition in control extract, and was stabilized by addition of Xnf7WT (Fig. 3 D). These results implicate Xnf7 as an APC regulator and suggest that it is the removal of Xnf7, not a codepleting protein, that leads to accelerated APC-mediated substrate degradation and mitotic exit.
To further confirm that Xnf7 inhibited mitotic exit at the level of ubiquitylation of APC substrates, not at the level of degradation by the 26S proteasome, we repeated the experiment described in the previous paragraph in the presence of methyl-ubiquitin to “trap” cyclin B in its ubiquitylated form. Although cyclin B ubiquitylation peaked at 10–12 min in the control extract, Xnf7WT addition delayed this peak (Fig. 3 E, 16–20 min). Moreover, cyclin B was eventually destroyed in the control extract (presumably due to ubiquitylation with endogenous ubiquitin following the action of deubiquitylating enzymes) but was stabilized by the addition of Xnf7WT.
Xnf7 is a ubiquitin ligase
In analyzing the Xnf7 sequence, Reddy et al. (1991) noted the presence of two putative zinc-binding domains and speculated that Xnf7 might be a transcription factor. However, as one zinc-binding region contains a consensus sequence typical of RING domains found in ubiquitin ligases, we suspected that Xnf7 might function as a ubiquitin ligase (Fig. 4 A). Accordingly, we performed in vitro ubiquitylation assays using purified E1, E2 (UbcH5a), ubiquitin, and recombinant Xnf7 as the only source of E3 activity. In addition to testing Xnf7WT, we constructed Xnf7 RING finger mutants in which conserved cysteine residues that coordinate zinc binding were replaced by alanine. Xnf7 mutant 3 (Xnf7m3) contained a mutation at position 3 of the RING finger (C160A) and Xnf7 mutant 36 (Xnf7m36) contained mutations at positions 3 and 6 (C160A and C168A; Fig. 4 A). As with many ubiquitin ligases in vitro, Xnf7WT was able to autoubiquitylate, converting the single electrophoretic form of Xnf7 into a polyubiquitylated form (Fig. 4 B). Mutation of the RING finger inhibited this activity. We note that Xnf7 was not able to ubiquitylate cyclin B or securin proteins directly (unpublished data), and that Xnf7 cannot use UbcH10, the ubiquitin-conjugating enzyme that functions with the APC, as a source of E2 in the autoubiquitylation reaction (Fig. 4 C, left). Note that all E2 enzymes were functional in E2-thioester assays (Fig. 4 C, right; unpublished data).
Figure 4.

Xnf7 is a ubiquitin ligase and its ligase activity is required for its effects on mitotic exit. (A) Xnf7 contains a RING finger consensus sequence. The eight amino acids in bold are the zinc-binding sites of the RING finger. Xnf7WT, Xnf7 mutant 3 (Xnf7m3) (C160A), and Xnf7 mutant 36 (Xnf7m36) (C160A and C168A) were produced in bacteria. (B) Xnf7 has ubiquitin ligase activity. Xnf7WT (lane 1), Xnf7m3 (lane 2), or Xnf7m36 (lane 3) were incubated with purified E1, UbcH5a, ubiquitin, and ATP. Lanes 4–7 contained Xnf7WT but lacked one component of the ubiquitylation reaction. Lane 4, −E1; lane 5, −UbcH5a; lane 6, −ATP; lane 7, −ubiquitin. Reactions were incubated at 37°C for 2 h and were analyzed by SDS-PAGE and immunoblotting with anti-ubiquitin or anti-Xnf7 antibodies. White line indicates that intervening lanes have been spliced out. (C) Xnf7 can function with several E2s but cannot function with UbcH10. (left) Xnf7WT was incubated as in B but with each reaction containing only the E2 indicated. The absence of E2 in the reaction is indicated (−). (right) E2-thioester assays demonstrate that E2s used are functional. The presence of the E2 alone (−) and the E2 following a 30-min thioester assay (+) are indicated. Similar results were obtained for all E2s used (unpublished data). (D) Xnf7's E3 ligase activity is required for its effects on mitotic exit. CSF extracts were treated with buffer, Xnf7WT, or Xnf7m36 and incubated at 23°C for 20 min. After CaCl2 addition, aliquots were removed at the indicated times and analyzed by SDS-PAGE and immunoblotting with anti–cyclin B2 antibodies. (E) Xnf7m36 blocks the function of endogenous Xnf7 protein. CSF extracts were treated with buffer or increasing amounts of Xnf7m36 and incubated at 23°C for 20 min. After CaCl2 addition, aliquots were removed and analyzed as in D. (F) Xnf7's ubiquitin ligase activity is required for its effect on APC substrate ubiquitylation. Methyl-ubiquitin was added to the experiment described in D. Aliquots were removed at the indicated times, analyzed by SDS-PAGE and autoradiography, and quantitated by Phosphorimager. White lines indicate that samples run on the same gel were reordered.
Given that Xnf7 has intrinsic ubiquitin ligase activity, we sought to determine whether or not this activity was required for its effects on mitotic exit. We therefore compared the effects of Xnf7WT or Xnf7m36 addition to extracts. As previously shown in Fig. 3 D, addition of excess Xnf7WT prevented cyclin B degradation and mitotic exit. Addition of Xnf7m36 did not stabilize cyclin B; rather, cyclin B was degraded (Fig. 4 D), demonstrating that Xnf7's ligase activity is required for its effects on mitotic exit. Indeed, addition of excess Xnf7m36 actually accelerated cyclin B degradation relative to buffer-treated control extracts, suggesting that this mutant protein may act in a dominant-negative fashion to antagonize the effects of endogenous Xnf7 (Fig. 4 E). In the presence of methyl-ubiquitin, cyclin B ubiquitylation peaked at 12–16 min after Ca2+ addition in the Xnf7m36-treated extract, whereas Xnf7WT addition slowed ubiquitylation of cyclin B, delaying the peak until 20 min (Fig. 4 F). As expected, cyclin B was destroyed eventually in the Xnf7m36-treated extract but was stabilized by Xnf7WT.
Xnf7 inhibits the APC
To determine if the inhibitory effect of Xnf7 was exerted directly on the APC, APC was immunopurified from mitotic egg extracts using anti-Cdc27 antibodies and treated with recombinant Xnf7WT. APC inhibitors Mad2 and Emi1 have been shown previously to inhibit the APC in this manner via their interactions with Cdc20 (Fang et al., 1998a; Reimann et al., 2001). As shown in Fig. 5 A, addition of Cdc20 to APC immunoprecipitates activated the APC, promoting ubiquitylation of cyclin B. Preincubation of Cdc20 with MBP-Emi1 significantly inhibited the APC; however, preincubation with recombinant Xnf7WT or Xnf7m36 had no discernible effect.
Figure 5.
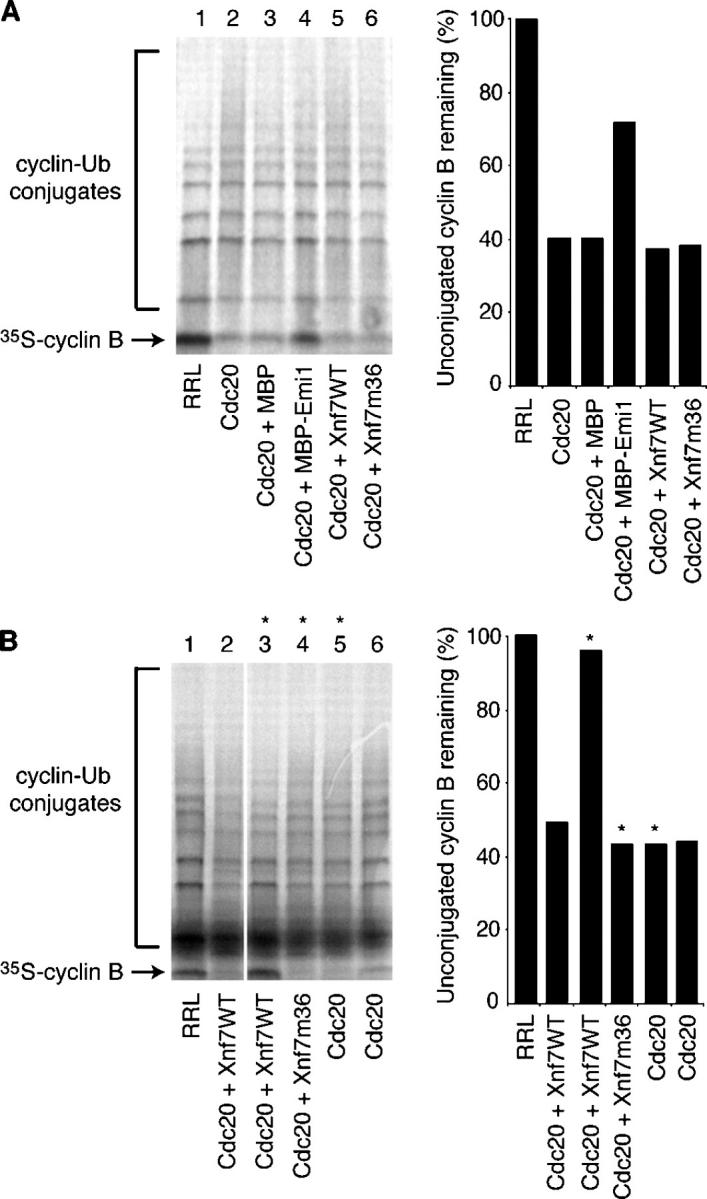
Xnf7 inhibits the APC. (A) RRL (lane 1) or Cdc20 (lanes 2–6) was incubated (30 min at 23°C) with buffer (lanes 1 and 2), 6 μM MBP (lane 3), 6 μM MBP-Emi1 (lane 4), 2 μM Xnf7WT (lane 5), or 2 μM Xnf7m36 (lane 6). APC was immunopurified from mitotic egg extracts with anti-Cdc27 beads and incubated with the RRL/Cdc20 mixtures (1 h at 23°C). APC beads were washed and assayed for cyclin ubiquitylation activity using a 35S-labeled in vitro transcribed-translated Xenopus cyclin B1 fragment (1–126 aa) as a substrate. Samples were run on a 4–15% Tris-HCl SDS-PAGE gel. Unconjugated cyclin B remaining was quantitated by Phosphorimager. (B) Similar to A, except that incubations included 5 μM Xnf7WT or Xnf7m36 and were performed under conditions permissive for Xnf7's ligase activity where noted (*). Note that more than doubling the concentration of Xnf7 (compared with that used in A) did not result in APC inhibition unless the assay was performed under ubiquitylating conditions. Samples were run on a 4–20% Tris-HCl SDS-PAGE gel. White line indicates that intervening lanes have been spliced out.
Because we had clearly observed an effect of Xnf7 on APC function in extracts (containing a full complement of cellular components) and Xnf7's enzymatic activity was required for its effect on mitotic exit (Fig. 4, D and F), we reasoned that the inhibitory effect of Xnf7 on the APC might be evident only under conditions that would allow Xnf7 to function as a ubiquitin ligase. (Note that the in vitro Xnf7 ubiquitylation assay was performed using UbcH5a and that the purified APC assay contained only UbcH10. Xnf7 does not function as a ligase with UbcH10 [Fig. 4 C, left].) To this end, UbcH5a was added during the incubation of Xnf7 with APC immunoprecipitates to create conditions permissive for Xnf7's enzymatic activity. Whereas Xnf7WT alone failed to inhibit the APC (Fig. 5 B, lane 2), Xnf7WT incubated under ubiquitylating conditions did, indeed, inhibit the purified APC (Fig. 5 B, lane 3). In contrast, Xnf7m36 protein did not inhibit the APC (Fig. 5 B, lane 4), consistent with a requirement for Xnf7's enzymatic activity in its direct inhibition of the APC. The presence of Xnf7 in the assay did not prevent Cdc20 from binding to the APC (unpublished data).
Xnf7 interacts with a core subunit of the APC
Given the inhibitory effects of Xnf7 on the activity of the APC, we sought to determine how Xnf7 might be exerting its effects. To investigate the potential association of Xnf7 with the APC, we separated CSF extracts on 10–40% sucrose gradients and examined the sedimentation of Xnf7, APC subunits Apc2 and Cdc27/Apc3, and the APC activator Cdc20. Xnf7 was found mainly in lower molecular mass fractions of ∼100–250 kD (Fig. 6 A, fractions 2–7) along with Cdc20 (Fig. 6 A, fractions 2–6), but peaked again with the ∼1.5-MD APC complex in fraction 11, where APC subunits Apc2 and Cdc27/Apc3 also sedimented (Fig. 6 A). Although a considerable amount of Xnf7 did not cosediment with APC components, Xnf7 immunoprecipitations from selected cosedimenting fractions (Fig. 6 A, *) revealed that Cdc27/Apc3 coimmunoprecipitated with Xnf7 from APC-containing fractions but that Cdc20 did not coimmunoprecipitate (Fig. 6 B), even in fractions where they were both abundantly represented. Similarly negative results were obtained when fractions were immunoprecipitated with anti-Cdc20 antibodies and immunoblotted with anti-Xnf7 antibodies (unpublished data). These data strongly suggest that Xnf7 acts in a manner distinct from previously reported APC regulators, most likely through direct interaction with a core APC subunit. That being said, we note that Xnf7 coimmunoprecipitated equally well with Cdc27 from CSF extracts before and after Ca2+ addition, suggesting that Xnf7 functions other than binding (possibly ubiquitin ligase activity) are required for Xnf7-mediated APC inhibition (Fig. 6 C).
Figure 6.

Xnf7 associates with the core APC. (A) Sucrose gradient cosedimentation of Xnf7 and APC subunits. CSF egg extract was fractionated on a 10–40% sucrose gradient. Fractions were collected and analyzed by SDS-PAGE and immunoblotting for the indicated proteins. Asterisks indicate the fractions used in the immunoprecipitations in B. (B) Xnf7 coimmunoprecipitates with Cdc27 from sucrose gradient fractions. Purified anti-Xnf7 antibodies or purified preimmune IgG were coupled to protein A–Sepharose and incubated with sucrose gradient fractions 4, 10, 11, and 18 (1 h at 4°C). Beads were washed and bound proteins were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies. (C) Xnf7 coimmunoprecipitates with Cdc27 during CSF arrest and during exit from arrest. Purified anti-Xnf7 antibodies were coupled to protein A–Sepharose and incubated with aliquots of CSF extracts removed at the indicated times after CaCl2 addition (1 h at 4°C). Beads were washed and bound proteins were analyzed by SDS-PAGE and immunoblotting with anti-Cdc27 antibodies.
Xnf7 is required for the spindle assembly checkpoint
Given the ability of Xnf7 to inhibit the APC and the previously reported localization of Xnf7 to the mitotic spindle (Li et al., 1994), we sought to determine if Xnf7 might play a role in the spindle assembly checkpoint. As described by Minshull et al. (1994), Xenopus extracts can be induced to undergo a spindle checkpoint arrest after the addition of high concentrations of demembranated sperm chromatin and nocodazole. To determine if Xnf7 might play a role in spindle checkpoint function, CSF extracts were preincubated with preimmune IgG or anti-Xnf7 antibodies for 15 min to functionally inhibit Xnf7. After incubation with high concentrations of sperm chromatin and nocodazole, Ca2+ was added. Although control extracts lacking nocodazole did not arrest, extracts containing nocodazole and preimmune IgG established a spindle checkpoint and therefore failed to degrade cyclin B (Fig. 7). In contrast, addition of anti-Xnf7 antibodies effectively overrode the spindle checkpoint, allowing cyclin B degradation and rapid reformation of interphase nuclei, even in the presence of nocodazole. These data demonstrate that Xnf7 is required for spindle assembly checkpoint function in Xenopus egg extracts and suggest that Xnf7 may provide a link between spindle status and the core APC machinery.
Figure 7.

Xnf7 is required for the spindle assembly checkpoint in egg extracts. CSF extracts were preincubated with preimmune IgG or anti-Xnf7 antibodies for 15 min at 23°C. Demembranated sperm chromatin (9,000/μl) and nocodazole (10 μg/ml) were added for 30 min, followed by the addition of CaCl2. Aliquots removed at the indicated times after CaCl2 addition were analyzed by SDS-PAGE and immunoblotting with anti–cyclin B2 antibodies. White line indicates samples run on the same gel were rearranged. Aliquots were also removed and visualized by Hoechst 33258 staining. Photographs of nuclear morphology at 60 min are shown.
Discussion
Activation of the APC is controlled in part by Cdc20/Cdh1 as well as their upstream regulators, including BubR1, Bub3, Mad2, and Emi1 (Zachariae and Nasmyth, 1999; Peters, 2002). Here, we have identified Xnf7 as a novel inhibitor of APC function unlikely to act via Cdc20. In addition, we have shown that Xnf7 itself is a ubiquitin ligase and that its ligase activity is required for its effects on APC-dependent ubiquitylation. Because Xnf7 cosedimented and coimmunoprecipitated with Cdc27, it is attractive to speculate that Xnf7 modifies either Cdc27 function or the function of other associated core APC subunits. Furthermore, we have shown that Xnf7 is required for spindle assembly checkpoint function in Xenopus egg extracts, suggesting that Xnf7 could be part of a mechanism for direct regulation of the APC by the spindle checkpoint (a model recently proposed by Acquaviva et al. [2004]).
Identification of Xnf7 as a cyclin B2–interacting protein
Although we identified Xnf7 as a cyclin B2–interacting protein, we have shown that Xnf7 also affects the degradation of cyclin B1 and securin, and, given its inhibitory effect on the APC, is likely to affect other APC substrates as well. Because Xnf7 is phosphorylated by Cdc2/cyclin B and mitogen-activated protein kinase during oocyte maturation (Reddy et al., 1991; El-Hodiri et al., 1997a), we speculate that the specific interaction with cyclin B2 might reflect an enzyme–substrate interaction due to phosphorylation of Xnf7 by Cdc2/cyclin B2.
Regulation of APC activation
Activation of the APC is controlled not only by the binding of activating proteins Cdc20 and Cdh1 but is also regulated by phosphorylation of its subunits by mitotic kinases including Cdc2/cyclin B, Plk1, and PKA (for review see Castro et al., 2005). One way in which mitotic phosphorylation of APC subunits promotes its activation is by facilitating the binding of Cdc20 (Kramer et al., 2000). In budding yeast, mutation of Cdk1 sites in APC subunits Cdc16, Cdc23, and Cdc27 inhibits APC phosphorylation, reduces Cdc20 binding, and delays mitotic exit, suggesting that phosphorylation of the APC is essential for its activation (Rudner and Murray, 2000). However, it is clear that the absence of APC subunit phosphorylation during interphase does not render the APC impervious to activation in that APC from interphase extracts can be activated in vitro by Cdc20 or Cdh1 (Fang et al., 1998b) or in vivo by overexpression of either activator at any stage in the cell cycle (Visintin et al., 1997). Thus, multiple mechanisms are likely to cooperate to ensure that the APC is not activated prematurely. Our data suggest that Xnf7 participates in one such mechanism.
Xnf7's ligase activity is required for its inhibition of the APC
Consistent with its role as an inhibitor of APC activity, excess Xnf7 was able to slow degradation of APC substrates and concomitantly delay mitotic exit. Intriguingly, this function of Xnf7 required that it be active as a ubiquitin ligase. Moreover, in our studies using mitotic APC immunoprecipitates to investigate direct inhibition of the APC, inhibition by Xnf7 required the presence of Xnf7WT in conditions permissive for Xnf7's enzymatic activity (E1, UbcH5a, and ubiquitin). It is not yet clear whether this reflects a requirement for Xnf7 autoubiquitylation in its ability to regulate the APC or the necessity for ubiquitylation of a substrate in the APC immunoprecipitates. Although it is formally possible that Xnf7 could promote ubiquitin-dependent degradation of APC components, the level of APC subunits examined has not been found to vary throughout the cell cycle. However, because ubiquitylation can modify substrate function without targeting it for degradation, it is possible that Xnf7 inhibits APC function by ubiquitylation of an APC subunit. Indeed, several ubiquitin ligases have been reported to modify substrate localization and/or protein–protein interactions through monoubiquitylation of targets (Hicke, 2001). With regard to the possibility that either autoubiquitylation or ubiquitylation of APC components might be important for Xnf7 function, it is interesting to note that Xnf7 binding to the APC appeared to be equal before and after release from CSF arrest (when the APC is differentially active). Moreover, direct association of Xnf7 with the APC was not measurably altered by the spindle checkpoint (unpublished data). These observations strongly suggest that binding alone is not sufficient for Xnf7-mediated inhibition of the APC. It is attractive to speculate that Xnf7's E3 ligase activity may be cell cycle regulated, and thus responsible for its cell cycle–specific actions. Alternatively, or additionally, cell cycle–specific modification of Xnf7 itself (e.g., phosphorylation or autoubiquitylation) may be required for it to effectively inhibit the APC.
Recently, it was reported that the core subunits of the APC, rather than Cdc20/Cdh1, are directly responsible for substrate recognition (Carroll and Morgan, 2002; Meyn et al., 2002; Passmore et al., 2003; Yamano et al., 2004). Given our finding that Xnf7 inhibits the APC via association with core APC subunits, it is plausible that this novel APC inhibitor exerts its effects by interfering with substrate recognition, perhaps through ubiquitylation of an APC subunit. Ideally, we would like to address this issue directly by determining if Xnf7 can ubiquitylate proteins present in the anti-Cdc27 precipitate. Unfortunately, this approach has been hampered thus far by the high level of ubiquitylation associated with the APC itself.
Although we have identified a role for Xnf7 in regulating APC activation, previously reported data have implicated Xnf7 as a regulator of dorsal–ventral polarity establishment in Xenopus (El-Hodiri et al., 1997b). Because ubiquitin ligases may have multiple substrates and therefore participate in several signaling pathways, it is possible that these two roles of Xnf7 are entirely distinct. However, it is interesting to note that reduction-of-function APC mutations in Caenorhabditis elegans can disrupt establishment of anterior–posterior polarity (Rappleye et al., 2002). Thus, Xnf7 could potentially regulate polarity establishment in Xenopus via its effects on the APC.
Collectively, the data reported here strongly support a role for Xnf7 in regulating APC activation and suggest that Xnf7 participates in the spindle checkpoint. Interestingly, it has recently been reported that the APC colocalizes with spindle checkpoint protein complexes at unattached kinetochores and that the spindle checkpoint machinery is required for the recruitment of the APC to these sites (Acquaviva et al., 2004). These data suggest that the APC can be regulated directly by the spindle checkpoint. Given our finding that Xnf7 is required for spindle checkpoint function in egg extracts, it is attractive to speculate that Xnf7 might be involved in such a mechanism. Future identification of direct Xnf7 ubiquitin ligase substrates should help to elucidate the mechanism of Xnf7-mediated APC inhibition while helping to further define the links between spindle function and APC activation.
Materials and methods
Recombinant protein expression
Wild-type Xnf7 was subcloned into the EcoRI and NcoI sites of pGexKG. Xnf7 RING-finger mutants Xnf7m3 (C160A) and Xnf7m36 (C160A and C168A) were prepared using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). To prepare pGexKG-Xnf7C, Xnf7 (257–609 aa) was PCR amplified and subcloned into the EcoRI and NcoI sites of pGex-KG. GST fusion proteins expressed in bacteria were purified on glutathione-Sepharose by standard protocols. Proteins were cleaved with thrombin and dialyzed against Xnf7 buffer (XB-Xnf7: 100 mM KCl, 25 mM sucrose, 10 mM Hepes, 1 mM MgCl2, and 1 mM DTT, pH 7.8). Uncleaved GST-Xnf7C was dialyzed against PBS.
CRS affinity chromatography
Glutathione-Sepharose coupled to GST, GST-cyclin B1 CRS, or GST-cyclin B2 CRS (Yang et al., 1998) were incubated with egg extracts. Bound material was biotinylated, resolved by SDS-PAGE, transferred to nitrocellulose, and probed with HRP-streptavidin. For Coomassie blue staining, bound material was washed and resolved by SDS-PAGE. The ∼80-kD band of interest was analyzed by mass spectrometry at Harvard Microchemistry.
Antibodies and immunodepletions
Rabbit polyclonal antibodies raised against Xnf7C were affinity purified on glutathione-Sepharose covalently coupled to GST-Xnf7C with dimethyl pimelimidate. Other antibodies used were as follows: anti–cyclin B1 and anti–cyclin B2 (Hochegger et al., 2001), anti-APC2 (a gift from G. Fang, Stanford University, Stanford, CA), anti-ubiquitin (Santa Cruz Biotechnology, Inc.), anti-Cdc27 (Santa Cruz Biotechnology, Inc.) for immunoprecipitation, anti-Cdc27 (Transduction Laboratories) for immunoblotting, and anti-Cdc20 (Santa Cruz Biotechnology, Inc.).
For immunodepletions, purified anti-Xnf7 antibodies or preimmune IgG coupled to protein A–Sepharose were washed in XB-CSF (100 mM KCl, 50 mM sucrose, 10 mM Hepes, pH 7.7, 5 mM EGTA, 2 mM MgCl2, and 100 μM CaCl2) or ELB (250 mM sucrose, 50 mM KCl, 10 mM Hepes, pH 7.7, 2.5 mM MgCl2, and 1 mM dithiothreitol), incubated with extracts (0.2 μl beads/μl extract, 30 min at 4°C), and removed by centrifugation (10,000 rpm, 1 min at 4°C).
Degradation assays
Xenopus egg extracts were prepared as described previously (Murray, 1991). Degradation assays were performed in CSF extracts by adding CaCl2 to 0.6 mM to initiate anaphase. Human securin (a gift from J. Pines, Wellcome Trust/Cancer Research UK, Cambridge, UK) was translated in vitro in the presence of 35S-methionine/cysteine. Aliquots were removed and analyzed by SDS-PAGE followed by autoradiography (35S-securin) or immunoblotting (cyclin B). In all degradation assays, the same amount of protein was loaded in each gel lane. For interphase extracts, 35S-labeled cyclin B1 or IAP was added, and aliquots were analyzed as 35S-securin. To stimulate IAP autoubiquitylation, Drosophila melanogaster reaper was added to extracts (37.5 μM final) with 100 μM zVAD-fmk (BIOMOL Research Laboratories).
For antibody addition to extracts, Xnf7 antibodies and preimmune IgG were concentrated to 3 μg/μl. Blocked Xnf7 antibodies were prepared by incubating with GST-Xnf7C. After antibody addition (0.3 μg antibody/μl extract), extracts were incubated at 23°C for 15 min before addition of CaCl2. For Xnf7 protein addition, buffer, Xnf7WT, or Xnf7m36 was added (0.06 μg protein/μl extract), followed by 15 min incubation at 23°C before addition of CaCl2. Methyl-ubiquitin (3 μg/μl final) and 35S-labeled Xenopus cyclin B1 fragment (1–126 aa) were added in the indicated experiments. For Xnf7 protein addition to depleted extracts, buffer or Xnf7WT was added (0.09 μg protein/μl extract), followed by 30 min incubation at 23°C before CaCl2 addition.
Spindle assembly checkpoints were formed as described previously (Minshull et al., 1994). DNA was stained with Hoechst 33258 and nuclear morphology was examined using a microscope (model Axioskop; Carl Zeiss MicroImaging, Inc.) with a 40× Plan-Neofluar objective (NA 0.75) or a 100× Plan-Apochromat objective (NA 1.4). Images were captured using a Pentamax cooled charge-coupled device camera (Princeton Instruments) interfaced with MetaMorph software (Universal Imaging Corp.).
Xnf7 ubiquitylation assay
4 μg Xnf7WT, Xnf7m3, or Xnf7m36 were incubated (2 h at 37°C) with 50 ng of rabbit E1, 1 μg UbcH5a, 10 μg ubiquitin, and 3 mM ATP brought to 50 μl with buffer (25 mM Hepes, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 10 mM DTT, and 0.05% Triton X-100). Reactions were stopped with SDS sample buffer, resolved by SDS-PAGE, and analyzed by immunoblotting.
E2-Thioester assay
2 μg of each E2 were incubated (30 min at 37°C) with 0.5 μg of rabbit E1, 40 μg ubiquitin, 10 mM ATP, and 10 mM MgCl2 in a 10-μl total volume. Reactions stopped with SDS sample buffer were resolved on 4–20% Tris-HCl SDS-PAGE gels and visualized by silver stain.
In vitro APC assay
Anti-Cdc27 antibodies coupled to protein A Dynabeads were washed in ELB and incubated (2.5 h at 4°C) with mitotic extracts. APC beads were washed with 500 mM KCl, 20 mM Hepes, pH 7.7, and 0.5% NP-40 followed by 20 mM Hepes, pH 7.7. Beads were incubated (1 h at 23°C) with RRL or in vitro translated human Cdc20 that had been preincubated with buffer, 6 μM MBP, 6 μM MBP-Emi1, or 2–5 μM Xnf7WT or Xnf7m36. The incubation also included 80 ng of rabbit E1, 120 ng UbcH5a, 10 μg of bovine ubiquitin, 3 mM ATP, and 3 mM MgCl2 where indicated. Beads were washed with 20 mM Hepes, pH 7.7, and assayed for cyclin ubiquitylation activity with 1.25 μg/μl of bovine ubiquitin, 60 ng/μl E1, 40 ng/μl UbcH10, an energy regenerating system, 2 μl of 35S-labeled in vitro translated Xenopus cyclin B1 fragment (1–126 aa) as a substrate, and 3 μl APC beads. Reactions were incubated at 23°C for 1 h, quenched with SDS sample buffer, analyzed by SDS-PAGE and autoradiography, and quantitated by Phosphorimager.
Sucrose gradient centrifugation
CSF extract diluted 1:3 in XB-CSF without sucrose was cleared at 40,000 rpm and resolved on a 12-ml 10–40% wt/vol sucrose gradient by centrifugation in an SW40 rotor (30,000 rpm, 18 h at 4°C). 20 0.5-ml fractions were collected and analyzed by SDS-PAGE and immunoblotting.
Acknowledgments
We thank Guowei Fang for anti-Xenopus APC2 antibodies and Jon Pines for the human securin construct. We are also grateful to Adam Eldridge for helpful discussions and to Christopher Freel for assistance with figure preparation.
This research was supported by a grant from the National Institutes of Health to S. Kornbluth (RO1 GM067225). J.B. Casaletto is a predoctoral fellow of the Howard Hughes Medical Institute.
Abbreviations used in this paper: APC, anaphase-promoting complex/cyclosome; CRS, cytoplasmic retention sequence; CSF, cytostatic factor; IAP, inhibitor of apoptosis; RRL, rabbit reticulocyte lysate; Xnf7, Xenopus nuclear factor 7.
References
- Acquaviva, C., F. Herzog, C. Kraft, and J. Pines. 2004. The anaphase promoting complex/cyclosome is recruited to centromeres by the spindle assembly checkpoint. Nat. Cell Biol. 6:892–898. [DOI] [PubMed] [Google Scholar]
- Amon, A. 1999. The spindle checkpoint. Curr. Opin. Genet. Dev. 9:69–75. [DOI] [PubMed] [Google Scholar]
- Burton, J.L., and M.J. Solomon. 2001. D box and KEN box motifs in budding yeast Hsl1p are required for APC-mediated degradation and direct binding to Cdc20p and Cdh1p. Genes Dev. 15:2381–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, C.W., and D.O. Morgan. 2002. The Doc1 subunit is a processivity factor for the anaphase-promoting complex. Nat. Cell Biol. 4:880–887. [DOI] [PubMed] [Google Scholar]
- Castro, A., C. Bernis, S. Vigneron, J.C. Labbe, and T. Lorca. 2005. The anaphase-promoting complex: a key factor in the regulation of cell cycle. Oncogene. 24:314–325. [DOI] [PubMed] [Google Scholar]
- den Elzen, N., and J. Pines. 2001. Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol. 153:121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hodiri, H.M., S. Che, M. Nelman-Gonzalez, J. Kuang, and L.D. Etkin. 1997. a. Mitogen-activated protein kinase and cyclin B/Cdc2 phosphorylate Xenopus nuclear factor 7 (xnf7) in extracts from mature oocytes. Implications for regulation of xnf7 subcellular localization. J. Biol. Chem. 272:20463–20470. [DOI] [PubMed] [Google Scholar]
- El-Hodiri, H.M., W. Shou, and L.D. Etkin. 1997. b. xnf7 functions in dorsal-ventral patterning of the Xenopus embryo. Dev. Biol. 190:1–17. [DOI] [PubMed] [Google Scholar]
- Fang, G. 2002. Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol. Biol. Cell. 13:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, G., H. Yu, and M.W. Kirschner. 1998. a. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12:1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, G., H. Yu, and M.W. Kirschner. 1998. b. Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell. 2:163–171. [DOI] [PubMed] [Google Scholar]
- Funabiki, H., and A.W. Murray. 2000. The Xenopus chromokinesin Xkid is essential for metaphase chromosome alignment and must be degraded to allow anaphase chromosome movement. Cell. 102:411–424. [DOI] [PubMed] [Google Scholar]
- Geley, S., E. Kramer, C. Gieffers, J. Gannon, J.M. Peters, and T. Hunt. 2001. Anaphase-promoting complex/cyclosome–dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol. 153:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan, A., Y. Yudkovsky, and A. Hershko. 2002. The cyclin-ubiquitin ligase activity of cyclosome/APC is jointly activated by protein kinases Cdk1-cyclin B and Plk. J. Biol. Chem. 277:15552–15557. [DOI] [PubMed] [Google Scholar]
- Hagting, A., M. Jackman, K. Simpson, and J. Pines. 1999. Translocation of cyclin B1 to the nucleus at prophase requires a phosphorylation-dependent nuclear import signal. Curr. Biol. 9:680–689. [DOI] [PubMed] [Google Scholar]
- Harper, J.W., J.L. Burton, and M.J. Solomon. 2002. The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev. 16:2179–2206. [DOI] [PubMed] [Google Scholar]
- Hershko, A., and A. Ciechanover. 1998. The ubiquitin system. Annu. Rev. Biochem. 67:425–479. [DOI] [PubMed] [Google Scholar]
- Hicke, L. 2001. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2:195–201. [DOI] [PubMed] [Google Scholar]
- Hochegger, H., A. Klotzbucher, J. Kirk, M. Howell, K. le Guellec, K. Fletcher, T. Duncan, M. Sohail, and T. Hunt. 2001. New B-type cyclin synthesis is required between meiosis I and II during Xenopus oocyte maturation. Development. 128:3795–807. [DOI] [PubMed] [Google Scholar]
- Holley, C.L., M.R. Olson, D.A. Colon-Ramos, and S. Kornbluth. 2002. Reaper eliminates IAP proteins through stimulated IAP degradation and generalized translational inhibition. Nat. Cell Biol. 4:439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, L.H., L.F. Lau, D.L. Smith, C.A. Mistrot, K.G. Hardwick, E.S. Hwang, A. Amon, and A.W. Murray. 1998. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 279:1041–1044. [DOI] [PubMed] [Google Scholar]
- Irniger, S., S. Piatti, C. Michaelis, and K. Nasmyth. 1995. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 81:269–278. [DOI] [PubMed] [Google Scholar]
- Jackman, M., M. Firth, and J. Pines. 1995. Human cyclins B1 and B2 are localized to strikingly different structures: B1 to microtubules, B2 primarily to the Golgi apparatus. EMBO J. 14:1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang, Y.L., J. Huang, J.M. Peters, M.E. McLaughlin, C.Y. Tai, and D. Pellman. 1997. APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science. 275:1311–1314. [DOI] [PubMed] [Google Scholar]
- King, R.W., J.M. Peters, S. Tugendreich, M. Rolfe, P. Hieter, and M.W. Kirschner. 1995. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 81:279–288. [DOI] [PubMed] [Google Scholar]
- Kotani, S., S. Tugendreich, M. Fujii, P.M. Jorgensen, N. Watanabe, C. Hoog, P. Hieter, and K. Todokoro. 1998. PKA and MPF-activated polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol. Cell. 1:371–380. [DOI] [PubMed] [Google Scholar]
- Kraft, C., F. Herzog, C. Gieffers, K. Mechtler, A. Hagting, J. Pines, and J.M. Peters. 2003. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 22:6598–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, E.R., N. Scheuringer, A.V. Podtelejnikov, M. Mann, and J.M. Peters. 2000. Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell. 11:1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X., W. Shou, M. Kloc, B.A. Reddy, and L.D. Etkin. 1994. The association of Xenopus nuclear factor 7 with subcellular structures is dependent upon phosphorylation and specific domains. Exp. Cell Res. 213:473–481. [DOI] [PubMed] [Google Scholar]
- Lorca, T., F.H. Cruzalegui, D. Fesquet, J.C. Cavadore, J. Mery, A. Means, and M. Doree. 1993. Calmodulin-dependent protein kinase II mediates inactivation of MPF and CSF upon fertilization of Xenopus eggs. Nature. 366:270–273. [DOI] [PubMed] [Google Scholar]
- Lorca, T., A. Castro, A.M. Martinez, S. Vigneron, N. Morin, S. Sigrist, C. Lehner, M. Doree, and J.C. Labbe. 1998. Fizzy is required for activation of the APC/cyclosome in Xenopus egg extracts. EMBO J. 17:3565–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyn, M.A., III, P.G. Melloy, J. Li, and S.L. Holloway. 2002. The destruction box of the cyclin Clb2 binds the anaphase-promoting complex/cyclosome subunit Cdc23. Arch. Biochem. Biophys. 407:189–195. [DOI] [PubMed] [Google Scholar]
- Minshull, J., H. Sun, N.K. Tonks, and A.W. Murray. 1994. A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell. 79:475–486. [DOI] [PubMed] [Google Scholar]
- Murray, A.W. 1991. Cell cycle extracts. Methods Cell Biol. 36:581–605. [PubMed] [Google Scholar]
- Papin, C., C. Rouget, T. Lorca, A. Castro, and E. Mandart. 2004. XCdh1 is involved in progesterone-induced oocyte maturation. Dev. Biol. 272:66–75. [DOI] [PubMed] [Google Scholar]
- Passmore, L.A., E.A. McCormack, S.W. Au, A. Paul, K.R. Willison, J.W. Harper, and D. Barford. 2003. Doc1 mediates the activity of the anaphase-promoting complex by contributing to substrate recognition. EMBO J. 22:786–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, J.M. 2002. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell. 9:931–943. [DOI] [PubMed] [Google Scholar]
- Pfleger, C.M., E. Lee, and M.W. Kirschner. 2001. Substrate recognition by the Cdc20 and Cdh1 components of the anaphase-promoting complex. Genes Dev. 15:2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines, J., and T. Hunter. 1994. The differential localization of human cyclins A and B is due to a cytoplasmic retention signal in cyclin B. EMBO J. 13:3772–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappleye, C.A., A. Tagawa, R. Lyczak, B. Bowerman, and R.V. Aroian. 2002. The anaphase-promoting complex and separin are required for embryonic anterior-posterior axis formation. Dev. Cell. 2:195–206. [DOI] [PubMed] [Google Scholar]
- Reddy, B.A., M. Kloc, and L. Etkin. 1991. The cloning and characterization of a maternally expressed novel zinc finger nuclear phosphoprotein (xnf7) in Xenopus laevis. Dev. Biol. 148:107–116. [DOI] [PubMed] [Google Scholar]
- Reimann, J.D., E. Freed, J.Y. Hsu, E.R. Kramer, J.M. Peters, and P.K. Jackson. 2001. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell. 105:645–655. [DOI] [PubMed] [Google Scholar]
- Rudner, A.D., and A.W. Murray. 2000. Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol. 149:1377–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, A., P.I. Duncan, N.R. Rauh, G. Sauer, A.M. Fry, E.A. Nigg, and T.U. Mayer. 2005. Xenopus polo-like kinase Plx1 regulates XErp1, a novel inhibitor of APC/C activity. Genes Dev. 19:502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab, M., M. Neutzner, D. Mocker, and W. Seufert. 2001. Yeast Hct1 recognizes the mitotic cyclin Clb2 and other substrates of the ubiquitin ligase APC. EMBO J. 20:5165–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, J.V., and D.W. Cleveland. 2000. Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell. 103:997–1000. [DOI] [PubMed] [Google Scholar]
- Shirayama, M., W. Zachariae, R. Ciosk, and K. Nasmyth. 1998. The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J. 17:1336–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin, V., D. Ganoth, A. Dahan, H. Heller, J. Hershko, F.C. Luca, J.V. Ruderman, and A. Hershko. 1995. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell. 6:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin, V., G.K. Chan, and T.J. Yen. 2001. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154:925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z., R. Bharadwaj, B. Li, and H. Yu. 2001. Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev. Cell. 1:227–237. [DOI] [PubMed] [Google Scholar]
- Tunquist, B.J., and J.L. Maller. 2003. Under arrest: cytostatic factor (CSF)-mediated metaphase arrest in vertebrate eggs. Genes Dev. 17:683–710. [DOI] [PubMed] [Google Scholar]
- Vigneron, S., S. Prieto, C. Bernis, J.C. Labbe, A. Castro, and T. Lorca. 2004. Kinetochore localization of spindle checkpoint proteins: who controls whom? Mol. Biol. Cell. 15:4584–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin, R., S. Prinz, and A. Amon. 1997. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 278:460–463. [DOI] [PubMed] [Google Scholar]
- Yamano, H., J. Gannon, H. Mahbubani, and T. Hunt. 2004. Cell cycle-regulated recognition of the destruction box of cyclin B by the APC/C in Xenopus egg extracts. Mol. Cell. 13:137–147. [DOI] [PubMed] [Google Scholar]
- Yang, J., E.S. Bardes, J.D. Moore, J. Brennan, M.A. Powers, and S. Kornbluth. 1998. Control of cyclin B1 localization through regulated binding of the nuclear export factor CRM1. Genes Dev. 12:2131–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., S. Fang, J.P. Jensen, A.M. Weissman, and J.D. Ashwell. 2000. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 288:874–877. [DOI] [PubMed] [Google Scholar]
- Yoshitome, S., N. Furuno, and N. Sagata. 1998. Overexpression of the cytoplasmic retention signal region of cyclin B2, but not of cyclin B1, inhibits bipolar spindle formation in Xenopus oocytes. Biol. Cell. 90:509–518. [PubMed] [Google Scholar]
- Yu, H. 2002. Regulation of APC-Cdc20 by the spindle checkpoint. Curr. Opin. Cell Biol. 14:706–714. [DOI] [PubMed] [Google Scholar]
- Zachariae, W., and K. Nasmyth. 1999. Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev. 13:2039–2058. [DOI] [PubMed] [Google Scholar]
- Zachariae, W., M. Schwab, K. Nasmyth, and W. Seufert. 1998. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 282:1721–1724. [DOI] [PubMed] [Google Scholar]