

Abstract
Viral fusion proteins of classes I and II differ radically in their initial structures but refold toward similar conformations upon activation. Do fusion pathways mediated by alphavirus E1 and influenza virus hemagglutinin (HA) that exemplify classes II and I differ to reflect the difference in their initial conformations, or concur to reflect the similarity in the final conformations? Here, we dissected the pathway of low pH–triggered E1-mediated cell–cell fusion by reducing the numbers of activated E1 proteins and by blocking different fusion stages with specific inhibitors. The discovered progression from transient hemifusion to small, and then expanding, fusion pores upon an increase in the number of activated fusion proteins parallels that established for HA-mediated fusion. We conclude that proteins as different as E1 and HA drive fusion through strikingly similar membrane intermediates, with the most energy-intensive stages following rather than preceding hemifusion. We propose that fusion reactions catalyzed by all proteins of both classes follow a similar pathway.
Introduction
Membrane fusion reactions mediated by diverse fusion proteins are crucial for eukaryotic cells and for development of multicellular organisms (Jahn et al., 2003; Shemer and Podbilewicz, 2003). Recent studies on the diversity of fusion proteins have focused on proteins that mediate fusion by which enveloped viruses deliver their genome into host cells. Influenza and Sindbis viruses are among the best-studied prototypes of fusion machinery. For both viruses, fusion is triggered by acidification of the virus-containing endosome. In the case of influenza virus, low pH triggers restructuring in a homotrimeric glycoprotein HA (Skehel and Wiley, 2000; Tamm, 2003; Earp et al., 2005). In the case of Sindbis virus (SIN), a 1:1:1 arrangement of three structural proteins (the fusogenic envelope glycoprotein E1, the accessory envelope glycoprotein E2, and the capsid protein C) forms a double-shelled icosahedron (Paredes et al., 1998). Low pH releases SIN E1 from its heterodimeric interaction with E2 and induces homotrimerization of E1. The final lowest-energy forms of E1, HA, and many other fusion proteins share an important motif, two sequences that interact with membranes: the fusion peptide and the transmembrane domain relocate to the same end of the rodlike molecule (Weber et al., 1998; Skehel and Wiley, 2000; Gibbons et al., 2003, 2004b; Bressanelli et al., 2004; Modis et al., 2004). Restructuring of HA and E1 under fusion conditions involves early reversible conformations (Leikina et al., 2002; Gibbons et al., 2004a) and lateral interactions between adjacent proteins (Markovic et al., 2001; Gibbons et al., 2004b).
In spite of the similarities, HA and E1 differ radically in their initial structures and have come to represent two divergent classes of viral fusion proteins (Lescar et al., 2001). Class I proteins (exemplified by HA and HIV gp120/gp41) are oriented perpendicularly to the envelope surface and feature α-helical coiled-coil domains. A highly conserved and critical for fusion “fusion peptide” sequence is located at or near the NH2 terminus of the fusion protein. Class II proteins (for instance, the E1 protein of alphaviruses such as SIN and Semliki Forest virus [SFV] and the E protein of flaviviruses) lie tangential to the virus membrane and have an “internal” rather than “terminal” fusion peptide. Class II proteins contain predominantly β-strand secondary structures and are not predicted to form coiled-coils.
Restructuring that brings proteins of classes I and II from dissimilar initial conformations to similar final structures drive membrane fusion. Fusion pathway mediated by class I proteins has been dissected in experiments in which fusion was slowed down or blocked at different stages by genetically modifying fusion proteins or decreasing their numbers and by using specific inhibitors (Kemble et al., 1994; Chernomordik et al., 1998; Kozerski et al., 2000; Melikyan et al., 2000; Russell et al., 2001; Borrego-Diaz et al., 2003; Park et al., 2003). For HA, progress through the fusion pathway toward the opening of an expanding fusion pore connecting an HA-expressing cell and a bound RBC is controlled by the surface density of HA (for review see Chernomordik and Kozlov, 2003). Upon an increase in the numbers of activated HAs, there is a shift in the observed fusion phenotypes from restricted hemifusion (RH), in which lipid flow through the hemifusion connections is restricted by the proteins surrounding the fusion site, to unrestricted hemifusion (UH), defined as lipid mixing without content mixing. Only at very high densities of activated HAs does the fusion reaction reach an irreversible stage of fusion pore expansion.
Although, in contrast to the pathway mediated by class I proteins, the fusion pathway for class II proteins has not been explored, fusion mediated by alphaviruses and flaviviruses has been systematically characterized using mainly an experimental system of viral particles fusing with liposomes (White and Helenius, 1980; Bron et al., 1993; Nieva et al., 1994; Kielian et al., 1996; Corver et al., 1997; Smit et al., 1999, 2002; McInerney et al., 2004). Fusion mediated by class II proteins is significantly faster, less sensitive to lowering of the temperature, and less leaky than fusion reactions mediated by viruses with class I fusion proteins (for instance, influenza virus) (Shangguan et al., 1996; Corver et al., 2000; Smit et al., 2002). These differences along with dissimilarities between the starting conformations of the class I and II proteins might indicate that protein- and membrane-restructuring for these two classes proceed by distinct pathways. On the other hand, the similarity between the post-fusion structures has suggested the similarity between the fusion pathways (Gibbons et al., 2004b; Modis et al., 2004).
Do proteins of different classes catalyze fusion via radically different pathways? For instance, very robust fusion mediated by class II proteins might proceed through the entirely proteinaceous fusion pore as proposed for some fusion events (Lindau and Almers, 1995; Peters et al., 2001; Han et al., 2004) rather than through hemifusion intermediates similar to those in HA fusion (Kemble et al., 1994; Melikyan et al., 1997a; Chernomordik and Kozlov, 2003). Within the fusion-through-hemifusion pathway, rate-limiting stages in fusion progression and, thus, “job description” of fusion proteins might vary: proteins might be driving the entire pathway, as is the case for HA, or, alternatively, act only to catalyze hemifusion with subsequent fusion stages to proceed spontaneously.
In this work, we found that cell fusion mediated by E1 of SIN and SFV proceeds through intermediates strikingly similar to those identified earlier for HA fusion. Only a fraction of all low pH–activated E1s needed for fusion is required in order to locally merge the contacting monolayers of two membranes into RH intermediates. Additional activated E1s drive transition from RH into UH phenotype that allows lipid mixing between the membranes. Still greater numbers of activated E1s advance fusion beyond transient hemifusion intermediates to an opening and then to expansion of a fusion pore. The dependence of the fusion phenotype on the number of activated E1s indicates that the most demanding part of the job for fusion proteins of class II, as for those of class I, follows rather than precedes a local membrane merger.
Results
Unrestricted hemifusion, small fusion pores, and expanding fusion pores in SIN E1-mediated fusion
To study the pathway of E1-fusion for HAb2 cells that were used in the work on HA-fusion (Danieli et al., 1996; Chernomordik et al., 1998) we infected these cells with SIN. HAb2 cells express the immature HA0 form of the HA. HA0, although competent to mediate binding with sialic acid receptors, acquires fusogenic activity only if cleaved by trypsin into the mature HA1-HA2 form. SIN-infected HAb2 cells (referred to below as E1-HAb2 cells), used without trypsin pretreatment and thus having only the fusion-incompetent HA0 form, were incubated with RBCs labeled with the lipid probe PKH26 and different aqueous probes.
Application of pH 5.6 to E1-HAb2/RBC pairs caused robust lipid and content mixing (Fig. 1). No fusion was observed at neutral pH (C1) or for noninfected HA0-expressing HAb2 cells treated with pH 5.6 (C2) or pH 4.9 (not depicted). Note that E1 fusion is triggered at less acidic pH than HA fusion. Even for HAb2 cells with fully cleaved HA, pH 5.6 application gave no measurable fusion (not depicted; see also Chernomordik et al., 1998). These controls confirmed that fusion between E1-HAb2 cells and RBCs is mediated by low pH forms of SIN E1.
Figure 1.

Fusion phenotypes at different pH. Fusion of SIN E1-HAb2 cells with bound PKH26- and CF- or FD-labeled RBCs was triggered by a 5-min application of different pH: pH 5.6, 6.0, 6.2, and 6.5. Here and in other figures final extents of lipid mixing and CF and FD transfer assayed at neutral pH are shown by red, green, and blue bars, respectively. In the control experiments we either did not apply low pH (C1) or replaced SIN E1-HAb2 cells with uninfected HAb2 cells, pH 5.6 (C2). Here and in the subsequent figures bars are mean ± SD, n ≥ 3. The inset shows the relative frequency of UH phenotype among all lipid mixing events and NEP phenotype among all CF mixing events (see Materials and methods) for different pH.
The less acidic the pH applied, the lower is the percentage of alphavirus E1 that undergoes conformational restructuring into functional E1 homotrimers (Sjoberg and Garoff, 2003). By shifting pH applied to E1-HAb2/RBC pairs to pH >5.6 we gradually decreased fusion extents and, more importantly, detected two phenotypes of partial fusion. The difference between fusion extents detected as transfer of 6-carboxyfluorescein (CF) and transfer of a larger aqueous probe, 70-kD fluorescent dextran (FD) (Fig. 1, green vs. blue bars), indicated that some cell pairs developed small, nonexpanding fusion pores (NEP). Similarly, the difference between lipid and CF mixing extents (Fig. 1, red vs. green bars) indicated that some cell pairs developed lipid mixing without content mixing, i.e., UH.
To test whether UH connections are stable or dissociate with time we analyzed the kinetics and final extents of lipid mixing. Fusion events yielding both lipid and CF transfer resulted in complete lipid mixing defined by lack of a distinct circular shape of the RBC at the end of the experiment (Fig. 2). In contrast, most of the E1-HAb2/RBC pairs that demonstrated UH presented a “partial lipid mixing” phenotype (Mittal et al., 2003), where lipid dye transfer remains incomplete at the end of the lipid mixing process. Because UH yields partial rather than complete lipid mixing and, thus, lipid flow through UH connections stops before its completion, we conclude that these connections spontaneously dissociate.
Figure 2.

Partial lipid mixing in E1 fusion. (A) Bright-field and fluorescent images for two SIN E1-HAb2 cell/RBC pairs represent the two phenotypes of lipid mixing. Bright-field images with RBCs marked by arrows were taken after low pH application. Full lipid mixing (top) is defined by lack of a distinct circular shape of the RBC at the end of the experiment, when dye transfer reaches the saturation (t = Final). Partial lipid mixing (bottom) is defined by incomplete dye transfer with a clear boundary of the initial RBC membrane seen at the time when lipid mixing reaches its saturation. PKH26 fluorescence spreads from RBCs (bright spot at the time the pH is lowered to 6.0, t = 0) to the E1-HAb2 cell (dark at t = 0 and fluorescent at t = Final) upon completion of the lipid mixing event. Right images show CF fluorescence at t = Final. (B) The percentages of E1-HAb2/RBC pairs showing at pH 6.0 any lipid mixing, full lipid mixing, and content (CF) mixing were measured. Cell pairs that show some but not full lipid mixing presented partial lipid mixing phenotype. The percentage of cells with full lipid mixing phenotype is close to the percentage of cells showing CF transfer. The data were collected in four independent experiments. The total number of cell pairs (=100%) in the analysis was 250.
Note that in the experiments shown in Fig. 2 fusion was not only triggered but also was observed in the low pH medium. Thus, a return to neutral pH after low pH application was not required for fusion in our experimental system. This finding contradicts the conclusions reached in Paredes et al. (2004), but is consistent with the conclusion reported in Waarts et al. (2002).
Transient restricted hemifusion
Both UH and RH connections can be transformed into complete fusion by a short-term application of chlorpromazine (CPZ). This cationic amphipath preferentially partitions into and destabilizes the inner membrane monolayers, which form the hemifusion structure (Melikyan et al., 1997a). To test whether E1 is capable of forming RH, E1-HAb2/RBC pairs were first treated with a low pH pulse and then, already at neutral pH, with a 1-min CPZ pulse (Fig. 3). CPZ pulse caused a significant increase in the extent of both lipid and content mixing in comparison with controls observed after the low pH pulse without CPZ application. CPZ-induced fusion promotion was less profound for lower pH (Fig. 3 B vs. Fig. 3 A) because fusion has proceeded further at the lower pH.
Figure 3.

RH in SIN E1 fusion. Fusion of SIN E1-HAb2 cells with bound PKH26- and CF-labeled RBCs was triggered by a 5-min pulse of pH 6.5 (A) or pH 6.0 (B). Bars 1: the final extents of lipid and CF mixing. Bars 2 and 3: hemifusion connections were transformed into complete fusion by a 1-min pulse of 0.5 mM CPZ applied immediately after the low pH pulse (bars 2) or with a 40 min delay (bars 3). No lipid mixing was observed when CPZ was applied to E1-HAb2/RBC pairs not exposed to acidic pH (C1) or to low-pH–treated noninfected HAb2 cells with bound RBCs (C2).
RH intermediates were formed by the low-pH–activated E1. Neither lipid nor content mixing was promoted when CPZ was applied to E1-HAb2 /RBC pairs not exposed to low pH or to low-pH–treated noninfected HAb2 cells with bound RBCs. The significant decrease in the fusion promotion, when CPZ was applied 40 min after the low-pH pulse (Fig. 3), indicates that RH is a transient intermediate that either dissociates (Leikina and Chernomordik, 2000) or shifts to an yet unidentified non-CPZ sensitive forms of hemifusion.
To summarize, at suboptimal pH, when the number of activated E1 proteins is insufficient to form an expanding fusion pore, E1 establishes less advanced fusion phenotypes, two types of transient hemifusion intermediates (RH and UH) and small fusion pores. Even when fusion did not reach irreversible stage of expanding fusion pore, RBCs remained associated with HA-cells via HA1-receptor binding (Chernomordik et al., 1998).
E1-mediated fusion in the absence of HA0
The fusion phenotypes observed for E1-HAb2 fusion can be observed in the absence of any forms of HA. Labeled RBCs were allowed to settle onto attached SIN-infected BHK21 cells. In contrast to E1-HAb2 cells, infected BHK21 cells have no mechanism of RBC binding. However, gentle replacement of the medium with RBC-free low pH buffer allowed us to observe fusion between infected BHK21 cells and loosely attached RBCs. Although the lack of tight RBC binding hindered quantification of E1 fusion in this system, we did observed complete fusion (transfer of both PKH26 and FITC-tagged FD [FFD]), the NEP phenotype (transfer of CF but not of rhodamine-tagged FD [RFD]), UH, and RH after application of suboptimal pH (unpublished data).
Fusion phenotypes formed by SFV E1
The fusion phenotypes described above for cells infected with SIN were also observed for cells transfected with a plasmid encoding the structural proteins of SFV. Raising the pH applied to trigger fusion between transfected HAb2 cells (SFV E1-HAb2 cells) and bound RBCs from pH 5.6 to pH 6.0 allowed us to identify UH (Fig. 4 A, bottom), NEP (Fig. 4 B), and RH (Fig. 4 C) phenotypes.
Figure 4.

RH and UH phenotypes and nonexpanding fusion pores in SFV E1 fusion. (A) Bright-field and fluorescent images for two SFV E1-HAb2 cell/RBC pairs after 5-min pulse of pH 6.0 represent the two phenotypes of lipid mixing: full lipid mixing correlated with content mixing (“fusion” in top panel) and partial lipid mixing correlated with UH (bottom panel). Spreading of the PKH26 and CF fluorescence from RBCs (marked by arrow in bright-field image) to the E1-HAb2 cell is shown upon completion of the lipid mixing event. (B) NEP phenotype observed in fusion between RBCs doubly labeled with CF and 70-kD RFD and bound SFV E1-HAb2 cell. Aqueous connection between the cells allows transfer of CF but not 70-kD RFD. (C) Low magnification images (bright field, PKH26- and CF-fluorescence) of SFV E1-HAb2 cells with bound RBCs treated with 5-min pulse of pH 5.6 or 6.0 (top and middle panels, respectively). Bottom panel shows the field of view for SFV E1-HAb2 cells with bound RBCs treated with 5-min pulse of pH 6.0 and then treated with a CPZ pulse to transform RH into complete fusion.
LPC-arrested stage
Additional phenotype of E1 fusion was identified using fusion inhibitor lysophosphatidylcholine (LPC), a lipid that inhibits diverse fusion reactions (Chernomordik et al., 1993; for review see Chernomordik and Kozlov, 2003). To test LPC activity for E1 fusion, E1-HAb2/RBC pairs were incubated with lauroyl LPC for 5 min and then, still in the presence of LPC, treated with a pH 6.0 pulse (Fig. 5). In contrast to the control with no LPC present, we observed significant inhibition of fusion (Fig. 5 A, bars 2 vs. bars 1). Fusion commenced upon LPC removal (Fig. 5 A, bars 3 vs. bars 1). Thus, the LPC-arrested fusion stage (LAS) in E1 fusion precedes lipid and CF mixing, but follows low pH–dependent E1 activation.
Figure 5.

LAS that follows all low pH–dependent stages of E1 restructuring and precedes actual membrane fusion. (A) Cartoon to the right illustrates the experimental protocol and our finding that LPC reversibly arrested fusion downstream low pH–dependent stages. Fusion between SIN E1-HAb2 cells and bound RBCs was triggered by a 5-min pulse of pH 6.0 and assayed as lipid and CF mixing. Bars 1: No LPC added; Bars 2: starting 5 min before the low pH pulse and throughout the entire experiment, cell pairs were incubated in the presence of 70 μM lauroyl LPC. Bars 3: LPC was washed out by LPC-free PBS 15 min after the end of low pH application. In the control experiments we applied and then removed LPC to E1-HAb2/RBC pairs that had not been treated with a low pH pulse (C1); or applied a low pH pulse in the presence of LPC to uninfected HAb2 cells with bound RBCs and then washed out LPC (C2). (B) SIN-infected BHK21 cells with loosely attached RBCs were treated with a 5-min pH 6.0 pulse in the absence (bars 1) or in the presence of 70 μM lauroyl LPC (bars 2 and 3). Low pH buffer was replaced with LPC-free PBS (bars 3) or with PBS supplemented with 70 μM LPC (bars 2). In parallel with fusion assay we counted the number of RBCs that remained associated with the E1-expressing cells after robust washes (shown by light blue bars). This low-pH– and E1-dependent binding most likely reflects insertion of an E1 FL into the RBC membrane. Bars C1 and C2: control experiments similar to the experiments shown by bars 1 but for noninfected BHK21 cells (C1) and for SIN-infected BHK21 cells with omitted low pH application (C2). These controls show the level of binding in the absence of the low pH–activated E1 insertion. (C) Stearoyl LPC added exclusively to E1-expressing membrane inhibits its fusion with RBCs. Bars 2: in the control experiment with no LPC added fusion between SIN E1-HAb2 cells and bound RBCs was triggered by a 5-min application of pH 6.0 buffer. Bars 3: SIN E1-HAb2 cells were pretreated with 5 μM stearoyl LPC. After washing out unbound LPC the cells were brought in contact with RBCs. As in (2) fusion was triggered by a 5-min application of pH 6.0 buffer. Bars 1: control experiment similar to that shown by bars 3 but with omitted low pH application.
As for E1-HAb2/RBC fusion, LPC inhibited fusion of SIN-infected BHK21 cells with RBCs (Fig. 5 B). Importantly, although there was no fusion when BHK21 cells with loosely attached RBCs were treated with a low pH pulse in the presence of LPC, after the pulse RBCs became tightly attached to the infected cells. This low pH–dependent binding was mediated by SIN glycoproteins, as evidenced by the lack of such RBC binding in the controls with SIN-infected cells untreated with low pH and low pH–treated noninfected BHK21. Low pH–induced binding between E1-expressing membrane and receptorless target membrane is known to be mediated by an insertion of an E1 fusion loop (FL) into the target membrane, a prerequisite for E1 homotrimerization and for fusion (Kielian et al., 1996; Smit et al., 1999; Gibbons et al., 2004a). Our finding that low pH–dependent binding between SIN-infected BHK21 cells and RBCs is not affected in the presence of LPC indicates that fusion inhibition by LPC does not involve inhibition of the FL insertion.
In the experiments discussed above, LPC was added to both fusing membranes. To further test whether fusion inhibition by LPC involves its effects on interactions between E1 and RBC membrane, we added stearoyl LPC to only E1-HAb2 cells (Fig. 5 C). Because of a low solubility in water, stearoyl LPC, in contrast to lauroyl LPC, remains in the cell membrane even when cells are washed with LPC-free medium (Chernomordik et al., 1997). Stearoyl LPC treatment of E1-HAb2 cells before bringing them into contact with RBCs was sufficient to inhibit subsequent low pH–triggered fusion, confirming that LPC inhibition does not involve disturbance of E1–target membrane interactions.
Zn-arrested fusion stage
In contrast to LPC, which inhibits diverse fusion reactions, Zn is a specific inhibitor of alphavirus fusion (Corver et al., 1997). Although, as expected, 5 mM Zn did not inhibit HA fusion of trypsin-treated HAb2 cells with bound RBCs (unpublished data), E1-HAb2/RBC pairs treated with a low pH buffer containing 5 mM Zn gave almost no fusion (Fig. 6 A, bars 2 vs. bars 1). To inhibit fusion, Zn had to be present during the low pH application. A 5-min preincubation of the cell pairs with Zn, followed by washing of the cells with Zn-free EDTA-containing buffer before a low pH pulse, had no effect on fusion.
Figure 6.
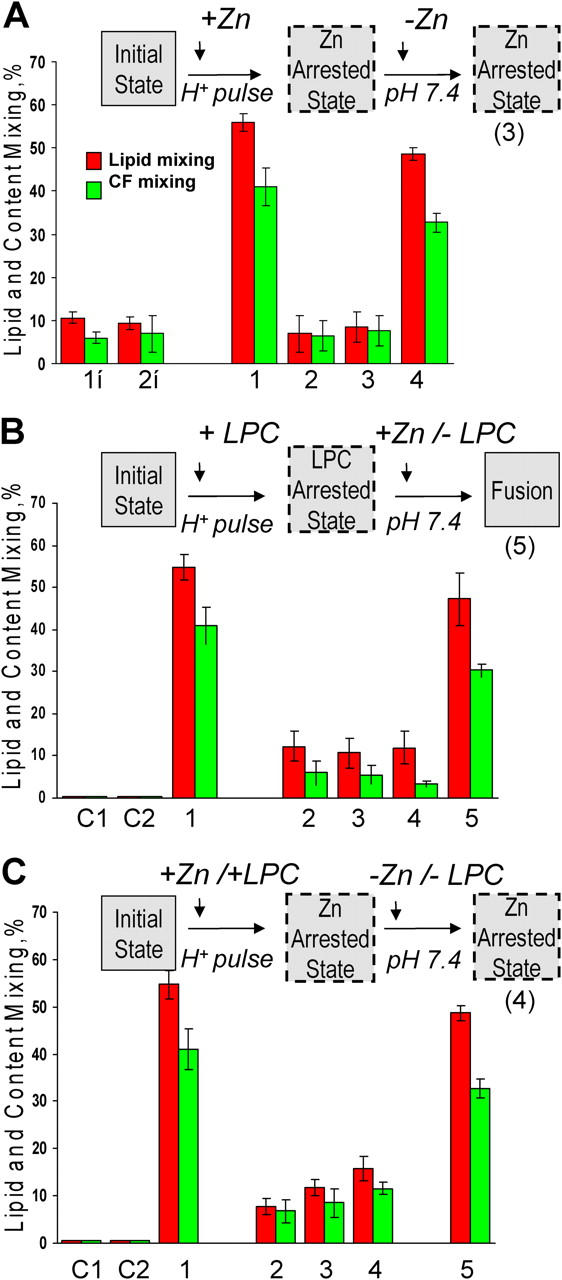
ZnAS precedes LAS. In these experiments we took advantage of the rapidness with which E1 fusion reaches its final extents under given conditions. Collecting video microscopy images for subsequent analysis and scoring fusion at several points in the experimental protocol (for instance, before and after removal of an inhibitor) allowed us to assay fusion extents for E1-HAb2 cells with bound RBCs under several conditions in the same dish rather than in independent experiments. Cartoons above A, B, and C illustrate the main finding of each figure. (A) Low pH application in the presence of Zn yields an arrested fusion stage, where E1 is neither irreversibly inactivated nor committed to fusion upon Zn removal. In the control experiment E1-HAb2 cells with bound RBCs were treated by a 5-min pH 6.0 pulse in the absence of Zn (bars 1). In the experiment on Zn inhibition (bars 2–4) fusion was first measured at the end of a 5-min pH 6.0 application in the presence of 5 mM Zn (bars 2). Fusion was next assayed after the cells were reneutralized and Zn removed (bars 3). The cells were treated by a second 5-min pH 6.0 pulse, and fusion was measured again (bars 4). The lack of E1 inactivation observed in this experiment is contrasted with the E1 inactivation observed when low pH was applied in the absence of the target membrane with or without Zn (bar 1i and 2i, respectively). SIN E1-HAb2 cells were treated with a 5-min pulse of pH 6.0, reneutralized, and incubated with RBCs for 15 min. Then, a second 5-min pulse of pH 6.0 was applied. Functional inactivation of E1 due to the first low pH pulse lowered fusion extents observed after the second pulse. (B) Fusion downstream of the LPC-arrested state is already insensitive to Zn. In the control experiment (bars 1) E1-HAb2 cells with bound RBCs were treated by a 10-min pH 6.0 pulse in the absence of fusion inhibitors. In the experiment shown in bars 2–5, E1-HAb2 cells with bound RBCs were incubated with lauroyl LPC for 5 min. Then, still in the presence of 70 μM lauroyl LPC, the cells were incubated for 5 min at pH 6.0. After taking video microscopy images for subsequent quantification (bars 2) the buffer was replaced by the pH 6.0 buffer supplemented with LPC and Zn. Thus the cells, while still kept at acidic pH and in the presence of LPC, were now exposed to Zn. After 5 min incubation we again measured fusion (bars 3). The cells were reneutralized still in the presence of LPC and Zn and fusion was measured again (bars 4). Finally, we removed LPC and assayed fusion at pH 7.4 still in the presence of Zn (bars 5). The control experiments shown in bars C1 and C2 were similar to that shown by bars 5, but we either omitted low pH application (C1) or replaced E1-HAb2 cells with noninfected HAb2 cells (C2). (C) SIN E1-HAb2 cells with bound RBCs acidified in the presence of both Zn and LPC do not reach LPC-arrested stage. In the control experiment E1-HAb2 cells with bound RBCs were treated by a 5-min pH 6.0 pulse in the absence of fusion inhibitors (bars 1). In the experiment shown in bars 2–5, E1-HAb2 cells with bound RBCs were incubated with both lauroyl LPC and Zn for 5 min at neutral pH. After taking video microscopy images for subsequent quantification, the cells were treated with a 5-min pulse of pH 6.0 still in the presence of both LPC and Zn. Fusion was quantified (bars 2) and then again assayed after reneutralization of the cells (bars 3) and removal of both LPC and Zn (bars 4). Bars 5: the cells were treated with a second 5-min pH 6.0 pulse, and fusion was measured again. The control experiments shown in bars C1 and C2 were similar to that shown by bars 4 but we either omitted low pH application (C1) or replaced E1-HAb2 cells with noninfected HAb2 cells (C2).
Low pH pretreatment of E1-expressing membrane in the absence of a target membrane inactivates E1 as evidenced by the lack of fusion after the application of an additional low pH pulse, in the presence of a target membrane (Smit et al., 1999). In agreement with an earlier work on SFV/liposome fusion (Corver et al., 1997), we found that Zn did not inhibit E1 inactivation (Fig. 6 A, bars 1i). In the control, we treated E1-HAb2 cells with a low pH pulse, reneutralized the cells, added RBCs, and applied a second low pH pulse. The functional inactivation of E1 after the first low pH pulse resulted in a significant decrease in fusion (bars 2i). Thus, under our conditions, the number of functional SIN glycoproteins delivered to the surface of the infected cell within the 25-min time interval between the two low pH pulses was insufficient to produce measurable fusion. Similar E1 inactivation was observed when the first low pH pulse was applied to E1-HAb2 cells in the presence of Zn and then, after reneutralization of the cells, Zn was removed by washes with Zn-free EDTA-containing PBS before addition of RBCs and the second low pH pulse (bars 1i).
Low pH application to E1-HAb2/RBC pairs in the presence of Zn did not inactivate E1. Although a low pH pulse applied in the presence of Zn did not yield fusion even after Zn removal (Fig. 6 A, bars 3), a second low pH pulse gave significant fusion (bars 4). Thus, SIN E1s that were activated by low pH in the presence of Zn and RBCs were neither irreversibly committed to fuse upon Zn removal nor irreversibly inactivated.
Because Zn, like LPC, inhibits fusion after low pH–dependent activation of E1 but before lipid and content mixing, we then tested whether Zn-arrested fusion stage (ZnAS) comes before or after the LAS. We established the LAS by treating E1-HAb2/RBC pairs with a low pH pulse in the presence of LPC (Fig. 6 B). Then, still at low pH and in the presence of LPC, we incubated the cells with Zn. Subsequent removal of LPC, still in the presence of Zn yielded significant fusion (bars 5), indicating that fusion stages downstream of the LAS are already insensitive to Zn. In a complementary experiment, when we treated E1-HAb2/RBC pairs with a low pH pulse in the presence of both LPC and Zn, the cells never reached the LAS, as evidenced by the lack of fusion upon subsequent removal of both Zn and LPC (Fig. 6 C, bars 4). Fusion observed after application of a second low pH pulse (bars 5) confirmed that the first low pH pulse applied in the presence of Zn and LPC did not inactivate E1. These findings indicate that ZnAS precedes the LAS.
Discussion
Although proteins that mediate diverse fusion reactions perform apparently similar jobs, the pathways and the underlying mechanisms might be radically different for different proteins. Here, to evaluate the generality of the fusion pathways, we have focused on alphavirus E1 and influenza HA, proteins of class II and class I that differ drastically in their initial conformations but refold toward similar final conformations upon activation. To characterize early intermediates in E1 fusion, we slowed down and/or arrested fusion by using suboptimal pH and fusion inhibitors. We found E1 to establish the same set of fusion intermediates as HA (LAS, transient hemifusion connections, RH and UH, and a small and then expanding fusion pore; Fig. 7). The only fusion phenotype specific for E1 fusion is the ZnAS. Although HA is homotrimeric already in the initial form, E1 forms functional homotrimers only after acidification. Zn, an inhibitor of this homotrimerization (Corver et al., 1997), inhibits E1 fusion and does not affect HA fusion.
Figure 7.

The progression of fusion phenotypes upon an increase in the number of activated fusion proteins. In the schematic diagram of the fusion site, the top and bottom membranes represent a section of a fusion protein-expressing membrane and a section of a prelabeled target cell membrane, respectively. Acidification initiates restructuring of E1 and HA from very different initial conformations (see the text). Upon increase in the number of low pH–activated fusion proteins the observed fusion phenotype shifts from an initial state with two apposing membranes, to restricted hemifusion, RH, and then to unrestricted hemifusion, UH. The transfer of the lipid probe from the target cell membrane through the fusion site is first detected at UH. At still higher numbers of activated proteins E1* and HA* fusion phenotype advances beyond transient hemifusion phenotypes to an opening of a small fusion pore and, finally, to an irreversible expansion of the fusion pore. Transfer of small and large aqueous probes preloaded into target cell proceeds only upon formation of small and expanding fusion pores, respectively. LAS follows a trigger-dependent activation of fusion proteins and precedes the actual membrane merger. This sequence of fusion phenotypes is conserved between class I and class II fusion proteins. ZnAS, specific for E1-fusion, precedes formation of functional E1 homotrimers (E1* HT) and LPC-arrested stage.
For E1 fusion, as for fusion mediated by HA, less acidic pH and thus fewer low pH–activated fusion proteins are required to hemifuse the membranes rather than to open an expanding fusion pore. This finding indicates that fusion proteins of both classes (I and II) drive the entire fusion pathway, rather than merely catalyze the merger of the contacting monolayers of two membranes.
Fusion phenotypes
LAS
LAS in E1 fusion precedes membrane merger and follows all low pH–dependent stages, including the ZnAS. Depending on concentrations and conditions, LPC might have diverse effects on biological fusion. LPC inhibits many fusion reactions by opposing the monolayer curvature that dominates in an early stalk-like local hemifusion intermediate (for review see Chernomordik and Kozlov, 2003). Alternatively, LPC might block the fusion protein interactions with the target membrane and subsequent protein restructuring required for fusion (Stiasny and Heinz, 2004). Our results support the former stalk-inhibiting mechanism in our system and are consistent with the finding of Wilschut's laboratory that LPC blocks actual membrane merger in E1 fusion rather than E1 homotrimerization (Wilschut, J., personal communication).
Importantly, LPC added to contacting membrane monolayers blocked not only their merger and resulting lipid mixing but also content mixing. This finding suggests that hemifusion is a stage of a productive fusion pathway yielding an expanding fusion pore rather than a side product of the reaction; and also indicates that E1 fusion, as HA fusion, proceeds through hemifusion rather than through an entirely proteinaceous fusion pore.
Aborted fusion intermediates
Similarly to earlier studies on HA fusion (Chernomordik et al., 1998; Leikina and Chernomordik, 2000), E1 forms RH and UH intermediates. The latter was documented earlier using an electrophysiological approach for fusion between cells expressing SFV fusion machinery and a planar lipid bilayer (Samsonov et al., 2002). We found that both RH and UH connections in E1 fusion, as in HA fusion, dissociate with time. Thus, until the numbers of activated E1s are high enough to form expanding fusion pore(s), fusion intermediates remain energy intensive and depend on protein machinery for their stabilization.
As for HA, low pH pretreatment of alphavirus glycoproteins in the absence of a target membrane causes inactivation of the fusion machinery. The limited lifetime of RH and UH indicates that the conformational energy of E1 gradually discharges even in the presence of the target membrane. However, in this case the inactivation is slower. Low pH–treated E1-cell/RBC pairs, with fusion inhibited by either LPC or Zn, retain their fusogenic potential for tens of minutes. Insertion of the E1 FL into the RBC membrane might slow down the transition of the E1 homotrimer from an extended conformation with FL in the target membrane to the discharged lowest-energy form, in which the FL and the transmembrane domain are anchored in the same membrane (Gibbons et al., 2003, 2004b).
Long-living conformations of E1 at the LAS did not need an additional low pH application to ensure fusion after LPC removal. In contrast, for ZnAS, E1 monomers with target-membrane–inserted FL (Corver et al., 1997) after Zn removal need a second low pH pulse for fusion, suggesting that E1 trimerization downstream of the monomer insertion is still low pH–dependent.
Diverse proteins with similar final structures drive fusion-through-hemifusion pathways
A number of important human diseases including Dengue fever are caused by viruses that enter the cells by using the class II fusion proteins. Here, we report that these proteins catalyze the membrane fusion pathway similar to that driven by class I influenza virus HA. Fusion can be mechanistically coupled to transition of fusion proteins from an “extended” conformation of an individual homotrimeric form anchored in both membranes into the hairpin post-fusion form, and/or it can be coupled to lateral interactions between adjacent fusion proteins that might already have achieved a hairpin form. Lateral interactions between fusion proteins have indeed been documented, and the possible importance of these interactions in fusion has been discussed (Blumenthal et al., 1996; Danieli et al., 1996; Gaudin et al., 1996; Plonsky and Zimmerberg, 1996; Chernomordik et al., 1998; Markovic et al., 1998, 2001; Bentz, 2000; Kozlov and Chernomordik, 2002; Roche and Gaudin, 2002; Chernomordik and Kozlov, 2003; Gibbons et al., 2003, 2004b). Interactions between activated fusion proteins assembled around the fusion site might explain the restriction of lipid mixing at RH (Chernomordik et al., 1998; this paper). The hypothesis that fusion depends on the energy released by interactions between restructured proteins would explain the fusogenic activity of the large HA2 ectodomain-based polypeptide that has a conformation similar to the post-fusion conformation of the HA2 subunit of HA (Epand et al., 1999; Leikina et al., 2001). This hypothesis is also consistent with a recent finding that HA mutations that are expected to affect only the latest stages of HA restructuring inhibit early fusion intermediates (Park et al., 2003).
E1 and HA fusion reactions share the transient hemifusion intermediates that precede an irreversible commitment to fusion completion. We hypothesize that the early stage of enveloped virus infection involves these intermediates. The number of activated viral fusion proteins within the endosome likely rises gradually upon progressive acidification of the endosome content (Roederer et al., 1987). Because the opening of a fusion pore requires more fusion proteins than does hemifusion, the conditions for hemifusion should develop earlier than those for fusion pores, suggesting that hemifusion intermediates are present in the membrane contact zone at the time of the opening of the first fusion pore. As discussed above, interactions with the target membrane slow down protein inactivation, thus allowing additional fusion proteins to join the proteins assembled around a developing fusion site. In this scenario, a rise in the number of activated proteins, and thus an increase in the conformational energy they might release upon fusion, drives the transition from hemifusion to an expanding fusion pore.
Our data support the hypothesis that the final hairpin structure shared by diverse viral fusion proteins is more important for fusion than the initial metastable conformations of these proteins. Because proteins involved in intracellular fusion assemble into similar hairpin structures (Weber et al., 1998), our results have important implications for the ongoing debate about the pathway of intracellular fusion (Chernomordik and Kozlov, 2003; Han et al., 2004; Szule and Coorssen, 2004). Several studies have concluded that intracellular fusion, in contrast to HA-mediated fusion, starts with an opening of an entirely proteinaceous fusion pore (Peters et al., 2001; Han et al., 2004). An alternative stalk-pore hypothesis suggests that intracellular fusion as HA-mediated fusion and other biological fusion reactions proceeds via hemifusion intermediates (for review see Chernomordik and Kozlov, 2003). This hypothesis is supported by the similarities between the effects of nonbilayer lipids on intracellular fusion, viral fusion, and fusion of protein-free lipid bilayers (Chernomordik et al., 1993; Vogel et al., 1993), and by studies on exocytotic fusion in yeasts (Grote et al., 2000) and insulin granule exocytosis in the pancreatic islet (Takahashi et al., 2002). Our finding that dissimilar viral fusion proteins catalyze the fusion-through-hemifusion pathway points to universality of this mechanism of biological fusion. Thus, the fusion pathway dissected here for E1-mediated fusion might be shared by disparate fusion reactions driven by diverse proteins with fundamentally similar final hairpin structures.
Materials and methods
Cells
We labeled human RBCs, freshly isolated from whole blood, with fluorescent lipid PKH26 (Sigma-Aldrich), and with the aqueous dyes CF, FFD, and RFD (Invitrogen) (as described in Melikyan et al., 1997b; Chernomordik et al., 1998). RBCs were doubly labeled with PKH26 and CF; or with PKH26 and FFD; or with CF and RFD. Neither of these probes changed fusion efficiency versus that observed in control without given probe.
HAb2 cells expressing A/Japan/305/57 HA (Doxsey et al., 1985) and BHK21 cells (American Type Culture Collection, Rockville, MD) were grown in DME containing 10% FCS, 10 mM Hepes, penicillin, and streptomycin at 37°C.
HA is expressed in HAb2 cells in the immature HA0 form of the HA. HA0, although competent to mediate binding with sialic acids on RBC surface, acquires fusogenic activity only if cleaved by trypsin into mature HA1-HA2 form. If not stated otherwise, in this study we omitted the trypsin treatment of HAb2 cells in order to leave HA in the fusion-incompetent HA0 form.
In a few experiments aimed at studying HA fusion, HAb2 cells were treated with 10 μg/ml trypsin (Fluka) for 10 min at 22°C to cleave HA0 into its fusion-competent HA1-S-S-HA2 form.
Infection and transfection
The stock of SIN strain TE was kept at −80°C and used for infection of BHK21 cells with low multiplicity to propagate the working virus stocks with titer between 108 and 109 plaque-forming units (pfu)/ml. Working virus stocks were kept at 4°C for less than two weeks. HAb2 and BHK21 cells were infected with SIN from working virus stocks with multiplicity 50–100 pfu per cell to ensure all cells were infected. The cells were used at 7–8 h after infection.
Plasmid pCB3 (Lu et al., 2001), a gift from Dr. Margaret Kielian (Albert Einstein College of Medicine, Bronx, NY), encodes all SFV structural proteins, capsid, p62, 6K, and E1. To transiently express the SFV proteins in HAb2 cells, the cells were plated at ∼70% confluence in 35-mm plates and transfected with 2 μg plasmid DNA using LipofectAMINE 2000 (Invitrogen) according to the manufacturer's protocol. The cells were used for fusion assay at 30 h after transfection.
Fusion assay
All experiments were performed at 22°C. After two washings with PBS, HAb2 cells were incubated for 10 min with a 1-ml suspension of RBCs (0.01% hematocrit). HAb2 cells with 0–2 bound RBCs per cell were washed three times with PBS to remove unbound RBCs.
To trigger fusion, E1-HAb2-cells with bound RBCs were incubated in PBS titrated with citrate to acidic pH for 5 min, if not stated otherwise. The low pH pulse was ended by replacement of the acidic solution with PBS. Fusion was quantified as the ratio of dye-redistributed bound RBCs to the total number of bound RBCs (Chernomordik et al., 1998). To characterize relative frequency of UH phenotype for different pH, we normalized the number of cell pairs that demonstrated PKH26 mixing without CF mixing to the total number of the cell pairs that demonstrated PKH26 mixing. Similarly, for NEP we plotted the difference between the number of cell pairs that demonstrated CF and FD transfer normalized by the number of the cell pairs that demonstrated CF transfer.
SIN-infected BHK21 cells do not bind RBCs. To study fusion between these cells, we first allowed RBCs to settle down and loosely attach onto BHK21 cells. Then we gently and slowly replaced PBS with low pH solution or overlay normal PBS with pretitrated acidic solution.
Even when fusion is inhibited, low pH–dependent insertion of E1 FL into the target membrane provides an effective binding mechanism between E1-expressing membrane and target membrane (Corver et al., 1997). To study this binding, SIN-infected BHK21 cells and RBCs were treated with a low pH pulse in the presence of LPC. Unbound RBCs were removed by three intensive washes with PBS. We screened at least 200 cells in six randomly selected areas of each plate. For each plate, the average number of RBCs bound to BHK21 cells was normalized by the number of RBCs loosely associated with the cells before the washes (∼300 RBCs).
Fusion extents reached the final levels within 1–2 min after low pH application and were assayed by analysis of the collected video microscopy images taken using a microscope (model IX70; Olympus) and a CCD camera (Photometrics CoolSNAP-fx; Roper Scientific) as the ratio of dye-redistributed bound RBCs to the total number of bound RBCs (Chernomordik et al., 1998). In all experiments, with the exception of those shown in Fig. 2, we assayed fusion already after reneutralization. Presented data were averaged from at least three experiments.
In our analysis of the patterns of lipid mixing to establish the completion of the lipid mixing we digitized the recorded fluorescence microscopy video and quantified the fluorescent area for each individual RBC/E1-HAb2 cell pair at each time point. Full- and partial-lipid mixing phenotypes were easily classified by eye (Fig. 2). The distinct patterns of the spreading of the lipid probe in the full- and partial-lipid mixing phenotypes were confirmed by the analyzing surface scans of five E1-cell/RBC pairs for each phenotype. Because peak pixel value of final image divided by the peak pixel value in the initial image for full lipid mixing is 55.01 ± 9.32% vs. 95.35 ± 7.41% for partial lipid mixing, we conclude that although for partial lipid mixing the highest intensity in the RBC matches that of the initial RBC, the highest intensity for cell–RBC pairs scored as full lipid mixing is significantly lowered by the spreading of the lipid probe. In addition, although for cells scored as full lipid mixing no pixel in the final image had value within 80% of peak pixel value of initial image, for partial mixing 56.67 ± 28.77% of pixels in the final image had value within 80% of the maximum pixel intensity of initial image (n = 5 for each phenotype). Analysis of only final images for full versus partial lipid mixing confirms that former phenotype presents much broader spreading of the lipid probe in E1-cell. Percentage of pixels within 80% of maximum fluorescence intensity in the final image (number of pixels within 80% of maximum pixel intensity in the final image divided by the total number of pixels in the final image) was 19.53 ± 13.04 in full lipid mixing vs. 1.97 ± 0.88 in partial lipid mixing.
Application of LPC, CPZ, and ZnCl2
70-μM and 5-μM solutions of lauroyl LPC and stearoyl LPC (Avanti Polar Lipids, Inc.), respectively, in PBS were freshly prepared. In the experiments with lauroyl LPC, PBS bathing the plastic-attached E1-HAb2 cells with bound RBCs was replaced by 1 ml of lauroyl LPC-supplemented PBS (Chernomordik et al., 1997). 5 min later, we triggered fusion by applying low pH buffer supplemented with the same concentration of the lipid. After 5-min incubation, low pH buffer was replaced with “normal” pH PBS with the same concentration of LPC. To study the reversibility of lauroyl LPC inhibition, we washed low pH-treated cells three times within 15 min by LPC-free normal pH PBS.
In the experiments with stearoyl LPC we pretreated E1-HAb2 cells with LPC and then removed unbound lipid by three washes with LPC-free PBS. Then the cells were brought into contact with untreated RBCs. Fusion was triggered by a 5-min application of LPC-free buffer, pH 6.0.
CPZ (Sigma-Aldrich) was prepared as a 0.5-mM solution in PBS. E1-HAb2-cells with bound RBCs were treated by low pH buffer, returned to neutral pH and immediately or 40 min later exposed to CPZ-containing solution for 60 s. Fusion analysis was performed in CPZ-free PBS.
A working solution of 5 mM ZnCl2 was prepared from 1 M stock in the buffer HNE (25 mM Hepes, 150 mM NaCl, and 0.1 mM EDTA) and titrated to normal or acid pH with citric acid. To remove Zn we washed the cells with HNE supplemented with 6 mM EDTA for 1 min and replaced it with PBS.
Acknowledgments
We are very grateful to Dr. M. Kielian for the gift of the SFV construct and to Dr. J. Wilschut for communicating results before publication. We are indebted to Drs. E. Leikina, M. Kozlov, H. Delanoe, K. Melikov, G. Melikyan, E. Zaitsev, and J. Zimmerberg for comments on the manuscript.
D.E. Griffin acknowledges funding support from National Institutes of Health grant NS18596.
Abbreviations used in this paper: CF, 6-carboxyfluorescein; CPZ, chlorpromazine; FD, 70-kD fluorescent dextran; FFD, FITC-tagged FD; FL, fusion loop, LPC, lysophosphatidylcholine; LAS, LPC-arrested fusion stage; NEP, nonexpanding fusion pores; RH, restricted hemifusion; RFD, rhodamine-tagged FD; SFV, Semliki Forrest virus; SIN, Sindbis virus; UH, unrestricted hemifusion; ZnAS, Zn-arrested fusion stage.
References
- Bentz, J. 2000. Minimal aggregate size and minimal fusion unit for the first fusion pore of influenza hemagglutinin-mediated membrane fusion. Biophys. J. 78:227–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal, R., D.P. Sarkar, S. Durell, D.E. Howard, and S.J. Morris. 1996. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell–cell fusion events. J. Cell Biol. 135:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrego-Diaz, E., M.E. Peeples, R.M. Markosyan, G.B. Melikyan, and F.S. Cohen. 2003. Completion of trimeric hairpin formation of influenza virus hemagglutinin promotes fusion pore opening and enlargement. Virology. 316:234–244. [DOI] [PubMed] [Google Scholar]
- Bressanelli, S., K. Stiasny, S.L. Allison, E.A. Stura, S. Duquerroy, J. Lescar, F.X. Heinz, and F.A. Rey. 2004. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 23:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron, R., J.M. Wahlberg, H. Garoff, and J. Wilschut. 1993. Membrane fusion of Semliki Forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J. 12:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik, L.V., and M.M. Kozlov. 2003. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72:175–207. [DOI] [PubMed] [Google Scholar]
- Chernomordik, L.V., S.S. Vogel, A. Sokoloff, H.O. Onaran, E.A. Leikina, and J. Zimmerberg. 1993. Lysolipids reversibly inhibit Ca2+-, GTP- and pH-dependent fusion of biological membranes. FEBS Lett. 318:71–76. [DOI] [PubMed] [Google Scholar]
- Chernomordik, L.V., E. Leikina, V. Frolov, P. Bronk, and J. Zimmerberg. 1997. An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J. Cell Biol. 136:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik, L.V., V.A. Frolov, E. Leikina, P. Bronk, and J. Zimmerberg. 1998. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 140:1369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corver, J., R. Bron, H. Snippe, C. Kraaijeveld, and J. Wilschut. 1997. Membrane fusion activity of Semliki Forest virus in a liposomal model system: specific inhibition by Zn2+ ions. Virology. 238:14–21. [DOI] [PubMed] [Google Scholar]
- Corver, J., A. Ortiz, S.L. Allison, J. Schalich, F.X. Heinz, and J. Wilschut. 2000. Membrane fusion activity of tick-borne encephalitis virus and recombinant subviral particles in a liposomal model system. Virology. 269:37–46. [DOI] [PubMed] [Google Scholar]
- Danieli, T., S.L. Pelletier, Y.I. Henis, and J.M. White. 1996. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J. Cell Biol. 133:559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxsey, S.J., J. Sambrook, A. Helenius, and J. White. 1985. An efficient method for introducing macromolecules into living cells. J. Cell Biol. 101:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earp, L.J., S.E. Delos, H.E. Park, and J.M. White. 2005. The many mechanisms of viral membrane fusion proteins. Curr. Top. Microbiol. Immunol. 285:25–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epand, R.F., J.C. Macosko, C.J. Russell, Y.K. Shin, and R.M. Epand. 1999. The ectodomain of HA2 of influenza virus promotes rapid pH dependent membrane fusion. J. Mol. Biol. 286:489–503. [DOI] [PubMed] [Google Scholar]
- Gaudin, Y., H. Raux, A. Flamand, and R.W. Ruigrok. 1996. Identification of amino acids controlling the low-pH-induced conformational change of rabies virus glycoprotein. J. Virol. 70:7371–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons, D.L., E. Erk, B. Reilly, J. Navaza, M. Kielian, F.A. Rey, and J. Lepault. 2003. Visualization of the target-membrane-inserted fusion protein of Semliki Forrest virus by combined electron microscopy and crystallography. Cell. 114:573–583. [DOI] [PubMed] [Google Scholar]
- Gibbons, D.L., A. Ahn, M. Liao, L. Hammar, R.H. Cheng, and M. Kielian. 2004. a. Multistep regulation of membrane insertion of the fusion peptide of Semliki Forest virus. J. Virol. 78:3312–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons, D.L., M.C. Vaney, A. Roussel, A. Vigouroux, B. Reilly, J. Lepault, M. Kielian, and F.A. Rey. 2004. b. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature. 427:320–325. [DOI] [PubMed] [Google Scholar]
- Grote, E., M. Baba, Y. Ohsumi, and P.J. Novick. 2000. Geranylgeranylated SNAREs are dominant inhibitors of membrane fusion. J. Cell Biol. 151:453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, X., C.T. Wang, J. Bai, E.R. Chapman, and M.B. Jackson. 2004. Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science. 304:289–292. [DOI] [PubMed] [Google Scholar]
- Jahn, R., T. Lang, and T.C. Sudhof. 2003. Membrane fusion. Cell. 112:519–533. [DOI] [PubMed] [Google Scholar]
- Kemble, G.W., T. Danieli, and J.M. White. 1994. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 76:383–391. [DOI] [PubMed] [Google Scholar]
- Kielian, M., M.R. Klimjack, S. Ghosh, and W.A. Duffus. 1996. Mechanisms of mutations inhibiting fusion and infection by Semliki Forest virus. J. Cell Biol. 134:863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozerski, C., E. Ponimaskin, B. Schroth-Diez, M.F. Schmidt, and A. Herrmann. 2000. Modification of the cytoplasmic domain of influenza virus hemagglutinin affects enlargement of the fusion pore. J. Virol. 74:7529–7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov, M.M., and L.V. Chernomordik. 2002. The protein coat in membrane fusion: lessons from fission. Traffic. 3:256–267. [DOI] [PubMed] [Google Scholar]
- Leikina, E., and L.V. Chernomordik. 2000. Reversible merger of membranes at the early stage of influenza hemagglutinin-mediated fusion. Mol. Biol. Cell. 11:2359–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikina, E., D.L. LeDuc, J.C. Macosko, R. Epand, Y.K. Shin, and L.V. Chernomordik. 2001. The 1-127 HA2 construct of influenza virus hemagglutinin induces cell-cell hemifusion. Biochemistry. 40:8378–8386. [DOI] [PubMed] [Google Scholar]
- Leikina, E., C. Ramos, I. Markovic, J. Zimmerberg, and L.V. Chernomordik. 2002. Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. EMBO J. 21:5701–5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescar, J., A. Roussel, M.W. Wien, J. Navaza, S.D. Fuller, G. Wengler, and F.A. Rey. 2001. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell. 105:137–148. [DOI] [PubMed] [Google Scholar]
- Lindau, M., and W. Almers. 1995. Structure and function of fusion pores in exocytosis and ectoplasmic membrane fusion. Curr. Opin. Cell Biol. 7:509–517. [DOI] [PubMed] [Google Scholar]
- Lu, Y.E., C.H. Eng, S.G. Shome, and M. Kielian. 2001. In vivo generation and characterization of a soluble form of the Semliki forest virus fusion protein. J. Virol. 75:8329–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic, I., H. Pulyaeva, A. Sokoloff, and L.V. Chernomordik. 1998. Membrane fusion mediated by baculovirus gp64 involves assembly of stable gp64 trimers into multiprotein aggregates. J. Cell Biol. 143:1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic, I., E. Leikina, M. Zhukovsky, J. Zimmerberg, and L.V. Chernomordik. 2001. Synchronized activation and refolding of influenza hemagglutinin in multimeric fusion machines. J. Cell Biol. 155:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney, G.M., J.M. Smit, P. Liljestrom, and J. Wilschut. 2004. Semliki Forest virus produced in the absence of the 6K protein has an altered spike structure as revealed by decreased membrane fusion capacity. Virology. 325:200–206. [DOI] [PubMed] [Google Scholar]
- Melikyan, G.B., S.A. Brener, D.C. Ok, and F.S. Cohen. 1997. a. Inner but not outer membrane leaflets control the transition from glycosylphosphatidylinositol-anchored influenza hemagglutinin-induced hemifusion to full fusion. J. Cell Biol. 136:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan, G.B., H. Jin, R.A. Lamb, and F.S. Cohen. 1997. b. The role of the cytoplasmic tail region of influenza virus hemagglutinin in formation and growth of fusion pores. Virology. 235:118–128. [DOI] [PubMed] [Google Scholar]
- Melikyan, G.B., R.M. Markosyan, H. Hemmati, M.K. Delmedico, D.M. Lambert, and F.S. Cohen. 2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151:413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal, A., E. Leikina, L.V. Chernomordik, and J. Bentz. 2003. Kinetically differentiating influenza hemagglutinin fusion and hemifusion machines. Biophys. J. 85:1713–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis, Y., S. Ogata, D. Clements, and S.C. Harrison. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature. 427:313–319. [DOI] [PubMed] [Google Scholar]
- Nieva, J.L., R. Bron, J. Corver, and J. Wilschut. 1994. Membrane fusion of Semliki Forest virus requires sphingolipids in the target membrane. EMBO J. 13:2797–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes, A.M., H. Heidner, P. Thuman-Commike, B.V. Prasad, R.E. Johnston, and W. Chiu. 1998. Structural localization of the E3 glycoprotein in attenuated Sindbis virus mutants. J. Virol. 72:1534–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes, A.M., D. Ferreira, M. Horton, A. Saad, H. Tsuruta, R. Johnston, W. Klimstra, K. Ryman, R. Hernandez, W. Chiu, and D.T. Brown. 2004. Conformational changes in Sindbis virions resulting from exposure to low pH and interactions with cells suggest that cell penetration may occur at the cell surface in the absence of membrane fusion. Virology. 324:373–386. [DOI] [PubMed] [Google Scholar]
- Park, H.E., J.A. Gruenke, and J.M. White. 2003. Leash in the groove mechanism of membrane fusion. Nat. Struct. Biol. 10:1048–1053. [DOI] [PubMed] [Google Scholar]
- Peters, C., M.J. Bayer, S. Buhler, J.S. Andersen, M. Mann, and A. Mayer. 2001. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature. 409:581–588. [DOI] [PubMed] [Google Scholar]
- Plonsky, I., and J. Zimmerberg. 1996. The initial fusion pore induced by baculovirus GP64 is large and forms quickly. J. Cell Biol. 135:1831–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche, S., and Y. Gaudin. 2002. Characterization of the equilibrium between the native and fusion-inactive conformation of rabies virus glycoprotein Indicates that the fusion complex Is made of several trimers. Virology. 297:128–135. [DOI] [PubMed] [Google Scholar]
- Roederer, M., R. Bowser, and R.F. Murphy. 1987. Kinetics and temperature dependence of exposure of endocytosed material to proteolytic enzymes and low pH: evidence for a maturation model for the formation of lysosomes. J. Cell. Physiol. 131:200–209. [DOI] [PubMed] [Google Scholar]
- Russell, C.J., T.S. Jardetzky, and R.A. Lamb. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsonov, A.V., P.K. Chatterjee, V.I. Razinkov, C.H. Eng, M. Kielian, and F.S. Cohen. 2002. Effects of membrane potential and sphingolipid structures on fusion of Semliki Forest virus. J. Virol. 76:12691–12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangguan, T., D. Alford, and J. Bentz. 1996. Influenza-virus-liposome lipid mixing is leaky and largely insensitive to the material properties of the target membrane. Biochemistry. 35:4956–4965. [DOI] [PubMed] [Google Scholar]
- Shemer, G., and B. Podbilewicz. 2003. The story of cell fusion: big lessons from little worms. Bioessays. 25:672–682. [DOI] [PubMed] [Google Scholar]
- Sjoberg, M., and H. Garoff. 2003. Interactions between the transmembrane segments of the alphavirus E1 and E2 proteins play a role in virus budding and fusion. J. Virol. 77:3441–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel, J.J., and D.C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569. [DOI] [PubMed] [Google Scholar]
- Smit, J.M., R. Bittman, and J. Wilschut. 1999. Low-pH-dependent fusion of Sindbis virus with receptor-free cholesterol- and sphingolipid-containing liposomes. J. Virol. 73:8476–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit, J.M., G. Li, P. Schoen, J. Corver, R. Bittman, K.C. Lin, and J. Wilschut. 2002. Fusion of alphaviruses with liposomes is a non-leaky process. FEBS Lett. 521:62–66. [DOI] [PubMed] [Google Scholar]
- Stiasny, K., and F.X. Heinz. 2004. Effect of membrane curvature-modifying lipids on membrane fusion by tick-borne encephalitis virus. J. Virol. 78:8536–8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szule, J.A., and J.R. Coorssen. 2004. Comment on “Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis”. Science. 306:813. [DOI] [PubMed] [Google Scholar]
- Takahashi, N., T. Kishimoto, T. Nemoto, T. Kadowaki, and H. Kasai. 2002. Fusion pore dynamics and insulin granule exocytosis in the pancreatic islet. Science. 297:1349–1352. [DOI] [PubMed] [Google Scholar]
- Tamm, L.K. 2003. Hypothesis: spring-loaded boomerang mechanism of influenza hemagglutinin-mediated membrane fusion. Biochim. Biophys. Acta. 1614:14–23. [DOI] [PubMed] [Google Scholar]
- Vogel, S.S., E.A. Leikina, and L.V. Chernomordik. 1993. Lysophosphatidylcholine reversibly arrests exocytosis and viral fusion at a stage between triggering and membrane merger. J. Biol. Chem. 268:25764–25768. [PubMed] [Google Scholar]
- Waarts, B.L., R. Bittman, and J. Wilschut. 2002. Sphingolipid and cholesterol dependence of alphavirus membrane fusion. Lack of correlation with lipid raft formation in target liposomes. J. Biol. Chem. 277:38141–38147. [DOI] [PubMed] [Google Scholar]
- Weber, T., B.V. Zemelman, J.A. McNew, B. Westermann, M. Gmachl, F. Parlati, T.H. Sollner, and J.E. Rothman. 1998. SNAREpins: minimal machinery for membrane fusion. Cell. 92:759–772. [DOI] [PubMed] [Google Scholar]
- White, J., and A. Helenius. 1980. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc. Natl. Acad. Sci. USA. 77:3273–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]