

Abstract
The c-Myc oncoprotein is strongly induced during the G0 to S-phase transition and is an important regulator of cell cycle entry. In contrast to c-Myc, the putative Myc antagonist Mnt is maintained at a constant level during cell cycle entry. Mnt and Myc require interaction with Max for specific DNA binding at E-box sites, but have opposing transcriptional activities. Here, we show that c-Myc induction during cell cycle entry leads to a transient decrease in Mnt–Max complexes and a transient switch in the ratio of Mnt–Max to c-Myc–Max on shared target genes. Mnt overexpression suppressed cell cycle entry and cell proliferation, suggesting that the ratio of Mnt–Max to c-Myc–Max is critical for cell cycle entry. Furthermore, simultaneous Cre-Lox mediated deletion of Mnt and c-Myc in mouse embryo fibroblasts rescued the cell cycle entry and proliferative block caused by c-Myc ablation alone. These results demonstrate that Mnt-Myc antagonism plays a fundamental role in regulating cell cycle entry and proliferation.
Introduction
The oncogenic activity of Myc family proteins is initiated when their transcriptional regulation is disrupted by events such as gene amplification and gene translocation, which typically lead to elevated cellular Myc levels. Under normal conditions, high c-Myc protein levels are produced only transiently when cells enter the cell cycle and subsequently decline to low, but detectable levels (Persson et al., 1984; Hann et al., 1985). The significance of the transient burst of c-Myc expression early during cell cycle entry is revealed by experiments showing that ectopic c-Myc expression, by itself, can trigger quiescent fibroblasts to enter the cell cycle (Eilers et al., 1991). However, high Myc levels and its effects on cell proliferation are typically not sustainable, as many cell types respond to this situation by undergoing apoptosis. This is particularly the case when the growth and survival factor environment is, or becomes, limiting (Askew et al., 1991; Evan et al., 1992). Thus, both the up-regulation of c-Myc and its subsequent down-regulation, function as critical elements in the normal transition of quiescent cells to proliferating cells.
Consistent with a key role for Myc proteins in stimulating cell cycle entry, primary mouse embryo fibroblasts (MEFs) lacking c-Myc accumulate in G0 of the cell cycle and are rendered incompetent to proliferate in response to mitogenic stimulation (de Alboran et al., 2001; Trumpp et al., 2001). In contrast to primary cells, deletion of c-Myc in the “immortal” Rat1A fibroblast cell line significantly slows, but does not abrogate cell proliferation (Mateyak et al., 1997). The slowed proliferation in the latter cells appears to be the result of both a decreased rate at which cells traverse the cell cycle and a failure of some cells to enter a productive cell cycle (Holzel et al., 2001; Schorl and Sedivy, 2003). From these and many other results, it can be concluded that deregulated and elevated Myc production, so common in tumors, has the consequence of both stimulating cells to enter the cell cycle and preventing cells from properly exiting the cell cycle.
The ability of Myc family proteins to promote cell proliferation and contribute to tumor formation is dependent on its bHLHZip domain, which mediates heterodimerization with Max and DNA binding (Blackwood and Eisenman, 1991). The Myc–Max heterodimer, but not Myc alone, is able to bind DNA at the E-box consensus sequence CANNTG and activate transcription (Amati et al., 1992; Kretzner et al., 1992). In logarithmically proliferating cells it appears that most, if not all newly synthesized c-Myc enters into a complex with Max and that the rapid turnover of c-Myc protein (half-life of approximately 15 min) is not affected by heterodimerization with Max (Blackwood et al., 1992). Together with results showing that Max is a highly stable protein with a half-life in excess of 24 h (Blackwood et al., 1992), these results support the idea that a pool of Max is always available for interaction with newly synthesized c-Myc. However, in addition to Myc family proteins, Max interacts with a number of additional bHLHZip proteins that have the potential to limit the supply of Max for Myc heterodimerization. Moreover, these additional Max-interacting proteins, which include Mad family proteins (Mad1, Mxi1, Mad3, and Mad4), Mnt, and Mga (Zhou and Hurlin, 2001), are transcriptional repressors that have been demonstrated to antagonize Myc-dependent cell transformation in cell culture experiments.
Mnt is unique among these putative Myc antagonists in that it is expressed ubiquitously and like Max, Mnt levels do not fluctuate during the G0 to S-phase transition (Hurlin et al., 1997a, 2003). Importantly, Mnt appears to play a role in cell cycle entry, as cells lacking Mnt were found to exhibit an accelerated G0 to S-phase transition (Hurlin et al., 2003). These results, together with the finding that deletion of Mnt can predispose cells in vivo to apoptosis and tumorigenesis (Hurlin et al., 2003, Nilsson et al., 2004), suggest that Mnt–Max regulation of Myc activity during cell cycle entry may endow it with a unique role as a cellular Myc antagonist. In this study, we investigate the role Mnt plays in cell cycle entry and provide evidence that a transient switch in the ratio of Mnt–Max to c-Myc–Max underlies the basic decision of cells to enter the cell cycle and proliferate.
Results
Complex switching between Mnt–Max and c-Myc–Max during cell cycle entry
The accelerated G0 to S-phase transition exhibited by Mnt null MEFs (Hurlin et al., 2003) suggested that the balance between Mnt–Max and c-Myc–Max complexes may regulate proper cell cycle entry. Therefore we examined the relationship between Mnt and c-Myc steady-state levels and complex formation with Max during cell cycle entry of primary human fibroblasts. Fibroblasts were driven into quiescence by a combination of confluence arrest and serum starvation, then stimulated to enter the cell cycle by the addition of 10% FCS. Tritiated thymidine incorporation was used to confirm entry into S-phase (unpublished data). Consistent with our previous results using primary MEFs, Mnt levels were maintained at a near constant level during cell cycle entry (Fig. 1 a). As expected, c-Myc levels were initially present at extremely low levels in quiescent cells and rapidly, but transiently, induced (Fig. 1 a). To examine Mnt–Max and c-Myc–Max complex formation during cell cycle entry, low stringency anti-Max immunoprecipitations were performed. Low stringency anti-Max immunoprecipitations were treated with high stringency buffer to elute bound proteins and secondary immunoprecipitations were performed on the eluates with anti–c-Myc and Mnt antibodies (Hurlin et al., 1997a). Whereas Max remained at near constant levels during cell cycle entry, its interaction with Mnt and c-Myc fluctuated as a function of c-Myc levels (Fig. 1 b). Although c-Myc and c-Myc–Max levels increased from a very low level in unstimulated cells to high levels at 4 h, a corresponding drop in Mnt–Max levels occurred, despite near constant Mnt levels (Fig. 1 a). As c-Myc and c-Myc–Max levels subsequently declined at 8 and 24 h after serum stimulation, levels of Mnt–Max increased (Fig. 1 b). These results suggest that induction of c-Myc to high levels during the G0 to S-phase transition sets up a competition between c-Myc and Mnt for Max, that results in a limited supply of Max.
Figure 1.
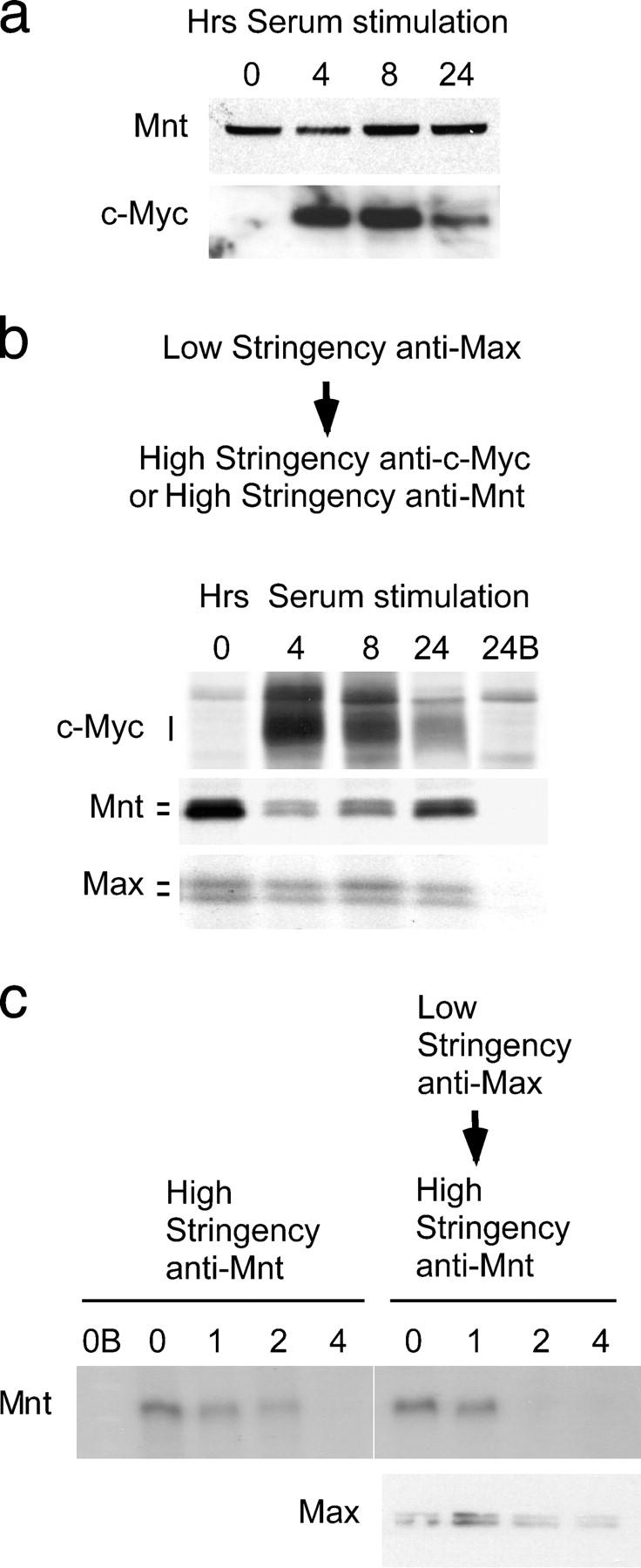
Complex switching between Mnt–Max and c-Myc–Max during cell cycle entry. (a) Western blot showing Mnt and c-Myc levels at 0, 4, 8, and 24 h after serum stimulation of quiescent MEFs. (b) Levels of Mnt and c-Myc found in low stringency anti-Max immunoprecipitations during cell cycle entry. Note reduction in Mnt (and Mnt–Max complexes) at times of high c-Myc levels. (c) Pulse-chase analysis of Mnt turnover when complexed to Max. Cells were metabolically labeled with medium containing [35S]methionine (pulse) then “chased” with medium containing unlabeled methionine. Mnt was immunoprecipitated under high stringency conditions (representing total Mnt) or from low stringency Max immunoprecipitated material at 0, 1, 2, and 4 h, as indicated, during the chase period. B, immunogen blocked antibody.
The presence of Mnt–Max complexes before c-Myc synthesis during cell cycle entry suggested that the availability of Max for newly synthesized c-Myc is, at least in part, a function of the stability of the Mnt–Max complex. Although we previously showed that Mnt is a very short-lived protein (Hurlin et al., 1997b), it was unknown whether the half-life of Mnt was affected by interaction with Max. To address this question, we used pulse-chase analysis combined with low stringency anti-Max immunoprecipitations to examine the half-life of Mnt when bound to Max. Consistent with previous results (Hurlin et al., 1997b), endogenous Mnt protein exhibited a half-life of ∼20–30 min (Fig. 1 c). The profile of Mnt turnover when complexed to Max was very similar to that observed under high stringency conditions (Fig. 1 c). These results suggest that the rapid turnover of Mnt, like that of c-Myc (Blackwood et al., 1992), occurs while in a complex with the long-lived Max protein and determines the stability of Mnt–Max complexes. Moreover, because the intensity of labeled Mnt immunoprecipitated under high stringency conditions was similar to that obtained from Max complexes (Fig. 1 c), these results suggest that most newly synthesized Mnt enters into a complex with Max in logarithmically growing cells.
Switching between Mnt–Max and c-Myc–Max binding at shared target genes during cell cycle entry
To directly examine the consequences of complex switching between Mnt–Max and c-Myc–Max during cell cycle entry, chromatin immunoprecipitation (ChIP) experiments were performed. Early passage Mnt null and wild-type MEFs were made quiescent and stimulated to enter the cell cycle as described above. c-Myc expression was monitored by Western blot (Fig. 2 a) and tritiated thymidine incorporation assays performed in parallel to confirm cells were arrested and entered into the cell cycle (not depicted). Binding to specific sites in a number of known or suspected Myc target genes, including Cdk4, Cyclin D2 (ccnd2), ODC, E2F2, Nucleolin, Tert, and CAD (for review see Cole and McMahon, 1999) was determined in quiescent cells and at 4 and 24 h after serum stimulation. One of several E-box–containing regions within the Cyclin E1 (ccne1) gene where no c-Myc or Mnt binding was observed was used as a negative control.
Figure 2.
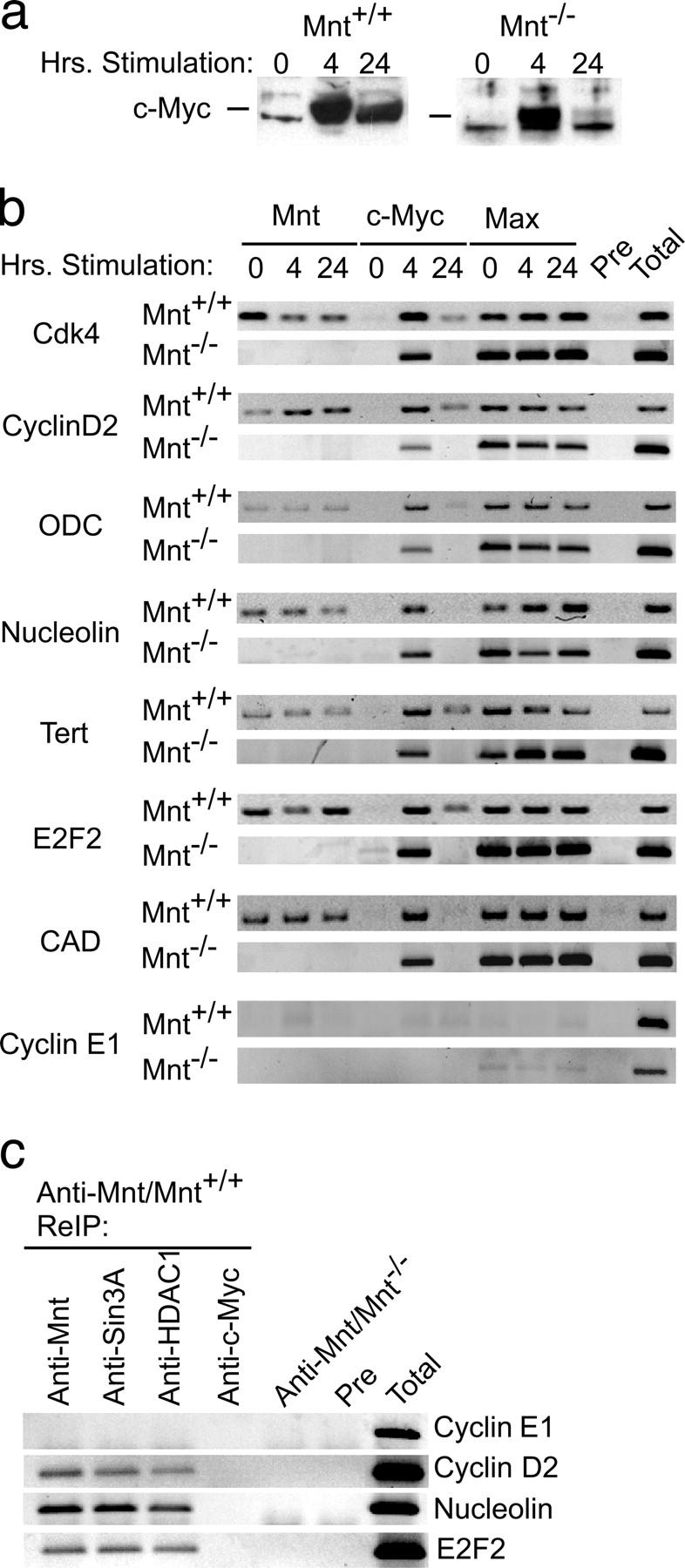
Mnt–Max and Myc–Max complex switching on shared target genes. (a) Western blot showing c-Myc expression in wild-type (Mnt+/+) and Mnt null (Mnt−/−) MEFs at 0, 4, and 24 h after serum stimulation of quiescent cells. (b) ChIP analysis, performed in parallel with the Western blot shown in panel a, examining Mnt, c-Myc, and Max binding to specific E-box–containing regions in the Cdk4, Cyclin D2, ODC, Nucleolin, Telomerase (Tert) E2F2, CAD, and Cyclin E1 genes at 0, 4, and 24 h after serum stimulation. (c) Chromatin reimmunoprecipitation (ReIP) assays showing the presence of Sin3A and HDAC1 at several Mnt-Myc target genes. Pre, preimmune serum; Total, total input.
As shown in Fig. 2 b, Mnt was bound at the Cdk4, Cyclin D2, ODC, Nucleolin, E2F2, Tert, Nucleolin, and CAD genes in quiescent cells (0 h) and at 4 and 24 h after serum stimulation. No signal was observed with either preimmune serum or in Mnt null MEFs (Fig. 2 b). Consistent with the strong induction of c-Myc observed in cells 4 h after serum stimulation (Fig. 2 b), c-Myc binding at the same sites was strongest at 4 h (Fig. 2 a). This was true in both wild-type and Mnt null MEFs. Very little signal for c-Myc binding was present at 0 and 24 h, when c-Myc levels were very low (Fig. 2 a). Like Mnt, signal for Max binding was consistently observed in both quiescent and serum-stimulated cells at the promoter sites in Myc target genes (Fig. 2 b). Signal for Max binding at these promoter regions was also readily detected in Mnt null MEFs, even at times when c-Myc was not detected. The latter results suggest that either Max homodimers and/or heterodimers with Max-interacting proteins other than c-Myc and Mnt occupy the same promoter sites at these times.
Previous resulted showed that Mnt interacts with Sin3 in vitro and that deletion of Mnt's Sin3 interaction domain abolished its ability to repress transcription (Hurlin et al., 1997a; Meroni et al., 1997). To examine endogenous Mnt-Sin3 interactions as a function of cell cycle entry, low stringency anti-Mnt immunoprecipitations were performed, followed by immunoblotting with anti-Sin3A and Max antibodies. These results showed that Mnt interacts with Sin3 in vivo and that Mnt-Sin3 interaction is not regulated during the cell cycle (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200411013/DC1). Furthermore, ChIP assays performed by first immunoprecipitating with Mnt antibody, then reimmunoprecipitating (Metivier et al., 2003) with either anti-Sin3A or anti-HDAC1 (an Sin3A associated protein; Ayer, 1999) demonstrated association of Sin3A and HDAC1 with Mnt at Mnt-Myc binding regions within the Cyclin D2, Nucleolin, and TERT genes (Fig. 2 c). These results suggest that Mnt–Sin3–HDAC complexes are present at Mnt binding sites in chromatin.
Mnt/c-Myc target gene regulation in the absence of Mnt
To determine how loss of Mnt affects transcription of c-Myc/Mnt target genes, real-time RT-PCR was used to analyze transcript abundance for genes identified above or previously (Hurlin et al., 2003; Nilsson et al., 2004) in the context of cell cycle entry. RNA was extracted from serum starved, quiescent Mnt null and wild-type cells and at 4, 12, 16, 20, and 24 h after serum stimulation. Levels of ARBP P0 RNA, encoded by a gene whose transcription is not regulated during cell cycle entry (Fig. 3; Humbert et al., 2000), was used to normalize all RT-PCR values obtained. As expected, c-Myc RNA levels were strongly induced in MEFs after serum stimulation (Fig. 3). However, the dynamics of induction were different between wild-type and Mnt null MEFs, with the latter cells showing higher levels at 12 h, but lower levels at other time points. Mnt and Max RNA levels, like their respective encoded proteins (Hurlin et al., 2003), were maintained at a near constant level. In contrast, expression levels of Nucleolin, E2F2, and CAD RNA were consistently altered, with each showing markedly increased levels in Mnt null MEFs, especially 12 h after serum stimulation (Fig. 3). The E1F4E gene, a putative Myc target gene (Jones et al., 1996) encoding elongation factor 4E, was also up-regulated at the 12-h time point (Fig. 3), although we have yet to identify Myc-Mnt binding sites in the mouse version of this gene. In contrast, ODC levels were not significantly affected by loss of Mnt. Interestingly, although the amplitude of Cyclin D2 and CDK4 expression did not appear to be significantly affected by Mnt loss, the expression patterns of Cyclin D2 and CDK4 in Mnt null MEFs appeared to reflect the accelerated cell cycle entry profile of these cells compared with wild-type MEFs (Fig. 5; Hurlin et al., 2003).
Figure 3.

Regulation of c-Myc/Mnt target genes during cell cycle entry in the absence of Mnt. RNA from wild-type and Mnt null primary MEFs was harvested at the indicated times after serum stimulation and quantitative real-time RT-PCR assays performed in triplicate for the indicated genes. The data are representative of two or more independent experiments. RNA levels of the ARBP P0 gene, which do not change during cell cycle entry (not depicted), were used to standardize samples. Fold expression values were calculated relative to the 0-h time point in wild-type cells for each gene as previously described (Livak and Schmittgen, 2001). SDs are shown.
Overexpression of Mnt suppresses cell cycle entry
The defective cell cycle entry of Mnt null MEFs (Hurlin et al., 2003), together with the above findings, suggest that the ratio of Mnt to c-Myc is key to proper cell cycle entry. To better define the relationship between levels of c-Myc and Mnt during cell cycle entry, primary wild-type and Mnt null MEFs were infected with Mnt-expressing retrovirus (pBabeMnt-HA), briefly selected to remove noninfected cells, and cell proliferation and cell cycle entry experiments performed. For proliferation assays, cells were plated at 1.5 × 104 cells per 60-mm dish and counted on four consecutive days at 24-h intervals. As shown in Fig. 4 (a and b), Mnt overexpression caused a significant decline in the proliferation rate of wild-type and Mnt−/− MEFs.
Figure 4.
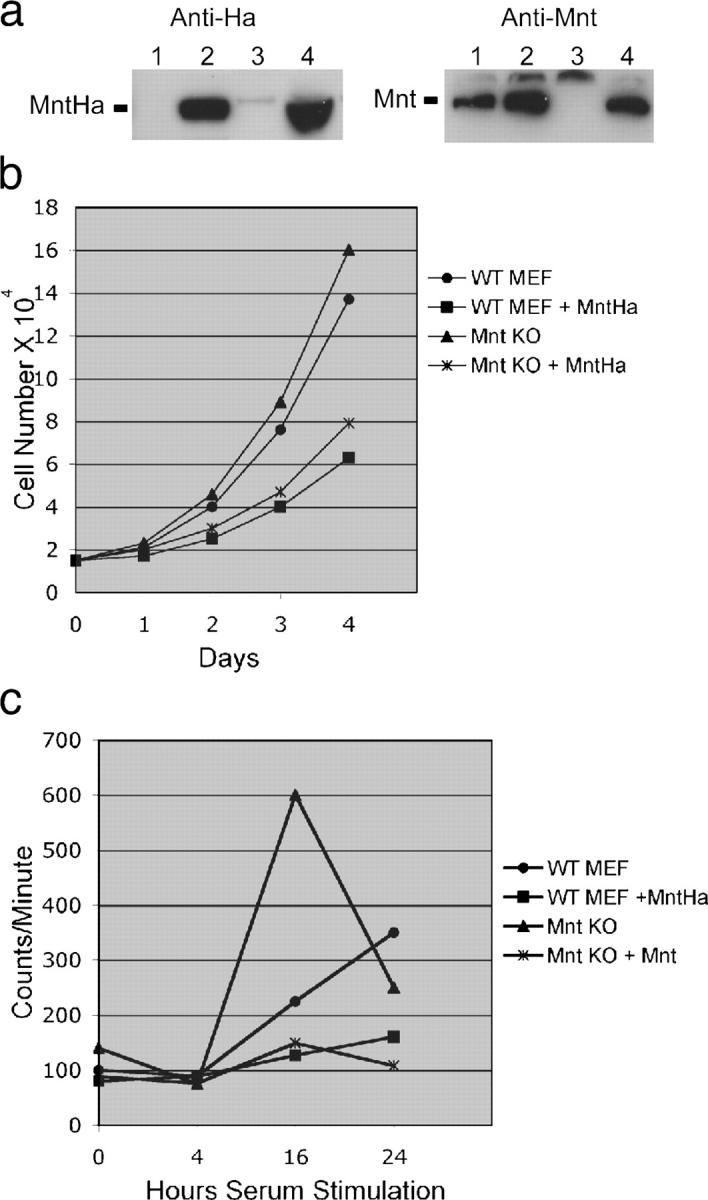
Mnt overexpression slows cell proliferation and impedes cell cycle entry. (a) Western blot showing expression of Ha-tagged Mnt (anti-Ha set) in pBabeMntHa-infected MEFs (lanes 2 and 4) and endogenous Mnt (anti-Mnt set) in MEFs infected with empty virus (lanes 1 and 3). (b) Proliferation curve conducted for 4 d showing decline in proliferation rate caused by Mnt overexpression in wild-type and Mnt null MEFs. Each value is the average number of cells counted from three different dishes in two separate experiments. (c) Analysis of cell cycle (S-phase) entry in wild-type and Mnt null MEFs overexpressing Mnt. Tritiated thymidine incorporation was measured at the indicated number of hours after serum stimulation of MEFs made quiescent by confluence arrest and serum starvation. Experiments were performed at least twice in triplicate and averages are shown.
To examine how Mnt overexpression affects the G0 to S-phase transition, tritiated [3H]thymidine incorporation assays were performed after serum stimulation of confluence arrested and serum starved cells. Consistent with previous results (Hurlin et al., 2003), Mnt null MEFs showed elevated tritiated thymidine incorporation 16 h after serum stimulation indicating robust entry into S-phase (Fig. 4 c). In contrast, overexpression of Mnt in both Mnt null and wild-type MEFs strongly suppressed tritiated thymidine incorporation over the time period analyzed (Fig. 4 c). However, these cells did eventually reenter the cell cycle and proliferate, albeit at a slowed rate (Fig. 4 b and not depicted). Thus, Mnt overexpression impeded, but did not completely prevent cell cycle reentry.
Mnt deficiency rescues proliferation arrest caused by c-Myc deletion in primary MEFs
To better define the relationship between Mnt and c-Myc in cell proliferation and cell cycle entry, we examined the consequences of acute, simultaneous deletion of both Mnt and c-Myc in primary MEFs. Crosses between Mnt flox/flox (Hurlin et al., 2003) and c-Myc floxN/floxN mice (Trumpp et al., 2001) were performed to generate mice containing homozygous floxed alleles for both Mnt and c-Myc (Mnt/c-Myc dCKO). MEFs were then isolated from the latter mice and from Mnt flox/flox mice and c-Myc flox/flox mice. MEFs at passage 1 were infected with adenovirus expressing Cre recombinase and GFP. GFP expression indicated that >95% of cells were infected and PCR genotyping and Western blot analysis confirmed that a very high percentage of floxed alleles were successfully targeted (Fig. 5, a and b, not depicted). Proliferation assays performed 48 h after infection cells confirmed previous results (de Alboran et al., 2001; Trumpp et al., 2001) showing that deletion of c-Myc leads to rapid cessation of primary MEF proliferation (Fig. 5 c). In contrast, primary MEFs in which c-Myc and Mnt were simultaneously ablated continued to proliferate (Fig. 5 c). Very similar results were achieved after AdCre infection of immortal (i) Mnt flox/flox, ic-Myc flox/flox, and iMnt-c-Myc dCKO MEF cell lines (Fig. 5 d). The latter cell lines were developed by continuously passaging primary cell populations until they escaped senescence (Todaro and Green, 1963).
Figure 5.
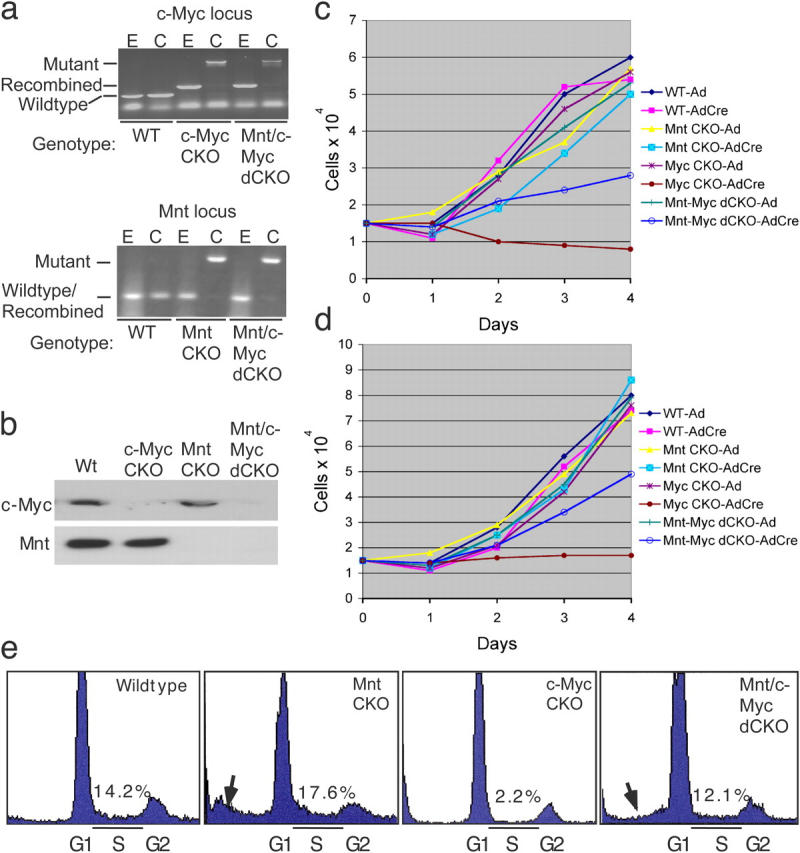
Deletion of Mnt rescues proliferation arrest caused by loss of c-Myc. (a) PCR genotyping of MEFs of the indicated genotypes after infection with empty (E) adenovirus or Cre (C) recombinase-expressing adenovirus. Note the PCR primers used to detect deleted Mnt alleles do not discriminate from wild-type and recombined Mnt alleles. (b) Western blot examining Mnt and c-Myc expression after AdCre infection of the indicated genotypes. (c and d) MEF proliferation curves after deletion of Mnt, c-Myc, and Mnt plus c-Myc in primary MEFs (c) and immortal MEFs (d). Values shown in c and d are the average of cell counts obtained from triplicate plates and are representative of results obtained from two independent experiments. (e) Cell cycle profiles generated from FAC sorting of propidium iodide stained cells 48 h after AdCre infection of the indicated cell lines. % of cells in S-phase (S) is shown. Arrows highlight sub G1 (growth phase 1) fractions consistent with apoptosis.
The ability of Mnt deficiency to rescue proliferation arrest was further examined by fluorescent cell sorting of propidium iodide stained primary cells after AdCre infection (Fig. 5 e). Consistent with the proliferation assays, the fraction of cells in S-phase was dramatically reduced by deletion of c-Myc, but increased to near wild-type levels in MEFs lacking both c-Myc and Mnt. A substantial elevation of a sub-G1 fraction of cells, which is consistent with apoptosis, was present in Mnt flox/flox MEF populations and, to a lesser extent, in Mnt-c-Myc dCKO MEFs, but not c-Myc flox/flox MEFs (Fig. 5 e, arrows). These results are consistent with previous results showing that Mnt deficiency triggers apoptosis (Hurlin et al., 2003; Nilsson et al., 2004).
To further characterize how Mnt deletion rescued proliferation arrest caused by loss of c-Myc, a series of Western blots were performed examining the expression of proteins that perform critical functions in cell cycle control (Fig. 6). From this analysis, it was observed that several proteins up-regulated by loss of Mnt, including Cyclin D2, Cyclin A, Cyclin B, Cyclin E, and p107, were also up-regulated in Mnt/c-Myc dCKO MEFs, relative to their expression in c-Myc–deficient MEFs (Fig. 6). In addition, pRB was hyperphosphorylated by Mnt deletion, both in the presence and absence of c-Myc. These results suggest that Mnt deletion rescues the proliferation arrest caused by c-Myc deletion at least partly through up-regulation of critical cell cycle regulatory proteins.
Figure 6.
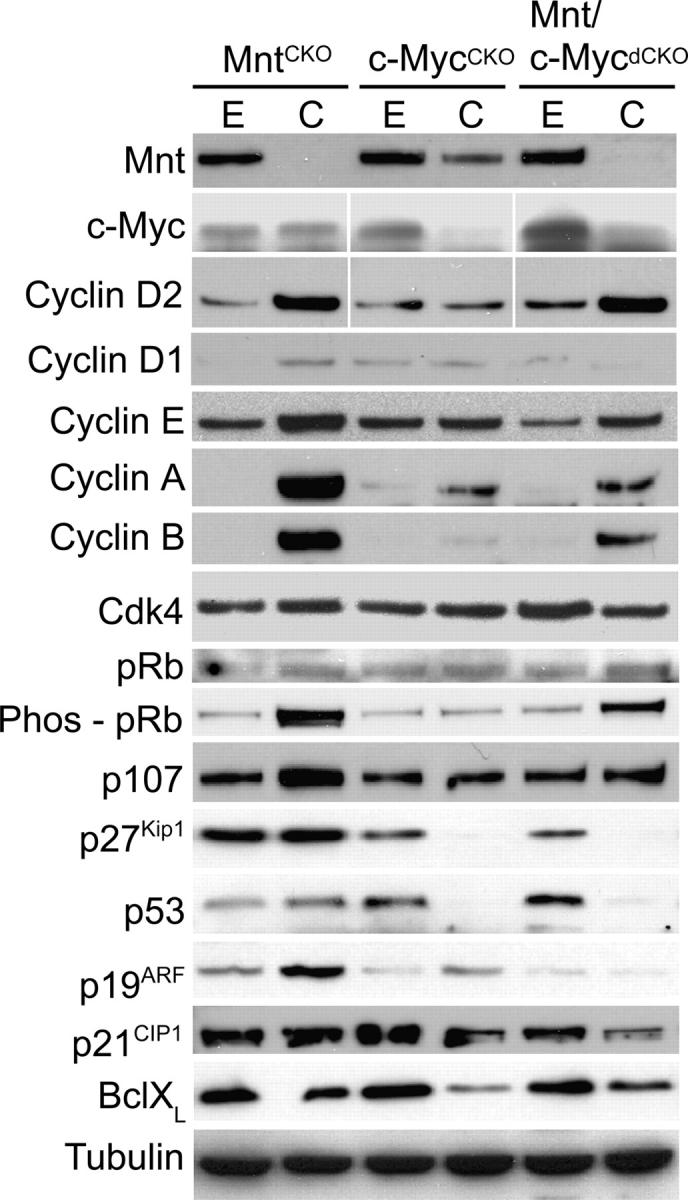
Effect of acute Mnt, c-Myc, and Mnt plus c-Myc deletion on the expression levels of proteins involved in the regulation of cell proliferation. Cell extracts were obtained from primary MEFs after AdCre-mediated deletion of Mnt, c-Myc, or both Mnt and c-Myc as shown in Fig. 5 (a and b). Western blots were performed with antibodies against the indicated proteins.
Interestingly, we found that c-Myc deletion caused down-regulation of p53, p19ARF, and p27Kip1, but that these affects were not reversed by simultaneous deletion of Mnt (Fig. 6). Moreover, the acute effect of Mnt deletion on p53, p19ARF, and BclXL levels was minimal. This is in contrast to previous results, which showed strong up-regulation of p53 and P19ARF, as well as BclXL down-regulation, after siRNA knockdown of Mnt, even in absence of c-Myc (Nilsson et al., 2004).
Finally, the relationship between Mnt and c-Myc specifically in the context of cell cycle entry was examined after AdCre-mediated deletion of Mnt, c-Myc, and Mnt plus c-Myc in primary MEF cell lines. Whereas ablation of c-Myc completely blocked the ability of MEFs to enter S-phase, MEFs in which c-Myc and Mnt were simultaneously ablated incorporated tritiated thymidine with kinetics similar to control AdCre-infected MEFs (Fig. 7 a). Consistent with the observed rescue of cell cycle entry kinetics, deletion of Mnt rescued the depressed RNA levels of the Cyclin D2, E2F2, ODC, and Cyclin E caused by c-Myc deletion (Fig. 7 b). Interestingly, Gadd45a, a target of c-Myc repression was up-regulated during cell cycle entry in the absence of c-Myc, but its regulation MEFs deficient for both Mnt and c-Myc was similar to that observed in wild-type cells (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200411013/DC1). However, other putative targets of Myc repression were not similarly affected (Fig. S2).
Figure 7.

Analysis of cell cycle (S-phase) entry after acute deletion of Mnt and Mnt plus c-Myc. (a) Tritiated thymidine incorporation (counts/minute) were determined at the indicated times after serum stimulation of immortal MEFs made quiescent by confluence arrest and serum deprivation. (b) Quantitative real-time RT-PCR analysis of Cyclin D2, E2F2, ODC, and Cyclin E1 gene expression after serum stimulation. Both tritiated thymidine and RT-PCR experiments were performed in triplicate and SDs are shown.
Discussion
In this study, we provide evidence that complex switching between Myc–Max and Mnt–Max governs proper entry into the cell cycle. Whereas c-Myc expression is strongly, but transiently, up-regulated during the transition of quiescent MEFs into S-phase, Mnt is constitutively expressed throughout cell cycle entry and in proliferating cells (Hurlin et al., 2003). However, Mnt–Max complexes were found to decrease, whereas c-Myc–Max levels increased during times of maximal Myc expression (Fig. 1). We interpret these data to suggest that as c-Myc levels rise above a critical threshold during cell cycle entry, competition between c-Myc and Mnt for binding to Max leads to a limiting supply of Max. A limiting supply of Max is significant in this setting because it theoretically accelerates a switch from repressive Mnt–Max complexes to c-Myc–Max complexes at shared target genes. Importantly, although the constitutive expression of Mnt during cell cycle entry is postulated to have a restrictive influence on c-Myc–Max complex formation, the short half-life of the Mnt protein, even when complexed with Max (Fig. 1 c), appears to ensure that a continuous, but measured supply of Max is available for complex formation with c-Myc or potentially other Max partners. As c-Myc levels increase, it is also possible that limiting amounts of Max may lead to Mnt engaging in alternative complexes such as with Mlx (Meroni et al., 2000). However, we have not observed Mnt–Mlx complexes in vivo (unpublished data).
The importance of maintaining a proper ratio of Myc to Mnt during cell cycle entry is suggested by results showing that overexpression of Mnt retards cell cycle entry and cell proliferation (Fig. 4) and loss of Mnt accelerates cell cycle entry (Figs. 4 and 5; Hurlin et al., 2003). Moreover, the notion that Mnt and c-Myc function as a binary system to regulate cell cycle entry and proliferation is firmly supported by the ability of Mnt deletion to rescue the proliferative arrest caused by c-Myc ablation in primary and immortal MEFs (Fig. 5). It is important to stress however, that Mnt ablation in primary and immortal MEFs does not completely rescue the proliferative block caused by loss of c-Myc (Fig. 5). Although increased apoptosis after Mnt deletion (Fig. 5; Hurlin et al., 2003; Nilsson et al., 2004) may be responsible for the partial, instead of full rescue of primary MEFs, it is also possible that antagonism of Myc by Mad family proteins or other unknown mechanisms may contribute as well.
While this work was in progress, it was reported that siRNA-mediated knockdown of Mnt in Myc-deficient immortal Rat1A fibroblasts (Mateyak et al., 1997) rescued the slow proliferation of these cells (Nilsson et al., 2004). However, whereas deletion of c-Myc in the Rat1A cell line causes slowed proliferation, deletion of c-Myc in primary and in immortal c-Myc flox/flox MEFs caused a complete block in proliferation (MEFs; Fig. 5, c and d; de Alboran et al., 2001; Trumpp et al., 2001). These results suggest that Rat1A cells acquired defects over the course of their extensive growth in culture that partially substitute for loss of c-Myc. The possibility and perhaps likelihood that such acquired defects affect cell cycle control, argue that primary or “freshly” immortalized Mnt/c-Myc dCKO MEFs are better suited to rigorously establish the relationship between c-Myc and Mnt in governing cell cycle entry and cell proliferation.
It should also be noted that there are significant differences observed between siRNA-mediated knockdown of Mnt (Nilsson et al., 2004) and Cre-mediated Mnt gene deletion in the regulation of gene targets and downstream effector protein levels. Whereas siRNA-mediated knockdown of Mnt is associated with a dramatic down-regulation of antiapoptotic BclXL and strong up-regulation of pro-apoptotic p19ARF and p53 protein levels and ODC RNA and protein levels (Nilsson et al., 2004), we find that chronic Mnt deletion or acute Mnt deletion in primary MEFs has only a modest or no effect on their expression levels. (Figs. 3, 6, and 7; Hurlin et al., 2003). Moreover, siRNA-mediated Mnt knockdown was found to dramatically down-regulate p27Kip protein levels in Myc −/− Rat1A fibroblasts, NIH3T3s, and MEFs (Nilsson et al., 2004), but we surprisingly find that p27Kip1 levels are up-regulated as a result of both chronic and acute deletion of Mnt, as well as in tumors caused by Mnt deletion in vivo (Fig. 6; Fig. S2; and not depicted). It is not clear why these differences are seen, but they are likely due to nonspecific effects associated with the different methods used to remove Mnt.
The displacement of Mnt–Max complexes in favor of c-Myc–Max complexes at shared target genes during cell cycle entry (Fig. 2) is consistent with the proposal that Myc–Max complexes operate, at least in part, by derepressing Mnt–Max bound target genes (Hurlin et al., 1997a, 2003; Nilsson et al., 2004). Although it is unlikely that Mnt–Max and c-Myc–Max complexes bind to and regulate exactly the same set of target genes, we find that they do bind and regulate at least a subset of overlapping target genes (Figs. 2, 3, and 7). What remains unclear is the precise relationship between c-Myc and Mnt binding and transcriptional status. For example, we have been unable to establish a clear link between the presence of Mnt at different target genes and local histone acetylation status (not depicted), despite being able to detect Mnt–Sin3 and presumably Mnt–Sin3–HDAC interactions at target gene binding sites (Fig. 2). However, chromatin deacetylation at the Cyclin D2 gene does appear to correspond to Mnt binding (unpublished data). Whatever the mechanism of Mnt repression, the finding that acute Mnt deletion rescues, at least partially, the severe down-regulation of Myc targets such as Cyclin D2 and E2F2 caused by acute c-Myc deletion in primary MEFs (Fig. 7 b) is consistent with Mnt and c-Myc functioning as a repression-activation system to regulate the expression of critical target genes involved in cell cycle progression. Although we observe strong down-regulation of some Myc target genes after c-Myc deletion, others such as ODC were only modestly down-regulated (Fig. 7 b). The slight affect on ODC levels are consistent with studies showing that most Myc target genes are only modestly affected by loss of c-Myc in immortal Rat1A cells (Bush et al., 1998; Cole and McMahon, 1999). Furthermore, transcript levels of most of the various Myc target genes examined in this and previous studies were only modestly affected by loss of Mnt (Fig. 3; Hurlin et al., 2003; Nilsson et al., 2004). One potential explanation for these findings is that the transcriptional activity of Mnt and Myc is inherently weak (Eisenman, 2001) and that their biological impact derives from eliciting small changes in the expression levels of perhaps thousands of transcriptional targets (for reviews see Fernandez et al., 2003; Orian et al., 2003; Cawley et al., 2004; see http//www.myc-cancer-gene.org). Alternatively, transcriptional regulation of Myc and Mnt target genes may be constrained to specific physiological settings that have yet to be fully recognized. For example, during cell cycle entry in Mnt null MEFs, up-regulation of target genes was most apparent 12 h after serum stimulation (Fig. 3). Thus, because c-Myc binding to target gene promoters was observed by 4 h after serum stimulation (Fig. 2 b), secondary events, perhaps related to cell cycle–regulated binding of other transcription factors to DNA or to Myc, may dictate the time and amplitude of Myc-dependent transcription activation (Cole and McMahon, 1999). The different responses in gene expression observed in Mnt-deficient cells may also reflect the existence of different classes of Myc target genes (Haggerty et al., 2003). The development of MEF cell lines containing conditional, floxed alleles of Mnt, c-Myc, and both Mnt and c-Myc (Fig. 5) should prove useful for more precisely defining the mechanisms that underlie the regulation of specific target genes by Myc and Mnt.
Although up-regulation of c-Myc expression plays an important role in stimulating cell cycle entry, it is of paramount importance that it is then rapidly down-regulated. Sustained high level Myc expression is not well tolerated and sensitizes cells to apoptosis and tumorigenesis (Pelengaris et al., 2002; Nilsson and Cleveland, 2003). Because both Myc–Max and Mnt–Max complexes are present in cells during the normal proliferative cell cycle (Hurlin et al., 1997a), we speculate that Myc-driven apoptosis and tumorigenesis results when the proper balance between Myc–Max and Mnt–Max in cells is breached. This idea is strongly supported by results showing that loss of Mnt triggers a “Myc-like” response of accelerated proliferation and apoptosis in fibroblasts and leads to tumor formation when deleted in breast epithelium (Hurlin et al., 2003) and T cells (unpublished data). Though it is not yet clear whether tumor formation caused by loss of Mnt is mechanistically equivalent to that caused by Myc, our results establish that Myc-Mnt antagonism underlies the basic decision of cells to enter the cell cycle and proliferate.
Materials and methods
Immunoprecipitation and Western blot studies
Low and high stringency immunoprecipitation conditions were performed as previously described (Hurlin et al., 1997a). Antibodies included anti-Mnt (affinity-purified H6823; Hurlin et al., 2003), anti-Max (Cruz C-17; Santa Cruz Biotechnology, Inc.), and anti c-Myc (N-262; Santa Cruz Biotechnology, Inc.). Max antibody was blocked using Gst-Max (Blackwood et al., 1992). In addition to the above antibodies, the following antibodies were used in Western blots: Cyclin D1, Cyclin D2, Cyclin E1, Cyclin A1, Cyclin B, Cdk4, p107, p53, tubulin (all obtained from Santa Cruz Biotechnology, Inc.), pRb, phospho-ser 807/811 Rb, (Cell Signaling), p27Kip1, p21Cip1, BclXL (BD Biosciences), and p19ARF (AbCam).
Generation of MEFs
MEFs were obtained from Mnt null and Mnt flox/flox (Hurlin et al., 2003; Toyo-oka et al., 2004), c-Myc flox/flox (Trumpp et al., 2001), and Mnt flox/flox − c-Myc FloxN/FloxN (or Mnt−/CKO − c-Myc−/CKO) embryos at embryonic day 13.5 as previously described (Hurlin et al., 2003). Immortal MEF cell lines were established by continuous passaging of cultures using a 3T9 protocol (Todaro and Green, 1963). Genotyping of cells was performed by PCR. Primer sets for the c-Myc locus were previously described (Trumpp et al., 2001).
Cell cycle entry and proliferation assays
For cell cycle entry experiments, MEFs or HFFs were grown to confluence and switched from medium containing 10% FCS to medium containing 0.1% FCS. Cells were maintained in 0.1% FCS for 2–3 d before stimulating with medium containing 10% FCS. Tritiated thymidine incorporation assays were performed in triplicate in 6-well dishes. The cells were labeled by incubating in 2 ml DME containing 1 μCi/ml methyl [3H]thymidine (ICN Biomedicals) for 2 h at 37°C, 5% CO2. Cells were washed twice with ice-cold PBS and incubated for 20 min at 4°C in 5% TCA to remove acid soluble radioactivity. The cells were washed twice with 70% EtOH and solubilized for 30 min at 37°C in 1 ml 0.1 M NaOH, 2% Na2CO3, and 1% SDS. 5 ml of Ecolume scintillation cocktail (ICN Biomedicals) was added to lysates and counts per minute determined using a scintillation counter (model LS5000TD; Beckman Coulter). Proliferation assays were performed as previously described (Hurlin et al., 2003).
ChIP analysis
For cell cycle entry assays, cells from four 150-mm tissue culture dishes (containing cells to grown to confluence, serum starved and serum stimulated) were washed once with PBS and fixed with 1.5% formaldehyde (in PBS) for 5 min at 37°C. Fixed cells were collected, lysed, sonicated to attain an average length of 750–1,200 bp, and immunoprecipitations were performed using equal amounts of sonicated lysates. Immunoprecipitations, washes, and elution of immunoprecipitated material were performed as previously described (Metivier et al., 2003). Preimmune serum was used as a negative control. Re-immunoprecipitations of primary immunoprecipitations were performed as described by Metivier et al. (2003) and PCR (30–35 cycles) was performed on equal amounts of eluate. Antibodies used for ChIP experiments included affinity-purified anti-Mnt (H6823), c-Myc (N-262), Max (C-17), Sin3A (AK-11) were obtained from Santa Cruz Biotechnology, Inc., and HDAC1 (2E10) was obtained from Upstate Biotechnology.
Real-time PCR
RNA was harvested using TRIzol reagent (Invitrogen), purified for mRNA using oligotex particles (QIAGEN), and quantified by spectrophotometer. 0.5 μg mRNA per sample was random primed using Superscript II reverse transcriptase (Invitrogen). Samples in triplicate were amplified using SYBR green I dye in an Applied Biosystems 7900HT sequence detection system. Analysis of data was performed using the 2−ΔΔCt method (Livak and Schmittgen, 2001) and quantitated relative to the ARBO PO gene. Gene expression was normalized to unstimulated (0 time point) wild-type samples, which provided an arbitrary constant for comparative fold expression.
Online supplemental material
Fig. S1 shows co-immunoprecipitation of Mnt and Sin3 in cells. Low stringency (LS) Mnt immunoprecipitates were separated by SDS-PAGE and Western blots performed using the antibodies against the indicated proteins. Max was used as a positive control for Mnt interaction and high stringency (HS) Mnt immunoprecipitates were used as a negative control. Fig. S2 shows expression of Myc repression targets during cell cycle entry of MEFs deficient in either Mnt, c-Myc, or both Mnt and c_Myc. RNA was harvested at the indicated times after serum stimulation and real-time RT-PCR performed for the indicated genes. Experiments were performed in triplicate and SDs are shown. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200411013/DC1.
Acknowledgments
This work was supported by National Institutes of Health grants CA 87788-02 and CA108855-01 to P.J. Hurlin.
Abbreviations used in this paper: ChIP, chromatin immunoprecipitation; MEF, mouse embryo fibroblast.
References
- Amati, B., S. Dalton, M.W. Brooks, T.D. Littlewood, G.I. Evan, and H. Land. 1992. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature. 359:423–426. [DOI] [PubMed] [Google Scholar]
- Askew, D.S., R.A. Ashmun, B.C. Simmons, and J.L. Cleveland. 1991. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene. 6:1915–1922. [PubMed] [Google Scholar]
- Ayer, D.E. 1999. Histone deacetylases: transcriptional repression with SINers and NuRDs. Trends Cell Biol. 9:193–198. [DOI] [PubMed] [Google Scholar]
- Blackwood, E.M., and R.N. Eisenman. 1991. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 251:1211–1217. [DOI] [PubMed] [Google Scholar]
- Blackwood, E.M., B. Luscher, and R.N. Eisenman. 1992. Myc and Max associate in vivo. Genes Dev. 6:71–80. [DOI] [PubMed] [Google Scholar]
- Bush, A., M. Mateyak, K. Dugan, A. Obaya, S. Adachi, J. Sedivy, and M. Cole. 1998. c-myc null cells misregulate cad and gadd45 but not other proposed c-Myc targets. Genes Dev. 12:3797–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawley, S., S. Bekiranov, H.H. Ng, P. Kapranov, E.A. Sekinger, D. Kampa, A. Piccolboni, V. Sementchenko, J. Cheng, A.J. Williams, et al., 2004. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 116:499–509. [DOI] [PubMed] [Google Scholar]
- Cole, M.D., and S.B. McMahon. 1999. The Myc oncoprotein: a critical evaluation of transactivation and target gene regulation. Oncogene. 18:2916–2924. [DOI] [PubMed] [Google Scholar]
- de Alboran, I.M., R.C. O'Hagan, F. Gartner, B. Malynn, L. Davidson, R. Rickert, K. Rajewsky, R.A. DePinho, and F.W. Alt. 2001. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity. 14:45–55. [DOI] [PubMed] [Google Scholar]
- Eilers, M., S. Schirm, and J.M. Bishop. 1991. The MYC protein activates transcription of the alpha-prothymosin gene. EMBO J. 10:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman, R.N. 2001. Deconstructing myc. Genes Dev. 15:2023–2030. [DOI] [PubMed] [Google Scholar]
- Evan, G.I., A.H. Wyllie, C.S. Gilbert, T.D. Littlewood, H. Land, M. Brooks, C.M. Waters, L.Z. Penn, and D.C. Hancock. 1992. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 69:119–128. [DOI] [PubMed] [Google Scholar]
- Fernandez, P.C., S.R. Frank, L. Wang, M. Schroeder, S. Liu, J. Greene, A. Cocito, and B. Amati. 2003. Genomic targets of the human c-Myc protein. Genes Dev. 17:1115–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggerty, T.J., K.I. Zeller, R.C. Osthus, D.R. Wonsey, and C.V. Dang. 2003. A strategy for identifying transcription factor binding sites reveals two classes of genomic c-Myc target sites. Proc. Natl. Acad. Sci. USA. 100:5313–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann, S.R., C.B. Thompson, and R.N. Eisenman. 1985. c-myc oncogene protein synthesis is independent of the cell cycle in human and avian cells. Nature. 314:366–369. [DOI] [PubMed] [Google Scholar]
- Holzel, M., F. Kohlhuber, I. Schlosser, D. Holzel, B. Luscher, and D. Eick. 2001. Myc/Max/Mad regulate the frequency but not the duration of productive cell cycles. EMBO Rep. 2:1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert, P.O., R. Verona, J.M. Trimarchi, C. Rogers, S. Dandapani, and J.A. Lees. 2000. E2f3 is critical for normal cellular proliferation. Genes Dev. 14:690–703. [PMC free article] [PubMed] [Google Scholar]
- Hurlin, P.J., C. Queva, and R.N. Eisenman. 1997. a. Mnt, a novel Max-interacting protein is coexpressed with Myc in proliferating cells and mediates repression at Myc binding sites. Genes Dev. 11:44–58. [DOI] [PubMed] [Google Scholar]
- Hurlin, P.J., C. Queva, and R.N. Eisenman. 1997. b. Mnt: a novel Max-interacting protein and Myc antagonist. Curr. Top. Microbiol. Immunol. 224:115–121. [DOI] [PubMed] [Google Scholar]
- Hurlin, P.J., Z.Q. Zhou, K. Toyo-Oka, S. Ota, W.L. Walker, S. Hirotsune, and A. Wynshaw-Boris. 2003. Deletion of Mnt leads to disrupted cell cycle control and tumorigenesis. EMBO J. 22:4584–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, R.M., J. Branda, K.A. Johnston, M. Polymenis, M. Gadd, A. Rustgi, L. Callanan, and E.V. Schmidt. 1996. An essential E box in the promoter of the gene encoding the mRNA cap-binding protein (eukaryotic initiation factor 4E) is a target for activation by c-myc. Mol. Cell. Biol. 16:4754–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzner, L., E.M. Blackwood, and R.N. Eisenman. 1992. Myc and Max proteins possess distinct transcriptional activities. Nature. 359:426–429. [DOI] [PubMed] [Google Scholar]
- Livak, K.J., and T.D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 25:402–408. [DOI] [PubMed] [Google Scholar]
- Mateyak, M.K., A.J. Obaya, S. Adachi, and J.M. Sedivy. 1997. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 8:1039–1048. [PubMed] [Google Scholar]
- Meroni, G., A. Reymond, M. Alcalay, G. Borsani, A. Tanigami, R. Tonlorenzi, C.L. Nigro, S. Messali, M. Zollo, D.H. Ledbetter, et al., 1997. Rox, a novel bHLHZip protein expressed in quiescent cells that heterodimerizes with Max, binds a non-canonical E box and acts as a transcriptional repressor. EMBO J. 16:2892–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meroni, G., S. Cairo, G. Merla, S. Messali, R. Brent, A. Ballabio, and A. Reymond. 2000. Mlx, a new Max-like bHLHZip family member: the center stage of a novel transcription factors regulatory pathway? Oncogene. 19:3266–3277. [DOI] [PubMed] [Google Scholar]
- Metivier, R., G. Penot, M.R. Hubner, G. Reid, H. Brand, M. Kos, and F. Gannon. 2003. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 115:751–763. [DOI] [PubMed] [Google Scholar]
- Nilsson, J.A., and J.L. Cleveland. 2003. Myc pathways provoking cell suicide and cancer. Oncogene. 22:9007–9021. [DOI] [PubMed] [Google Scholar]
- Nilsson, J.A., K.H. Maclean, U.B. Keller, H. Pendeville, T.A. Baudino, and J.L. Cleveland. 2004. Mnt loss triggers Myc transcription targets, proliferation, apoptosis, and transformation. Mol. Cell. Biol. 24:1560–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orian, A., B. van Steensel, J. Delrow, H.J. Bussemaker, L. Li, T. Sawado, E. Williams, L.W. Loo, S.M. Cowley, C. Yost, et al., 2003. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 17:1101–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelengaris, S., M. Khan, and G. Evan. 2002. c-MYC: more than just a matter of life and death. Nat. Rev. Cancer. 2:764–776. [DOI] [PubMed] [Google Scholar]
- Persson, H., L. Hennighausen, R. Taub, W. DeGrado, and P. Leder. 1984. Antibodies to human c-myc oncogene product: evidence of an evolutionarily conserved protein induced during cell proliferation. Science. 225:687–693. [DOI] [PubMed] [Google Scholar]
- Schorl, C., and J.M. Sedivy. 2003. Loss of protooncogene c-Myc function impedes G1 phase progression both before and after the restriction point. Mol. Biol. Cell. 14:823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro, G.J., and H. Green. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17:299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyo-oka, K., S. Hirotsune, M.J. Gambello, Z.Q. Zhou, L. Olson, M.G. Rosenfeld, R.N. Eisenman, P.J. Hurlin, and A. Wynshaw-Boris. 2004. Loss of the Max-interacting protein Mnt contained within the murine Miller-Dieker syndrome region results in defective embryonic growth and craniofacial defects. Hum. Mol. Genet. 13:1057–1067. [DOI] [PubMed] [Google Scholar]
- Trumpp, A., Y. Refaeli, T. Oskarsson, S. Gasser, M. Murphym, G.R. Martin, and J.M. Bishop. 2001. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature. 414:768–773. [DOI] [PubMed] [Google Scholar]
- Zhou, Z.Q., and P.J. Hurlin. 2001. The interplay between Mad and Myc in proliferation and differentiation. Trends Cell Biol. 11:S10–S14. [DOI] [PubMed] [Google Scholar]