

Abstract
MCAK is a member of the kinesin-13 family of microtubule (MT)-depolymerizing kinesins. We show that the potent MT depolymerizer MCAK tracks (treadmills) with the tips of polymerizing MTs in living cells. Tip tracking of MCAK is inhibited by phosphorylation and is dependent on the extreme COOH-terminal tail of MCAK. Tip tracking is not essential for MCAK's MT-depolymerizing activity. We propose that tip tracking is a mechanism by which MCAK is preferentially localized to regions of the cell that modulate the plus ends of MTs.
Introduction
MCAK is a member of the kinesin-13 (Lawrence, 2004) family of microtubule (MT)-depolymerizing kinesins. Previous studies have implicated MCAK in the regulation of MT dynamics during interphase and mitosis and in ensuring proper chromosome segregation during mitosis (for review see Moore and Wordeman, 2004b). MCAK is a potent depolymerase of stabilized MTs (Desai et al., 1999; Hunter et al., 2003) and increases the catastrophe frequency of dynamic MTs in vitro 10-fold (Newton et al., 2004). MCAK is the most potent cellular MT depolymerizer that has yet been identified (Tournebize et al., 2000; Ogawa et al., 2004). MCAK displays extremely high affinity for both plus and minus MT ends, yet its localization to MT ends in fixed cells is intermittent at best. For these reasons we imaged live cells transfected with levels of GFP or RFP-MCAK that do not appreciably disturb the overall MT polymer level. When imaged in this manner, MCAK is observed to track with polymerizing MT tips.
A number of proteins exhibit the unusual property of targeting and treadmilling (tracking) along polymerizing MT ends. The two most well characterized of these are members of the Clip-170 and EB1 families (for review see Carvalho et al., 2003). Thus far, all known treadmilling tip proteins in mammalian cells exhibit MT-stabilizing activity. For this reason, it is remarkable that the potent MT depolymerase, MCAK, tracks the tips of polymerizing MTs. MCAK's tip tracking is dependent on phosphorylation, which may regulate the association of MCAK with the MT lattice or with recruiting proteins, thus influencing the ability to localize to MT plus ends. The closely related kinesin-13 family member Kif2A does not tip track in mammalian cells and its activity appears to dominate at MT minus ends (Gaetz and Kapoor, 2004; Ganem and Compton, 2004; Rogers et al., 2004) by virtue of its dynein-dependent preferential localization to centrosomes. Hence, tip tracking may represent a mechanism to preferentially localize MCAK's activity to regions of the cell, such as the distal face of the centromere, that interact with MT plus ends.
Results and discussion
When fluorescent MCAK is expressed in live cultured cells at levels that do not significantly alter MT polymer levels, MCAK can be detected on MT lattice and in obvious densities on MT tips (Fig. 1 A). Time-lapse imaging shows tips polymerizing toward the cell edge (tracking) in a manner similar to that which has been previously reported for GFP-EB1 (Fig. 1 B; Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200411089/DC1). In living HeLa cells the MCAK tip densities are coincident with GFP-EB1 (Fig. 1 C). Tracking of MCAK to MT tips is not dependent on a functional motor domain, as it can be seen clearly in cells transfected with GFP-ML-MCAK (Fig. 2 B; Video 3), a construct in which the 50-kD motor domain has been removed and the NH2-terminal domain of MCAK is fused directly to the COOH-terminal domain (Maney et al., 1998). Tracking tips of GFP-ML-MCAK are consistently longer than those seen with GFP-wt-MCAK (Fig. 2, A′ and B′), suggesting that the motor may influence the off-rate from the lattice. Another mutant of MCAK that contains three point mutations in the motor domain rendering the protein inactive with respect to depolymerizing activity (GFP-hypir-MCAK) also exhibits tip tracking (Fig. 2 C, Table I; Video 4). Tip localization and tracking is seen in both interphase and mitotic cells. This is a striking observation because MCAK has been previously shown to be a potent depolymerizer of MTs (Desai et al., 1999; Hunter et al., 2003). Therefore, the presence of MCAK on the tips of polymerizing MTs in interphase and mitotic cells suggests that MCAK's depolymerizing activity is transiently inhibited on tips.
Figure 1.
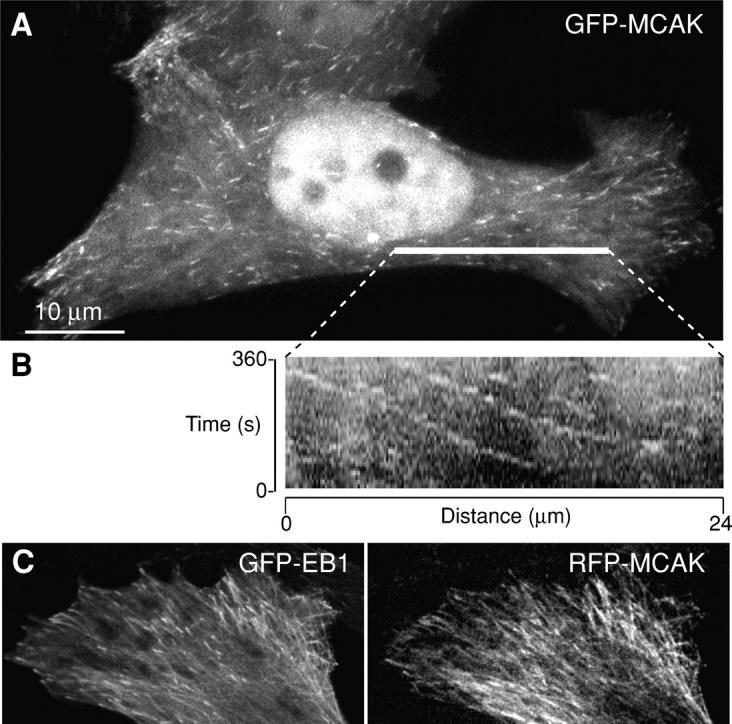
MCAK exhibits tip tracking behavior in living cells. (A) GFP-MCAK can be detected at the ends of MTs in an interphase HeLa cell. (B) Kymograph of tips traveling from the centrosomal region to the edge of the cell. (C) RFP-MCAK roughly colocalizes with GFP-EB1 in live HeLa cells cotransfected with both constructs.
Figure 2.
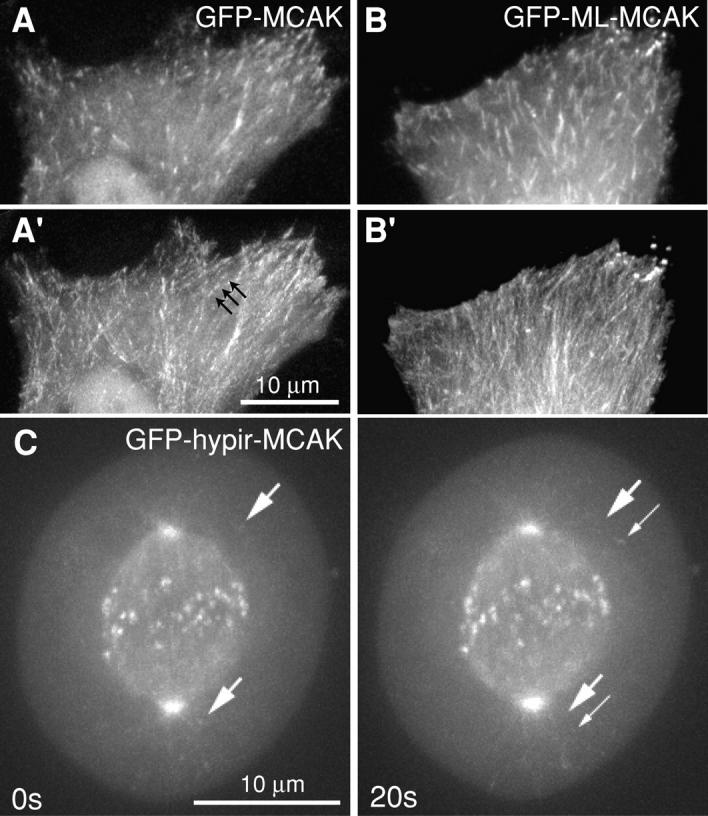
Inactive MCAK tip tracks in interphase and mitotic cells. (A) The edge of a cell transfected with GFP-MCAK. (A′) Three-dimensional reconstruction of 30 frames of MCAK tip tracks. Successive moving tips are visible (black arrows). (B) GFP-ML-MCAK tracks at the edge of an interphase cell. (B′) Reconstruction of 30 frames of motorless MCAK show uniform lines (rather than successive tips) because the tips are consistently longer than is seen with GFP-MCAK. (C) Mitotic cell transfected with GFP-hypir-MCAK. Tip tracking is evident in astral MTs (white arrows).
Table I. MCAK is the only MT depolymerizer that tip tracks.
| Construct | Tips | Tip tracking |
|---|---|---|
| GFP-MCAK, RFP-MCAK | Yes | Yes |
| GFP-ML-MCAK | Yes | Yes |
| GFP-hypir-MCAK | Yes | Yes |
| GFP-Kif2Aα | No | No |
| GFP-Kif2Aβ | No | No |
| GFP-EB1 | Yes | Yes |
| GFP-APC | Yes | No |
| GFP-Stathmin | No | No |
| GFP-ch-Tog | No | No |
To determine whether other known MT depolymerizing proteins are present on MT tips, we assayed two splice variants of another kinesin-13 family member: Kif2Aα and Kif2Aβ. Both proteins exhibited significant depolymerizing activity when overexpressed (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200411089/DC1). However, these proteins were not found tracking on MT tips (Table I; Fig. S1 B and Video 8). This is unexpected because the putative Drosophila melanogaster Kif2A orthologue, Klp10A, does track on polymerizing tips, whereas the putative MCAK orthologue, Klp59C, does not (Mennella et al., 2005). Furthermore, neither GFP-stathmin (Marklund et al., 1996) nor GFP-ch-TOG (Charrasse et al., 1998) was found to be tracking on polymerizing MT tips. This suggests that MCAK is the major MT depolymerizer in mammalian cells that tracks on the tips of polymerizing MTs. GFP-APC decorated MT tips but did not exhibit the same distribution or dynamics as those decorated with MCAK or EB1. Instead, tips exhibited aperiodic bending previously described in Dayanandan et al. (2003).
We mapped the domains outside of the core motor domain required for tip tracking, and the results are shown in Table II. Neither the core motor domain nor the neck+motor domain was capable of tip tracking. The neck+motor domain is a significantly more potent MT depolymerizer than full-length wtMCAK because it is missing the COOH-terminal negative regulatory domain (Moore and Wordeman, 2004a). Tip tracking is exquisitely sensitive to the presence of the COOH terminus. Loss of 9 amino acids from the COOH terminus eliminated tip tracking (MCAK-Q710), and loss of even 2–5 amino acids significantly impaired tip tracking (Table II).
Table II.

Referred to in the text as “MCAK-Q710.”
Weak.
Because phosphorylation has been invoked as a mechanism for inactivating MCAK's depolymerizing activity (Andrews et al., 2004; Lan et al., 2004; Ohi et al., 2004), we tested whether tip-associated MCAK was inhibited by phosphorylation. We found that our more active (Fig. S1 A) phospho-mutant of MCAK (GFP-AAAAA-MCAK) exhibited robust tip tracking (Table III, Fig. 3 A; Video 5, available at http://www.jcb.org/cgi/content/full/jcb.200411089/DC1). In this mutant all of the sites known to be phosphorylated by Aurora B kinase have been mutated to alanine (Andrews et al., 2004). In contrast, a less active (Fig. 3 A) phospho-mimic mutant of MCAK (GFP-EEEEE-MCAK) showed no tip tracking (Table III, Fig. 3 A; Video 6). The fact that active GFP-AAAAA-MCAK tip tracks suggests that phosphorylation is not the means by which MCAK is inhibited on tips of polymerizing MTs.
Table III. Tip tracking of MCAK is negatively regulated by phosphorylation.
| Construct | Tips | Tip tracking |
|---|---|---|
| GFP-MCAK, RFP-MCAK | Yes | Yes |
| GFP-AAAAA-MCAK | Yes | Yes |
| GFP-EEEEE-MCAK | No | No |
| GFP-S196E-MCAK | Yes | Yes |
| GFP-S92E-MCAK | Yesa | Yesa |
Tip length is much shorter than wt-MCAK.
Figure 3.
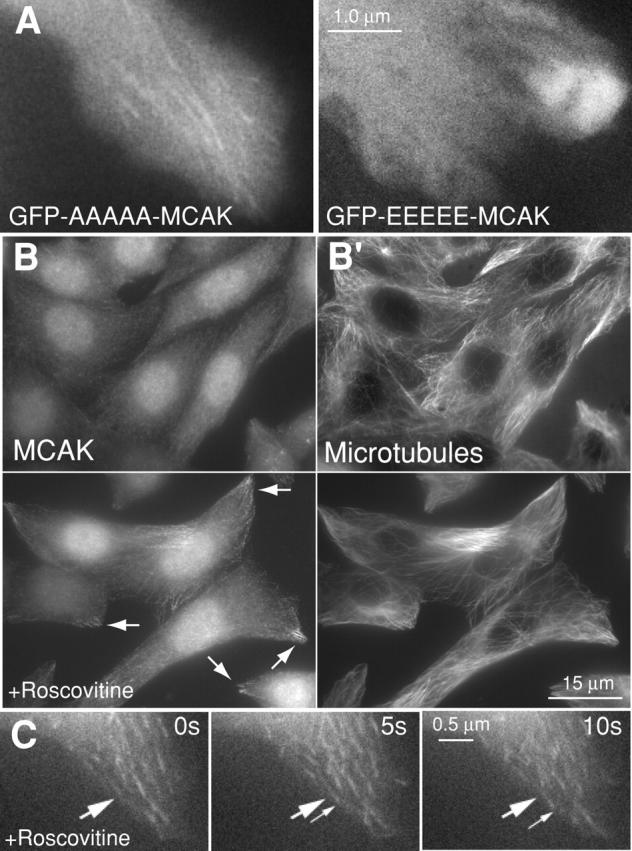
Tip tracking of MCAK protein is dependent on phosphorylation. (A) GFP-AAAAA-MCAK binds to MT tips (left), whereas GFP-EEEEE-MCAK is not found on tips (right). See also Videos 4 and 5 (available at http://www.jcb.org/cgi/content/full/jcb.200411089/DC1). (B) Two fields of CHO cells labeled for endogenous MCAK (B) and MTs (B′). The top field of CHO cells are control cells. The bottom field of CHO cells were cultured for 5 h in 10 μM roscovitine and then fixed. Increased association of endogenous MCAK with distal ends of MTs is evident (B, bottom; arrows). (C) MCAK protein tracks on tips (arrows) in a HeLa cell transfected with RFP-MCAK and cultured subsequently in 10 μM roscovitine (Video 6).
Although the neck domain is required for maximally efficient MT depolymerization activity the S196E mutant of MCAK, which mimics phosphorylation within the neck domain, had no effect on tip tracking. This is consistent with our data suggesting that tip tracking is not essential for maximal MT depolymerizing activity. However, a single substitution of S92E, NH2-terminal to the neck domain, significantly reduced the length and intensity of the tracking tips (Table III). This suggests that the Aurora B sites NH2-terminal to the neck are most critical for the regulation of tip tracking. It is significant that although the Aurora B consensus phosphorylation sites appear to be conserved between all the KinI/kinesin-13 family members (Andrews et al., 2004), the protein sequence surrounding the conserved phosphorylation sites are quite divergent between MCAK and Kif2Aα/β. This implicates the NH2 terminus of MCAK, particularly the region surrounding the Aurora B/Ipl1 phosphorylation sites in associating preferentially with MT ends.
Because the tip-tracking behavior of MCAK was negatively regulated by phosphorylation, we observed the behavior of endogenous MCAK in the presence of the kinase inhibitor roscovitine. CHO cells were incubated in either DMSO or 10 μM roscovitine in DMSO for 5 h, and then were fixed and labeled for endogenous MCAK and MTs (Fig. 3 B, top and bottom, respectively). After 5 h of roscovitine treatment endogenous MCAK exhibited significantly greater association with MT tips. The level of roscovitine used is capable of inhibiting a number of kinases (Meijer et al., 1997). Because the assay was performed on interphase cells, the inhibited kinase is not Aurora B. However, we do find that inhibition of Aurora B with small molecule inhibitors or a kinase-dead mutant promotes greater association of MCAK with MT tips in mitotic cells (unpublished data). The redistribution of endogenous MCAK was striking (Fig. 3 B, bottom). We also performed time-lapse imaging of GFP-MCAK transfected cells in order to confirm that MCAK was tracking with tips in the presence of roscovitine (Fig. 3 C; Video 7). To summarize, MCAK's ability to tip track is negatively dependent on phosphorylation of the NH2-terminal domain and positively mediated by the extreme COOH-terminal tail.
Presently, it is not known how proteins track with MT ends. Both copolymerization with tubulin and preferential affinity for MT end structures have been proposed. MCAK has been previously shown to possess an affinity for both tubulin and MT ends. To distinguish between these two mechanisms, we compared tip-tracking (wt-unphosphorylated) and nontip-tracking (wt-phosphorylated and Q710) MCAK proteins for their ability to be competed off MT lattice by free tubulin. If tubulin copolymerization were the mechanism by which MCAK preferentially associates with MT ends, then one might expect versions of MCAK that tip track would interact with free tubulin to a greater degree than versions that do not. We have previously shown that tubulin inhibits lattice association of MCAK (Moore and Wordeman, 2004a). We compared the extent to which unphosphorylated and Ipl1/Sli15-phosphorylated wild-type MCAK bind to assembled MTs, and the extent to which that binding is limited by excess tubulin monomer. The former tip tracks, whereas the latter presumably does not. Under conditions in which 100% of nonphosphorylated MCAK is bound to lattice, only about two thirds of Ipl1-phosphorylated MCAK is found in the pellet (Fig. 4 A, lanes 1). However, both unphosphorylated and phosphorylated MCAK can be competed off lattice to a similar extent by addition of excess free tubulin: 19% in the former case, and 13% (9% out of 68%) of the bound fraction in the latter case (Fig. 4 A, lanes 2). Therefore phosphorylation, which is expected to reduce tip tracking, changes the affinity of MCAK for lattice but not for free tubulin.
Figure 4.

Tip tracking is positively correlated with apparent lattice affinity rather than tubulin affinity. (A) Wild-type MCAK binds MT lattice in the absence of ATP (left, lane 1). Excess free tubulin dimer can compete off 13–15% of lattice-associated wtMCAK (left, lanes 2 and 3). Phosphorylation of MCAK significantly decreases the apparent affinity of wtMCAK for stabilized MTs (right, lane 1). Excess free tubulin competes off a further 2–9% of lattice-bound phosphorylated MCAK (right lanes 2 and 3). (B) MCAK-Q710 binds MT lattice in the absence of ATP (left, lane 1). Excess tubulin dimer can compete off 30–45% of lattice-bound MCAK-Q710 (left, lanes 2 and 3). Phosphorylation of MCAK-Q710 decreases the apparent affinity of MCAK-Q710 for MT lattice to a similar extent as is seen with wtMCAK (right, lane 1). Excess free tubulin competes off a further 9–11% of lattice-bound phosphorylated MCAK-Q710 (right lanes 2 and 3).
We performed the same experiment using MCAK-Q710, which does not tip track but exhibits a higher apparent affinity for MT polymer than wt-MCAK (Moore and Wordeman, 2004a). We found that this mutant form can be competed away from MT lattice to a much greater extent than either wt-MCAK or phosphorylated wt-MCAK (Fig. 4 B, lanes 2 and 3). However, phosphorylation of MCAK-Q710 reduced lattice binding by approximately the same level as did phosphorylation of wt-MCAK. Curiously, phosphorylated MCAK-Q710 was only competed away from lattice by free tubulin to approximately the same extent as wt-MCAK (8% of 63% = 13% of the bound fraction). These experiments show that tip tracking is correlated positively with lattice affinity but negatively with free tubulin affinity, contrary to the prediction of a simple model for tip tracking by copolymerization.
Instead, tip tracking may simply depend on the ability of MCAK to see a higher affinity-binding site at the end of the MT relative to the lattice. As the MT polymerizes, this higher affinity site becomes a lower affinity site, leading to increased loss of MCAK from the lattice and the characteristic comet tail appearance. Because tip tracking does not depend on the motor domain we propose that this binding site is distinctly different from the very high affinity binding of MCAK to deformable tubulin dimers at both ends of the MT in AMP-PNP described in structural and kinetic studies (Desai et al., 1999; Moores et al., 2002; Hunter et al., 2003; Moore and Wordeman, 2004a; Ogawa et al., 2004). We have previously hypothesized that the COOH-terminal tail domain “sees” a site at the end of the MT that relieves the inhibition of the tail and promotes lattice binding (Moore and Wordeman, 2004a). This site could be the exposed β-tubulin subunit end. In that case, tip tracking would be plus end dependent and might be antagonized by free tubulin (Fig. 4 B). If tip tracking could be reconstituted in vitro this hypothesis could be tested.
A remaining question is why a MT tip that is enriched for bound MCAK would polymerize at all. MCAK's MT depolymerizing activity must be inhibited. We propose two models, which are not mutually exclusive, to explain the inhibition of MCAK on MT tips (Fig. 5). In the first case, the MT tip clearly associates with both MT stabilizers (such as EB1) and MT destabilizers (MCAK). MT stabilizers, such as EB1, may be capable of successfully competing with MCAK on MT tips by preventing MCAK from reaching a critical concentration at the end of the MT. We coexpressed EB1 and MCAK in live cells and found that excess EB1 can indeed antagonize the MT-depolymerizing activity of MCAK in living cells as long as the excess MCAK levels are relatively low (Fig. 5 A). We call this the competition model (Fig. 5 B). Another possibility is that MCAK's activity is inhibited during initial encounters with living MT ends. Because this binding site is independent of the motor domain, it presumably does not involve stabilizing a curved tubulin conformation. The COOH-terminal tail and NH2 terminus must prefer a binding site unique to the MT end (relative to lattice), such as the extreme end of a dimer (Fig. 5 C, higher affinity site). A subsequent conformational change would then bring the motor into contact with the lattice (Fig. 5 C, low affinity site). This model is also compatible with recruitment of MCAK to MT ends in an inactive form by EB1 (Mennella et al., 2005), although for simplicity we are assuming a direct association of MCAK with MTs. If this conformational change occurs further down on the lattice it will have a neutral effect on MT depolymerization. Thus, if the polymerization rate of MT ends exceeds that of the conformational change of the motor, depolymerization will not occur. It is important to note that the loss of tip tracking by phosphorylation of the NH2 terminus suggests that the NH2 terminus of MCAK is also necessary but not sufficient for tip tracking.
Figure 5.
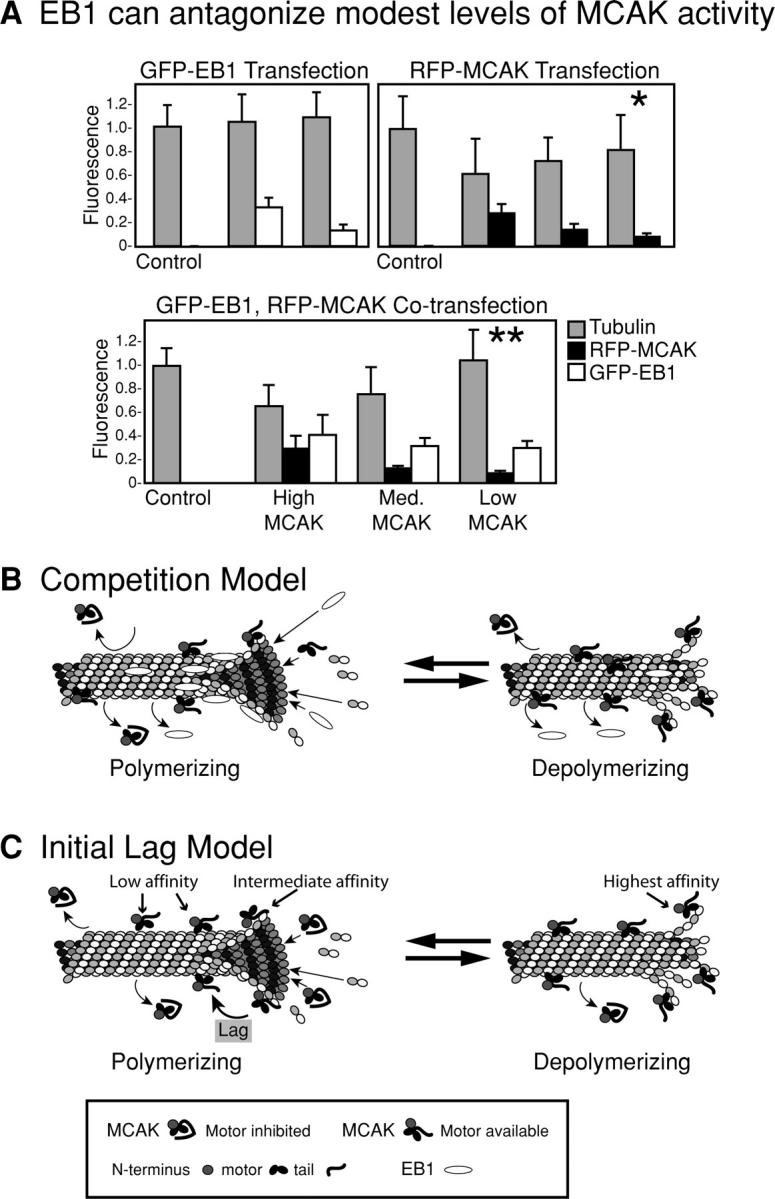
Models to explain the presence of MCAK on polymerizing MT tips. (A) EB1 is capable of antagonizing modest levels of MCAK's depolymerizing activity. Cultured cells were transfected with GFP-EB1, RFP-MCAK, or both. Cells expressing low levels of MCAK have a significantly reduced amount of MT polymer relative to control cells (*, T = 0. 0042, P = 0.05). In contrast, there is no significant difference between control levels of MT polymer and low MCAK+EB1 (**, T = 0.4345, P = 0.05). Thus, at low levels of MCAK expression, overexpressed EB1 is capable of restoring bulk MT polymer to control levels. (B) Competition model: EB1 and MCAK compete for high affinity binding sites at the end of the MT. EB1 prevents MCAK from achieving a quorum of occupied protofilaments. (C) The NH2 and COOH terminus interact with MT ends, relieving the MCAK motor domain from inhibition by the COOH terminus and putting it in a state in which the motor becomes receptive to lattice interactions.
MCAK is the sole identified MT depolymerizer that tracks on MT tips in mammalian cells. Neither stathmin nor the splice variants of Kif2A are able to track on MT tips in HeLa cells. We have shown, using hyperactive deletion constructs, that tip tracking is not required for robust MT depolymerization. What then, is its role in the cell? The activity of the nontip tracking kinesin, Kif2A, has been shown to predominate at MT minus ends. Thus, tip tracking may be a mechanism for preferentially targeting MCAK's activity to the plus ends of MTs. Previously, it has been shown that MCAK's activity and subcentromeric distribution is regulated by phosphorylation by Aurora kinase (Andrews et al., 2004). Phosphorylated MCAK is preferentially localized to the inner centromere, whereas de-phosphorylated MCAK is preferentially localized to the distal face of the centromere. Tip tracking along kinetochore MTs by de-phosphorylated MCAK may be the mechanism by which active de-phosphorylated MCAK “offloads” to the outer centromere during mitosis. This mechanism has been proposed for the plus end tip-tracking behavior of the minus end–directed motor, dynein (Lee et al., 2005) Thus, even though tip tracking is not involved in MCAK's mechanism of depolymerization, in the context of the living cell, plus end targeting may affect MT depolymerization kinetics with subtle dynamics that are not reflected in whole-cell assays of bulk polymer. We are presently using high resolution imaging to directly test this hypothesis.
Materials and methods
Constructs
EGFP-cgMCAK (pYOY152) was prepared by pasting a fragment of pYOY71 (Ovechkina et al., 2002) into pEGFPC1 (CLONTECH Laboratories, Inc.). EGFP-ML-cgMCAK (pYOY154) was prepared from GFP-ML-MCAK (Maney et al., 1998). EGFP-hypir-cgMCAK (pYOY153) consists of H530A, R534A, and K537A mutations in pEGFPC1. EGFP-mmStathmin (pMX666) was subcloned from IMAGE clone 314147. EGFP-hsch-TOG (pMX114) was subcloned from ch-TOGp, a gift of Dr. Christian Larroque (INSERM, Montpellier, France). Kif2Aα (GenBank/EMBL/DDBJ accession no. D12644) and Kif2Aβ (GenBank/EMBL/DDBJ accession no. Y15894) cDNA plasmids were a gift of N. Santama (University of Cyprus, Nicosia, Cyprus). The coding regions were amplified by PCR, and were cloned into pEGFP-C1 to generate pMX155 and pMX156, which were sequenced to confirm identity. EGFP-APC was a gift of Dr. Rina Arbesfeld (Tel Aviv University, Tel Aviv, Israel). EGFP-EEEEE-MCAK and EGFP-AAAAA-MCAK are described in Andrews et al. (2004). RFP was copied from pRSETB-mRFP1 (a gift of Roger Tsien; University of San Diego, San Diego, CA) by PCR, cloned into pYOY152, and ligated to generate RFP-MCAK (pMX188). EGFP-EB1 was a gift of Lynne Cassimeris (Lehigh University, Bethlehem, PA). GFP-Neck+Motor and GFP-motor alone are described in Ovechkina et al. (2002). COOH-terminal deletions are described in Moore and Wordeman (2004a).
Cell transfection and immunofluorescence
HeLa cells or CHO cells were plated onto either glass-bottomed Petri dishes (MatTek Corp.) for live filming or 12-mm round coverslips and transfected using LipofectAMINE (Invitrogen) for 4 h. HeLa cells were imaged 48 h after transfection, whereas CHO cells were imaged 24 h after transfection. Fixation was performed as described in Maney et al. (1998) and immunofluorescence staining using DM1α (Sigma-Aldrich) or sheep anti-MCAK antibodies was completed at RT for 1 h. Purified cgMCAK protein was prepared as described in Maney et al. (1998) and used to immunize sheep (Pocono). Serum was affinity purified using cgMCAK protein linked to affi-gel (Bio-Rad Laboratories).
Quantitation of MT levels in fixed cells
Quantitation of the depolymerization efficacy of Kif2Aα/β, 5AMCAK, and 5EMCAK were performed as described in Ovechkina et al. (2002). For the GFP-EB1/RFP-MCAK cotransfections, immunofluorescent staining of MTs was performed using DM1α (Sigma-Aldrich) and appropriate secondary antibodies. Digital images were acquired using a microscope (model FX-A; Nikon) with a Sensys cooled CCD camera (Photometrics) controlled by QED camera software (QED Imaging). MT levels were evaluated by measuring the average pixel intensity of tubulin. Fluorescent images of transfected cells were normalized against untransfected cells on the same plate. The values presented are in percentages of the average value of untransfected cells. MCAK and EB1 expression levels are presented in percentages of the total possible fluorescent value (i.e., 256 for an 8-bit image).
Live imaging
Transfected cells were imaged using a Nikon inverted microscope with either a Photometrics cooled CCD camera or a CARV (Kinetic Imaging) spinning disk unit and ORCA ER (Hamamatsu) camera. Images were acquired using MetaMorph (Universal Imaging Corp.). Cells were maintained in CO2-independent media (GIBCO BRL) with 10% FBS during imaging. Temperature was maintained at 36°C using a thermoelectric stage. Interphase cells were imaged at 1 frame per 5 s and mitotic cells were imaged at 1 frame per 20 s. Subsequent analysis was performed with Image J. EB1/MCAK cotransfected cells were imaged live on a Radian confocal microscope (Bio-Rad Laboratories).
In vitro phosphorylation and pelleting assays
MCAK expression and purification were performed as previously described (Maney et al., 1998). Ipl1p and Sli15p GST fusion plasmids were gifts of Dr. Sue Biggins (Fred Hutchinson Cancer Research Center, Seattle, WA). Bovine brain tubulin was acquired from Cytoskeleton, Inc. 50 μM tubulin was added to BRB80 (80 mM Pipes, pH 6.8, 1 mM EGTA, and 1 mM MgCl2) with 4 mM MgCl2, 1.8 mM GMP-CPP (a nonhydrolyzable form of GTP), and 5% DMSO and incubated for 20 min at 37°C. After the incubation, MTs were diluted 1:12.5 in 37°C BRB80. Phosphorylation of MCAK was performed by incubating 50 nM of active MCAK with 1 μl Ipl1p and 0.5 μl Sli15p in kinase buffer (50 mM Tris-HCl, pH 7.5, 0.1 mM EGTA, and 5 mM MgCl2) with 1 mM DTT, 0.2 mM ATP, 1 μM Microcystin-LR, and 0.3% Triton X-100 for 1 h at RT (25 ± 1°C). 8 μl of the phosphorylation reaction was incubated with 30 μl of MTs with 1 mM DTT and 75 mM KCl at RT for 5 min and centrifuged in an airfuge at 149,000 g for 10 min. For experiments with tubulin preincubation, motor was first incubated with 25 μM or 12.5 μM tubulin with 1 mM DTT and 75 mM KCl for 30 min at RT. Supernatants and pellets were assayed for the presence of tubulin and motor on Coomassie-stained SDS-polyacrylamide gels (Invitrogen). Bands were quantified using NIH Image 1.62.
Online supplemental material
Fig. S1: relative MT-depolymerizing activity of Kif2A splice variants and MCAK phospho-mutants versus wt-MCAK. Kif2Aβ does not tip track (see also Video 8). Fig. S2: Phosphorylated MCAK exhibits impaired MT-depolymerizing activity. There is no difference between the extent of phosphorylation of MCAK that is in the supernatant versus the pellet in these experiments. Video 1: interphase HeLa cell and GFP-MCAK. Collected at 1 frame per 12 s. Video 2: interphase HeLa cell and GFP-MCAK. Collected at 1 frame per 12 s. Video 3: interphase HeLa cell and GFP-ML-MCAK. Collected at 1 frame per 12 s. Video 4: mitotic HeLa cell and GFP-hypir-MCAK. Collected at 1 frame per 20 s. Video 5: interphase HeLa cell and GFP-AAAAA-MCAK. Collected at 1 frame per 5 s. Video 6: interphase HeLa cell and GFP-EEEEE-MCAK. Collected at 1 frame per 5 s. Video 7: interphase HeLa cell, GFP-MCAK, and 10 μm roscovitine 6 h. Collected at 1 frame per 5 s. Video 8: interphase HeLa cell, GFP-Kif2Aβ. 1 frame per 5 s. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200411089/DC1.
Acknowledgments
We are indebted to Greg Martin and Paulette Brunner for invaluable assistance with live imaging. We thank Ryan Littlefield for valuable advice.
This work is supported by a National Institutes of Health (NIH) Predoctoral Fellowship (GM65061) to A.T. Moore and an NIH grant (GM69429) to L. Wordeman. K.E. Rankin was supported in part by predoctoral NIH fellowship GM07270. The Center for Cell Dynamics is an NIH Center for Excellence (GM066050).
Abbreviation used in this paper: MT, microtubule.
References
- Andrews, P.D., Y. Ovechkina, N. Morrice, M. Wagenbach, K. Duncan, L. Wordeman, and J.R. Swedlow. 2004. Aurora B regulates MCAK at the mitotic centromere. Dev. Cell. 6:253–268. [DOI] [PubMed] [Google Scholar]
- Carvalho, P., J.S. Tirnauer, and D. Pellman. 2003. Surfing on microtubule ends. Trends Cell Biol. 13:229–237. [DOI] [PubMed] [Google Scholar]
- Charrasse, S., M. Schroeder, C. Gauthier-Rouviere, F. Ango, L. Cassimeris, D.L. Gard, and C. Larroque. 1998. The TOGp protein is a new human microtubule-associated protein homologous to the Xenopus XMAP215. J. Cell Sci. 111:1371–1383. [DOI] [PubMed] [Google Scholar]
- Dayanandan, R., R. Butler, P.R. Gordon-Weeks, A. Matus, S. Kaech, S. Lovestone, B.H. Anderton, and J.M. Gallo. 2003. Dynamic properties of APC-decorated microtubules in living cells. Cell Motil. Cytoskeleton. 54:237–247. [DOI] [PubMed] [Google Scholar]
- Desai, A., S. Verma, T.J. Mitchison, and C.E. Walczak. 1999. Kin I kinesins are microtubule-destabilizing enzymes. Cell. 96:69–78. [DOI] [PubMed] [Google Scholar]
- Gaetz, J., and T.M. Kapoor. 2004. Dynein/dynactin regulate metaphase spindle length by targeting depolymerizing activities to spindle poles. J. Cell Biol. 166:465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem, N.J., and D.A. Compton. 2004. The KinI kinesin Kif2a is required for bipolar spindle assembly through a functional relationship with MCAK. J. Cell Biol. 166:473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter, A.W., M. Caplow, D.L. Coy, W.O. Hancock, S. Diez, L. Wordeman, and J. Howard. 2003. The kinesin-related protein MCAK is a microtubule depolymerase that forms an ATP-hydrolyzing complex at microtubule ends. Mol. Cell. 11:445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan, W., X. Zhang, S.L. Kline-Smith, S.E. Rosasco, G.A. Barrett-Wilt, J. Shabanowitz, D.F. Hunt, C.E. Walczak, and P.T. Stukenberg. 2004. Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr. Biol. 14:273–286. [DOI] [PubMed] [Google Scholar]
- Lawrence, C.J. 2004. A standardized kinesin nomenclature. J. Cell Biol. 167:19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W.L., M.A. Kaiser, and J.A. Cooper. 2005. The offloading model for dynein function: differential function of motor subunits. J. Cell Biol. 168:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maney, T., A.W. Hunter, M. Wagenbach, and L. Wordeman. 1998. Mitotic centromere-associated kinesin is important for anaphase chromosome segregation. J. Cell Biol. 142:787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund, U., N. Larsson, H.M. Gradin, G. Brattsand, and M. Gullberg. 1996. Oncoprotein 18 is a phosphorylation-responsive regulator of microtubule dynamics. EMBO J. 15:5290–5298. [PMC free article] [PubMed] [Google Scholar]
- Meijer, L., A. Borgne, O. Mulner, J.P. Chong, J.J. Blow, N. Inagaki, M. Inagaki, J.G. Delcros, and J.P. Moulinoux. 1997. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 243:527–536. [DOI] [PubMed] [Google Scholar]
- Mennella, V., G.C. Rogers, S.L. Rogers, D.W. Buster, R.D. Vale, and D.J. Sharp. 2005. Functionally distinct kinesin-13 family members cooperate to regulate microtubule dynamics during interphase. Nat. Cell Biol. 7:235–245. [DOI] [PubMed] [Google Scholar]
- Moore, A.T., and L. Wordeman. 2004. a. C-terminus of mitotic centromere-associated kinesin (MCAK) inhibits its lattice-stimulated ATPase activity. Biochem J. 383:227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, A.T., and L. Wordeman. 2004. b. The mechanism, function and regulation of depolymerizing kinesins during mitosis. Trends Cell Biol. 14:537–546. [DOI] [PubMed] [Google Scholar]
- Moores, C.A., M. Yu, J. Guo, C. Beraud, R. Sakowicz, and R.A. Milligan. 2002. A mechanism for microtubule depolymerization by KinI kinesins. Mol Cell. 9:903–909. [DOI] [PubMed] [Google Scholar]
- Newton, C.N., M. Wagenbach, Y. Ovechkina, L. Wordeman, and L. Wilson. 2004. MCAK, a Kin I kinesin, increases the catastrophe frequency of steady-state HeLa cell microtubules in an ATP-dependent manner in vitro. FEBS Lett. 572:80–84. [DOI] [PubMed] [Google Scholar]
- Ogawa, T., R. Nitta, Y. Okada, and N. Hirokawa. 2004. A common mechanism for microtubule destabilizers-M type kinesins stabilize curling of the protofilament using the class-specific neck and loops. Cell. 116:591–602. [DOI] [PubMed] [Google Scholar]
- Ohi, R., T. Sapra, J. Howard, and T.J. Mitchison. 2004. Differentiation of cytoplasmic and meiotic spindle assembly MCAK functions by Aurora B-dependent phosphorylation. Mol. Biol. Cell. 15:2895–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovechkina, Y., M. Wagenbach, and L. Wordeman. 2002. K-loop insertion restores microtubule depolymerizing activity of a “neckless” MCAK mutant. J. Cell Biol. 159:557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, G.C., S.L. Rogers, T.A. Schwimmer, S.C. Ems-McClung, C.E. Walczak, R.D. Vale, J.M. Scholey, and D.J. Sharp. 2004. Two mitotic kinesins cooperate to drive sister chromatid separation during anaphase. Nature. 427:364–370. [DOI] [PubMed] [Google Scholar]
- Tournebize, R., A. Popov, K. Kinoshita, A.J. Ashford, S. Rybina, A. Pozniakovsky, T.U. Mayer, C.E. Walczak, E. Karsenti, and A.A. Hyman. 2000. Control of microtubule dynamics by the antagonistic activities of XMAP215 and XKCM1 in Xenopus egg extracts. Nat. Cell Biol. 2:13–19. [DOI] [PubMed] [Google Scholar]