

Abstract
fA amily of aspartic proteases, the plasmepsins (PMs), plays a key role in the degradation of hemoglobin in the Plasmodium falciparum food vacuole. To study the trafficking of proPM II, we have modified the chromosomal PM II gene in P. falciparum to encode a proPM II–GFP chimera. By taking advantage of green fluorescent protein fluorescence in live parasites, the ultrastructural resolution of immunoelectron microscopy, and inhibitors of trafficking and PM maturation, we have investigated the biosynthetic path leading to mature PM II in the food vacuole. Our data support a model whereby proPM II is transported through the secretory system to cytostomal vacuoles and then is carried along with its substrate hemoglobin to the food vacuole where it is proteolytically processed to mature PM II.
Keywords: protease; protein trafficking; brefeldin A; endoplasmic reticulum; hemoglobin
Introduction
The pathology of severe malaria is caused by the intraerythrocytic form of the human parasite Plasmodium falciparum. Within the erythrocyte, P. falciparum inhabits a parasitophorous vacuole and from there, directs an extensive reorganization of the host cell which includes major alterations to the permeability, rigidity, and surface characteristics of the erythrocyte membrane (Deitsch and Wellems, 1996). To support its growth and asexual replication, the parasite undertakes a feeding process whereby it endocytoses a large quantity of erythrocyte cytosol and digests its primary constituent, hemoglobin, in an acidic degradative organelle known as the food vacuole or digestive vacuole (Banerjee and Goldberg, 2000). Because of the continuing high levels of morbidity and mortality attributed to this protozoan parasite, much effort has been directed toward understanding the cell biology of its intraerythrocytic cycle, which consists of three morphologically distinct stages: ring, trophozoite, and schizont. During the ring stage, which lasts 22–24 h from the time of merozoite invasion, metabolic activity is low. The trophozoite stage, 10–12 h in duration, is characterized by an acceleration of metabolic processes that include the ingestion and digestion of large amounts of host hemoglobin. Formation and release of up to 32 daughter merozoites per infected cell occur during the schizont stage, which lasts 8–10 h.
The food vacuole is a lysosome-like organelle unique to the genus Plasmodium. It is the site of massive hemoglobin degradation during the trophozoite stage (Banerjee and Goldberg, 2000) and is the target of several important antimalarial drugs (Ziegler et al., 2001). Endocytosis of erythrocyte cytoplasm in trophozoites occurs mainly via invagination of the parasite plasma membrane and the parasitophorous vacuolar membrane through a specialized opening called the cytostome (Aikawa et al., 1966; Slomianny et al., 1985; Slomianny, 1990). In P. falciparum, double-membrane hemoglobin-containing transport vesicles pinch off from the cytostome and fuse with the food vacuole, releasing a single-membrane vesicle into the lumen (Yayon et al., 1984; Slomianny, 1990). Hemoglobin catabolism in the food vacuole is achieved through a semi-ordered sequence of proteolytic events involving the aspartic proteases plasmepsins (PMs) I, II, and IV (Francis et al., 1994; Gluzman et al., 1994; Banerjee et al., 2002; Wyatt and Berry, 2002), histo-aspartic protease (HAP; Banerjee et al., 2002), cysteine proteases (Shenai et al., 2000; Sijwali et al., 2001), and a metalloprotease (Eggleson et al., 1999). Cytosolic aminopeptidases may participate in the terminal steps of degradation (Kolakovich et al., 1997). Residual heme is sequestered as a crystalline pigment called hemozoin (Hempelmann and Egan, 2002).
The means by which newly synthesized proteins reach the food vacuole are poorly understood. Several observations suggest that some resident food vacuole proteins traverse the ‘classical’ secretory pathway. Maturation of PM I, II, and HAP is blocked by treatment of parasites with brefeldin A (BFA), an inhibitor of anterograde protein traffic from the ER (Francis et al., 1997a; Banerjee et al., 2003). PM I has been detected at the parasite plasma membrane and in cytostomal vacuoles and hemoglobin-containing transport vesicles, which suggests a trafficking route that takes advantage of the hemoglobin endocytic pathway (Francis et al., 1994). Histidine-rich protein II is transported to the erythrocyte cytosol in a BFA-sensitive manner (Akompong et al., 2002), and small amounts may be transported along with hemoglobin to the food vacuole via the cytostome (Sullivan et al., 1996); alternatively, a direct trafficking route from the ER to the food vacuole has been proposed (Akompong et al., 2002). The reporter GFP, when targeted through the secretory pathway to the parasitophorous vacuole, is thought to be transported to the food vacuole in small packets of parasitophorous vacuole lumen that are incorporated into cytostomal vacuoles (Wickham et al., 2001; Adisa et al., 2003). It is unclear whether any endogenous plasmodial proteins traffic to the food vacuole in this fashion.
A better understanding of PM trafficking and maturation may have practical application in the development of novel antimalarial strategies, because disruption of PM function in the food vacuole is expected to impair parasite survival (Coombs et al., 2001). PM I and II are synthesized as unglycosylated type II integral membrane proenzymes (Francis et al., 1997a), with the membrane-spanning domain in the proregion. Removal of the proregion and release of soluble mature PM (mPM) in vivo and in vitro has been shown to require an acidic environment and an activity blocked by the cysteine protease inhibitor N-acetyl-l-leucyl-l-leucyl-l-norleucinal (ALLN) but not by the aspartic protease inhibitor pepstatin (Francis et al., 1997a; Banerjee et al., 2003). These observations have led to the idea that PM maturation is not autocatalytic, but rather that the proenzymes are processed by a “PM convertase” activity in the food vacuole (Francis et al., 1997b; Banerjee et al., 2003).
Here, we have analyzed the trafficking and maturation of a proPM II–GFP chimera. These studies were conducted with a transgenic parasite line in which the chromosomal copy of the PM II gene was modified to encode proPM II–GFP. First, we demonstrated that the GFP tag does not interfere with the transport and maturation of PM II, and then we characterized the route taken by proPM II–GFP to the food vacuole.
Results
Generation of transgenic parasites
The single-copy PM II gene of P. falciparum strain 3D7 was altered to encode a proPM II–GFP fusion by modifying an established gene disruption procedure (Crabb et al., 1997a). Plasmid pPM2GT was constructed with a targeting sequence of 1 kb of the 3′ end of the PM II coding region fused in-frame to a sequence encoding a linker and the enhanced GFP variant GFPmut2 (Fig. 1 A). Parasites transfected with pPM2GT were selected with the antifolate drug WR99210 and subjected to two rounds of drug cycling to enrich the population for parasites that had integrated pPM2GT into the PM II gene (Fig. 1, B and C). Single-cell cloning of the twice-cycled culture was undertaken to obtain parasites of defined genotype. The clone studied extensively here, designated B7, contains a single copy of pPM2GT integrated into the PM II gene (Fig. 1 C).
Figure 1.
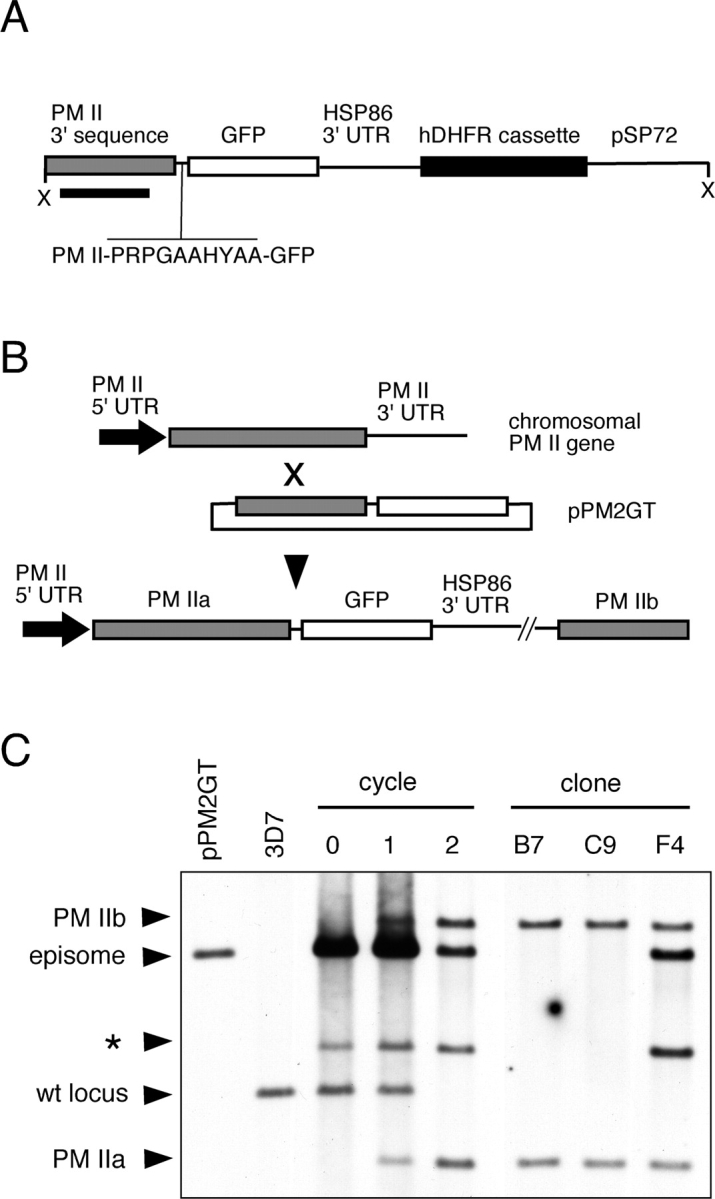
Creation of a chromosomal PM II–GFP chimera. (A) Schematic diagram of the integration plasmid pPM2GT linearized at the unique XhoI site (X). 1 kb of the 3′ end of the PM II coding sequence (gray box) was fused in frame to a linker sequence followed by the GFPmut2 open reading frame (white box). The amino acid sequence of the linker is shown. A WR99210-resistant variant of human dihydrofolate reductase (Fidock and Wellems, 1997) was incorporated as a selectable marker. The black bar indicates the PM II sequence used for probing Southern blots. Elements are not drawn to scale. (B) Schematic representation of events leading to a chromosomal PM II–GFP chimera. Single-site homologous recombination between the episomal pPM2GT target sequence and the chromosomal PM II locus produces the PM II–GFP chimera. The integration event produces a full-length PM II ORF (designated PM IIa) fused to GFP and a downstream promoterless copy of the PM II target sequence (PM IIb). Some elements of the plasmid, including the drug-resistance cassette, have been omitted for clarity. (C) Southern blot of StuI–NotI digested total DNA from untransfected parasites (3D7), stably transfected parasites before cycling (cycle 0) and after one and two drug cycles, and from three clones (B7, C9, and F4). The identity of the StuI–NotI fragments is indicated at left. The band identified with an asterisk is of unknown origin and probably reflects a rearrangement of pPM2GT after transfection. This figure was assembled from two experiments. wt locus, wild-type PM II locus.
Effect of the GFP tag on the expression, maturation, and location of PM II
Before examining the trafficking of the proPM II–GFP fusion, it was necessary to demonstrate that the addition of the GFP tag did not interfere with transport to and maturation in the food vacuole. Like untagged proPM II, proPM II–GFP is synthesized as an integral membrane proenzyme (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200307147/DC1). To assess whether proPM II–GFP is appropriately processed to the mature protease (Fig. 2 A), trophozoite extracts were examined by immunoblotting with anti–PM II and anti-GFP antibodies (Fig. 2 B). Only mPM II was observed in parasites containing a wild-type copy of the PM II gene. mPM II was also the predominant form of the protein in B7 parasites. Along with the proregion, GFP appeared to be proteolytically cleaved from mPM II, as the mobility of mPM II in SDS-PAGE was only slightly slower than that from 3D7 parasites. This slight shift in mobility presumably derives from retention of some of the linker sequence at the COOH terminus of mPM II. Significantly, addition of the GFP tag had a relatively small effect on the steady-state levels of mPM II: densitometric quantitation of Fig. 2 B using BiP as a normalization reference indicated that the amount of mPM II in B7 extract was only 1.2-fold greater than that in transfected parasites before cycling (cycle 0) and 1.6-fold greater than that in 3D7 extract. In addition to mPM II, a small but significant amount of proPM II–GFP was observed in extracts of B7 parasites; in contrast, proPM II was not detected in 3D7 extracts. Immunoblotting with anti-GFP antibodies indicated that GFP is undetectable in stably transfected parasites before drug cycling, which demonstrates that integration of pPM2GT into the PM II gene is a prerequisite for GFP expression.
Figure 2.
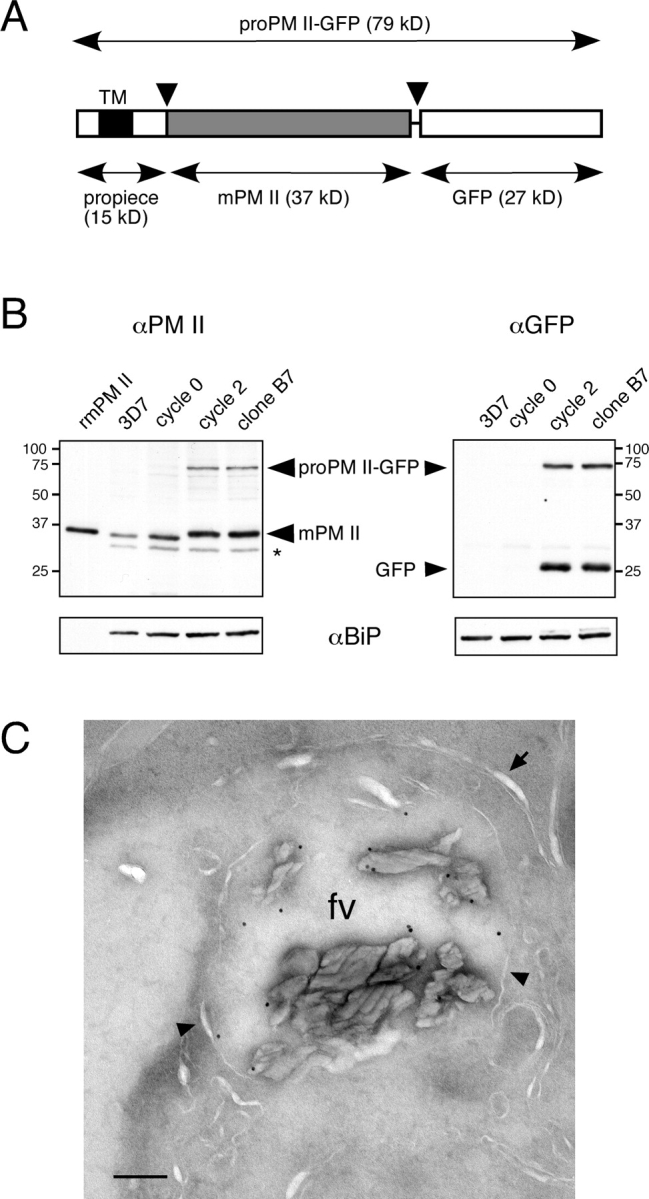
PM II–GFP expression in B7 parasites. (A) Schematic representation of the proPM II–GFP fusion. The propiece is shown with the single transmembrane domain (TM) in black. proPM II–GFP is proteolytically processed twice (arrowheads) to generate mPM II (gray box). (B) Immunoblot analysis of PM II and GFP expression in synchronized trophozoites before and after integration of pPM2GT. SDS-solubilized protein from 107 saponin-treated trophozoites was loaded in each lane. To assess relative amounts of protein loaded, blots were stripped and reprobed with anti-PfBiP antibody (bottom). Recombinant mature PM II (rmPM II) was used as a marker in the PM II blot. The species labeled “GFP” comigrated with recombinant GFPmut2 (not depicted). The band denoted with an asterisk reacted with secondary anti–rabbit Ig antibody alone (not depicted). Sizes of molecular mass markers are indicated in kD. (C) Immunoelectron micrograph illustrating labeling of the food vacuole of a B7 trophozoite with affinity-purified anti–PM II. The food vacuole membrane is indicated with arrowheads, and the closely apposed parasitophorous vacuole and parasite plasma membranes are indicated with an arrow. A low magnification image of this parasite is provided in Fig. S2, http://www.jcb.org/cgi/content/full/jcb.200307147/DC1. fv, food vacuole. Bar, 200 nm.
Direct evidence for the transport of PM II to the food vacuole was obtained by immunoEM with an affinity-purified anti–PM II antibody. Significant amounts of antibody label were detected in the food vacuole in B7 parasite sections (Fig. 2 C), a result consistent with previous observations in wild-type parasites (Banerjee et al., 2002).
The biosynthesis and maturation of proPM II and the closely related proteins proPM I, proPM IV, and proHAP have been shown to proceed relatively rapidly, with a half-time of ∼20 min (Francis et al., 1997a; Banerjee et al., 2003). To determine the effect of the GFP tag on the rate of maturation of proPM II, B7 parasites were pulse labeled with [35S]methionine and -cysteine, chased for various times, and polypeptides containing PM I, PM II, or GFP were immunoprecipitated (Fig. 3). The PM II and GFP immunoprecipitations indicated that proPM II–GFP was processed to mPM II and GFP without significant accumulation of the intermediate forms proPM II or mPM II–GFP, which suggests that PM II maturation and cleavage at the PM II–GFP linker occur essentially simultaneously (Fig. 3, A and B). The rate of disappearance of proPM II–GFP was considerably slower than that of proPM I. After 45 min, a significant amount of proPM II–GFP remained unprocessed (Fig. 3 B), whereas proPM I was completely converted to mPM I (Fig. 3 C). This slower rate of processing may be a factor in the elevation of steady-state levels of proPM II–GFP observed in the immunoblots.
Figure 3.

Pulse-chase analysis of proPM II–GFP maturation. B7 trophozoites were pulse labeled for 15 min with [35S]methionine and -cysteine and chased for the times indicated (min). (A) Immunoprecipitation of PM II–containing species. To indicate the position of proPM II (without GFP), this species was immunoprecipitated from labeled wild-type 3D7 parasites. In the B7 lanes, proPM II–GFP is partially obscured by high background in this region of the gel. (B) Immunoprecipitation of polypeptides containing GFP. The low intensity of the GFP band relative to proPM II–GFP is likely due to two factors: GFP contains one third of the label present in proPM II–GFP, and may be slowly degraded in the food vacuole. (C) Immunoprecipitation of pro- and mPM I. Parasites are from the same labeled populations as those in B.
proPM II–GFP traverses the ER and cytostomal vacuoles on the way to the food vacuole
The distribution of GFP in live B7 parasites was followed over the course of the ∼44 h intraerythrocytic asexual reproductive cycle by epifluorescence microscopy. The food vacuole of trophozoites and schizonts contained abundant GFP (Fig. 4), which indicates that it is transported along with PM II to this organelle. In some parasites, particularly younger trophozoites, GFP fluorescence was observed in a perinuclear ring-like structure adjacent to the food vacuole (Fig. 4 A). The perinuclear distribution of GFP suggested that it might be within the nuclear envelope. Further analysis of this compartment in the presence of BFA is described in the next section. Also in trophozoites, some GFP fluorescence was concentrated in a small number of spots or foci (2.4 ± 1.2 foci per trophozoite, range 0–5, n = 35) that often resided at the periphery of the parasite (Fig. 4 B). In parasites undergoing nuclear division (schizonts), fluorescence was limited to the food vacuole (Fig. 4 C). No fluorescence was observed in association with individual merozoites in segmented schizonts or in ring-stage parasites. Similar GFP distributions were observed with the cloned parasite lines C9 and F4 (unpublished data).
Figure 4.
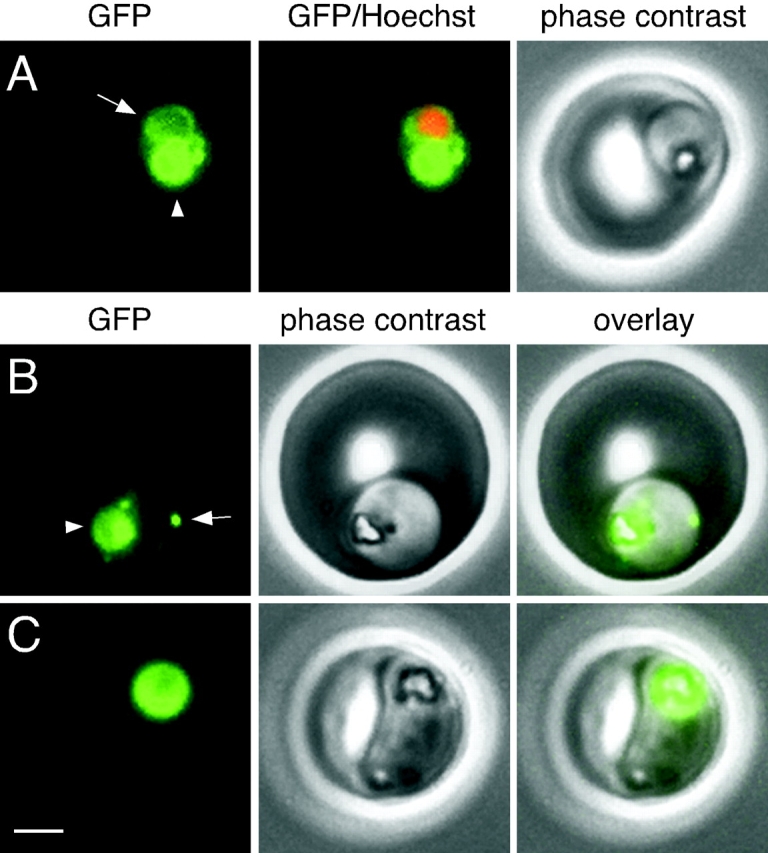
GFP fluorescence in live B7 parasites. (A) A trophozoite exhibiting GFP fluorescence in the food vacuole (arrowhead) and in a perinuclear ring (arrow). Fluorescence from the nuclear stain Hoechst 33342 is pseudocolored red. (B) A trophozoite with a fluorescent food vacuole (arrowhead) and a bright fluorescent spot (arrow) that lies at the periphery of the parasite. (C) A mature parasite displaying a large fluorescent food vacuole. Note the absence of fluorescence outside of the food vacuole. Bar, 2 μm.
To define the distribution of GFP at the ultrastructural level, B7 parasites were fixed and analyzed by immunoEM with an affinity-purified anti-GFP polyclonal antibody. To first assess the specificity of the antibody, untransfected 3D7 trophozoites were subjected to the labeling protocol. Approximately one colloidal gold particle per parasite was observed, with no clear labeling pattern in evidence. In contrast, sections of B7 trophozoites exhibited heavily labeled food vacuoles (Fig. 5 A). The membranes of cytostomal vacuoles were also frequently labeled (Fig. 5, B and C), an observation that provided the first indication that proPM II–GFP populates the cytostomal vacuolar membrane (presumably the outer membrane; see Discussion) en route to the food vacuole. Although the protein collar of the cytostomal pore itself is poorly visible in these sections, the neck of the cytostomal vacuole, the continuity between the erythrocyte cytosol and the vacuole lumen, and the two membranes making up the vacuole are all evident in Fig. 5 (B and C). Double-membrane structures that may be cross sections of the cytostomal vacuole or hemoglobin transport vesicles are shown in Fig. 5 (D and E). Given the location of cytostomes at the periphery of the parasite and the apparent paucity of immunogold label elsewhere in the vicinity of the parasite plasma membrane, we presume that the peripheral GFP foci seen in live parasites (Fig. 4 B) correspond to cytostomal vacuoles.
Figure 5.
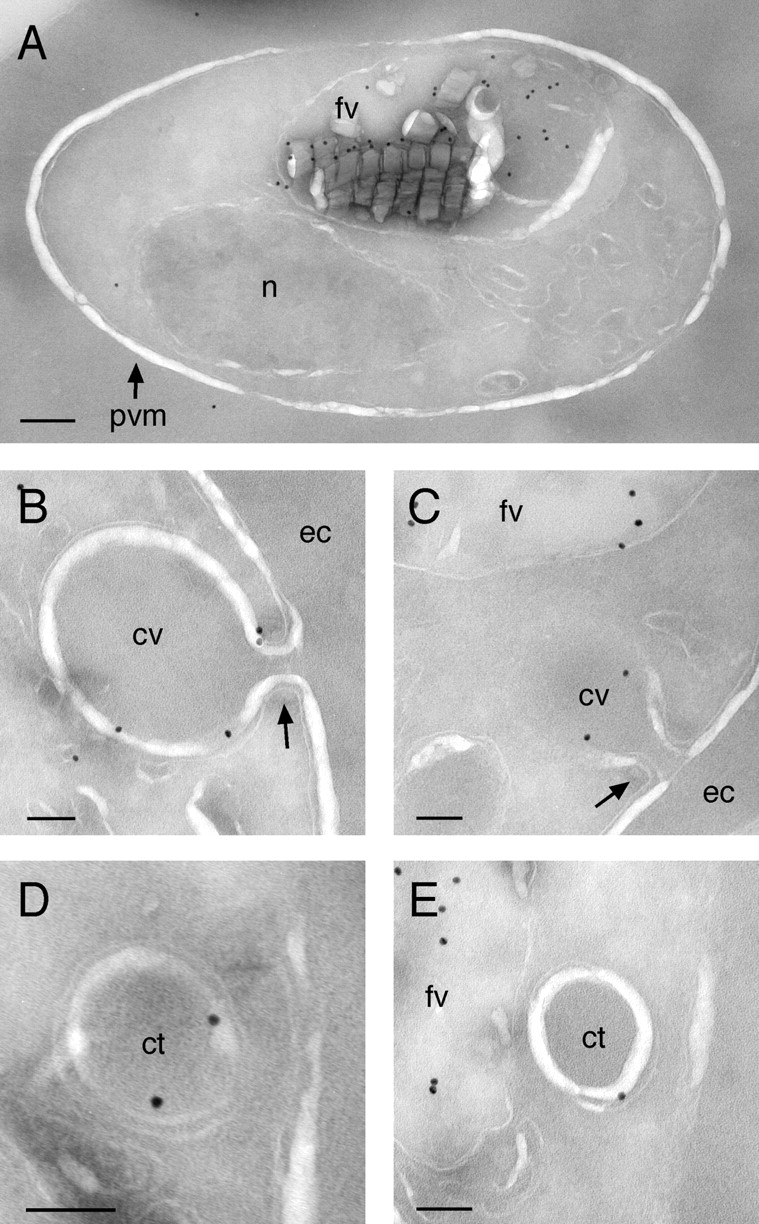
ImmunoEM localization of GFP to the food vacuole and cytostomal vacuoles. (A) A B7 trophozoite displaying prominent food vacuole labeling. Bar, 200 nm. (B and C) Cross sections of cytostomal vacuoles formed during the uptake of erythrocyte cytosol showing labeling of the vacuole membrane. In both panels, the “neck” of the cytostome (arrows) is clearly visible. (D and E) Cross sections of cytostomal vacuoles or hemoglobin transport vesicles displaying membrane labeling. Low magnification images of parasites in B–E are provided in Fig. S2. fv, food vacuole; n, nucleus; pvm, parasitophorous vacuole membrane; cv, cytostomal vacuole; ec, erythrocyte cytoplasm; ct, cytostomal vacuole or hemoglobin-containing transport vesicle. (B–E) Bars, 100 nm.
proPM II–GFP populates the nuclear envelope and the peripheral ER upon BFA treatment
BFA has enjoyed wide application due to its ability to inhibit anterograde protein trafficking from the ER; however, it has been shown to affect protein transport to and from organelles other than the ER, such as the endosomal–lysosomal system (Klausner et al., 1992). BFA inhibits processing of proPM II to mPM II (Francis et al., 1997a), presumably because proPM II cannot reach the food vacuole. To better understand the root cause of inhibition of PM maturation, and to characterize the morphology of the BFA-induced structure(s) in live parasites, we analyzed the distribution of GFP fluorescence in B7 parasites treated with 5 μg/ml BFA for 2 h. In trophozoites, a prominent perinuclear ring of fluorescence was observed (Fig. 6 A). The morphology of this structure closely resembled that observed in untreated trophozoites (Fig. 4 A), but the intensity of fluorescence was greatly increased by BFA treatment. The number of fluorescent foci was greatly diminished (0.14 ± 0.35 per trophozoite, range 0–1, n = 29), which suggests a depletion of cytostomal fluorescence upon BFA treatment. In early schizonts, the fluorescent compartment developed greater complexity and consisted of multiple perinuclear rings (Fig. 6 B). The perinuclear position of this compartment suggested that it was the nuclear envelope, a structure that in many eukaryotic cells is continuous with the ER lumen (Franke et al., 1981). To confirm this, B7 trophozoites were treated with BFA for 2 h and then fixed and sectioned for immunoEM. Both GFP and the resident ER protein BiP were localized in the same sections using secondary antibodies conjugated to 18- and 12-nm colloidal gold, respectively. Much of the GFP label was associated with the nuclear envelope, but some was also observed in elements of the peripheral ER, which consists of tubulovesicular structures extending away from the nucleus toward the parasite plasma membrane (Fig. 6 D). The bulk of the BiP label was associated with the peripheral ER, although it could also be detected in the nuclear envelope (Fig. 6 D and unpublished data). In some sections, continuity between the nuclear envelope and tubulovesicular elements of the peripheral ER could be observed (unpublished data).
Figure 6.

BFA induces a reversible accumulation of proPM II–GFP in the ER. (A and B) GFP fluorescence in live B7 parasites treated for 2 h with BFA: (A) a trophozoite and (B) a schizont undergoing nuclear division. The arrowhead in B indicates food vacuole fluorescence. (C) Redistribution of GFP 10 min after release of the BFA block. Fluorescent spots reappear at the periphery of the parasite (arrowheads). 100 μg/ml cycloheximide was present to inhibit protein synthesis after BFA washout. Similar results were obtained in the absence of cycloheximide. In A–C, fluorescence from the nuclear stain Hoechst 33342 is pseudocolored red. Bar, 2 μm. (D) Cryosection of a BFA treated B7 trophozoite double-labeled with an antibody against GFP (18-nm colloidal gold) and an antibody recognizing the ER marker BiP (12-nm colloidal gold). Most of the 18-nm gold label is associated with the nuclear envelope (arrowhead), whereas the 12-nm gold label is associated with the peripheral ER (asterisk) extending away from the nucleus. A low magnification image of this parasite is provided in Fig. S2. n, nucleus. Bar, 200 nm. (E) B7 trophozoites were 35S-labeled for 2 h in the presence of 5 μg/ml BFA (“BFA” lane). Both BFA and unincorporated 35S were washed out either in the absence (no inhib) or presence (ALLN) of an inhibitor of PM II maturation. proPM II–GFP and GFP were immunoprecipitated with an anti-GFP antibody. The low intensity of the GFP band in “no inhib” lane relative to proPM II–GFP in the “BFA” lane is likely due to two factors: GFP contains one third of the label present in proPM II–GFP, and may be slowly degraded in the food vacuole. Sizes of molecular mass markers are indicated in kD.
Transport of proPM II–GFP downstream of the ER was followed in live parasites by washing out BFA in the presence of cycloheximide, an inhibitor of protein synthesis. 10 min after release of the BFA block, fluorescence was once again visible in cytostomal vacuoles before the appearance of significant fluorescence in the food vacuole (Fig. 6 C). These observations are consistent with a pathway for proPM II–GFP trafficking that proceeds from the ER/nuclear envelope to cytostomal vacuoles and then to the food vacuole.
To confirm that proPM II–GFP synthesized during BFA treatment was not proteolytically processed and thus retained the GFP tag, B7 trophozoites were 35S-labeled for 2 h in medium containing 5 μg/ml BFA. When GFP-containing polypeptides were immunoprecipitated, full-length proPM II–GFP but not free GFP was observed (Fig. 6 E). Upon washout of BFA and a further 2 h incubation at 37°C, nearly all of the labeled proPM II–GFP disappeared and 35S-labeled GFP appeared (Fig. 6 E), a result consistent with release of the secretion block, transport of labeled proPM II–GFP to the food vacuole, and proteolytic cleavage of GFP from mPM II. Washout of BFA after 2 h followed by culturing for 96 h in the absence of BFA provided confirmation that the parasites were viable throughout the BFA treatment period.
proPM II–GFP accumulates in the food vacuole membrane upon treatment with the PM maturation inhibitor ALLN
ALLN is a tripeptide aldehyde inhibitor of cysteine proteases (Sasaki et al., 1990) that prevents the maturation of proPM II in vivo and in vitro (Francis et al., 1997a; Banerjee et al., 2003). Because ALLN does not inhibit PM II activity, this result has been interpreted as evidence for an ALLN-inhibitable PM convertase in the food vacuole (Francis et al., 1997a; Banerjee et al., 2003); however, an indirect effect of ALLN treatment on PM II processing, for example by interfering with transport to the food vacuole, has been difficult to rule out. To better define the effect of ALLN, we localized proPM II–GFP in B7 trophozoites in which PM maturation had been blocked by this inhibitor.
To better visualize proPM II–GFP fluorescence against a background of GFP in the food vacuole, B7 parasites were first treated with BFA for 2 h to accumulate proPM II–GFP, and then the BFA was washed out in the presence of 100 μM ALLN. A marked redistribution of proPM II–GFP was observed in live parasites upon replacement of BFA with ALLN. The perinuclear ring of fluorescence observed upon BFA treatment disappeared and was replaced by a fluorescent rim around the food vacuole (Fig. 7, A and B). In many parasites, regions of more intense fluorescence could be observed within this rim (Fig. 7 A). These may reflect a local concentration of proPM II–GFP in the food vacuole membrane, or may arise from the docking of transport vesicles to the food vacuole; an explanation for this phenomenon was not obvious from immunoEM experiments. In addition, peripheral fluorescent spots indicative of cytostomal vacuoles were seen (Fig. 7 A). The observation of fluorescence circumscribing the food vacuole suggested that the integral membrane protein proPM II–GFP had been transported to the food vacuole and, in the absence of cleavage of proPM II or GFP, remained anchored in the food vacuole membrane. To confirm this idea, B7 parasites treated sequentially with BFA and ALLN were analyzed by immunoEM with either anti-GFP or anti–PM II antibody. In contrast to the lumenal labeling observed with untreated parasites (Fig. 2 C and Fig. 5 A), BFA/ALLN-treated parasites exhibited accumulation of gold label primarily at the food vacuole membrane (Fig. 7, C and D). Control experiments confirmed that parasites remained viable throughout the BFA and ALLN treatments.
Figure 7.
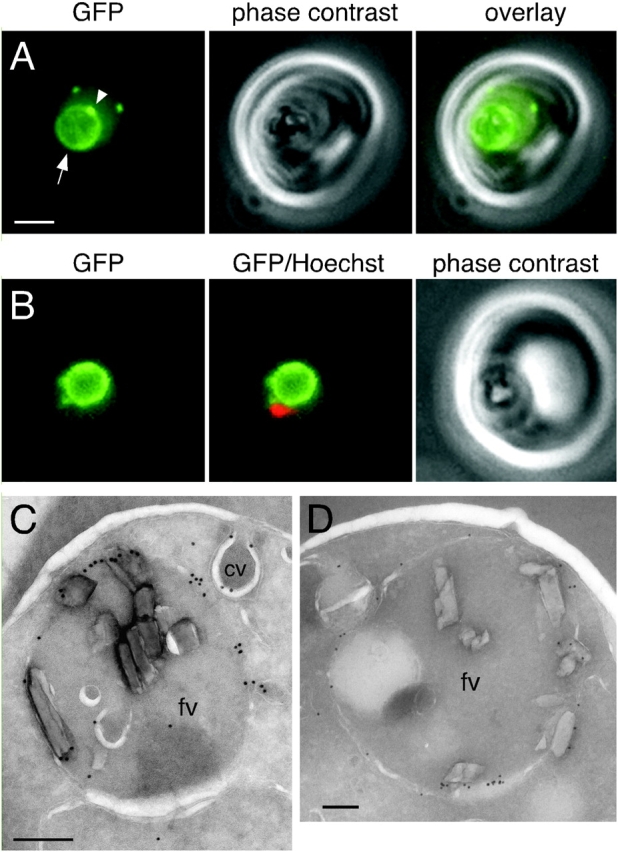
ALLN treatment results in accumulation of proPM II–GFP in the food vacuole membrane. (A) GFP fluorescence in a live B7 trophozoite treated with BFA for 2 h followed by replacement of BFA with 100 μM ALLN for a further 2 h. A bright rim of fluorescence circumscribes the food vacuole (arrow). A local concentration of fluorescence on the food vacuole membrane is indicated with an arrowhead. Two cytostomal vacuoles above the food vacuole are also visible. Bar, 2 μm. (B) A trophozoite treated as in A in which the Hoechst 33342–stained nucleus is pseudocolored red. (C and D) Trophozoites treated as in A and labeled with either (C) anti-GFP or (D) anti–PM II antibody. Low magnification images of these parasites are provided in Fig. S2. Abbreviations are given in the legend to Fig. 5. Bars, 200 nm.
To ensure that GFP was covalently associated with proPM II after washout of BFA in the presence of ALLN, B7 trophozoite proteins were 35S-labeled in the presence of BFA to accumulate [35S]-proPM II–GFP, and both BFA and 35S-label were washed out in the presence of 100 μM ALLN. No diminution of the amount of labeled proPM II–GFP was observed after 2 h incubation with ALLN and no labeled GFP appeared (Fig. 6 E), which indicates that ALLN inhibited both the maturation of proPM II and cleavage of GFP from PM II.
Discussion
We have tagged the chromosomal copy of the P. falciparum PM II coding sequence with that of GFP in order to facilitate analysis of PM trafficking. The GFP tag had a relatively small effect on steady-state mPM II levels in trophozoites. Although GFP did not remain covalently linked to mPM II, cleavage of GFP from PM II occurred simultaneously with conversion of proPM II to the mature form of the enzyme. Thus, GFP was considered to be a faithful reporter of the location of proPM II in the parasite up to the point of maturation. Small amounts of proPM II–GFP accumulated in B7 parasites, whereas proPM II in wild-type parasites was undetectable. The difference in proenzyme levels is likely attributable to the slower rate of maturation of proPM II–GFP, which in turn probably derives from a decreased rate of transport to the food vacuole. The GFP tag may delay folding of proPM II in the ER, or may delay recruitment of proPM II to ER exit sites. We anticipate that the strategy described here for the fusion of tags to the COOH terminus of proteins will be a useful addition to the P. falciparum genetic tool box.
Trafficking of PMs begins in the ER, where they are inserted as type II integral membrane proteins (Francis et al., 1997a). We have shown that BFA treatment induces accumulation of proPM II–GFP in a perinuclear compartment in live parasites. This compartment was identified as the nuclear envelope and the peripheral ER by immunoEM. This is, to our knowledge, the first description of the morphology of the ER in live trophozoites after a relatively brief exposure to BFA. The prominent perinuclear distribution of the ER is reminiscent of that observed in other unicellular organisms (in the absence of BFA) such as Saccharomyces cerevisiae and Toxoplasma gondii (Preuss et al., 1991; Hager et al., 1999). Our results, along with a recent report of colocalization of PM IV and BiP in fixed BFA treated parasites (Banerjee et al., 2003), indicate that the previously observed BFA-induced block in PM maturation derives from an inability to exit the ER. There is some evidence to suggest that ER exit sites (transitional ER) may be located in the nuclear envelope of P. falciparum. In developing P. falciparum merozoites, vesicle budding from the apical face of the nuclear envelope has been documented (Langreth et al., 1978; Bannister and Mitchell, 1995; Ward et al., 1997), and budding of coated vesicles from the nuclear envelope of a Taxol-treated trophozoite has been observed (Taraschi et al., 1998).
In the absence of BFA, weak GFP fluorescence was observed in the nuclear envelope in live parasites during a narrow window of intraerythrocytic development: young trophozoites, but not rings or mature trophozoites (distinguished by the lack of or abundance of hemozoin, respectively), exhibited such fluorescence. Because the steady-state level of mPM II increases as parasites mature (Banerjee et al., 2002), a decrease in the rate of proPM II–GFP biosynthesis in more mature parasites is unlikely to account for the disappearance of fluorescence from the ER. A plausible explanation for this phenomenon is that transport of proPM II–GFP out of the nuclear envelope is relatively inefficient in young trophozoites and becomes more efficient in older parasites.
The presence of proPM II–GFP in the membranes of cytostomal vacuoles points to a role for a major hemoglobin endocytic pathway in proPM II trafficking. The number of cytostomal vacuoles visible by fluorescence microscopy dropped dramatically when proPM II–GFP trafficking was blocked with BFA, and fluorescent cytostomal vacuoles reappeared when the BFA block was relieved. These observations are consistent with trafficking of proPM II–GFP from the ER to cytostomal vacuoles, and in cytostomal vacuoles to the food vacuole. It is presently not known whether proPM II–GFP transits a post-ER compartment such as the Golgi apparatus. A Golgi complex with a classic stacked cisternal morphology has not yet been reported in P. falciparum trophozoites, although several proteins associated with the Golgi apparatus in mammalian cells have been identified in this stage (Elmendorf and Haldar, 1993; de Castro et al., 1996; Van Wye et al., 1996). Characterization of the Golgi apparatus in trophozoites with antibodies against ERD2 has revealed that it takes the form of a single, small, slightly elongated perinuclear structure (Elmendorf and Haldar, 1993; Van Wye et al., 1996). As this differs from the number and location of fluorescent foci that we attribute to cytostomal vacuoles in proPM II–GFP expressing trophozoites, it is unlikely that these foci are Golgi structures.
Maturation of proPM II has been demonstrated to require a proteolytic activity that is sensitive to inhibition by ALLN (Banerjee et al., 2003; Francis et al., 1997b), a potent inhibitor of cathepsin L-type cysteine proteases and calpains (Sasaki et al., 1990). Because proPM II–GFP synthesized in the presence of BFA is not converted to mPM II, processing must occur downstream of the ER. Upon washout of BFA in the presence of ALLN, proPM II–GFP exited from the ER and populated the food vacuole membrane. This result demonstrates that inhibition of proPM II–GFP processing does not stem from interference with trafficking to the food vacuole. Our observations are consistent with the notion that a PM convertase activity is responsible for PM activation in an acidic environment, namely the food vacuole (Francis et al., 1994; Banerjee et al., 2003). We cannot exclude the possibility that proteolytic maturation occurs in transport vesicles on the way to the food vacuole (Hempelmann et al., 2003); however, this would appear to require cotrafficking of the PM convertase and acidification of the transport vesicles, events for which there is currently no evidence.
Together, these results suggest a trafficking pathway for proPM II that takes advantage of the parasite's nutritional requirement for hemoglobin degradation (Fig. 8). We propose that, after insertion into the ER, proPM II is transported to the cytostome, where it accumulates and is brought to the food vacuole along with its substrate, hemoglobin. In the acidic environment of the food vacuole, the proenzyme is cleaved and mature, active, soluble PM II is released. The half-time for biosynthesis and maturation of untagged PMs has been estimated to be 20 min (Francis et al., 1997a; Banerjee et al., 2003). If PM II is trafficked exclusively through the cytostome, we can estimate that the half-time for the delivery of cytostomal vacuolar contents to the food vacuole is less than 20 min; however, our data do not exclude the possibility of alternate PM II trafficking pathways such as direct transport from the ER to the food vacuole.
Figure 8.

Model for trafficking of proPM II–GFP to the food vacuole. This model is based on data presented here and elsewhere (Francis et al., 1994, 1997a). proPM II–GFP is inserted as a type II membrane protein into the ER/nuclear envelope (NE). Transport vesicles containing proPM II–GFP bud from ER exit sites in a BFA-sensitive manner. The vesicles migrate to the cytostome (possibly via a Golgi-like compartment) and fuse with the outer membrane of the cytostomal vacuole, which is topologically contiguous with the parasite plasma membrane. This event would place proPM II–GFP in the space between the two vacuole membranes. The double-membrane hemoglobin transport vesicle pinches off from the cytostome, migrates to the food vacuole, and its outer membrane fuses with that of the food vacuole, leaving proPM II–GFP anchored in the food vacuole membrane. The proregion of PM II and GFP are proteolytically removed to yield mPM II, a process that is inhibited by ALLN. Black circles, PM II; white circles, GFP; PVM, parasitophorous vacuole membrane; PPM, parasite plasma membrane; Hb, hemoglobin.
Trafficking of food vacuole proteins to the cytostomal vacuole would offer a simple means for de novo production of food vacuoles. There is currently no evidence to suggest that hemoglobin-degrading vacuoles are inherited by daughter merozoites, so this organelle may undergo de novo genesis after each round of invasion. Rodent Plasmodium species appear to degrade hemoglobin in multiple cytostome-derived vacuoles that do not fuse into a large central food vacuole until late in the erythrocytic cycle (Slomianny, 1990), which further suggests that a mechanism exists for the de novo production of functional hemoglobin-degrading vacuoles. It will be of interest to determine whether other food vacuole proteins follow a similar route and whether nonfood vacuole proteins share components of this pathway.
The data for trafficking of proPM II–GFP to the food vacuole that we present here support some of the features of a model put forward by Francis et al. (1994) for the transport of proPM I albeit with important modifications. Experiments with PM I could not resolve whether this protein was membrane associated or lumenal in cytostomal vacuoles and transport vesicles (Francis et al., 1994). Our data suggest a model (Fig. 8) in which vesicular transport of PMs to the cytostomal vacuole places the proenzyme within the space between the two vacuole membranes. The detection of low levels of PM I at the parasite plasma membrane led Francis et al. (1994) to propose that PM I is first transported to the plasma membrane and then incorporated into the cytostomal vacuole, possibly by passive lateral diffusion in the plasma membrane. In contrast, we do not observe a similar accumulation of proPM II–GFP in the parasite plasma membrane either by fluorescence microscopy or immunoEM. Although the question of how proPM I and proPM II–GFP reach the cytostome is far from resolved, we currently favor a model that incorporates direct vesicular transport to the outer membrane of the cytostome.
Materials and methods
Vector construction
The gene for Aequorea victoria GFP allele GFPmut2 (Cormack et al., 1996) was PCR amplified with forward primer 5′-CTCGAGGGCCCACTAGTGGTCCTAGGCCAGGTGCAGCACATTATGCAGCAATGAGTAAAGGAGAAGAACTTTTC-3′ (XhoI, SpeI, and AvrII restriction sites are underlined) and reverse primer 5′-GTCGACGCGGCCGCTTATTTGTATAGTTCATCCATGCC-3′ (SalI and NotI restriction sites are underlined), and cloned as an XhoI–SalI fragment into the XhoI site of pHC1 (Crabb et al., 1997b) to produce pHCGFP. The last kilobase of the PM II ORF (omitting the stop codon) was PCR amplified from P. falciparum 3D7 genomic DNA with primers 5′-GCACGCTCGAGTAAAATTATTTAGGTAGTTCAAATGATAA-3′ (XhoI site underlined) and 5′-GCACGCCTAGGTAAATTCTTTTTAGCAAGAGCAATAC-3′ (AvrII site underlined), digested with XhoI and AvrII, and ligated into the same sites of pHCGFP to create pPM2GFP. An XhoI–HindIII fragment from pPM2GFP containing the PM II–GFP fusion sequence and the P. falciparum HSP86 3′ UTR was cloned into the same sites of pSP72 (Promega) to generate pSPGFP. A drug-resistance cassette containing human DHFR was excised from pHHT-TK (a gift of A. Cowman, Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia; Duraisingh et al., 2002) with BglII and EcoRI, and introduced into the same sites of pSPGFP to yield pPM2GT.
Parasite culture and transfection
P. falciparum 3D7 was cultured in human O+ erythrocytes at 37°C under 5% O2, 5% CO2, and 90% N2 in RPMI 1640 (GIBCO BRL), supplemented with 27 mM sodium bicarbonate, 11 mM glucose, 0.37 mM hypoxanthine, 10 μg/ml gentamicin, and either 5 g/l Albumax (for routine growth; GIBCO BRL) or 10% heat-inactivated human serum (for transfection and cycling) as described previously (Trager and Jensen, 1976). Parasite cultures were synchronized by sorbitol treatment (Lambros and Vanderberg, 1979). Ring-stage parasites were transfected by electroporation with 100 μg of supercoiled pPM2GT using low voltage/high capacitance conditions (Fidock and Wellems, 1997). Resistant parasites were selected with 10 nM WR99210 (a gift of D. Jacobus, Jacobus Pharmaceuticals, Princeton, NJ) and reached a parasitemia of 0.5% after 22 d. Selection for integration of the episome into the PM II gene was achieved by subjecting WR99210-resistant parasites to two drug cycles, each consisting of 21 d of growth in the absence of WR99210 followed by reselection of resistant parasites. Parasites were cloned by the method of limiting dilution. Genotypes were analyzed by probing Southern blots of NotI–StuI-digested total parasite DNA with a gel-purified PCR product extending from bases 496 to 1143 of the PM II gene. The signal was generated with an AlkPhos direct labeling and detection kit (Amersham Biosciences).
Western and pulse-chase analysis
For immunoblotting, synchronized trophozoites were separated from erythrocyte cytosol by treatment with cold 0.1% saponin in PBS for 15 min on ice followed by two washes in cold PBS. Saponin-treated parasites were counted with a hemocytometer and frozen at −80°C until use. Frozen parasite pellets were rapidly suspended in SDS-PAGE sample buffer at 106 parasites/μl, placed immediately in a boiling water bath for 5 min and centrifuged at 16,000 g for 5 min to remove insoluble material. Protein extracted from 107 parasites was resolved by 10% SDS-PAGE and blotted to nitrocellulose. Antibodies used for immunodetection were rabbit anti–PM II Ab 737 (1:5,000) raised against a peptide corresponding to the NH2 terminus of mPM II (Francis et al., 1997a), affinity-purified rabbit anti-GFP ab6556 (1:5,000; Abcam) or rabbit anti-BiP (1:10,000; Kumar et al., 1991). The signal was developed with an HRP-conjugated anti–rabbit Ig secondary antibody and ECL Western blotting detection reagents (Amersham Biosciences). Recombinant PM II was a gift from E. Istvan (Washington University, St. Louis, MO).
For pulse-chase analysis, a synchronized trophozoite culture (10–15% parasitemia) was washed twice with RPMI 1640 without cysteine, methionine, and glutamine (Sigma-Aldrich), and incubated at 4% hematocrit for 15 min at 37°C in the same medium supplemented as described previously (Banerjee et al., 2003) but with 180 μCi/ml [35S]methionine and -cysteine (EXPRE35S35S, NEN). The chase period was initiated either by replacing the labeling medium with complete RPMI (PM II immunoprecipitations) or by addition of an equal volume of complete RPMI (GFP and PM I immunoprecipitations), and the parasites were incubated at 37°C in a 5% CO2 environment. At the desired time, a 12-ml aliquot of the culture was removed and placed on ice. Culture aliquots were washed once in cold PBS, treated with 1.5 pellet volumes of 0.15% saponin in PBS for 10 min on ice, washed once with cold PBS, and stored at −80°C. PMs were immunoprecipitated from denatured parasite lysates as described previously (Francis et al., 1997a) using anti–PM I Ab574 (1:200) or affinity-purified anti–PM II Ab737 (1:20). GFP was immunoprecipitated in the absence of SDS using living colors full-length A.v. polyclonal anti-GFP antibody (1:500; BD Biosciences). A cocktail of protease inhibitors (1 μM pepstatin, 10 μM bestatin, 10 μM ALLN, 1 μM leupeptin, 1 μM N-[trans-epoxysuccinyl]-l-leucine-4-guanidinobutylamide, and 0.5 mM 4-[2-aminoethyl]-benzenesulfonylfluoride) was added to the GFP immunoprecipitations.
The effect of BFA and ALLN on proPM II–GFP maturation was assayed by modifying the pulse-chase protocol as follows: 5 μg/ml BFA/0.1% DMSO was added to the 35S-containing labeling media, hematocrit was reduced to 2% and the labeling time was extended to 2 h, at which point an aliquot was placed on ice (BFA sample). The remaining culture was split into two aliquots, washed three times with complete media containing either 100 μM ALLN/0.1% DMSO or 0.1% DMSO, and incubated for an additional 2 h at 37°C. Parasites were released by saponin treatment and GFP-containing species were immunoprecipitated as described in the preceding paragraph.
Fluorescence and immunoEM
Fluorescence and phase contrast images were collected using a Plan-Neo-fluar 100x/1.3 NA objective on an Axioscope microscope (Carl Zeiss MicroImaging, Inc.) equipped with a Axiocam CCD camera and Axiovision 3.1 software (Carl Zeiss MicroImaging, Inc.). For imaging of live parasites, cultures were maintained in RPMI 1640 media without phenol red (GIBCO BRL) supplemented as described in parasite culture and transfection. 10 μl of culture was placed underneath a coverslip and viewed at ambient temperature for a maximum of 10 min. Nuclei were visualized by adding 5 μM Hoechst 33342 (Molecular Probes) to aliquots of parasite cultures immediately before mounting. Quantitation of GFP-fluorescent foci was done by visual examination while adjusting the focus to sample all focal planes through the specimen. For analysis of the effects of BFA, parasites were cultured in media containing 5 μg/ml BFA/0.1% DMSO for 2 h before observation. BFA was removed from the culture by washing twice and resuspending to 2% hematocrit with BFA-free media containing 100 μg/ml cycloheximide/0.1% DMSO. ALLN-treated cultures were first incubated with 5 μg/ml BFA/0.1% DMSO for 2 h, the BFA was removed by washing three times with media containing 100 μM ALLN/0.1% DMSO, and the parasites were cultured for an additional 2 h. Images were imported into Adobe Photoshop for contrast enhancement, pseudocoloring, and creation of overlays.
For immunolocalization by EM, infected RBCs were separated from culture media by centrifuging at 860 g for 3 min, and were fixed and prepared for labeling as described previously (Banerjee et al., 2002). BFA- and BFA/ALLN-treated parasites were cultured as described for fluorescence microscopy, and the fix solution was supplemented with 5 μg/ml BFA and 100 μM ALLN, respectively. 70-nm sections were incubated with either affinity-purified rabbit anti–PM II Ab737 (1:100) or rabbit anti-GFP ab6556 (1:1,000 or 1:2,000) followed by 18-nm colloidal gold-conjugated anti–rabbit IgG (Jackson ImmunoResearch Laboratories). For double-labeling experiments, rat anti-BiP antibody (1:400; Malaria Research and Reference Reagent Resource Center, American Type Culture Collection) was used with a 12-nm colloidal gold-conjugated anti–rat IgG secondary antibody (Jackson ImmunoResearch Laboratories). Sections were stained with 0.3% uranyl acetate/2% polyvinyl alcohol and viewed on a transmission electron microscope (model 1200EX; JEOL).
Online supplemental material
Fig. S1 shows that proPM II–GFP is an integral membrane protein. Fig. S2 contains low magnification immunoEM images of the parasites depicted in Figs. 2 C, 5 (B–E), 6 D, and 7 (C and D). Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200307147/DC1.
Acknowledgments
We are grateful to Eva Istvan for providing recombinant mature plasmepsin II, Alan Cowman for pHHT-TK, and Phyllis Hanson for helpful discussions. We thank E. Istvan and Mark Drew for critical reading of the manuscript. WR99210 was a gift of David Jacobus.
This work was supported by National Institutes of Health grant AI41718. D.E. Goldberg is also a recipient of a Burroughs Wellcome Fund Scholar Award in Molecular Parasitology.
The online version of this article contains supplemental material.
Abbreviations used in this paper: ALLN, N-acetyl-l-leucyl-l-leucyl-l-norleucinal; BFA, brefeldin A; HAP, histo-aspartic protease; mPM, mature plasmepsin; PM, plasmepsin.
References
- Adisa, A., M. Rug, N. Klonis, M. Foley, A.F. Cowman, and L. Tilley. 2003. The signal sequence of exported protein-1 directs the green fluorescent protein to the parasitophorous vacuole of transfected malaria parasites. J. Biol. Chem. 278:6532–6542. [DOI] [PubMed] [Google Scholar]
- Aikawa, M., P.K. Hepler, C.G. Huff, and H. Sprinz. 1966. The feeding mechanism of avian malarial parasites. J. Cell Biol. 28:355–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akompong, T., M. Kadekoppala, T. Harrison, A. Oksman, D.E. Goldberg, H. Fujioka, B.U. Samuel, D. Sullivan, and K. Haldar. 2002. Trans expression of a Plasmodium falciparum histidine-rich protein II (HRPII) reveals sorting of soluble proteins in the periphery of the host erythrocyte and disrupts transport to the malarial food vacuole. J. Biol. Chem. 277:28923–28933. [DOI] [PubMed] [Google Scholar]
- Banerjee, R., and D.E. Goldberg. 2000. The Plasmodium food vacuole. Antimalarial Chemotherapy: Mechanisms of Action, Resistance, and New Directions in Drug Discovery. P.J. Rosenthal, editor. Humana Press, Totowa, NJ. 43–63.
- Banerjee, R., J. Liu, W. Beatty, L. Pelosof, M. Klemba, and D.E. Goldberg. 2002. Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc. Natl. Acad. Sci. USA. 99:990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee, R., S.E. Francis, and D.E. Goldberg. 2003. Food vacuole plasmepsins are processed at a conserved site by an acidic convertase activity in Plasmodium falciparum. Mol. Biochem. Parasitol. 129:157–165. [DOI] [PubMed] [Google Scholar]
- Bannister, L.H., and G.H. Mitchell. 1995. The role of the cytoskeleton in Plasmodium falciparum merozoite biology: an electron-microscopic view. Ann. Trop. Med. Parasitol. 89:105–111. [DOI] [PubMed] [Google Scholar]
- Coombs, G.H., D.E. Goldberg, M. Klemba, C. Berry, J. Kay, and J.C. Mottram. 2001. Aspartic proteases of Plasmodium falciparum and other parasitic protozoa as drug targets. Trends Parasitol. 17:532–537. [DOI] [PubMed] [Google Scholar]
- Cormack, B.P., R.H. Valdivia, and S. Falkow. 1996. FACS-optimized mutants of the green fluorescent protein (GFP). Gene. 173:33–38. [DOI] [PubMed] [Google Scholar]
- Crabb, B.S., B.M. Cooke, J.C. Reeder, R.F. Waller, S.R. Caruana, K.M. Davern, M.E. Wickham, G.V. Brown, R.L. Coppel, and A.F. Cowman. 1997. a. Targeted gene disruption shows that knobs enable malaria-infected red cells to cytoadhere under physiological shear stress. Cell. 89:287–296. [DOI] [PubMed] [Google Scholar]
- Crabb, B.S., T. Triglia, J.G. Waterkeyn, and A.F. Cowman. 1997. b. Stable transgene expression in Plasmodium falciparum. Mol. Biochem. Parasitol. 90:131–144. [DOI] [PubMed] [Google Scholar]
- de Castro, F.A., G.E. Ward, R. Jambou, G. Attal, V. Mayau, G. Jaureguiberry, C. Braun-Breton, D. Chakrabarti, and G. Langsley. 1996. Identification of a family of Rab G-proteins in Plasmodium falciparum and a detailed characterisation of pfrab6. Mol. Biochem. Parasitol. 80:77–88. [DOI] [PubMed] [Google Scholar]
- Deitsch, K.W., and T.E. Wellems. 1996. Membrane modifications in erythrocytes parasitized by Plasmodium falciparum. Mol. Biochem. Parasitol. 76:1–10. [DOI] [PubMed] [Google Scholar]
- Duraisingh, M.T., T. Triglia, and A.F. Cowman. 2002. Negative selection of Plasmodium falciparum reveals targeted gene deletion by double crossover recombination. Int. J. Parasitol. 32:81–89. [DOI] [PubMed] [Google Scholar]
- Eggleson, K.K., K.L. Duffin, and D.E. Goldberg. 1999. Identification and characterization of falcilysin, a metallopeptidase involved in hemoglobin catabolism within the malaria parasite Plasmodium falciparum. J. Biol. Chem. 274:32411–32417. [DOI] [PubMed] [Google Scholar]
- Elmendorf, H.G., and K. Haldar. 1993. Identification and localization of ERD2 in the malaria parasite Plasmodium falciparum: separation from sites of sphingomyelin synthesis and implications for organization of the Golgi. EMBO J. 12:4763–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock, D.A., and T.E. Wellems. 1997. Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc. Natl. Acad. Sci. USA. 94:10931–10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, S.E., I.Y. Gluzman, A. Oksman, A. Knickerbocker, R. Mueller, M.L. Bryant, D.R. Sherman, D.G. Russell, and D.E. Goldberg. 1994. Molecular characterization and inhibition of a Plasmodium falciparum aspartic hemoglobinase. EMBO J. 13:306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, S.E., R. Banerjee, and D.E. Goldberg. 1997. a. Biosynthesis and maturation of the malaria aspartic hemoglobinases plasmepsins I and II. J. Biol. Chem. 272:14961–14968. [DOI] [PubMed] [Google Scholar]
- Francis, S.E., D.J. Sullivan, Jr., and D.E. Goldberg. 1997. b. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu. Rev. Microbiol. 51:97–123. [DOI] [PubMed] [Google Scholar]
- Franke, W.W., U. Scheer, G. Krohne, and E.D. Jarasch. 1981. The nuclear envelope and the architecture of the nuclear periphery. J. Cell Biol. 91:39s–50s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluzman, I.Y., S.E. Francis, A. Oksman, C.E. Smith, K.L. Duffin, and D.E. Goldberg. 1994. Order and specificity of the Plasmodium falciparum hemoglobin degradation pathway. J. Clin. Invest. 93:1602–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager, K.M., B. Striepen, L.G. Tilney, and D.S. Roos. 1999. The nuclear envelope serves as an intermediary between the ER and Golgi complex in the intracellular parasite Toxoplasma gondii. J. Cell Sci. 112:2631–2638. [DOI] [PubMed] [Google Scholar]
- Hempelmann, E., and T.J. Egan. 2002. Pigment biocrystallization in Plasmodium falciparum. Trends Parasitol. 18:11. [DOI] [PubMed] [Google Scholar]
- Hempelmann, E., C. Motta, R. Hughes, S.A. Ward, and P.G. Bray. 2003. Plasmodium falciparum: sacrificing membrane to grow crystals? Trends Parasitol. 19:23–26. [DOI] [PubMed] [Google Scholar]
- Klausner, R.D., J.G. Donaldson, and J. Lippincott-Schwartz. 1992. Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116:1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakovich, K.A., I.Y. Gluzman, K.L. Duffin, and D.E. Goldberg. 1997. Generation of hemoglobin peptides in the acidic digestive vacuole of Plasmodium falciparum implicates peptide transport in amino acid production. Mol. Biochem. Parasitol. 87:123–135. [DOI] [PubMed] [Google Scholar]
- Kumar, N., G. Koski, M. Harada, M. Aikawa, and H. Zheng. 1991. Induction and localization of Plasmodium falciparum stress proteins related to the heat shock protein 70 family. Mol. Biochem. Parasitol. 48:47–58. [DOI] [PubMed] [Google Scholar]
- Lambros, C., and J.P. Vanderberg. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65:418–420. [PubMed] [Google Scholar]
- Langreth, S.G., J.B. Jensen, R.T. Reese, and W. Trager. 1978. Fine structure of human malaria in vitro. J. Protozool. 25:443–452. [DOI] [PubMed] [Google Scholar]
- Preuss, D., J. Mulholland, C.A. Kaiser, P. Orlean, C. Albright, M.D. Rose, P.W. Robbins, and D. Botstein. 1991. Structure of the yeast endoplasmic reticulum: localization of ER proteins using immunofluorescence and immunoelectron microscopy. Yeast. 7:891–911. [DOI] [PubMed] [Google Scholar]
- Sasaki, T., M. Kishi, M. Saito, T. Tanaka, N. Higuchi, E. Kominami, N. Katunuma, and T. Murachi. 1990. Inhibitory effect of di- and tripeptidyl aldehydes on calpains and cathepsins. J. Enzyme Inhib. 3:195–201. [DOI] [PubMed] [Google Scholar]
- Shenai, B.R., P.S. Sijwali, A. Singh, and P.J. Rosenthal. 2000. Characterization of native and recombinant falcipain-2, a principal trophozoite cysteine protease and essential hemoglobinase of Plasmodium falciparum. J. Biol. Chem. 275:29000–29010. [DOI] [PubMed] [Google Scholar]
- Sijwali, P.S., B.R. Shenai, J. Gut, A. Singh, and P.J. Rosenthal. 2001. Expression and characterization of the Plasmodium falciparum haemoglobinase falcipain-3. Biochem. J. 360:481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomianny, C. 1990. Three-dimensional reconstruction of the feeding process of the malaria parasite. Blood Cells. 16:369–378. [PubMed] [Google Scholar]
- Slomianny, C., G. Prensier, and P. Charet. 1985. Ingestion of erythrocytic stroma by Plasmodium chabaudi trophozoites: ultrastructural study by serial sectioning and 3-dimensional reconstruction. Parasitology. 90:579–588. [DOI] [PubMed] [Google Scholar]
- Sullivan, D.J., Jr., I.Y. Gluzman, and D.E. Goldberg. 1996. Plasmodium hemozoin formation mediated by histidine-rich proteins. Science. 271:219–222. [DOI] [PubMed] [Google Scholar]
- Taraschi, T.F., D. Trelka, T. Schneider, and I. Matthews. 1998. Plasmodium falciparum: characterization of organelle migration during merozoite morphogenesis in asexual malaria infections. Exp. Parasitol. 88:184–193. [DOI] [PubMed] [Google Scholar]
- Trager, W., and J.B. Jensen. 1976. Human malaria parasites in continuous culture. Science. 193:673–675. [DOI] [PubMed] [Google Scholar]
- Van Wye, J., N. Ghori, P. Webster, R.R. Mitschler, H.G. Elmendorf, and K. Haldar. 1996. Identification and localization of rab6, separation of rab6 from ERD2 and implications for an ‘unstacked’ Golgi, in Plasmodium falciparum. Mol. Biochem. Parasitol. 83:107–120. [DOI] [PubMed] [Google Scholar]
- Ward, G.E., L.G. Tilney, and G. Langsley. 1997. Rab GTPases and the unusual secretory pathway of Plasmodium. Parasitol. Today. 13:57–62. [DOI] [PubMed] [Google Scholar]
- Wickham, M.E., M. Rug, S.A. Ralph, N. Klonis, G.I. McFadden, L. Tilley, and A.F. Cowman. 2001. Trafficking and assembly of the cytoadherence complex in Plasmodium falciparum-infected human erythrocytes. EMBO J. 20:5636–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt, D.M., and C. Berry. 2002. Activity and inhibition of plasmepsin IV, a new aspartic proteinase from the malaria parasite, Plasmodium falciparum. FEBS Lett. 513:159–162. [DOI] [PubMed] [Google Scholar]
- Yayon, A., R. Timberg, S. Friedman, and H. Ginsburg. 1984. Effects of chloroquine on the feeding mechanism of the intraerythrocytic human malarial parasite Plasmodium falciparum. J. Protozool. 31:367–372. [DOI] [PubMed] [Google Scholar]
- Ziegler, J., R. Linck, and D.W. Wright. 2001. Heme aggregation inhibitors: antimalarial drugs targeting an essential biomineralization process. Curr. Med. Chem. 8:171–189. [DOI] [PubMed] [Google Scholar]