

Abstract
Lipid movement between organelles is a critical component of eukaryotic membrane homeostasis. Niemann Pick type C (NP-C) disease is a fatal neurodegenerative disorder typified by lysosomal accumulation of cholesterol and sphingolipids. Expression of yeast NP-C–related gene 1 (NCR1), the orthologue of the human NP-C gene 1 (NPC1) defective in the disease, in Chinese hamster ovary NPC1 mutant cells suppressed lipid accumulation. Deletion of NCR1, encoding a transmembrane glycoprotein predominantly residing in the vacuole of normal yeast, gave no phenotype. However, a dominant mutation in the putative sterol-sensing domain of Ncr1p conferred temperature and polyene antibiotic sensitivity without changes in sterol metabolism. Instead, the mutant cells were resistant to inhibitors of sphingolipid biosynthesis and super sensitive to sphingosine and C2-ceramide. Moreover, plasma membrane sphingolipids accumulated and redistributed to the vacuole and other subcellular membranes of the mutant cells. We propose that the primordial function of these proteins is to recycle sphingolipids and that defects in this process in higher eukaryotes secondarily result in cholesterol accumulation.
Keywords: transport; vacuole; lysosome; ceramide; neurodegeneration
Introduction
The function of eukaryotic membranes reflects their lipid composition as much as that of proteins, and yet the mechanisms by which lipid partitioning is maintained are largely unknown. Niemann Pick type C (NP-C) disease is a neurodegenerative lipid storage disorder, the accepted biochemical hallmark of which is the lysosomal accumulation of low density lipoprotein (LDL)–derived unesterified cholesterol. Consequently, the reactions responsible for sterol biosynthesis, uptake, and esterification are misregulated (Pentchev et al., 1997). Although the defect in cholesterol egress from the endosomal–lysosomal (E–L) system is the predominant cellular phenotype, NP-C disease is further typified by the subcellular accumulation of complex glycosphingolipids (Vanier, 1999). Moreover, fibroblasts from a “variant” subgroup of NP-C individuals with profound neurodysfunction exhibit severe sphingolipid transport aberrancies but modest cholesterol mislocalization (Sun et al., 2001). Subcellular protein transport does not appear to be altered in NP-C gene 1 (NPC1)–defective cells; however, markers of endocytosis exhibit retarded transport to extralysosomal sites (Neufeld et al., 1999).
A positional cloning approach identified the NPC1 gene as defective in 95% of patients with NP-C disease (Carstea et al., 1997). NPC1 predicts a glycosylated, transmembrane protein with a similarity to the morphogen receptor Patched (Ptc, 23% identity) and the sterol regulatory element-binding protein (SREBP) cleavage-activating protein (SCAP), a protein integral to sterol and fatty acid homeostasis (26% identity over 190 residues; Fig. 1). The latter conservation corresponds to a putative sterol-sensing domain (SSD). NPC1 is ubiquitously and constitutively expressed in mammalian tissues and is conserved from yeast to humans (Loftus et al., 1997). In contrast, many aspects of receptor-mediated uptake of sterol are absent over the same period of evolution, suggesting a primordial function of the Npc1 protein family that predates exogenous sterol transport. We demonstrate that the NP-C–related gene 1 (NCR1), the yeast orthologue of mammalian NPC1, performs a conserved function that is distinct from trafficking of sterol. We show that in mammalian cells the proteins are interchangeable with respect to cholesterol and ganglioside transport, and yet a null mutation in yeast NCR1 has no sterol-related phenotype. A dominant mutation in the SSD of yeast NCR1 confers pleiotropic phenotypes consistent with altered sphingolipid transport. We propose that in this ancestral eukaryote, the role of NPC1-like proteins is to recycle sphingolipids and that in multicellular organisms, one consequence of this is transport of cholesterol.
Figure 1.


Sequence comparisons of human Npc1 and yeast Ncr1p. (A) Conserved domains in NPC1 and Ncr1p. In addition to the NPC domain that distinguishes the NPC1 gene family, Ncr1p retains a putative sterol-sensing domain (SSD) and several regions of similarity to the morphogen receptor, Patched (Patched homology domain, PHD). (B) The putative SSD of NPC1, Ncr1p, and SCAP. Residues 599–789 of human NPC1, 538–721 of yeast Ncr1p, and 264–444 of human SCAP are aligned. Sequence identity is indicated by the vertical bar and similarity by colons and periods. Across this region the NPC1 and Ncr1p sequences are 43% identical. The NCR1Y 571C, NCR1Y 718D, and NCR1Y 718N variants are indicated.
Results
Yeast NCR1, an orthologue of human NPC1
The predominant sterol of Saccharomyces cerevisiae is ergosterol, which is synthesized, esterified, used, and regulated in a similar manner to cholesterol in mammals (Sturley, 2000). Subcellular sterol transport may also be conserved between yeast and mammalian cells; Ncr1p (YPL006w) is an uncharacterized 1,170-residue transmembrane protein with a signal peptide, eight N-glycosylation sites, and 35% identity to the human Npc1 protein. As for Npc1p, Ncr1p is structurally similar to Ptc and the SSD of SCAP (Fig. 1), and furthermore, identical to the normal NPC1 allele at many residues implicated in NP-C disease (Greer et al., 1999; Ribeiro et al., 2001). These alleles include G897 and I972 in Ncr1p, corresponding to two common NP-C disease alleles (G992W and I1061T, respectively). Overall, the molecules are compellingly similar.
Expression of yeast NCR1 complements loss of NPC1
To test the functional equivalence of the yeast and human proteins, we expressed NCR1 in CHO CT60 and NPC1-trap cells lacking Npc1 (Cruz et al., 2000; Higaki et al., 2001). Yeast proteins lack targeting information for mammalian endosomes. Because Npc1p localization, which is mediated by COOH-terminal motifs is required for full complementation (Watari et al., 1999; Ioannou, 2000), we fused the last 64 residues of human NPC1 to Ncr1p (NCR1-Cterm). Expression constructs for NCR1 and NCR1-Cterm were transfected into the CHO cells, and sterol transport was assessed by filipin staining and fluorescence microscopy. Expression of NCR1-Cterm restored cholesterol clearance to levels indistinguishable from cells expressing human NPC1 (Fig. 2 and Table I). A significant decrease in E–L filipin staining was also observed upon expression of NCR1. Although fewer filipin negative cells were observed, most transfected cells displayed “partial” clearance of cholesterol. This diminished suppression likely reflects mislocalization of the native yeast protein; a GFP fusion with Ncr1p localizes to the early endosome, whereas NCR1-Cterm–GFP was detected in the late endosomal compartment (unpublished data).
Figure 2.
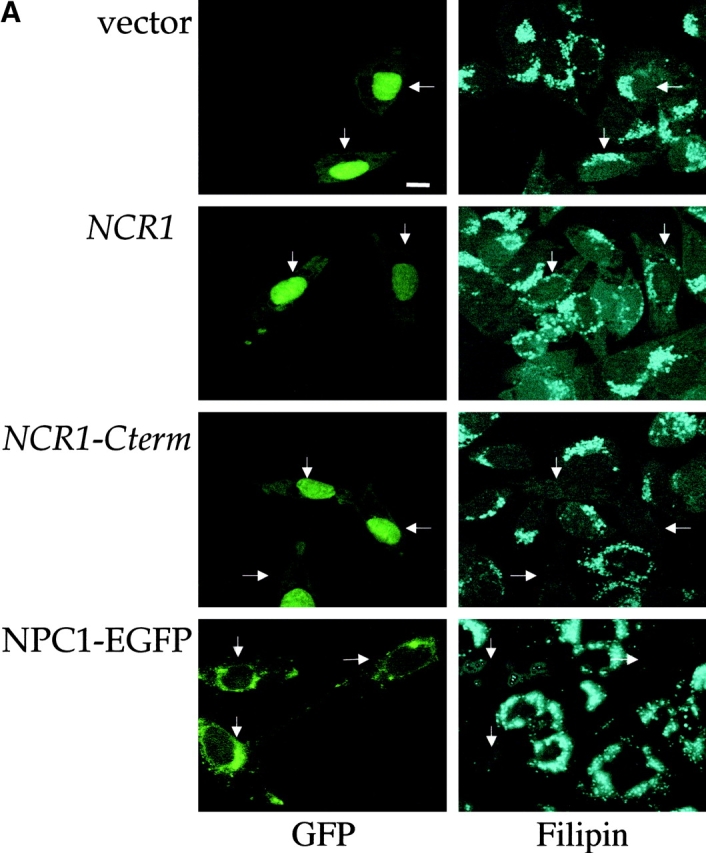

Expression of yeast NCR1 restores lipid trafficking in NPC1 mutant mammalian cells. CHO cell line CT60 was transiently transfected with a human NPC1–EGFP fusion or cotransfected with pEGFP and pCR3.1 (vector), pCR3.1-NCR1 (NCR1), or pCR3.1–NCR1-Cterm (NCR1-Cterm). The arrows indicate transfected cells based on fluorescence conferred by the pEGFP vector or the NPC1–EGFP fusion. Representative images are shown. (A) Cells were preincubated with LDLs and stained for cholesterol with filipin and imaged for GFPs. (B) Transfected cells were stained with fluorescent subunit B of cholera toxin (CTx-B) to detect the ganglioside GM1 and imaged for GFPs.
Table I. Restoration of cholesterol transport in npc1-deficient CHO cells by expression of yeast NCR1 .
| Percentage of corrected cells per GFP-positive cells
|
||||
|---|---|---|---|---|
| Partial
|
Complete
|
|||
| DNA | CT60 | NPC1-trap | CT60 | NPC1-trap |
| vector | 6.7 ± 2.1 | 3.0 ± 1.7 | 0.7 ± 0.5 | 2.3 ± 1.5 |
| NCR1 | 18.7 ± 4.5a | 17.3 ± 0.6a | 17.0 ± 2.6a | 15.3 ± 2.5a |
| NCR1-Cterm | 14.3 ± 2.5 | 10.7 ± 4.0 | 56.7 ± 2.1b | 61.3 ± 5.1b |
CHO cells were cotransfected with pEGFP and pCR3.1 (vector), pCR3.1-NCR1 (NCR1), or pCR3.1-NCR1-Cterm (NCR1-Cterm), incubated in the presence of low density lipoproteins, stained with filipin to detect unesterified cholesterol, and imaged for GFP. Partial restoration was defined as the reduction of intensity or the number of fluorescent vesicles compared with vector control. Cells lacking filipin fluorescence were classified as completely corrected. Means ± SEM from three independent experiments.
Significant differences from vector transfection, P < 0.01.
Significant differences from vector transfection, P < 0.001.
The subcellular accumulation of sphingolipids, particularly gangliosides, is another characteristic of the NP-C syndrome, and in the case of GM1 has been observed in NPC1-deficient CHO cells (Sugimoto et al., 2001). Expression of NCR1-Cterm or human NPC1 reversed the accumulation of GM1 in the E–L compartment of transfected CT60 cells as detected by fluorescent cholera toxin (subunit B) binding assays (Fig. 2 B). Thus, when provided with the correct localization motif, the yeast and human proteins function interchangeably, suggesting a conserved activity despite several billion years of divergence.
Null mutations in yeast NCR1 have no effect on sterol metabolism
In mammalian cells lacking NPC1, exogenous sterol is sequestered from the esterification reaction and sterol biosynthesis is misregulated. During anaerobic growth (Gollub et al., 1974) or in upc2-1 mutants (Lewis et al., 1988), sterol is absorbed by yeast in an ABC-transporter–dependent process (Wilcox et al., 2002) and esterified by the acylCoA cholesterol acyltransferase orthologues (Yang et al., 1996). Deletion of NCR1 had no effect on acetate incorporation into sterols, oleate incorporation into steryl esters, anaerobic viability, or esterification of exogenous [14C]cholesterol (Table II). Cell sterol composition can be exaggerated by deletion of ERG6 (C-24 sterol methyl transferase; Gaber et al., 1989) or ERG9 (squalene synthase; Fegueur et al., 1991) or treatment with zaragozic acid, an inhibitor of squalene synthase (Bergstrom et al., 1993). This exaggeration results in membrane perturbations and, in the latter two cases, sterol auxotrophy; however, loss of NCR1 in these contexts had no effect on cell viability or sterol uptake (unpublished data).
Table II.
Phenotypic characterization of ncr1 mutant strains
| Genotypes
|
|||||
|---|---|---|---|---|---|
| Phenotypes tested |
NCR1
(normal) |
ncr1 Δ | Y718D | Y718N | Y571C |
| Sterol synthesis ([3H]acetate), (P vs. NCR1) |
18.2 ± 2.9 | 19.7 ± 1.2 (0.4) |
13.2 ± 2.3 (0.07) |
13.2 ± 1.8 (0.07) |
16.2 ± 1.6 (0.37) |
| Sterol esterification ([3H]oleate), (P vs. NCR1) |
11.0 ± 2.8 | 11.9 ± 2.4 (>0.05) |
11.9 ± 0.9 (>0.05) |
10.5 ± 2.7 (>0.05) |
10.7 ± 1.7 (>0.05) |
| Exogenous sterol esterification [14C]cholesterol, (P vs. NCR1) |
74.3 ± 2.0 | 69.2 ± 6.3 (>0.05) |
72.2 ± 3.8 (>0.05) |
77.9 ± 3.6 (>0.05) |
78.1 ± 0.8 (>0.05) |
| 30°C (2% dextrose) | ++ | ++ | + | ++ | ++ |
| 38°C (2% dextrose) | ++ | ++ | – | ++ | ++ |
| Anaerobiosis | ++ | ++ | + | ++ | ++ |
| Nystatin (5 μg/ml) | ++ | ++ | – | ++ | ++ |
| Ethanol (3%) | ++ | ++ | – | ++ | ++ |
| Acetate (2%) | ++ | ++ | – | ++ | ++ |
| Sodium chloride (0.75 M) | ++ | ++ | – | ++ | ++ |
| Sorbitol (1.0 M) | ++ | ++ | + | ++ | ++ |
| Calcium chloride (0.8 M) | ++ | ++ | + | ++ | ++ |
| Hydroxyurea (40 mM) | ++ | ++ | + | ++ | ++ |
Yeast strains with the missense mutations Y718D, Y718N, and Y571C in the SSD of NCR (Fig. 1 B) were assessed for growth or metabolic incorporation (percent incorporation ± SEM) under the conditions described. The growth of the normal strain is considered ++ and the growth of the other strains is expressed relative to that scale.
In contrast to observations with NP-C mutant fibroblasts (Fig. 2 A), the majority of sterol in ncr1Δ and normal yeast strains detected by filipin fluorescence resides at the plasma membrane (Fig. 3 A). In validation of the assay, arv1 strains that accumulate sterols in subcellular membranes (Tinkelenberg et al., 2000) displayed significant subcellular filipin staining, whereas an erg6 mutant had barely detectable plasma membrane fluorescence. Moreover, ncr1Δ strains displayed no changes in sensitivity to polyene antibiotics (e.g., nystatin) that form lethal complexes with ergosterol. Finally, the relative distribution of sterols and phospholipids across a subcellular fractionation was unchanged in ncr1Δ mutants (Fig. 3 B), further suggesting that membrane composition, particularly with respect to sterol, was unaffected by loss of NCR1.
Figure 3.


Sterol distribution in yeast NCR1 mutants. (A) Yeast strains (normal, ncr1Δ, and NCR1Y 718D) were stained with filipin to detect ergosterol. Deletions in ERG6 (erg6Δ) block synthesis of ergosterol, whereas ARV1 deletions (arv1Δ) accumulate sterol in subcellular membranes. (B) Cell membrane preparations from normal and NCR1 deletion strains were subjected to subcellular fractionation, lipid extraction, and TLC analysis after [14C]oleate or [3H]acetate incorporation. Distributions of sterols relative to phospholipid are given as a ratio of [3H]acetate to [14C]oleate. Total [3H]acetate and [14C]oleate incorporation was 3.06 and 1.79 (normal) versus 2.77 and 1.18 (deletion) × 105 dpm/OD600, respectively. Fractions were characterized by immunoblotting with antisera to the plasma membrane ATPase (Pma1p, solid line, squares) and the vacuolar membrane H+-ATPase (Vph1p, dashed line, diamonds), followed by scanning densitometry (arbitrary units, representative data).
Normal and ncr1Δ strains grew comparably on a variety of carbon sources and at different temperatures (12°, 30°, 38°, and 40°C). Similarly, there was no growth differential due to supplementation with 1 M sorbitol or 0.75 M NaCl (Table II). A transmembrane permease activity with specificity for oleic acid and acriflavine has been suggested for Npc1p (Davies et al., 2000). However, we could detect no defect in uptake of [3H]oleate or in accumulation of acriflavine in ncr1 mutants (unpublished data). In a further attempt to reveal a phenotype due to loss of NCR1, we undertook a transcriptional profiling approach. Oligonucleotide arrays (Affymetrix) were used to compare normal and ncr1Δ strains. Consistent with the redundancy of NCR1, there were no significant (i.e., greater than twofold) transcriptional differences between these strains (unpublished data). In summation, the deletion of NCR1 had no detectable physiological consequences.
Expression and localization of Ncr1p
The NCR1 gene is transcribed (Fig. 4 A) and expressed in normal strains and growth conditions as evidenced by tagging the endogenous protein with the HA epitope NCR1-HA. The biological activity of this allele was confirmed in the NPC1 mutant CHO cells (unpublished data). Ncr1-HAp was detected as a membrane-associated, endoglycosidase H-sensitive glycoprotein (∼130 kD; Fig. 4 B), indicating its passage through the ER. In a further example of concordance between the yeast and human proteins, Ncr1-HAp cofractionated predominantly with normal yeast vacuoles, the equivalent organelle to the E–L system of mammalian cells (Fig. 4 C). Furthermore, a GFP-Ncr1 fusion protein localizes to the vacuolar membrane (Huh et al., 2003; unpublished data).
Figure 4.



NCR1 expression in yeast. (A) RNA hybridization of the indicated strains grown in YPD media at 30°C. The loading control of 28S ribosomal RNA is shown. No significant differences in NCR1 transcript levels for normal, NCR1Y 718D, NCR1Y 718N, or NCR1Y 571C (not depicted) strains were detected. (B) Expression of Ncr1-HAp. The chromosomal copy of NCR1 was tagged with HA at the COOH terminus by homologous recombination. Duplicate membrane extracts were solubilized with Triton X-100, deglycosylated (endo-H), and resolved by SDS-PAGE and immunoblotting (αHA 12CA5 mAb). Molecular mass markers (Bio-Rad Laboratories) are shown. (C) Subcellular localization of Ncr1-HAp. Membrane preparations from cells expressing Ncr1-HA were fractionated by ultracentrifugation in Renograffin 60. Fractions 1–14 were characterized by SDS-PAGE and immunoblotting with antibodies to Ncr1-HA (Anti-HA), the plasma membrane ATPase (Anti-Pma1p), and the vacuolar H+-ATPase (Anti-Vph1p).
A dominant mutation in the SSD of yeast NCR1
Ncr1p is biologically active when expressed in mammalian cells, and yet loss of Ncr1p is of no apparent consequence in yeast. To bypass this redundancy, we introduced mutations in NCR1 (Fig. 1 B) analogous to dominant mutations in the SSD of SCAP (Nohturfft et al., 1998). The presence of asparagine instead of aspartic acid at residue 443 in SCAP confers sterol-resistant SREBP cleavage; thus, we mutated the analogous residue (Tyr718) of Ncr1p to either aspartic acid (NCR1Y 718D) or asparagine (NCR1Y 718N). In addition, we changed an invariant tyrosine of the Ncr1p SSD (Tyr571) to cysteine (NCR1Y 571C). The mutations were introduced at the NCR1 locus by homologous recombination and did not affect NCR1 transcript levels (Fig. 4 A).
Because the NCR1Y 571C, NCR1Y 718D, and NCR1Y 718N mutations reside in a putative SSD, we anticipated an alteration in sterol metabolism. NCR1Y 718D strains, unlike the others, were hypersensitive to nystatin, suggestive of perturbed plasma membrane properties (Fig. 5 A). However, anaerobic growth, an indicator of exogenous sterol transport, was unaltered relative to aerobic growth (Table II). Furthermore, sterol synthesis, esterification (Table II), and localization (Fig. 3 A; and see Fig. 7 C) were not significantly different in these strains compared with normal strains. The NCR1Y 718D allele conferred poor growth at 30°C, inviability at 38°C (Fig. 5 A), and salt sensitivity and poor growth on carbon sources such as acetate and ethanol (Table II). The NCR1Y 718N and NCR1Y 571C, strains were modestly impaired at elevated temperatures (Fig. 5 A), but were otherwise indistinguishable from normal strains. The NCR1Y 718D strains were not uniformly sick; responses to sorbitol, calcium, hydroxyurea, and low pH were indistinguishable from growth on YPD at 30°C (Table II).
Figure 5.




Sphingolipid phenotypes in NCR1 mutants. (A) Fivefold dilutions of strains of the indicated genotypes were plated to the media shown and incubated at 30 or 38°C. (B) Haploid yeast of the indicated genotype were transformed with control or NCR1 expression plasmids, serially diluted, and plated at 30 and 38°C. (C) Growth of strains of the indicated genotypes in response to sphingosine or ABA were derived by continuous analysis of culture absorbance (600 nm). (D) Synthesis of ceramide and IPC after metabolic incorporation of [3H]DHS (2 h) in the presence of ABA. Sphingolipids were extracted and analyzed by TLC and radio-image scanning. The percent total incorporation into all lipids are shown. Means ± SEM; *, P < 0.01, relative to previous concentration of ABA. Absence of error bars indicates too small for scale.
Figure 7.

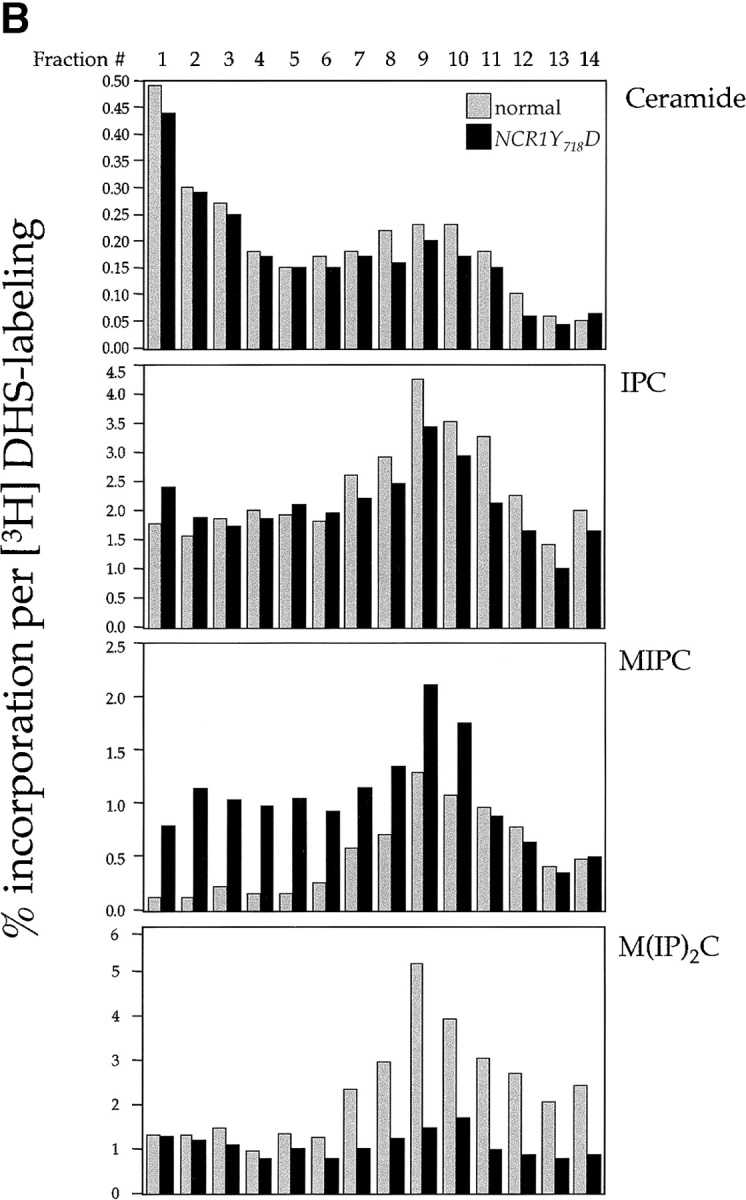


Subcellular distribution of lipids in NCR1 mutants. Lipid levels were assessed after metabolic labeling to steady state with [3H]DHS (sphingolipids), [14C]oleate (phospholipids), or [3H]acetate (sterols) at 30°C for 18 h. (A) Total cellular incorporation into sphingolipid pools (percentage of total extraction, means ± SEM; *, P < 0.01 relative to normal). Total incorporation of [3H]DHS for normal and NCR1Y 718D strains was 1.826 and 2.152 × 105 cpm per OD600, respectively. (B) Cell membrane preparations of identical cultures to those in A were subjected to subcellular fractionation after [3H]DHS incorporation. The distributions of IPC, MIPC, and M(IP)2C are presented as the percentage of total incorporation in normal (gray bars) and NCR1Y 718D (black bars) strains (representative data). (C) Membrane preparations after [14C]oleate or [3H]acetate incorporation were subjected to subcellular fractionation. Total [3H]acetate and [14C]oleate incorporation was 3.06 and 1.79 (normal) versus 6.09 and 2.43 (NCR1Y 718D) × 105 dpm/OD600, respectively. The distributions of sterol and phospholipids are presented as the percentage of total incorporation in normal (gray bars) and NCR1Y 718D (black bars) cells per fraction. (D) Immunoblotting of fractions with antibodies to plasma membrane (Pma1p), dolichol phosphate mannose synthase (Dpm1p, ER), and vacuoles (Vph1). Peak fractions for these markers were coincident between the strains, despite apparent differences in protein expression.
Yeast strains with the NCR1Y 718D mutation exhibit pleiotropic phenotypes, whereas the ncr1Δ strain showed no defects, predicting that this variant would act as a dominant allele. Normal, ncr1Δ, and NCR1Y 718D strains were transformed with vector control or a plasmid carrying NCR1. NCR1 expression was confirmed by RT-PCR assays (not depicted) but failed to rescue inviability at 38°C of the NCR1Y 718D strains (Fig. 5 B). In addition, heterozygous NCR1/NCR1Y 718D diploids retain the temperature sensitivity of the NCR1Y 718D allele, further indicating dominant inheritance (unpublished data).
The NCR1Y 718D allele alters sphingolipid metabolism
NP-C disease is characterized by sphingolipid accumulation. Furthermore, the sensitivity of yeast to temperature, high salt, or polyenes can result from modulations in sphingolipids (Dickson and Lester, 2002). Thus, in the absence of a sterol-related phenotype, we questioned whether or not sphingolipid metabolism was disturbed by mutations in NCR1. In S. cerevisiae, ceramide is synthesized from phytosphingosine and is the precursor for synthesis of inositolphosphorylceramide (IPC), mannosyl-IPC (MIPC), and mannosyl-di-IPC (M(IP)2C). These complex sphingolipids are analogous to mammalian sphingolipids in that they associate with sterols in the plasma membrane and perform a critical role in membrane function (Dickson and Lester, 2002). Normal yeast strains are sensitive to sphingosine and C2-ceramide (Nickels and Broach, 1996; Chung et al., 2001). The NCR1Y 718D mutant strain was super sensitive to 10 μM C2-ceramide (Fig. 5 A) and 5 μM sphingosine (Fig. 5 C) compared with the other strains. This phenotype was evident as a prolonged lag phase, which is consistent with retarded nutrient uptake, a trait common to alterations in sphingolipids (Chung et al., 2001).
Changes in sphingolipid status can also be monitored using antifungals such as aureobasidin A (ABA), which blocks synthesis of IPC causing ceramide accumulation and inviability (Nagiec et al., 1997). NCR1Y 718D strains were resistant to 500 ng/ml ABA, whereas normal and ncr1Δ strains were sensitive (Fig. 5 C). ABA resistance was associated with increased incorporation of [3H]dihydrosphingosine (DHS) into IPC at the expense of ceramide over a range of drug concentrations (Fig. 5 D), which is consistent with increased flux through the sphingolipid biosynthetic pathway of NCR1Y 718D strains.
The sensitivity of the NCR1Y 718D strain to sphingosine and C2-ceramide, and resistance to ABA, led us to assess metabolic incorporation of [3H]DHS into sphingolipids in the mutant strains over a 2-h labeling period (Fig. 6). The levels of ceramide were comparable between the strains. However, NCR1Y 718D strains accumulated 2.8-fold more MIPC (23.3% vs. 8.1%, P < 0.01%) and significantly less IPC and M(IP)2C (Fig. 6 A). No changes in sphingolipids were observed in the other NCR1 mutants (unpublished data). Given the elevated MIPC levels in NCR1Y 718D strains, we assessed the expression of CSG1, CSG2 (encoding components of MIPC synthase), and IPT1 (mediating the synthesis of M(IP)2C). CSG2 transcript levels were elevated in NCR1Y 718D strains (Fig. 6 B).
Figure 6.



Sphingolipid metabolism in NCR1 mutants. (A) The sphingolipid biosynthetic pathway and the metabolism of [3H]DHS in normal and NCR1Y 718D cells. Genes for the biosynthesis of ceramide, IPC, MIPC, and M(IP)2C are indicated. Long-chain fatty acylCoA (LCFA-CoA) is an essential substrate for the synthesis of ceramide. Sphingolipids were extracted from the indicated strains grown in the presence of [3H]DHS for 2 h and analyzed by TLC and radio-image scanning. The percent incorporation into all lipids are shown for sphingolipids. Means ± SEM; *, P < 0.01, relative to normal cells. Total incorporation of [3H]DHS for normal and NCR1Y 718D strains was 1.43 and 1.32 × 106 cpm/mg dry weight, respectively. (B) Quantification of RNA hybridization analysis for genes determining the synthesis of MIPC (CSG1 and CSG2). RNA was extracted, resolved, transferred to nitrocellulose membranes by conventional methods, hybridized with CSG1 or CSG2 probes, and analyzed using a phosphorimager (arbitrary units, means ± SEM). Deletion strains csg1Δ and csg2Δ, respectively, for CSG1 and CSG2 acted as hybridization controls. Actin message levels were comparable between strains (not depicted). (C) The stress induction of ceramide synthesis in NCR1 mutants. Cultures of the indicated genotype were grown to exponential phase, split, and grown for a further 2 h in the presence of [3H]DHS at 30 or 38°C. Incorporation of label was normalized to total incorporation into all lipids, which was equivalent at both temperatures (e.g., for normal strains, 2.53 and 2.19 × 106, cpm at 30 and 38°C, respectively). *, P < 0.01.
Upon heat shock, yeast elevate ceramide levels (Jenkins et al., 1997). To assess if the temperature sensitivity of the NCR1Y 718D strains was related to sphingolipid metabolism, we monitored the changes in ceramide levels of normal, ncr1Δ, and NCR1Y 718D strains upon shifting to 38°C and labeling with [3H]DHS. Ceramide levels increased fourfold in normal and ncr1Δ strains upon temperature shift, whereas NCR1Y 718D strains showed less than a twofold increase (Fig. 6 C). Complex sphingolipids were not altered by heat shock relative to growth at 30°C. The molecular mechanism or purpose for increased de novo synthesis of ceramide after heat stress is not known, however, the diminished response by the NCR1Y 718D strains may explain their compromised growth rates.
The NCR1Y 718D mutation causes sphingolipid mislocalization
In NPC1-deficient mammalian cells, the primary defect in sphingolipid metabolism relates to transport out of the E–L system (Choudhury et al., 2002). To assess whether the recycling of complex sphingolipids through the yeast endosomal pathway might be disturbed by mutations in NCR1, we labeled sphingolipids to steady state by incubation for 18 h with [3H]DHS and subjected membranes to subcellular fractionation to separate plasma membranes from subcellular organelles (Fig. 7). Before fractionation, it is clear that in NCR1Y 718D strains MIPC accumulates at the expense of M(IP)2C, which is consistent with diversion of the former molecule from a biosynthetic pool (Fig. 7 A). In normal strains, MIPC and M(IP)2C accumulate in the plasma membrane (Fig. 7, fractions 8–10), colocalizing with the plasma membrane ATPase (Pma1p). We could detect no differences in the distribution of DHS metabolites in ncr1Δ strains (unpublished data). However, in the NCR1Y 718D strains, MIPC, and to a lesser extent M(IP)2C, redistributed to subcellular organelles, particularly the vacuolar compartment as inferred by colocalization with the vacuolar H+-ATPase (Figs. 7, B and D, Vph1p). Indeed, if we consider fractions 4 and 5 (Fig. 7), in which Vph1p is predominant but Pma1p or Dpm1p are low, NCR1Y 718D causes a 6.5- and 7.2-fold accumulation of MIPC relative to controls. This lipid rearrangement in the NCR1Y 718D strains was specific to complex sphingolipids. The subcellular distribution of ceramide (Fig. 7 B), phospholipids, and sterols (Fig. 7 C) was similar between strains.
Discussion
Niemann Pick disease type C is a neurodegenerative disorder with a significant defect in subcellular transport of exogenous sterols. However, NP-C disease is not solely a cholesterol storage disorder, and the nature of the offending metabolite in this syndrome is contentious, particularly in the brain. Sterol accumulation is detectable in NPC1-deficient neurons, but is not as striking as at extra-neuronal sites. Several works in genetically modified murine models of this disease have concluded that sterol uptake has no role in symptom development (Xie et al., 2000a,b). In contrast, the metabolism and transport of sphingolipids is aberrant in most npc1 −/−cells, including neurons (Sugimoto et al., 2001; Zervas et al., 2001a; Zhang et al., 2001). Finally, pharmacological intervention in sphingolipid synthesis alleviates symptoms in animal models of NP-C disease, whereas reductions in plasma cholesterol give no benefit (Patterson et al., 1993; Zervas et al., 2001b).
What does the yeast system inform regarding the human syndrome? Despite evolutionary divergence, the yeast and mammalian proteins are functionally equivalent for sterol and ganglioside transport in mammalian cells. However, we failed to identify any defect in sterol metabolism conferred by mutations in NCR1. Thus, we hypothesize that Ncr1p performs a primordial function that underlies the sterol transport defects associated with NP-C disease. However, a quandary remains: what is the physiological role of this protein in yeast? The similarities of yeast Ncr1p and human Npc1p to the SREBP cleavage-activating protein led us to create constitutive mutations in the SSD of Ncr1p. One such allele, NCR1Y 718D, functioned as a dominant neomorph, its most salient phenotype being the errant subcellular accumulation of MIPC. The normal pathways of sphingolipid distribution (Fig. 8) commence with synthesis of ceramide in the ER and transport to the Golgi compartment where complex sphingolipid synthesis progresses (Funato et al., 2002). An asymmetry is then created; MIPC and M(IP)2C accumulate in the plasma membrane, whereas IPC localizes to the vacuole. In the NCR1Y 718D strains, the sequestration of MIPC in the vacuole limits its retrograde movement to the Golgi compartment resulting in reduced M(IP)2C synthesis. The same event likely results in up-regulation of biosynthesis, in part at the level of transcription of the CSG2 gene. This increased flux explains resistance to pathway inhibitors, sensitivity to substrates such as C2-ceramide and sphingosine, and inadequate response to heat shock in terms of ceramide accumulation.
Figure 8.

Pathways of subcellular sphingolipid transport in yeast. Ceramide is synthesized in the ER and transported by vesicular-dependent (1) and -independent (1a) pathways to the Golgi compartment. Conversion to complex sphingolipids (IPC, MIPC, and M(IP)2C) is followed by migration from the Golgi apparatus to the plasma membrane (2) or to vacuoles (3). In the sphingolipid recycling pathway, transport from the plasma membrane to vacuoles (4), or from vacuoles to the plasma membrane or the Golgi compartment (5 or 6), is selective between sphingolipids. Thus, in normal yeast, IPC accumulates in vacuoles, whereas M(IP)2C and to a lesser extent MIPC accumulate in the plasma membrane. In NCR1Y 718D strains, recycling is disturbed such that MIPC and M(IP)2C accumulate in the vacuole. The sequestration of MIPC results in diminished M(IP)2C synthesis and up-regulation of biosynthesis of MIPC from IPC.
Based on these observations, we propose that the primordial role of the Npc1 protein family lies in sphingolipid recycling with sterol movement as a consequence, which is consistent with studies of NPC1 in mammalian cells (Coxey et al., 1993; Sugimoto et al., 2001) and with hypotheses that neurodegeneration in NP-C disease results from ganglioside accumulation. There are many precedents for a link between sphingolipid and sterol homeostasis. Multiple sphingolipid storage disorders are associated with perturbations in cholesterol homeostasis (Pagano et al., 2000), perhaps because the two molecules readily form membrane microdomains (Bagnat et al., 2000; Simons and Ehehalt, 2002). Indeed, cholesterol accumulation was strikingly reduced when NPC1 deficiency was combined with loss of the β-1-4GalNAc transferase responsible for complex ganglioside synthesis (Liu et al., 2000; Gondre-Lewis et al., 2003).
The SCAPs of mammals and insects via the SSDs are proposed to respond to membrane perturbations rather than changes in a specific molecule (Seegmiller et al., 2002). It is clear that residue 443 of SCAP is a critical position in the SSD (Nohturfft et al., 1996). Our work confirms this in a different protein (Ncr1p) and illustrates a clear species and molecule specificity. In mammalian SCAP, insect SCAP (Seegmiller et al., 2002), or yeast Ncr1p, the normal residue is aspartic acid, asparagine, or tyrosine, respectively. In mammalian SCAP or yeast Ncr1p, substitution with asparagine or aspartic acid produce dominant phenotypes, which is likely due to the loss of specific protein–protein interactions. A critical component of this homeostasis is the interaction of SCAP with ER retention proteins such as INSIG1 and INSIG2. Although there is no SCAP orthologue in yeast, INSIG-like proteins are conserved (Hampton, R., personal communication). Although they have no reported mutant phenotype, it is tempting to speculate they may interact with Ncr1p.
One model for the failure to identify an ncr1 null phenotype could be that the protein functions in a redundant pathway for sphingolipid recycling. Accordingly, NCR1Y 718D acts as a dominant mutant that corrupts mutually redundant pathways of sphingolipid recycling, thus revealing a phenotype. The manipulation of these bypass pathways would be obvious targets to circumvent the transport defects and thus treat NP-C disease. Interestingly, overexpression of certain Rab proteins implicated in vesicular transport suppresses loss of NPC1 (Choudhury et al., 2002; Walter et al., 2003). Whether or not this is an avenue to therapy remains to be determined; however, the identification of such bypass pathways in yeast and humans will provide a significant step in this direction.
Materials and methods
General
Standard methods were used for yeast manipulation (Ausubel et al., 1998). Yeast strains are isogenic with W303-1A (Thomas and Rothstein, 1989). Cholesterol, ergosterol, and nystatin were added from stock solutions (5 and 2 mg/ml in 1:1, ethanol/tyloxapol [Sigma-Aldrich] and 10 mg/ml in propylene glycol, respectively). Zaragosic acid (provided by Y.S. Chao, Merck & Co., Rahway, NJ), sphingosine, C2-ceramide (BIOMOL Research Laboratories, Inc.), and ABA (Panvera) were used at the concentrations indicated. The BLAST alignment tool was used for sequence comparisons (Altschul et al., 1997).
Mutagenesis of NCR1
NCR1 deletions were constructed by allele replacement (Erdeniz et al., 1997) using a PCR product (oligos: 5′ KONP', 3′ KONP) homologous to NCR1 at its 5′ and 3′ ends and flanking the Kluyveromyces lactis URA3 gene. The locus was characterized by PCR with 5′ and 3′ NCR1 or URA3-specific oligos (5′NPSCR, 006INTSCR, 3′ NP-internal', and 3′NPSCR). Missense alleles were created similarly with oligos N25, D25, and C25 for the NCR1Y 718N, NCR1Y718D, and NCR1Y 571C alleles, respectively. Ncr1p was tagged at the COOH terminus with a triple HA epitope (18 residues) using a PCR-generated template (Schneider et al., 1995). All alleles were sequenced.
Expression of NCR1 in mammalian cells
NCR1 or NCR1-HA was amplified from yeast genomic DNA (oligos 5′NPSCR and 3′NPSCR) and incorporated into pCR3.1 digested with EcoRV (pCR3.1-NCR1 and pCR3.1-NCR1-HA). The COOH-terminal 53 residues of NCR1 were replaced with amino acids 1214–1278 of human NPC1 in pCR3.1-NCR1-Cterm after PCR amplification of the human cDNA (5′NCR1tail-BsmI and 3′NPC1tail), digestion with BsmI and XhoI, and ligation at the same sites in pCR3.1-NCR1.
CHO lines CT60 and NPC1-trap were cultured 3 d in Ham's F12 and 10% lipoprotein-deficient serum and transfected (FuGENE 6; Roche Molecular Biochemicals) with pCR3.1, pCR3.1-NCR1, or pCR3.1-NCR1-Cterm and a mammalian nuclear targeting-EGFP vector (Watari et al., 1999) to identify transfected cells. Cells were incubated with 10% lipoprotein-deficient serum and 50 μg/ml of human LDL for 24 h, fixed with 3% PFA, and stained with 50 μg/ml of filipin (Sigma-Aldrich). Cells were imaged by confocal fluorescence microscopy (model LSM510; Carl Zeiss MicroImaging, Inc.; excitation 488 nm for EGFP). For quantitative analysis, filipin-corrected cells were counted from multiple fields of EGFP-positive cells. For detection of GM1, cells were washed with Ham's F12, 25 mM Hepes, pH 7.4, and 0.01% BSA, incubated with 20 nM of Alexa 555–conjugated CtxB (Molecular Probes, Inc.) for 1 h at 37°C, and fixed with 4% PFA. Fluorescence images were collected after excitation at 568 nm using an microscope (model Axiomat 100; Carl Zeiss MicroImaging, Inc.).
Phenotypic analyses
The expression of NCR1-HA was confirmed in Triton X-100–soluble protein extracts (Guo et al., 2001) by immunoblotting with the 12CA5 mAb (Roche Molecular Biochemicals). Protein deglycosylation with endoglycosidase H was accomplished as directed (Roche Molecular Biochemicals). For growth in the presence of sphingosine (in ethanol) or ABA (in DMSO), cells were continuously monitored at 25°C at 600 nm using a microbiology reader (model Bioscreen C; ThermoLabsystems). BBL Gas Jars and GasPak Plus (Becton Dickinson) were used for anaerobic experiments (Tinkelenberg et al., 2000). Growth tests on 2% potassium acetate, 3% ethanol, 0.8 M calcium chloride, 0.75 M sodium chloride, C2-ceramide (in 0.1% ethanol/0.05% tergitol), or 40 mM hydroxyurea were performed on solid YP media. Total RNA was isolated from independent isolates of the ncr1Δ strain grown to OD600 = 0.5–0.6 in YEPD and analyzed in microarray or northern hybridizations (Wilcox et al., 2002).
Cell labeling (10 μCi [3H]acetic acid [NEN Life Science Products], 5.0 μCi [3H]oleic acid [NEN Life Science Products], or 2 μCi/ml [3H]DHS [American Radiolabeled Chemicals]; for 2–5 h), lysis, release of lipids, TLC, and data collection were performed as described previously (Yang et al., 1996). Sphingolipids were isolated from cells treated with 5% TCA (ice, 15 min) by extraction with diethylether/ethanol/water/pyridine/ammonia (5:15:15:1:0.18, vol/vol) for 60 min at 60°C, resuspension was performed in chloroform/methanol/water (16:16:5) and TLC was performed on silica gel 60 (EM Science) plates resolved with chloroform/methanol/4.2N ammonia (9:7:2). Ergosterol localization was visualized by filipin staining (20 μg/ml) as above.
Membrane preparations from cells grown for 18 h in the presence of [3H]DHS, [3H]acetic acid, or [3H]oleic acid were subjected to discontinuous gradient ultracentrifugation in Renografin-60 (Bracco Diagnostics; Tinkelenberg et al., 2000). 250-μl fractions were diluted 10-fold with Tris EDTA buffer and centrifuged at 100,000 g. Radiolabeled sphingolipids were extracted from the pellet and resolved by TLC as before. Marker proteins for the ER (Dpm1p [dolichol phosphate mannose synthase]), vacuoles (Vph1p [vacuolar H+-ATPase]), and plasma membrane (Pma1p [plasma membrane ATPase]; antisera provided by A. Chang, University of Michigan, Ann Arbor, MI) indicated peak fractions for these organelles.
Online supplemental material
All oligonucleotides were synthesized by Invitrogen and are described in Table S1. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200310046/DC1.
Acknowledgments
The work is dedicated to all NP-C–afflicted families and to the memory of Mrs. A.M.R. Sturley.
We thank Peter Pentchev for support and constructive criticism.
This work was supported by the Ara Parseghian Medical Research Foundation, the Hirschl/Weil-Caulier Trust, and the National Institutes of Health (NIH; grant DK54320). K. Higaki, A.H. Tinkelenberg, and L.J. Wilcox received fellowships from Bristol Myers Squibb-Mead Johnson, NIH (grant DK07715 in nutrition), and the Heart and Stroke Foundation of Canada, respectively.
K. Malathi and K. Higaki contributed equally to this work.
K. Malathi's present address is Lerner Research Institute, Cleveland Clinic Foundation, Cleveland, OH 44195.
K. Higaki's present address is Research Center for Bioscience and Technology, Tottori University, Japan.
Abbreviations used in this paper: ABA, aureobasidin A; DHS, dihydrosphingosine; E–L, endosomal–lysosomal; IPC, inositolphosphorylceramide; LDL, low density lipoprotein; MIPC, mannosyl-IPC; M(IP)2C, mannosyl-di-IPC; NP-C, Niemann Pick type C; NPC1, NP-C gene 1; NCR1, NP-C–related gene 1; SCAP, SREBP cleavage-activating protein; SREBP, sterol regulatory element-binding protein; SSD, sterol-sensing domain.
References
- Altschul, S.F., T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D.J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1998. Saccharomyces cerevisiae. Current Protocols in Molecular Biology. John Wiley & Sons Inc., New York. 13.0.1–13.13.9.
- Bagnat, M., S. Keranen, A. Shevchenko, and K. Simons. 2000. Lipid rafts function in biosynthetic delivery of proteins to the cell surface in yeast. Proc. Natl. Acad. Sci. USA. 97:3254–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom, J.D., M.M. Kurtz, D.J. Rew, A.M. Amend, J.D. Karkas, R.G. Bostedor, V.S. Bansal, C. Dufresne, F.L. VanMiddlesworth, O.D. Hensens, et al. 1993. Zaragozic acids: a family of fungal metabolites that are picomolar competitive inhibitors of squalene synthase. Proc. Natl. Acad. Sci. USA. 90:80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstea, E.D., J.A. Morris, K.G. Coleman, S.K. Loftus, D. Zhang, C. Cummings, J. Gu, M.A. Rosenfeld, W.J. Pavan, D.B. Krizman, et al. 1997. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 277:228–231. [DOI] [PubMed] [Google Scholar]
- Choudhury, A., M. Dominguez, V. Puri, D.K. Sharma, K. Narita, C.L. Wheatley, D.L. Marks, and R.E. Pagano. 2002. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Invest. 109:1541–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, N., C. Mao, J. Heitman, Y.A. Hannun, and L.M. Obeid. 2001. Phytosphingosine as a specific inhibitor of growth and nutrient import in Saccharomyces cerevisiae. J. Biol. Chem. 276:35614–35621. [DOI] [PubMed] [Google Scholar]
- Coxey, R.A., P.G. Pentchev, G. Campbell, and E.J. Blanchette-Mackie. 1993. Differential accumulation of cholesterol in Golgi compartments of normal and Niemann-Pick type C fibroblasts incubated with LDL: a cytochemical freeze-fracture study. J. Lipid Res. 34:1165–1176. [PubMed] [Google Scholar]
- Cruz, J.C., S. Sugii, C. Yu, and T.Y. Chang. 2000. Role of Niemann-Pick type C1 protein in intracellular trafficking of low density lipoprotein-derived cholesterol. J. Biol. Chem. 275:4013–4021. [DOI] [PubMed] [Google Scholar]
- Davies, J.P., F.W. Chen, and Y.A. Ioannou. 2000. Transmembrane molecular pump activity of Niemann-Pick C1 protein. Science. 290:2295–2298. [DOI] [PubMed] [Google Scholar]
- Dickson, R.C., and R.L. Lester. 2002. Sphingolipid functions in Saccharomyces cerevisiae. Biochim. Biophys. Acta. 1583:13–25. [DOI] [PubMed] [Google Scholar]
- Erdeniz, N., U.H. Mortensen, and R. Rothstein. 1997. Cloning-free PCR-based allele replacement methods. Genome Res. 7:1174–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegueur, M., L. Richard, A.D. Charles, and F. Karst. 1991. Isolation and primary structure of the ERG9 gene of Saccharomyces cerevisiae encoding squalene synthetase. Curr. Genet. 20:365–372. [DOI] [PubMed] [Google Scholar]
- Funato, K., B. Vallee, and H. Riezman. 2002. Biosynthesis and trafficking of sphingolipids in the yeast Saccharomyces cerevisiae. Biochemistry. 41:15105–15114. [DOI] [PubMed] [Google Scholar]
- Gaber, R.F., D.M. Copple, B.K. Kennedy, M. Vidal, and M. Bard. 1989. The yeast gene ERG6 is required for normal membrane function but is not essential for biosynthesis of the cell-cycle-sparking sterol. Mol. Cell. Biol. 9:3447–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollub, E.G., P. Trocha, P.K. Liu, and D.B. Sprinson. 1974. Yeast mutants requiring ergosterol as only lipid supplement. Biochem. Biophys. Res. Commun. 56:471–477. [DOI] [PubMed] [Google Scholar]
- Gondre-Lewis, M.C., R. McGlynn, and S.U. Walkley. 2003. Cholesterol accumulation in NPC1-deficient neurons is ganglioside dependent. Curr. Biol. 13:1324–1329. [DOI] [PubMed] [Google Scholar]
- Greer, W.L., M.J. Dobson, G.S. Girouard, D.M. Byers, D.C. Riddell, and P.E. Neumann. 1999. Mutations in NPC1 highlight a conserved NPC1-specific cysteine-rich domain. Am. J. Hum. Genet. 65:1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z., D. Cromley, J.T. Billheimer, and S.L. Sturley. 2001. Identification of potential substrate-binding sites in yeast and human acyl-CoA sterol acyltransferases by mutagenesis of conserved sequences. J. Lipid Res. 42:1282–1291. [PubMed] [Google Scholar]
- Higaki, K., H. Ninomiya, Y. Sugimoto, T. Suzuki, M. Taniguchi, H. Niwa, P.G. Pentchev, M.T. Vanier, and K. Ohno. 2001. Isolation of NPC1-deficient Chinese hamster ovary cell mutants by gene trap mutagenesis. J. Biochem. (Tokyo). 129:875–880. [DOI] [PubMed] [Google Scholar]
- Huh, W.K., J.V. Falvo, L.C. Gerke, A.S. Carroll, R.W. Howson, J.S. Weissman, and E.K. O'Shea. 2003. Global analysis of protein localization in budding yeast. Nature. 425:686–691. [DOI] [PubMed] [Google Scholar]
- Ioannou, Y.A. 2000. The structure and function of the Niemann-Pick C1 protein. Mol. Genet. Metab. 71:175–181. [DOI] [PubMed] [Google Scholar]
- Jenkins, G.M., A. Richards, T. Wahl, C. Mao, L. Obeid, and Y. Hannun. 1997. Involvement of yeast sphingolipids in the heat stress response of Saccharomyces cerevisiae. J. Biol. Chem. 272:32566–32572. [DOI] [PubMed] [Google Scholar]
- Lewis, T.L., G.A. Keesler, G.P. Fenner, and L.W. Parks. 1988. Pleiotropic mutations in Saccharomyces cerevisiae affecting sterol uptake and metabolism. Yeast. 4:93–106. [DOI] [PubMed] [Google Scholar]
- Liu, Y., Y.P. Wu, R. Wada, E.B. Neufeld, K.A. Mullin, A.C. Howard, P.G. Pentchev, M.T. Vanier, K. Suzuki, and R.L. Proia. 2000. Alleviation of neuronal ganglioside storage does not improve the clinical course of the Niemann-Pick C disease mouse. Hum. Mol. Genet. 9:1087–1092. [DOI] [PubMed] [Google Scholar]
- Loftus, S.K., J.A. Morris, E.D. Carstea, J.Z. Gu, C. Cummings, A. Brown, J. Ellison, K. Ohno, M.A. Rosenfeld, D.A. Tagle, et al. 1997. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 277:232–235. [DOI] [PubMed] [Google Scholar]
- Nagiec, M.M., E.E. Nagiec, J.A. Baltisberger, G.B. Wells, R.L. Lester, and R.C. Dickson. 1997. Sphingolipid synthesis as a target for antifungal drugs. Complementation of the inositol phosphorylceramide synthase defect in a mutant strain of Saccharomyces cerevisiae by the AUR1 gene. J. Biol. Chem. 272:9809–9817. [DOI] [PubMed] [Google Scholar]
- Neufeld, E.B., M. Wastney, S. Patel, S. Suresh, A.M. Cooney, N.K. Dwyer, C.F. Roff, K. Ohno, J.A. Morris, E.D. Carstea, et al. 1999. The Niemann-Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J. Biol. Chem. 274:9627–9635. [DOI] [PubMed] [Google Scholar]
- Nickels, J.T., and J.R. Broach. 1996. A ceramide-activated protein phosphatase mediates ceramide-induced G1 arrest of Saccharomyces cerevisiae. Genes Dev. 10:382–394. [DOI] [PubMed] [Google Scholar]
- Nohturfft, A., X. Hua, M.S. Brown, and J.L. Goldstein. 1996. Recurrent G-to-A substitution in a single codon of SREBP cleavage-activating protein causes sterol resistance in three mutant Chinese hamster ovary cell lines. Proc. Natl. Acad. Sci. USA. 93:13709–13714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohturfft, A., M.S. Brown, and J.L. Goldstein. 1998. Topology of SREBP cleavage-activating protein, a polytopic membrane protein with a sterol-sensing domain. J. Biol. Chem. 273:17243–17250. [DOI] [PubMed] [Google Scholar]
- Pagano, R.E., V. Puri, M. Dominguez, and D.L. Marks. 2000. Membrane traffic in sphingolipid storage diseases. Traffic. 1:807–815. [DOI] [PubMed] [Google Scholar]
- Patterson, M.C., A.M. Di Bisceglie, J.J. Higgins, R.B. Abel, R. Schiffmann, C.C. Parker, C.E. Argoff, R.P. Grewal, K. Yu, P.G. Pentchev, et al. 1993. The effect of cholesterol-lowering agents on hepatic and plasma cholesterol in Niemann-Pick disease type C. Neurology. 43:61–64. [DOI] [PubMed] [Google Scholar]
- Pentchev, P.G., E.J. Blanchette-Mackie, and L. Liscum. 1997. Biological implications of the Niemann-Pick C mutation. Subcell. Biochem. 28:437–451. [DOI] [PubMed] [Google Scholar]
- Ribeiro, I., A. Marcao, O. Amaral, M.C. Sa Miranda, M.T. Vanier, and G. Millat. 2001. Niemann-Pick type C disease: NPC1 mutations associated with severe and mild cellular cholesterol trafficking alterations. Hum. Genet. 109:24–32. [DOI] [PubMed] [Google Scholar]
- Schneider, B.L., W. Seufert, B. Steiner, Q.H. Yang, and A.B. Futcher. 1995. Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. Yeast. 11:1265–1274. [DOI] [PubMed] [Google Scholar]
- Seegmiller, A.C., I. Dobrosotskaya, J.L. Goldstein, Y.K. Ho, M.S. Brown, and R.B. Rawson. 2002. The SREBP pathway in Drosophila: regulation by palmitate, not sterols. Dev. Cell. 2:229–238. [DOI] [PubMed] [Google Scholar]
- Simons, K., and R. Ehehalt. 2002. Cholesterol, lipid rafts, and disease. J. Clin. Invest. 110:597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturley, S.L. 2000. Conservation of eukaryotic sterol homeostasis: new insights from studies in budding yeast. Biochim. Biophys. Acta. 1529:155–163. [DOI] [PubMed] [Google Scholar]
- Sugimoto, Y., H. Ninomiya, Y. Ohsaki, K. Higaki, J.P. Davies, Y.A. Ioannou, and K. Ohno. 2001. Accumulation of cholera toxin and GM1 ganglioside in the early endosome of Niemann-Pick C1-deficient cells. Proc. Natl. Acad. Sci. USA. 98:12391–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, X., D.L. Marks, W.D. Park, C.L. Wheatley, V. Puri, J.F. O'Brien, D.L. Kraft, P.A. Lundquist, M.C. Patterson, R.E. Pagano, and K. Snow. 2001. Niemann-Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1. Am. J. Hum. Genet. 68:1361–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, B.J., and R. Rothstein. 1989. Elevated recombination rates in transcriptionally active DNA. Cell. 56:619–630. [DOI] [PubMed] [Google Scholar]
- Tinkelenberg, A.H., Y. Liu, F. Alcantara, S. Khan, Z. Guo, M. Bard, and S.L. Sturley. 2000. Mutations in yeast ARV1 alter intracellular sterol distribution and are complemented by human ARV1. J. Biol. Chem. 275:40667–40670. [DOI] [PubMed] [Google Scholar]
- Vanier, M.T. 1999. Lipid changes in Niemann-Pick disease type C brain: personal experience and review of the literature. Neurochem. Res. 24:481–489. [DOI] [PubMed] [Google Scholar]
- Walter, M., J.P. Davies, and Y.A. Ioannou. 2003. Telomerase immortalization upregulates Rab9 expression and restores LDL cholesterol egress from Niemann-Pick C1 late endosomes. J. Lipid Res. 44:243–253. [DOI] [PubMed] [Google Scholar]
- Watari, H., E.J. Blanchette-Mackie, N.K. Dwyer, J.M. Glick, S. Patel, E.B. Neufeld, R.O. Brady, P.G. Pentchev, and J.F. Strauss III. 1999. Niemann-pick C1 protein: obligatory roles for N-terminal domains and lysosomal targeting in cholesterol mobilization. Proc. Natl. Acad. Sci. USA. 96:805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox, L.J., D.A. Balderes, B. Wharton, A.H. Tinkelenberg, G. Rao, and S.L. Sturley. 2002. Transcriptional profiling identifies two members of the ATP-binding cassette transporter superfamily required for sterol uptake in yeast. J. Biol. Chem. 277:32466–32472. [DOI] [PubMed] [Google Scholar]
- Xie, C., D.K. Burns, S.D. Turley, and J.M. Dietschy. 2000. a. Cholesterol is sequestered in the brains of mice with Niemann-Pick type C disease but turnover is increased. J. Neuropathol. Exp. Neurol. 59:1106–1117. [DOI] [PubMed] [Google Scholar]
- Xie, C., S.D. Turley, and J.M. Dietschy. 2000. b. Centripetal cholesterol flow from the extrahepatic organs through the liver is normal in mice with mutated Niemann-Pick type C protein (NPC1). J. Lipid Res. 41:1278–1289. [PubMed] [Google Scholar]
- Yang, H., M. Bard, D.A. Bruner, A. Gleeson, R.J. Deckelbaum, G. Aljinovic, T.M. Pohl, R. Rothstein, and S.L. Sturley. 1996. Sterol esterification in yeast: a two-gene process. Science. 272:1353–1356. [DOI] [PubMed] [Google Scholar]
- Zervas, M., K. Dobrenis, and S.U. Walkley. 2001. a. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J. Neuropathol. Exp. Neurol. 60:49–64. [DOI] [PubMed] [Google Scholar]
- Zervas, M., K.L. Somers, M.A. Thrall, and S.U. Walkley. 2001. b. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr. Biol. 11:1283–1287. [DOI] [PubMed] [Google Scholar]
- Zhang, M., N.K. Dwyer, E.B. Neufeld, D.C. Love, A. Cooney, M. Comly, S. Patel, H. Watari, J.F. Strauss, III, P.G. Pentchev, et al. 2001. Sterol-modulated glycolipid sorting occurs in Niemann-Pick C1 late endosomes. J. Biol. Chem. 276:3417–3425. [DOI] [PubMed] [Google Scholar]