

Abstract
Elevated coexpression of colony-stimulating factor receptor (CSF-1R) and its ligand, CSF-1, correlates with invasiveness and poor prognosis of a variety of epithelial tumors (Kacinski, B.M. 1995. Ann. Med. 27:79–85). Apart from recruitment of macrophages to the tumor site, the mechanisms by which CSF-1 may potentiate invasion are poorly understood. We show that autocrine CSF-1R activation induces hyperproliferation and a profound, progressive disruption of junctional integrity in acinar structures formed by human mammary epithelial cells in three-dimensional culture. Acini coexpressing receptor and ligand exhibit a dramatic relocalization of E-cadherin from the plasma membrane to punctate intracellular vesicles, accompanied by its loss from the Triton-insoluble fraction. Interfering with Src kinase activity, either by pharmacological inhibition or mutation of the Y561 docking site on CSF-1R, prevents E-cadherin translocation, suggesting that CSF-1R disrupts cell adhesion by uncoupling adherens junction complexes from the cytoskeleton and promoting cadherin internalization through a Src-dependent mechanism. These findings provide a mechanistic basis whereby CSF-1R could contribute to invasive progression in epithelial cancers.
Keywords: CSF-1 receptor; autocrine signaling; Src; cadherins; mammary neoplasms
Introduction
Glandular morphogenesis is directed by a convergence of diverse extracellular signals, including secreted trophic factors, interaction with the ECM, and contact with neighboring cells (Alford and Taylor-Papadimitriou, 1996; Niemann et al., 1998; Schmeichel et al., 1998). One essential class of soluble factors acts as ligands for receptor tyrosine kinases (RTKs), initiating signaling cascades that promote the proliferation and differentiation of epithelial cells and orchestrate the assembly of polarized glandular structures. Appropriate regulation of receptor activity is crucial for the establishment and maintenance of normal epithelial architecture, and aberrant activation of such signals can promote tumor progression.
Several RTKs, including c-Met and EGFR, have been implicated in the growth and morphogenesis of the mammary epithelium, an arborized ductal network composed of hollow tubules that terminate in secretory acinar units (Niemann et al., 1998). Unlike many quiescent adult tissues, the mammary epithelium must retain a broad spectrum of developmental capacities for the duration of the mammalian reproductive lifetime, to facilitate rapid lobuloaveolar outgrowth and lactogenic differentiation during pregnancy. Colony-stimulating factor receptor (CSF-1R) is an RTK required for outgrowth of the mammary ductal network and lactation (Dai et al., 2002). In the absence of CSF-1R or its ligand, CSF-1, expansion of the mammary epithelium into the surrounding fatty stroma is restricted, a defect that has been attributed to the loss of macrophage recruitment to the developing gland (Pollard and Hennighausen, 1994; Van Nguyen and Pollard, 2002). Elevated expression of CSF-1R has been described for a variety of malignancies of epithelial origin, including breast, prostate, and ovarian carcinomas, but the mechanisms by which the receptor contributes to tumor development and progression remain incompletely understood (Kacinski, 1995; Ide et al., 2002). While invasive tumors may rely on cooperation with inflammatory cells, both in vivo and in vitro data support a multifaceted role for CSF-1R in tumorigenesis. In addition to macrophages, CSF-1R is expressed in the mammary epithelium of pregnant and lactating women (Sapi et al., 1998a). Furthermore, elevated epithelial coexpression of CSF-1R and CSF-1 has been described for >50% of mammary tumors, and correlates with invasiveness in both tumors and several mammary carcinoma cell lines (Kacinski et al., 1991).
These data imply that CSF-1 may also act in an autocrine fashion to potentiate tumor cell invasion. In support of this, introduction of CSF-1R in noninvasive HC11 murine mammary epithelial cells, which express CSF-1, enhanced invasion through a Matrigel™ barrier by 100-fold (Sapi et al., 1996). In contrast, a complete inhibition of the invasive activity of BT20 breast tumor cells, which express high levels of both receptor and ligand, was achieved by expression of a dominant-negative mutant of Ets-2, a transcription factor that acts as a key mediator of the cellular response to CSF-1 (Langer et al., 1992; Sapi et al., 1998b). However, such studies do not assess the impact of autocrine CSF-1R signaling in the context of intact glandular structures, and little is known about the critical downstream signals that may be involved in potentiating invasive behavior.
While the essential role for CSF-1 in mammary development was revealed through mouse knockout studies, detection of the receptor in the murine gland has thus far been restricted to infiltrating macrophages. Interestingly, epithelial expression of the CSF-1R transcript in human mammary epithelium is hormonally regulated via a glucocorticoid response element that is absent from the murine locus (Flick et al., 2002). Thus, while mouse models have contributed much to an understanding of the role that macrophages, recruited to the mammary gland by CSF-1 secretion, can play in tumor progression, other model systems may be needed to reveal the mechanisms by which autocrine signaling can contribute to invasion in human tumors. Therefore, we have examined the consequences of CSF-1R signaling on epithelial cell behavior in the nontransformed human mammary cell line MCF-10A. To enable a careful dissection of specific alterations in epithelial physiology induced by CSF-1R, we have employed a three-dimensional (3D) basement membrane culture system in which MCF-10A cells assemble into spherical structures that recapitulate many features of lobular acini in vivo (Barcellos-Hoff et al., 1989). This system is tractable to biochemical and cell biological analysis, and has been used previously by our laboratory to analyze the biological activities of various oncogenes (Muthuswamy et al., 2001; Debnath et al., 2002).
Here we demonstrate that constitutive CSF-1R activation results in a striking and progressive disruption of cell–cell adhesion in MCF-10A acini. In cells expressing either a constitutively active form of the receptor, or coexpressing wild-type receptor and CSF-1 ligand, initial acinar development proceeds normally. However, in either context of CSF-1R hyperstimulation, structures fail to form lumen, become hyperproliferative, and progressively lose junctional stability and basement membrane integrity, culminating in the release of individual cells into the surrounding matrix. While chronic CSF-1R activation does not alter steady-state levels or synthesis of the major epithelial cell adhesion molecule E-cadherin, immunofluorescence studies indicate that E-cadherin is lost from the plasma membrane and adopts a punctate localization. In addition, interaction of adherens junction complexes with the Triton-insoluble cytoskeleton is disrupted, impeding the ability of MCF-10A cells to achieve adhesive compaction. Interfering with the activity of Src family kinases, either by pharmacological inhibition or by mutation of the Y561 docking site on CSF-1R, suppresses the adhesion phenotype. These results suggest that CSF-1R activation, possibly by an autocrine mechanism, can profoundly disrupt the junctional integrity and architecture of mammary epithelial acini in vitro via activation of a Src-dependent pathway. These consequences distinguish hyperstimulation of CSF-1R from other RTKs examined in this model, and provide insight into the mechanisms whereby CSF-1 and its receptor could potentiate invasive activity in vivo.
Results
CSF-1R activation supports EGF-independent proliferation and acinar development
The proliferation and 3D morphogenesis of MCF-10A cells is dependent on the inclusion of EGF in the growth medium. We initially sought to determine whether activation of CSF-1R could promote proliferation and acinar development in the absence of EGF. Significant expression of CSF-1R in the mammary gland is observed only during late pregnancy and lactation; MCF-10As were derived from the mammary tissue of a nonpregnant female, and do not express sufficient levels of CSF-1R to elicit a mitogenic response to CSF-1. Therefore, to achieve higher receptor levels, we infected these cells with a retroviral vector encoding wild-type CSF-1R. Receptor expression and CSF-1–dependent activation were confirmed by immunoblotting (Fig. 1 A). Cells expressing wild-type CSF-1R cultured as monolayers in the presence of physiological levels of CSF-1 (10 ng/ml) proliferated at a similar rate as cells cultured in EGF and maintained a cobblestone phenotype (Fig. 1 B). In addition, treatment of these cells with CSF-1 promoted the formation of acini indistinguishable from those induced by EGF in 3D Matrigel™ culture (Fig. 1 C) (Debnath et al., 2002).
Figure 1.
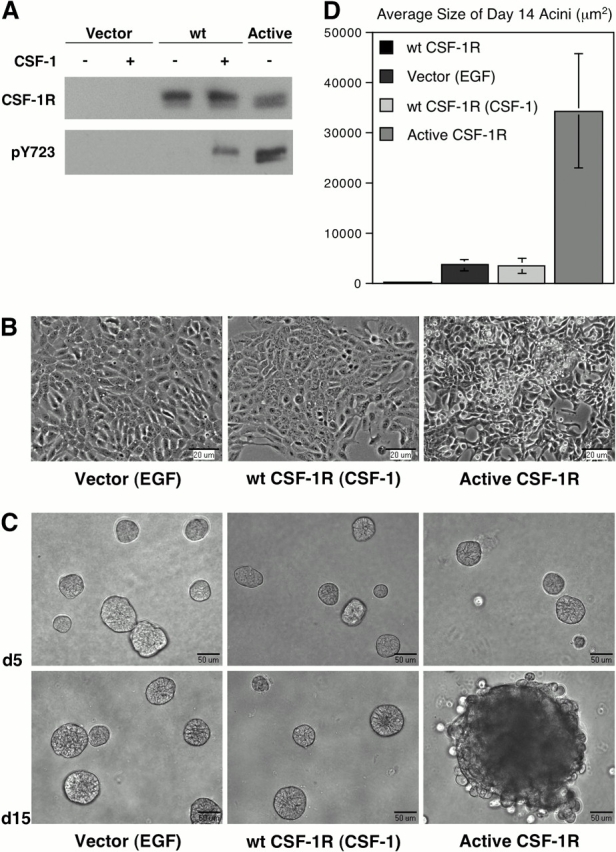
Constitutive CSF-1R activation alters MCF-10As monolayer morphology and acinar morphogenesis. (A) MCF-10A cells expressing wild-type or active CSF-1R were lysed in RIPA buffer and probed for receptor levels. Receptor activation was detected with a phospho-specific antibody to the Y723 autophosphorylation site. Cells were starved overnight in assay media, and where noted were treated with 10 ng/ml CSF-1 for 5 min before lysis. (B) Phase-contrast images of MCF-10A cells expressing wild-type or active CSF-1R cultured to confluence in assay media with growth factors noted in parentheses. (C) MCF-10A cells expressing wild-type or active CSF-1R were cultured in MatrigelTM, and assay media with growth factors was replaced every 4 d. Brightfield images of day 5 and day 15 acini are shown. (D) The average size of MCF-10A acini was determined by microscopic area measurements using MetaMorph software (n = 30).
Constitutive activation of CSF-1R disrupts MCF-10A acinar architecture
Previous studies have shown that constitutively active CSF-1R has potent transforming activity in fibroblasts (Roussel et al., 1987). To investigate whether deregulated CSF-1R signaling could similarly perturb the behavior of MCF-10As, we expressed an active variant of the receptor bearing two point mutations essential for the transforming capacity of the viral oncogene (v-fms). The mutation L301S in the extracellular domain permits ligand-independent receptor activation, and Y969F relieves an autoinhibitory regulatory mechanism that may involve down-regulation of the receptor via recruitment of Cbl (Roussel et al., 1988; Wilhelmsen et al., 2002). Monolayers of MCF-10As expressing active CSF-1R proliferated in the absence of ligand, adopted a more refractile appearance, and displayed reduced contact inhibition (Fig. 1 B). In 3D culture, the cells initiated morphogenesis in the absence of exogenous growth factors and initially formed spherical clusters with well-defined borders. However, as morphogenesis progressed, these structures exhibited enhanced proliferation and significantly increased size relative to wild-type controls (Fig. 1 D). Furthermore, after ∼8–9 d in culture, acini exhibited a progressive loss of structural integrity. The structures maintained a high proliferative rate, became visibly discohesive, and individual cells dissociated from the acini (Fig. 1 B). This phenotype is distinct from that induced by activation of ErbB2, which promotes the formation of filled, multiacinar structures but does not impact the integrity of the outer cell layer (Muthuswamy et al., 2001).
Autocrine or chronic stimulation of CSF-1R mimics the effects of constitutive activity on acinar integrity
While the presence of activating CSF-1R mutations in epithelial tumors has not been confirmed, coexpression of CSF-1R and CSF-1 is frequently observed in the epithelium of invasive mammary carcinomas (Kacinski et al., 1991). This suggests that autocrine CSF-1R activation may be sufficient to promote tumor progression. To determine whether autocrine signaling could disrupt acinar structure, we generated MCF-10As coexpressing CSF-1 and wild-type CSF-1R. Ligand expression was confirmed by ELISA (unpublished data). Monolayers of cells expressing both ligand and receptor exhibited a similar phenotype to those expressing constitutively active receptor (unpublished data). Furthermore, in 3D culture, these cells initiated morphogenesis normally, but were hyperproliferative; as a result, acini expanded greatly in size and failed to undergo growth arrest. Similar to those expressing active receptor, these acini also exhibited a progressive architectural disruption after 8–9 d, such that the integrity of the luminal cell layer was compromised and single cells were released into the matrix (Fig. 2 A; Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200309102/DC1). These results indicate that activation of CSF-1R in an autocrine fashion can have profound effects on in vitro acinar morphogenesis.
Figure 2.

Autocrine or chronic activation can mimic the active CSF-1R phenotype. (A) MCF-10As expressing CSF-1R and CSF-1 were cultured in MatrigelTM in assay media. Brightfield images of day 5 and day 19 acini are shown. (B) 3D cultures of cells expressing CSF-1R were stimulated daily with 10 ng/ml CSF-1. Phase-contrast images of day 5 and day 13 acini are shown.
The development of discohesive structures upon CSF-1R hyperstimulation suggested that constitutive receptor signaling might impact molecules involved in cell–cell adhesion. Consequently, we analyzed the localization of the adherens junction proteins during morphogenesis. Acini formed in response to exogenous CSF-1 exhibited well-defined cell–cell junctions, illustrated by membrane localization of E-cadherin, concentrated toward the basal pole of the lateral membrane (Fig. 3 A). Furthermore, acini formed lumen and displayed aspects of polarity, basally deposited the ECM protein laminin 5, and underwent proliferative arrest over a similar time course as with acini grown in the presence of EGF (Fig. 3, A and B) (Debnath et al., 2002).
Figure 3.

CSF-1 coexpression causes hyperproliferation and architectural disruption. MCF-10A cells expressing CSF-1R and vector control (CSF-1R/Vector) or CSF-1 (CSF-1R/CSF-1) were cultured in MatrigelTM in assay media with 10 ng/ml CSF-1 where noted (CSF-1). Confocal images of day 6 and day 17 acinar cultures are shown. Structures were stained with DAPI (blue) along with Ki67 (green) and E-cadherin (red) (A and C) or cleaved caspase-3 (green) and laminin 5 (red) (B and D).
Although E-cadherin initially exhibited a similar membrane localization in acini coexpressing CSF-1R and CSF-1, subsequently, it became diffusely localized, and immunostaining was diminished (Fig. 3 C). Loss of membrane staining correlated with the degree of architectural disruption. Concomitant with the relocalization of junctional components, we observed disruption of the integrity of the basal basement membrane and disordered deposition of laminin 5 in these structures (Fig. 3 D). Furthermore, the expansion in size of the autocrine structures correlated with significantly increased proliferation (Fig. 3 C). While activation of ErbB2 was shown to have similar effects on cell proliferation and lumen formation, it had no such impact on the distribution of cell adhesion molecules (Muthuswamy et al., 2001), suggesting that this may represent a distinct capacity of autocrine CSF-1R signaling.
These effects could reflect a unique capability of autocrine engagement of CSF-1R, such as activation of an internal pool of newly synthesized molecules during trafficking to the plasma membrane. Alternatively, the disruptive effects of ligand coexpression could simply be a consequence of chronic receptor stimulation. We addressed the latter possibility by modifying the feeding schedule of 3D cultures, which typically involves replenishment with fresh medium every 4 d. When cultures were instead supplemented with CSF-1 daily, structural disruption was perceptible by day 13 (Fig. 2 B). In contrast, daily treatment of parental MCF-10As with EGF did not elicit a discohesive phenotype (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200309102/DC1). This suggests that while intracellular activation of CSF-1R may occur in the autocrine context, it is not necessary to produce the observed phenotype. Rather, chronic exposure to ligand is sufficient to promote disruption of acinar architecture, whether provided exogenously or in an autocrine fashion.
Autocrine CSF-1R activation alters the motility of MCF-10A monolayers
Perturbation of cadherin-mediated adhesion has been shown to influence the movement of epithelial monolayers (Owens et al., 2000). To determine whether CSF-1R activation altered the motility of MCF-10A cells, we assessed the dynamics of wound healing in response to exogenous or coexpressed CSF-1. Assays were performed in the presence of mitomycin C to eliminate the contribution of proliferation to wound closure. Receptor-expressing cells treated with 10 ng/ml CSF-1 began to migrate into the wound space as an intact sheet from ∼6–8 h after wounding, and required a period of >48 h to completely seal the wound. In contrast, cells coexpressing CSF-1 rapidly broke away from the wound margin and migrated into the wound space as individual cells within 4 h after wounding. Complete closure was achieved in <24 h (Fig. 4).
Figure 4.

Autocrine CSF-1R activation alters wound healing dynamics in MCF-10As. Confluent monolayers of MCF-10As expressing CSF-1R and vector control (CSF-1R/Vector) or CSF-1 (CSF-1R/CSF-1) were starved overnight in assay media. A wound was introduced into each monolayer with a pipette tip, and cells were incubated in assay media with 10 ng/ml CSF-1 where noted (CSF-1) for 48 h. Phase-contrast images of wound clo-sure are representative of four separate experiments.
CSF-1R promotes cadherin internalization and dissociation from the actin cytoskeleton
We then examined whether the altered cell–cell adhesion and increased motility mediated by autocrine CSF-1R activation involved modulation of junctional complexes. Assessment of E-cadherin localization at the wound margin revealed that while discrete lateral staining was maintained in cells expressing CSF-1R alone, it accumulated in intracellular vesicles in cells coexpressing receptor and ligand (Fig. 5 A). Similarly punctate E-cadherin staining was detected in discohesive acini viewed at high magnification (Fig. 5 B). Although internalization of E-cadherin may promote its degradation, we did not observe a reduction in total E-cadherin in lysates from cells coexpressing CSF-1R and CSF-1 (Fig. 5 C), and no degradation products were detected (Fujita et al., 2002). In addition, both cell types exhibited a similar recovery of E-cadherin after trypsin treatment, suggesting that the adhesive defect is not a consequence of a decline in cadherin synthesis (Fig. 5 D). Rather the disappearance of E-cadherin from the membrane may be due to an increased rate of endocytosis or a defect in the recycling of internalized molecules (Le et al., 1999).
Figure 5.

Coexpression of CSF-1 with CSF-1R elicits relocalization of E-cadherin from the membrane to intracellular vesicles. (A) MCF-10As expressing CSF-1R and vector control (CSF-1R/Vector) or CSF-1 (CSF-1R/CSF-1) were fixed for E-cadherin immunofluorescence 8 h after wounding. (B) Confocal images of day 21 acini expressing wild-type or active CSF-1R, stained with DAPI (blue) and E-cadherin (green). (C) Total levels of E-cadherin from RIPA lysates of MCF-10As expressing CSF-1R and vector control (Vector) or CSF-1 (CSF-1). (D) MCF-10As expressing CSF-1R and vector control (lanes 1, 3, 5, and 7) or CSF-1 (lanes 2, 4, 6, and 8) were harvested by treatment with EDTA (1–2 and 5–6) or trypsin (3–4 and 7–8) and placed in suspension for 6 h before lysis in RIPA buffer and E-cadherin immunoblotting.
The finding that E-cadherin levels are not altered by autocrine CSF-1R activation raised the question as to which stage in the formation and maturation of cell junctions is affected. Adhesion is initiated by homophilic cadherin contact, which then consecutively triggers catenin binding, cadherin clustering, and recruitment of the actin cytoskeleton. Cytoskeletal association corresponds with a progression from relatively weak adhesion to the generation of strong adhesive forces and initiation of cell–cell compaction (Adams et al., 1998). This transition can be observed when epithelial cells are allowed to cluster in suspension. Initially, cells form loose chains in which the membranes of contacting cells remain distinct. After several hours, compaction occurs between adjacent cells such that individual cell borders are no longer discernible. Compaction of MCF-10As expressing wild-type CSF-1R was detected within 4 h in suspension in the presence of CSF-1, and by 8 h, few loosely associated clusters remained. In contrast, cells coexpressing CSF-1R and CSF-1 formed weak associations in suspension, but displayed limited progression to the compaction stage (Fig. 6). This suggests that these cells retain adequate transmembrane cadherin to initiate adhesive contacts; however, surface levels may nonetheless be insufficient to initiate clustering and promote cytoskeletal linkage. This contrasts with the effect of Ca2+ sequestration, which prevents productive homophilic cadherin interaction; MCF-10As placed in suspension in the presence of EGTA remained as single cells (unpublished data).
Figure 6.

Autocrine CSF-1R activation prevents adhesive compaction. MCF-10As expressing CSF-1R and vector control (CSF-1R/Vector) or CSF-1 (CSF-1R/CSF-1) were placed in suspension in assay media with 10 ng/ml CSF-1 where noted (CSF-1) for 6 h before harvest. Cells were resuspended in 0.5% agarose and spotted onto glass slides for DIC imaging.
Since autocrine CSF-1R activation does not result in cadherin loss, we evaluated whether it could compromise cell adhesion by modulating the composition of adherens junction complexes. Various members of the catenin family associate directly or indirectly with E-cadherin, and impact its adhesive capacity and association with the cytoskeleton (Herrenknecht et al., 1991; McCrea et al., 1991; Reynolds et al., 1994). However, autocrine cells did not exhibit changes in total levels of β-catenin, p120 catenin, or α-catenin (Fig. 7 A). Furthermore, when E-cadherin was immunoprecipitated from these cells, the levels of associated catenins were likewise unaltered (Fig. 7 B). This is consistent with previous data showing that adherens junction plaques are internalized as intact complexes (Boterberg et al., 2000). Following complex formation, interaction of α-catenin with the actin cytoskeleton is required for stabilization of adhesion and cellular compaction (Adams and Nelson, 1998). To determine if the proportion of E-cadherin associated with the cytoskeletal fraction was reduced in autocrine CSF-1R cells, solubility in the presence of a nonionic detergent was assessed. A significant decrease in E-cadherin in Triton-insoluble lysate fractions was observed in cells coexpressing CSF-1 (Fig. 7 C). This suggests that chronic CSF-1R signaling is capable of thwarting the progression from initial cell contact to strong adhesion, and provides a potential explanation for its ability to disrupt 3D architecture.
Figure 7.

Assembly of the cadherin–catenin complex is unaltered in CSF-1 coexpressing cells, but association with the cytoskeleton is disrupted. Parental MCF-10As and cells expressing CSF-1R and vector control (CSF-1R/Vector) or CSF-1 (CSF-1R/CSF-1) were grown to confluence in assay media before lysis. Media was supplemented with 5 ng/ml EGF for parental cells and 10 ng/ml CSF-1 for CSF-1R/Vector cells. (A) RIPA lysates were probed for total levels of E-cadherin and catenin proteins. (B) E-cadherin immunoprecipitates from Triton lysates were probed for associated catenins. (C) Levels of E-cadherin in the Triton-soluble (TS) and Triton-insoluble (TI) lysate fractions were assayed. Actin is shown as a control for equal protein loading.
Src kinases are required for CSF-1R disruption of cell–cell adhesion
Stimulation of CSF-1R leads to the activation of multiple signaling effectors, including PI3K and Src, which have been implicated in the regulation of adhesion (Warren and Nelson, 1987; Roussel, 1998; Conacci-Sorrell et al., 2002). To address which CSF-1R signaling pathways were responsible for the observed architectural disruption, we generated mutant variants of the constitutively active CSF-1R that encoded single tyrosine-to-phenylalanine substitutions at various autophosphorylation sites on the receptor (Y561, Y699, Y708, Y723, and Y923). MCF-10As infected with each mutant receptor were examined in 3D culture for their ability to recapitulate the discohesive phenotype. While the majority of these substitutions were insufficient to prevent perturbation of adhesion, mutation of Y561 significantly attenuated junctional disruption without impeding proliferation (Fig. 8 A). Mutation of the corresponding residue in the murine receptor substantially reduced binding and activation of Src upon receptor activation, and phosphopeptides spanning this region of the human receptor were shown to bind Src in vitro (Alonso et al., 1995; Marks et al., 1999). In agreement with these findings, we observed a decreased association of Src with the Y561F mutant receptor (Fig. 8 B). In addition, using an antibody specific for the activation loop phosphorylation site (Y419 in human Src), we observed that at least two Src family kinases are activated by CSF-1R: Src (Mr60K) and Lyn (Mr53 and 56K). Phosphorylation of both proteins was reduced to basal levels in cells expressing the Y561F mutant receptor (Fig. 8 C), while autophosphorylation of CSF-1R was unaffected (not depicted). This suggested that the ability of constitutive CSF-1R signaling to perturb cell–cell adhesion might be dependent on Src family kinases.
Figure 8.
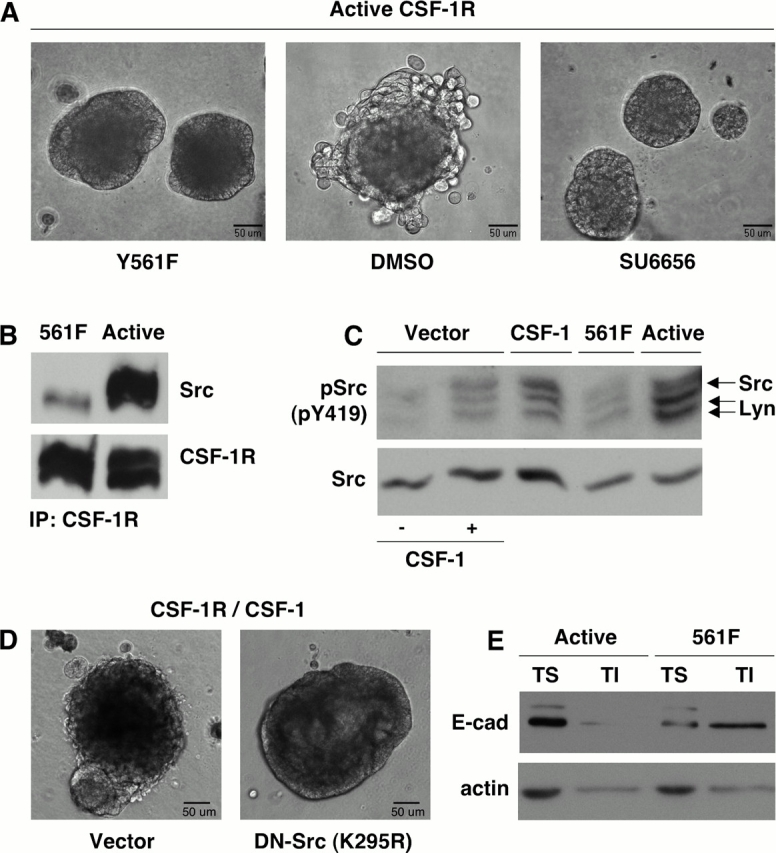
The ability of CSF-1R to promote junctional disruption is Src dependent. (A) MCF-10A cells expressing active CSF-1R (right and center), or active CSF-1R including a point mutation at the Src binding site (Y561F, left), were cultured in MatrigelTM. Cultures were treated every 48 h with Src inhibitor SU6656 (2 μm) where noted. Brightfield images of day 15 acini are shown. (B) CSF-1R immunoprecipitates from MCF-10As expressing active CSF-1R or active CSF-1R with the Y561F mutation were probed for Src association with the receptor. (C) MCF-10As expressing CSF-1R and vector control (CSF-1R/Vector) or CSF-1 (CSF-1R/CSF-1), active CSF-1R, or active CSF-1R with the Y561F mutation were starved overnight in assay media. Cells were treated with 10 ng/ml CSF-1 before lysis where noted. Lysates were probed for Src tyrosine 419 phosphorylation and total Src levels. (D) MCF-10As coexpressing CSF-1R and CSF-1 along with vector control or DN-Src (K295R) were cultured in MatrigelTM. Phase-contrast images of day 16 acini are shown. (E) Levels of E-cadherin in the Triton-soluble (TS) and Triton-insoluble (TI) lysate fractions of MCF-10As expressing active CSF-1R or active CSF-1R with the Y561F mutation were assayed. Actin is shown as a control for equal protein loading.
To determine whether Src catalytic activity is necessary for this perturbation, 3D cultures of cells expressing active CSF-1R were treated from day 6 with SU6656, which selectively inhibits Src at concentrations under 5 μM and does not impact CSF-1R kinase activity (Blake et al., 2000). While structures treated with vehicle exhibited a progressive disruption of cell adhesion, periodic treatment with the Src inhibitor preserved acinar integrity (Fig. 8 A). Furthermore, overexpression of a kinase inactive, dominant-interfering variant of Src (K295R) in MCF-10As coexpressing CSF-1R and CSF-1 also prevented junctional disruption in acinar cultures (Fig. 8 D). Finally, levels of E-cadherin in the Triton-insoluble fraction were restored in Y561F cells (Fig. 8 E). Collectively, these data suggest that Src activity is essential for the destabilization of cell adhesion induced by chronic CSF-1R signaling.
Constitutive c-Met stimulation does not persistently disrupt adhesion despite comparable levels of Src activation
Several RTKs have been implicated in regulation of adhesive contacts through mechanisms that involve Src activity (Conacci-Sorrell et al., 2002). Activation of c-Met, which can alter the composition of adherens junction complexes, induces the extension of hollow tubules from MDCK cysts (Pollack et al., 1998). To determine whether the discohesive phenotype elicited by constitutive CSF-1R activation in MCF-10A acini was a consequence of chronic activation of other RTKs, we assessed the morphology of acinar structures formed by MCF-10As overexpressing c-Met alone or in conjunction with HGF. When cells expressing c-Met were grown in 3D culture in the presence of exogenous HGF (40 ng/ml), a small proportion of structures developed multiple branching tubules (Fig. 9 A). Both the frequency and extent of tubule formation were increased by HGF coexpression; however, no additional alterations in acinar architecture were observed. Notably, despite chronic activation of c-Met, membrane localization of E-cadherin remained intact (Fig. 9 B). Thus the discohesive phenotype elicited by CSF-1R activation is a relatively distinct feature of this receptor. To determine whether this distinction correlated with differing capacities of c-Met and CSF-1R to activate Src family kinases, we assessed the level of Src activation in response to acute or autocrine activation of each receptor. Acute stimulation of c-Met or CSF-1R resulted in equivalent levels of Src phosphorylation; upon chronic stimulation of either receptor, levels of Src activation were again comparable (Fig. 9 C).
Figure 9.

Autocrine stimulation of c-Met does not persistently disrupt acinar adhesion despite comparable activation of Src. (A) MCF-10A cells expressing c-Met and vector control (c-Met/Vector) or HGF (c-Met/HGF) were cultured in MatrigelTM in assay media with 40 ng/ml HGF where noted (HGF). Brightfield images of day 5 and day 17 acini are shown. (B) 3D cultures of MCF-10As expressing c-Met and HGF were stained with DAPI (blue) and E-cadherin (green). Confocal image of day 19 structure is shown. (C) MCF-10As expressing CSF-1R and vector control or CSF-1, or c-Met and vector control or HGF were starved overnight in assay media. Cells were treated with 10 ng/ml CSF-1 or 40 ng/ml HGF before lysis where noted. Lysates were probed for Src tyrosine 419 phosphorylation and total Src levels.
Discussion
Despite significant evidence supporting a role for CSF-1R in epithelial tumorigenesis, little is known about the mechanisms by which autocrine CSF-1 activation may contribute to tumor progression. We have used the MCF-10A mammary epithelial cell line to assess the effects of ligand and receptor coexpression on epithelial motility and morphogenesis. We have found that constitutive CSF-1R signaling causes uncontrolled proliferation, allowing these cells to subvert growth-suppressive mechanisms typically operative in monolayer and 3D cultures. Furthermore, we observed a profound perturbation of cell–cell adhesion, resulting in a striking disintegration of 3D acinar structures and likely contributing to enhanced motility in MCF-10A monolayers.
Using immunolocalization and fractionation studies, we have demonstrated that autocrine CSF-1R signaling can dramatically disrupt adherens junction–mediated adhesion. The formation of adherens junctions, directed by homophilic interactions between the extracellular domains of E-cadherin molecules on neighboring cells, is essential for epithelial cell–cell adhesion; other adhesive interactions are limited in their absence. The cytoplasmic tail of E-cadherin interacts with β-catenin, which strengthens adhesive contacts by tethering E-cadherin to the actin cytoskeleton via interaction with α-catenin (Herrenknecht et al., 1991; McCrea et al., 1991). Loss of cadherin function has been associated with migratory behavior in vitro, and is a common hallmark of invasive carcinoma in vivo (Kowalski et al., 2003). While cellular remodeling involved in the formation of glandular structures requires transient disruption of adhesive contacts, the preservation of tissue architecture relies on stable intercellular interaction. Tubule formation in response to the activation of c-Met in MDCK cysts involves local redistribution of cadherin complexes without dissolution of cell–cell contacts (Pollack et al., 1998). In contrast to this discrete morphogenetic response to an appropriate growth factor signal, we have found that autocrine CSF-1R activation can dramatically and persistently disrupt cell–cell adhesion in mammary epithelial structures in vitro. The resulting disintegration of acinar integrity culminates in the release of individual cells into the surrounding matrix.
Cell–cell adhesion functions not only to maintain the physical integrity of the epithelial cell layer as a barrier between glandular and stromal compartments, but also contributes to the maintenance of cell polarity and suppression of proliferation (Watabe et al., 1994). Loss of membrane E-cadherin in MCF-10A acini consistently correlated with increased Ki67 staining, suggesting that compromised cell adhesion may influence the uncontrolled proliferation observed. Furthermore, perturbations of cell–cell contact have been shown to promote migratory behavior, and appear to play a role in tumor progression (Nollet et al., 1999). In agreement with this, autocrine CSF-1R also accelerated MCF-10A wound closure, not only increasing the rate of cell motility, but also disrupting the sheet-like character of epithelial cell movement. Enhanced motility likewise correlated with reduced membrane E-cadherin staining. Despite the capacity of CSF-1 to promote migration in this context, it was unable to elicit invasive behavior through intact basement membrane, as individual cells did not actively disperse from disrupted acini. This suggests that cooperation with additional factors may be required for CSF-1R activation to promote invasion within the tumor microenvironment.
The disruption of adhesion resulting from autocrine CSF-1R activation correlates with the translocation of E-cadherin to intracellular vesicles and loss of Triton insolubility rather than a decrease in total E-cadherin. These findings are consistent with the possibility that the adhesive defect results from dissociation of junctional complexes from the actin cytoskeleton and their concomitant internalization. Recent data suggests that multiple regulators of cytoskeletal dynamics impinge upon the assembly and maturation of adherens junctions, and this interface may play as important a role in regulation of adhesion as modulation of the composition of junctional complexes (Braga et al., 1997). Association of adherens junction complexes with the cytoskeleton might further act as an important determinant in controlling the rate of cadherin endocytosis and recycling.
The effects of autocrine CSF-1R signaling on cell–cell adhesion are exquisitely dependent upon the sustained activation of Src kinases. Interfering with Src activity by pharmacological inhibition or expression of a dominant-inhibitory Src mutant, or by mutation of the Src binding site on the receptor, resulted in a complete preservation of acinar integrity. Furthermore, Src inhibition restored the ability of cells in suspension to undergo adhesive compaction, as well as the controlled, sheet-like migration of cells in a wounded monolayer (unpublished data). Src can phosphorylate β-catenin in vitro, and v-Src has been shown to destabilize the association of β-catenin with E-cadherin, an effect that correlated with phosphorylation of both proteins (Behrens et al., 1993; Roura et al., 1999). In addition, p120 catenin was initially identified as a Src substrate and was shown to be phosphorylated after RTK activation (Reynolds et al., 1989; Downing and Reynolds, 1991). However, v-Src retained the ability to disrupt cadherin cytoskeletal association in cells expressing an E-cadherin/α-catenin fusion protein, excluding β-catenin as the relevant target and suggesting that Src activity may also influence adherens junction stability indirectly (Takeda et al., 1995). Src-mediated junctional disruption in MCF-10As is likely to occur via such an indirect mechanism, as we were unable to detect catenin phosphorylation unless cells were treated with phosphatase inhibitors before lysis, and levels were not enhanced by CSF-1R activation (unpublished data).
Candidate Src targets include proteins that are localized to cell junctions and influence but do not directly participate in the adherens junction complex, such as ZO-1 or ezrin (Pujuguet et al., 2003). Alternatively, Src could exert its effects by targeting a regulator of cadherin endocytosis, which would account for the punctate accumulation of E-cadherin observed upon autocrine CSF-1R activation. PKCδ has been shown to stabilize adhesion by limiting cadherin endocytosis, and the ability of Src to mediate its degradation could play a role (Blake et al., 1999; Le et al., 2002). Moreover, the GTPase ARF6 has been shown to act downstream of v-Src to promote cadherin endocytosis, and CSF-1 stimulation of macrophages induces its translocation to the plasma membrane (Zhang et al., 1999; Palacios et al., 2001). The inability of c-Met to elicit a similar disruption of acinar architecture, despite its capacity to induce comparable levels of Src activity, suggests that additional factors are involved in CSF-1R–mediated perturbation of adhesion. Accessory pathways activated by the receptor may be required to collaborate with Src; alternatively, CSF-1R may be uniquely poised to colocalize active Src and its relevant target(s). Studies to address this distinction and the aforementioned potential targets are currently underway.
Coexpression of CSF-1R and CSF-1 is most often detected in invasive tumors, and elevated serum levels of CSF-1 correlate with metastatic progression and poor prognosis (Kacinski et al., 1991; Price et al., 1993). The ability of CSF-1R to disrupt cell–cell adhesion, coupled with its capacity to promote cell motility and proliferation, corroborates that constitutive receptor signaling can coordinately promote multiple biological activities associated with tumor progression and invasion. Furthermore, our findings, in conjunction with those of Ide et al. (2002), suggest that inhibition of epithelial CSF-1R signaling may prove to be a valuable strategy in the treatment of invasive tumors. This may be of particular relevance to malignancies that exhibit a discohesive phenotype. Although often attributable to loss or silencing of the CDH1 locus, a loss of membrane E-cadherin in tumors has also been observed in the absence of a genetic lesion, suggesting that in vivo perturbations of cell adhesion may also involve posttranslational mechanisms (Kowalski et al., 2003).
Materials and methods
Cell culture and reagents
MCF-10A cells were obtained from American Type Culture Collection and cultured in DME/F12 (Invitrogen) supplemented with 5% donor horse serum, 20 ng/ml EGF, 10 μg/ml insulin, 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, 50 U/ml penicillin, and 50 mg/ml streptomycin. Growth factor–reduced MatrigelTM (BD Biosciences) with protein concentrations between 9 and 11 mg/ml was used. Antibodies to CSF-1R and c-Met were obtained from Santa Cruz Biotechnology, Inc.; laminin 5 from Chemicon; E-cadherin, β-catenin, α-catenin, and p120ctn from BD Transduction Labs; Ki-67 from Zymed Laboratories; phospho-Src(Y416) from New England Biolabs, Inc.; and cleaved caspase-3 and phospho-CSF-1R(Y723) from Cell Signaling Technologies. Src Mo327 has been previously described (Lipsich et al., 1983). Src inhibitor SU6656 (Sugen, Inc.) was used at 2 μM.
Retroviral vectors
pBabe-puro CSF-1 was constructed using an EcoRI fragment of pSMX human CSF-1 4.0 (Roussel et al., 1987). pMSCV human CSF-1R and CSF-1R L301S/Y969F were provided by O. Witte (University of California, Los Angeles, CA). The Y561F mutant receptor was generated by QuickChange (Stratagene) site-directed mutagenesis using the following primers: forward, 5′-gagggcaacagttTtactttcatcgaccc-3′; reverse, 5′-gggtcgatgaaagtaAaactgttgccctc-3′. pBabe-puro c-Met was constructed from pSP64 c-Met (Iyer et al., 1990). pBabe-puro HGF was obtained from the Harvard Institute of Proteomics FLEX library. pLNCX DN-Src(K295R) has been previously described (Mukhopadhyay et al., 1995). Ecotropic retroviruses were produced by transfection of the PhoenixTM ECO packaging line (Swift et al., 1999) with 15 μg of DNA using Lipofectamine 2000TM (Invitrogen). Viral supernatants were collected on day 4 after transfection and passed through a 0.45-μm filter before storage at −80°C.
Generation of MCF-10A cell lines
MCF-10A cells expressing the ecotropic receptor (derived by infection with pWZLneo mEcoR retrovirus, obtained from G. Daley, Harvard Medical School, Boston, MA) were plated at 4 × 105 cells per 10-cm dish and infected with the above retroviruses. Stable populations were obtained by selection with 2 μg/ml puromycin or 200 μg/ml G418 (Sigma-Aldrich). CSF-1R, c-Met, and Src overexpression were confirmed by immunoblotting. Conditioned media from cells expressing CSF-1 was assayed for ligand activity using the Quantikine CSF-1 ELISA Kit (R&D Systems).
Immunoprecipitation and immunoblotting
Monolayers were washed once in PBS and lysed in 1 ml Triton lysis buffer (0.5% Triton X-100, 25 mM Tris pH 7.5, 150 mM NaCl, 2 mM CaCl2) or RIPA buffer (1% Triton X-100, 1% NaDOC, 0.1% SDS, 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA) supplemented with protease and phosphatase inhibitors (1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 10 μg/ml PMSF, 1 mM NaVO4, 1 mM NaF) for 15 or 30 min at 4°C, respectively. Lysates were cleared 15 min at 13,000 rpm and supernatants were collected. Triton-insoluble (TI) pellets were resuspended in 2× SDS sample buffer.
For immunoprecipitations, 500 μg total protein in 500 μl total volume was incubated with 1 μg primary antibody for 1 h at 4°C with rocking. 40 μl of a protein G Sepharose slurry (Amersham Biosciences) was added for an additional 40 min. Immune complexes were washed four times in lysis buffer before elution in 60 μl 2× SDS sample buffer. Immunoprecipitates or cell lysates were resolved by 8–10% SDS-PAGE and immunoblotted using standard techniques. For fractionation experiments, equivalent amounts of supernatant (TS) protein were loaded along with a proportionate volume of each TI sample.
Morphogenesis assay
The 3D culture of MCF-10A cells on basement membrane was performed as previously described (Debnath et al., 2003). Assay media (DME/F12 supplemented with 2% donor horse serum, 10 μg/ml insulin, 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, 50 U/ml penicillin, and 50 mg/ml streptomycin) containing 5 ng/ml EGF, 10 ng/ml CSF-1 (R&D Systems), or 40 ng/ml HGF (Van Andel Institute) was replaced every 4 d unless otherwise noted. Where noted, SU6656 (2 μM) was added every 48 h without replacement of media.
Cell compaction assay
Monolayers were harvested by treatment with 0.05% trypsin or 5 mM EDTA in PBS for 30 min. 3 × 105 cells were resuspended in 1.5 ml assay media ± 10 ng/ml CSF-1 and incubated for up to 12 h in 35-mm dishes precoated with polyHEMA (6 mg/ml in 95% ethanol). Cell aggregates were harvested by centrifugation at 200 rpm and washed once in PBS before suspension in molten 0.5% agarose. Aliquots of resuspended aggregates were dropped onto 1-mm glass slides and allowed to solidify. DIC imaging was performed on a Nikon TE300 equipped with a CCD camera; images were acquired using MetaMorph Version 4.6r6.
Wound healing assay
MCF-10A cells were seeded on glass coverslips at 5 × 104 cells per well and grown to confluence before overnight starvation in assay media. A single wound was introduced in the monolayer using a micropipette tip, and the media replaced with assay media ± 10 ng/ml CSF-1 containing 1 μg/ml mitomycin C (Sigma-Aldrich). Wound closure was monitored for 48 h.
Immunofluorescence analysis and image acquisition
Monolayers were fixed 8 h after wounding for 20 min in 2% formalin, permeabilized 5 min with 0.2% Triton at 4°C, and washed three times in PBS. Coverslips were blocked for 30 min in PBS + 10% goat serum and incubated 45 min each in primary and secondary antibody diluted 1:100 in block, followed by three PBS washes. Cells were stained with DAPI, washed, and mounted on 1-mm glass slides with ProLong Antifade (Molecular Probes). Images were acquired on a Nikon TE300 using MetaMorph Version 4.6r6. Immunostaining of acinar structures was performed as previously described (Debnath et al., 2002). Confocal analyses were performed using either the Nikon E800 Bio-Rad Laboratories or Carl Zeiss MicroImaging, Inc. LSM410 confocal microscopy systems. Composite images were generated using MetaMorph Version 4.6r6 software.
Online supplemental material
The online supplemental material (Figs. S1 and S2) is available at http://www.jcb.org/cgi/content/full/jcb.200309102/DC1. Fig. S1 shows images that illustrate the progressive nature of the architectural disruption of MCF-10A acini expressing CSF-1 and CSF-1R. Fig. S2 shows images that illustrate that daily EGF treatment does not elicit the discohesive phenotype induced by similar treatment with CSF-1.
Acknowledgments
We are grateful to Owen Witte for the CSF-1R retroviral constructs, Mina Bissell for contributions to the establishment of 3D acinar culture, and Charles J. Sherr for pioneering work on CSF-1R autocrine signaling in fibroblasts. We also thank George Vande Woude and Margaret Gustafson (Van Andel Research Institute, Grand Rapids, MI) for the generous gift of the c-Met cDNA and discussions of c-Met biology, and Barry Kacinski and Jeffrey Pollard for helpful discussions of CSF-1R signaling.
This research was supported by grants from the American Cancer Society and the Department of Defense (J.S. Brugge), grants from the National Cancer Institute (J.S. Brugge and M.F. Roussel), a gift from the Virginia and D.K. Ludwig Fund for Cancer Research (InterVivos) to Harvard Medical School (J.S. Brugge), contributions from the American Lebanese Syrian Associated Charities of St. Jude Children's Research Hospital (M.F. Roussel), a Howard Hughes Medical Institute (HHMI) Predoctoral Fellowship (C.N. Wrobel), and an HHMI Physician Postdoctoral Fellowship (J. Debnath).
The online version of this article contains supplemental material.
Abbreviations used in this paper: 3D, three dimensional; CSF-1R, colony-stimulating factor receptor; RTK, receptor tyrosine kinase.
References
- Adams, C.L., and W.J. Nelson. 1998. Cytomechanics of cadherin-mediated cell-cell adhesion. Curr. Opin. Cell Biol. 10:572–577. [DOI] [PubMed] [Google Scholar]
- Adams, C.L., Y.T. Chen, S.J. Smith, and W.J. Nelson. 1998. Mechanisms of epithelial cell–cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin–green fluorescent protein. J. Cell Biol. 142:1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford, D., and J. Taylor-Papadimitriou. 1996. Cell adhesion molecules in the normal and cancerous mammary gland. J. Mammary Gland Biol. Neoplasia. 1:207–218. [DOI] [PubMed] [Google Scholar]
- Alonso, G., M. Koegl, N. Mazurenko, and S.A. Courtneidge. 1995. Sequence requirements for binding of Src family tyrosine kinases to activated growth factor receptors. J. Biol. Chem. 270:9840–9848. [DOI] [PubMed] [Google Scholar]
- Barcellos-Hoff, M.H., J. Aggeler, T.G. Ram, and M.J. Bissell. 1989. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development. 105:223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens, J., L. Vakaet, R. Friis, E. Winterhager, F. Van Roy, M.M. Mareel, and W. Birchmeier. 1993. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/β-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J. Cell Biol. 120:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake, R.A., P. Garcia-Paramio, P.J. Parker, and S.A. Courtneidge. 1999. Src promotes PKCdelta degradation. Cell Growth Differ. 10:231–241. [PubMed] [Google Scholar]
- Blake, R.A., M.A. Broome, X. Liu, J. Wu, M. Gishizky, L. Sun, and S.A. Courtneidge. 2000. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol. Cell. Biol. 20:9018–9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boterberg, T., K.M. Vennekens, M. Thienpont, M.M. Mareel, and M.E. Bracke. 2000. Internalization of the E-cadherin/catenin complex and scattering of human mammary carcinoma cells MCF-7/AZ after treatment with conditioned medium from human skin squamous carcinoma cells COLO 16. Cell Adhes. Commun. 7:299–310. [DOI] [PubMed] [Google Scholar]
- Braga, V.M., L.M. Machesky, A. Hall, and N.A. Hotchin. 1997. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell–cell contacts. J. Cell Biol. 137:1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell, M., J. Zhurinsky, and A. Ben-Ze'ev. 2002. The cadherin-catenin adhesion system in signaling and cancer. J. Clin. Invest. 109:987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, X.M., G.R. Ryan, A.J. Hapel, M.G. Dominguez, R.G. Russell, S. Kapp, V. Sylvestre, and E.R. Stanley. 2002. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 99:111–120. [DOI] [PubMed] [Google Scholar]
- Debnath, J., K.R. Mills, N.L. Collins, M.J. Reginato, S.K. Muthuswamy, and J.S. Brugge. 2002. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 111:29–40. [DOI] [PubMed] [Google Scholar]
- Debnath, J., S.K. Muthuswamy, and J.S. Brugge. 2003. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 30:256–268. [DOI] [PubMed] [Google Scholar]
- Downing, J.R., and A.B. Reynolds. 1991. PDGF, CSF-1, and EGF induce tyrosine phosphorylation of p120, a pp60src transformation-associated substrate. Oncogene. 6:607–613. [PubMed] [Google Scholar]
- Flick, M.B., E. Sapi, and B.M. Kacinski. 2002. Hormonal regulation of the c-fms proto-oncogene in breast cancer cells is mediated by a composite glucocorticoid response element. J. Cell. Biochem. 85:10–23. [PubMed] [Google Scholar]
- Fujita, Y., G. Krause, M. Scheffner, D. Zechner, H.E. Leddy, J. Behrens, T. Sommer, and W. Birchmeier. 2002. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 4:222–231. [DOI] [PubMed] [Google Scholar]
- Herrenknecht, K., M. Ozawa, C. Eckerskorn, F. Lottspeich, M. Lenter, and R. Kemler. 1991. The uvomorulin-anchorage protein alpha catenin is a vinculin homologue. Proc. Natl. Acad. Sci. USA. 88:9156–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide, H., D.B. Seligson, S. Memarzadeh, L. Xin, S. Horvath, P. Dubey, M.B. Flick, B.M. Kacinski, A. Palotie, and O.N. Witte. 2002. Expression of colony-stimulating factor 1 receptor during prostate development and prostate cancer progression. Proc. Natl. Acad. Sci. USA. 99:14404–14409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, A., T.E. Kmiecik, M. Park, I. Daar, D. Blair, K.J. Dunn, P. Sutrave, J.N. Ihle, M. Bodescot, and G.F. Vande Woude. 1990. Structure, tissue-specific expression, and transforming activity of the mouse met protooncogene. Cell Growth Differ. 1:87–95. [PubMed] [Google Scholar]
- Kacinski, B.M. 1995. CSF-1 and its receptor in ovarian, endometrial and breast cancer. Ann. Med. 27:79–85. [DOI] [PubMed] [Google Scholar]
- Kacinski, B.M., K.A. Scata, D. Carter, L.D. Yee, E. Sapi, B.L. King, S.K. Chambers, M.A. Jones, M.H. Pirro, E.R. Stanley, and L.R. Rohrschneider. 1991. FMS (CSF-1 receptor) and CSF-1 transcripts and protein are expressed by human breast carcinomas in vivo and in vitro. Oncogene. 6:941–952. [PubMed] [Google Scholar]
- Kowalski, P.J., M.A. Rubin, and C.G. Kleer. 2003. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 5:R217–R222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer, S.J., D.M. Bortner, M.F. Roussel, C.J. Sherr, and M.C. Ostrowski. 1992. Mitogenic signaling by colony-stimulating factor 1 and ras is suppressed by the ets-2 DNA-binding domain and restored by myc overexpression. Mol. Cell. Biol. 12:5355–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, T.L., A.S. Yap, and J.L. Stow. 1999. Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J. Cell Biol. 146:219–232. [PMC free article] [PubMed] [Google Scholar]
- Le, T.L., S.R. Joseph, A.S. Yap, and J.L. Stow. 2002. Protein kinase C regulates endocytosis and recycling of E-cadherin. Am. J. Physiol. Cell Physiol. 283:C489–C499. [DOI] [PubMed] [Google Scholar]
- Lipsich, L.A., A.J. Lewis, and J.S. Brugge. 1983. Isolation of monoclonal antibodies that recognize the transforming proteins of avian sarcoma viruses. J. Virol. 48:352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks, D.C., X.F. Csar, N.J. Wilson, U. Novak, A.C. Ward, V. Kanagasundarum, B.W. Hoffmann, and J.A. Hamilton. 1999. Expression of a Y559F mutant CSF-1 receptor in M1 myeloid cells: a role for Src kinases in CSF-1 receptor-mediated differentiation. Mol. Cell Biol. Res. Commun. 1:144–152. [DOI] [PubMed] [Google Scholar]
- McCrea, P.D., C.W. Turck, and B. Gumbiner. 1991. A homolog of the armadillo protein in Drosophila (plakoglobin) associated with E-cadherin. Science. 254:1359–1361. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, D., L. Tsiokas, X.M. Zhou, D. Foster, J.S. Brugge, and V.P. Sukhatme. 1995. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature. 375:577–581. [DOI] [PubMed] [Google Scholar]
- Muthuswamy, S.K., D. Li, S. Lelievre, M.J. Bissell, and J.S. Brugge. 2001. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol. 3:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann, C., V. Brinkmann, E. Spitzer, G. Hartmann, M. Sachs, H. Naundorf, and W. Birchmeier. 1998. Reconstitution of mammary gland development in vitro: requirement of c-met and c-erbB2 signaling for branching and alveolar morphogenesis. J. Cell Biol. 143:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nollet, F., G. Berx, and F. van Roy. 1999. The role of the E-cadherin/catenin adhesion complex in the development and progression of cancer. Mol. Cell Biol. Res. Commun. 2:77–85. [DOI] [PubMed] [Google Scholar]
- Owens, D.W., G.W. McLean, A.W. Wyke, C. Paraskeva, E.K. Parkinson, M.C. Frame, and V.G. Brunton. 2000. The catalytic activity of the Src family kinases is required to disrupt cadherin-dependent cell-cell contacts. Mol. Biol. Cell. 11:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios, F., L. Price, J. Schweitzer, J.G. Collard, and C. D'Souza-Schorey. 2001. An essential role for ARF6-regulated membrane traffic in adherens junction turnover and epithelial cell migration. EMBO J. 20:4973–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack, A.L., R.B. Runyan, and K.E. Mostov. 1998. Morphogenetic mechanisms of epithelial tubulogenesis: MDCK cell polarity is transiently rearranged without loss of cell-cell contact during scatter factor/hepatocyte growth factor-induced tubulogenesis. Dev. Biol. 204:64–79. [DOI] [PubMed] [Google Scholar]
- Pollard, J.W., and L. Hennighausen. 1994. Colony stimulating factor 1 is required for mammary gland development during pregnancy. Proc. Natl. Acad. Sci. USA. 91:9312–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, F.V., S.K. Chambers, J.T. Chambers, M.L. Carcangiu, P.E. Schwartz, E.I. Kohorn, E.R. Stanley, and B.M. Kacinski. 1993. Colony-stimulating factor-1 in primary ascites of ovarian cancer is a significant predictor of survival. Am. J. Obstet. Gynecol. 168:520–527. [DOI] [PubMed] [Google Scholar]
- Pujuguet, P., L. Del Maestro, A. Gautreau, D. Louvard, and M. Arpin. 2003. Ezrin regulates E-cadherin–dependent adherens junction assembly through Rac1 activation. Mol. Biol. Cell. 14:2181–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, A.B., D.J. Roesel, S.B. Kanner, and J.T. Parsons. 1989. Transformation-specific tyrosine phosphorylation of a novel cellular protein in chicken cells expressing oncogenic variants of the avian cellular src gene. Mol. Cell. Biol. 9:629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, A.B., J. Daniel, P.D. McCrea, M.J. Wheelock, J. Wu, and Z. Zhang. 1994. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol. Cell. Biol. 14:8333–8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roura, S., S. Miravet, J. Piedra, A. Garcia de Herreros, and M. Dunach. 1999. Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J. Biol. Chem. 274:36734–36740. [DOI] [PubMed] [Google Scholar]
- Roussel, M.F. 1998. Key effectors of signal transduction and G1 progression. Adv. Cancer Res. 74:1–24. [DOI] [PubMed] [Google Scholar]
- Roussel, M.F., T.J. Dull, C.W. Rettenmier, P. Ralph, A. Ullrich, and C.J. Sherr. 1987. Transforming potential of the c-fms proto-oncogene (CSF-1 receptor). Nature. 325:549–552. [DOI] [PubMed] [Google Scholar]
- Roussel, M.F., J.R. Downing, C.W. Rettenmier, and C.J. Sherr. 1988. A point mutation in the extracellular domain of the human CSF-1 receptor (c-fms proto-oncogene product) activates its transforming potential. Cell. 55:979–988. [DOI] [PubMed] [Google Scholar]
- Sapi, E., M.B. Flick, S. Rodov, M. Gilmore-Hebert, M. Kelley, S. Rockwell, and B.M. Kacinski. 1996. Independent regulation of invasion and anchorage-independent growth by different autophosphorylation sites of the macrophage colony-stimulating factor 1 receptor. Cancer Res. 56:5704–5712. [PubMed] [Google Scholar]
- Sapi, E., M.B. Flick, S. Rodov, D. Carter, and B.M. Kacinski. 1998. a. Expression of CSF-I and CSF-I receptor by normal lactating mammary epithelial cells. J. Soc. Gynecol. Investig. 5:94–101. [DOI] [PubMed] [Google Scholar]
- Sapi, E., M.B. Flick, S. Rodov, and B.M. Kacinski. 1998. b. Ets-2 transdominant mutant abolishes anchorage-independent growth and macrophage colony-stimulating factor-stimulated invasion by BT20 breast carcinoma cells. Cancer Res. 58:1027–1033. [PubMed] [Google Scholar]
- Schmeichel, K.L., V.M. Weaver, and M.J. Bissell. 1998. Structural cues from the tissue microenvironment are essential determinants of the human mammary epithelial cell phenotype. J. Mammary Gland Biol. Neoplasia. 3:201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift, S., J. Lorens, P. Achacoso, and G.P. Nolan. 1999. Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. In Current Protocols in Immunology. Unit 10.28 (Suppl. 31). [DOI] [PubMed]
- Takeda, H., A. Nagafuchi, S. Yonemura, S. Tsukita, J. Behrens, and W. Birchmeier. 1995. V-src kinase shifts the cadherin-based cell adhesion from the strong to the weak state and beta catenin is not required for the shift. J. Cell Biol. 131:1839–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nguyen, A., and J.W. Pollard. 2002. Colony stimulating factor-1 is required to recruit macrophages into the mammary gland to facilitate mammary ductal outgrowth. Dev. Biol. 247:11–25. [DOI] [PubMed] [Google Scholar]
- Warren, S.L., and W.J. Nelson. 1987. Nonmitogenic morphoregulatory action of pp60v-src on multicellular epithelial structures. Mol. Cell. Biol. 7:1326–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe, M., A. Nagafuchi, S. Tsukita, and M. Takeichi. 1994. Induction of polarized cell–cell association and retardation of growth by activation of the E-cadherin–catenin adhesion system in a dispersed carcinoma line. J. Cell Biol. 127:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen, K., S. Burkhalter, and P. van der Geer. 2002. C-Cbl binds the CSF-1 receptor at tyrosine 973, a novel phosphorylation site in the receptor's carboxy-terminus. Oncogene. 21:1079–1089. [DOI] [PubMed] [Google Scholar]
- Zhang, Q., J. Calafat, H. Janssen, and S. Greenberg. 1999. ARF6 is required for growth factor- and rac-mediated membrane ruffling in macrophages at a stage distal to rac membrane targeting. Mol. Cell. Biol. 19:8158–8168. [DOI] [PMC free article] [PubMed] [Google Scholar]