

Abstract
The Myosin heavy chain (Mhc) locus encodes the muscle-specific motor mediating contraction in Drosophila. In a screen for temperature-sensitive behavioral mutants, we have identified two dominant Mhc alleles that lead to a hypercontraction-induced myopathy. These mutants are caused by single point mutations in the ATP binding/hydrolysis domain of Mhc and lead to degeneration of the flight muscles. Electrophysiological analysis in the adult giant fiber flight circuit demonstrates temperature-dependent seizure activity that requires neuronal input, as genetic blockage of neuronal activity suppresses the electrophysiological seizure defects. Intracellular recordings at the third instar neuromuscular junction show spontaneous muscle movements in the absence of neuronal stimulation and extracellular Ca2+, suggesting a dysregulation of intracellular calcium homeostasis within the muscle or an alteration of the Ca2+ dependence of contraction. Characterization of these new Mhc alleles suggests that hypercontraction occurs via a mechanism, which is molecularly distinct from mutants identified previously in troponin I and troponin T.
Keywords: myosin; neuromuscular; dystrophy; cardiomyopathy; Drosophila
Introduction
Muscle contraction is a highly complex and coordinated process, involving a molecular machine tightly regulated to provide ATP-dependent motion in response to neuronal stimulation. Activation of the muscle from the innervating neuron results in a postsynaptic action potential that stimulates release of Ca2+ from internal stores. Ca2+ binds to regulatory proteins on the thin filament of the muscle. This leads to coordinated conformational changes in a large number of proteins, allowing myosin to shorten the length of the sarcomere in an ATP-dependent process. In addition to proteins required during contraction, numerous secondary proteins are required for its continued maintenance, structural support, and force transduction (Huxley, 2000; Lamb, 2000; Pollard, 2000; Poage and Meriney, 2002; Ruff, 2003). Characterization of this complex system may lead to molecular understanding of human diseases such as cardiomyopathies, which arise from perturbations in several known cardiac muscle proteins (Seidman and Seidman, 2001).
The genetic tractability of Drosophila has made it an ideal system to characterize mutations affecting neuromuscular function. Many of these mutants have been identified by screening for temperature-sensitive (TS) behavioral defects, allowing the identification of gene products important for neuromuscular function (Ganetzky and Wu, 1983; Littleton et al., 1998). Additionally, the Drosophila flight system has provided an efficient genetic model for hypertrophic cardiomyopathies. Mutated genes leading to flightless behavior in Drosophila are also disrupted in many forms of primary hypertrophic cardiomyopathies, including Myosin heavy chain (Mhc), Tropomyosin2 (Tm2), wings up A (wupA [troponin I]), actin88F (act88F), and upheld (troponin T). Mutations in these genes also show extensive genetic interactions, providing key insights into the regulatory pathways underlying muscle function (Ferrus et al., 2000; Vigoreaux, 2001).
Flightless behavior in Drosophila can arise from mutations that lead to muscle hypercontraction. Hypercontraction of the Drosophila indirect flight muscles (IFM) occurs due to specific mutations of the contractile machinery that lead to either decreased structural integrity of the sarcomere, or dysregulation of the contractile process in vivo. These mutations result in a degeneration of the IFM of adult flies after muscle differentiation and development (Fekete and Szidonya, 1979; Deak et al., 1982; Homyk and Emerson, 1988; Beall and Fyrberg, 1991). Hypercontraction mutants can be genetically suppressed by specific mutant alleles of Mhc. The mechanism of suppression has been suggested to be a potential Mhc–Troponin I direct interaction (Kronert et al., 1999). However, additional data suggests that an overall decrease in actomyosin force is sufficient to explain suppression of hypercontraction by mutant Mhc (Nongthomba et al., 2003). The identification of Mhc alleles that directly cause hypercontraction and enhance the hypercontraction defects of other mutants may facilitate defining the role of myosin in the regulation of contraction.
To further understand the molecular and cellular processes underlying neuromuscular function, we performed a screen for Drosophila TS behavioral mutants. One complementation group isolated in our screens, Samba, disrupts the Mhc locus, leading to hypercontraction and muscle degeneration. Characterization of the Samba mutants has revealed potential molecular mechanisms that lead to muscle degeneration through hypercontraction via distinct mechanisms from hypercontraction mutants characterized previously. In addition, these mutants give insight into the role of Mhc in the regulation of the contractile process in addition to its role in ATP-dependent motor function.
Results
Isolation and characterization of the Samba mutants, MhcS1 and MhcS2
The Samba (Samba 1 and Samba 2) mutants were isolated in an ethyl methanesulfonate mutagenesis screen for X-linked and autosomal-dominant TS behavioral mutants. Samba adults exhibit dominant TS behavioral defects that include a rapid onset of seizure-like behavior and TS loss of mesothoracic leg function. This behavior is readily evident in all flies by 1 min of exposure to 38°C (Fig. 1 A). Samba mutants also display defects at permissive temperatures. Samba/+ flies are flightless, with thoracic indentations similar to those found in ether-ago-go 1, Shaker133 (eag 1, Sh133) mutant flies (Fig. 1 B). Thoracic indentations in eag 1, Sh133 flies occur through hypercontraction of the IFM thought to be induced by excessive neurotransmitter release in the presynaptic neuron, which is caused by loss of voltage-gated potassium channels and subsequent reduction in repolarization (Ganetzky and Wu, 1983; Wu et al., 1983). Although homozygous Samba mutants are lethal, rare Samba 1 escapers with femur hypercontration defects can be found.
Figure 1.

Characterization of the Samba mutants, MhcS1 and MhcS2. (A) Temperature-sensitivity curves of several mutant strains that affect muscle function. TS behavioral defects are found only in hypercontraction mutants. (B) Thoraces of CantonS (left), eag 1, Sh133/Y (center), and Samba 1/+ (right) adult male flies. (C) Schematic of a normal fly leg (left), a leg dissected from CantonS (center), and a leg showing femur hypercontraction from a Swg X118/+;Brkd J29/+ double heterozygote (right). Arrows indicate femoral segment. (D) Schematic depicting the genetic interactions of the Samba locus. (E–G) Polarized light micrographs showing hypercontraction of the IFM are shown in sagittal views of the thorax from adult flies of (E) CantonS, (F) Samba 1/+, and (G) Samba 1/+;Tm2 D53. Partial suppression is observed in Samba 1/+;Tm2 D53, as IFM are found less degraded despite the presence of thoracic indentations (arrows).
In addition to Samba, two other complementation groups, Swing (Swg X118) and Breakdance (Brkd J29), were isolated in our screens that exhibited similar dominant TS behavioral defects and showed genetic interactions with Samba. Double heterozygotes of Swg X118 and Samba 1 or Samba 2 and double heterozygotes of Swg X118and Brkd J29 are semi-lethal with escapers having hypercontracted femurs (Fig. 1 C). Double heterozygotes of Samba 1 or Samba 2 and Brkd J29 are synthetic lethal (Fig. 1 D). These genetic interactions and similarities to eag 1, Sh133 double mutants suggest that Samba, Swing, and Breakdance define a genetic pathway required in the regulation of membrane excitability, and when disrupted, lead to abnormal muscle hypercontraction.
Segregation analysis of Samba revealed an autosomal-dominant mutation on the second chromosome, refined to 2–52 cM by recombination mapping. Deficiency mapping by lethality narrowed the cytological interval between 36A8 and 36C4 on the left arm of chromosome 2. To help identify the Samba locus, we screened for revertants of TS seizure behavior in a γ-irradiation reversion screen in order to isolate potential loss of function mutations in the Samba locus. Three revertants were identified by loss of TS behavioral defects. These revertants were embryonic lethal with normal morphological development, but showed complete loss of muscle wave propagation in late stage embryos (unpublished results). Noncomplementation to Mhc 1 by both the TS mutants and the three revertants identified the Samba mutations as new alleles of the Mhc locus (Mogami and Hotta, 1981). We designated the Samba 1 and Samba 2 alleles Mhc S1 and Mhc S2, respectively, and the revertants Mhc rv1, Mhcrv2, and Mhc rv3.
The Mhc locus is complex, encoding all muscle-specific isoforms through the use of extensive alternative splice patterns (Rozek and Davidson, 1983; Bernstein et al., 1983). The locus contains 19 coding exons, 5 of which are alternatively spliced, and one that is either included or excluded (Wassenberg et al., 1987; George et al., 1989; Collier et al., 1990; Hess and Bernstein, 1991; Zhang and Bernstein, 2001). An allele isolated previously, Mhc 5 (G200D), causes similar hypercontraction defects to the Samba mutants. Mhc 5 introduces a point mutation in exon 4 of the Mhc locus, disrupting the ATPase domain of Mhc (Homyk and Emerson, 1988). Due to the phenotypic similarities with Mhc 5, exon 4 of these new alleles was sequenced. Both Mhc S1 and Mhc S2 were found to be point mutations (V235E and E187K, respectively) mapping to the ATP binding and hydrolysis site of the protein (Fig. 2, A–E).
Figure 2.
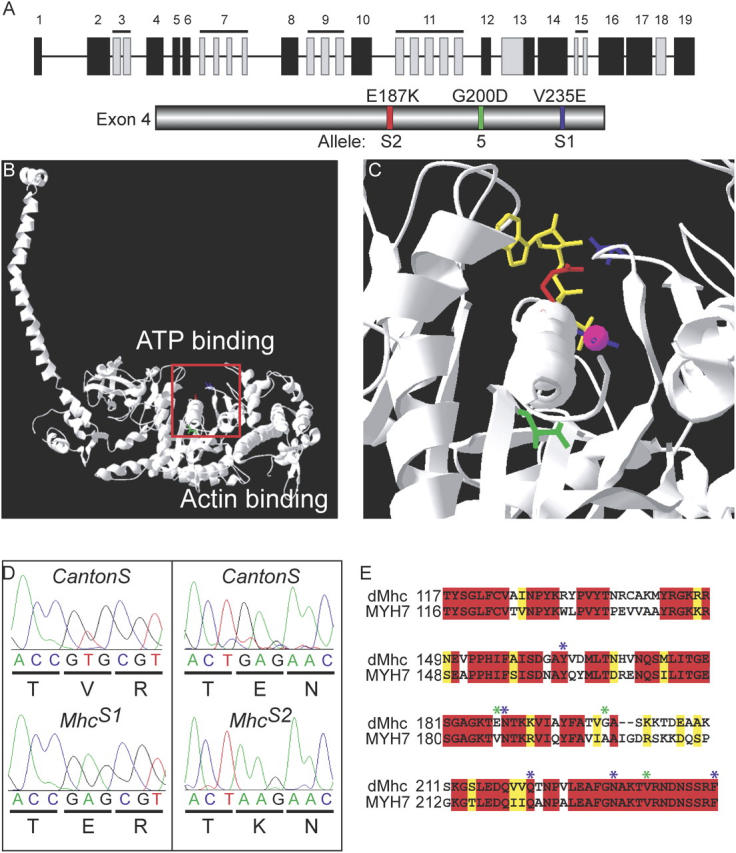
Localization of the MhcS1 and MhcS2 mutations onto the crystal structure. (A) Schematic of the Mhc locus. Constitutive exons are depicted in black, and alternative exons in gray. Exon 4 has been expanded to show the relative positions of the point mutations in Mhc 5, MhcS1, and Mhc S2. (B) A view of the crystal structure of chicken Mhc depicting the mutations Mhc 5 (green), Mhc S1 (blue), and Mhc S2 (red). (C) The region corresponding to the red box has been expanded from a crystal structure from the motor domain of Dictyostelium Mhc showing the mutation positions in relation to the ATP analogue (yellow) and bound Mg2+ (magenta). (D) Sequence data showing the base pair changes in Mhc S1 and Mhc S2 and the corresponding amino acid point mutations. (E) An alignment of exon 4 of Drosophila Mhc with β-cardiac Mhc (MYH7) from humans. Red corresponds to identical amino acids, and yellow identifies similar amino acids. Positions of Drosophila mutations are indicated in green relative to amino acids that are mutated in several primary hypertrophic cardiomyopathies, shown in blue.
Seizure activity in Samba flies is dependent upon neuronal activity
Extracellular dorsal longitudinal muscle (DLM) recordings were used to characterize the behavioral seizures at restrictive temperatures (Engel and Wu, 1992). Electrical activity was observed in adult flies at room temperature, shifted to 38°C, and returned to room temperature. Correlating with the behavioral defects, abnormal spiking activity in the DLMs was recorded at restrictive temperatures that was not observed at permissive temperatures, nor in Canton-S at 38°C (Fig. 3, A, B, D, and F). Similar seizure activity was observed in Swg X118 and Brkd J29 (Fig. 3, H and J). This abnormal activity could be either muscle autonomous or dependent upon synaptic input. To address these possibilities, we used a mutation in the voltage-gated Na+ channel, paralytic (para ts1), which is required for action potential propagation in the motor neuron (Suzuki et al., 1971). This mutation specifically abolishes neuronal action potentials at elevated temperatures, as para expression is not detected in muscles (Hong and Ganetzky, 1994). In para ts1/Y;MhcX/+ (X indicating 5, S1, or S2) flies, we observed a suppression of the seizure activity at 38°C (Fig. 3, C, E, and G). Similarly, suppression of activity was observed with para ts1/Y;SwgX118/+ and para ts1/Y;BrkdJ29/+ (Fig. 3, I and K). These data suggest that mutant muscles are hyperexcitable at restrictive temperatures. This hyperexcitable state, however, cannot lead to autonomous muscle firing, but must be triggered by an initial input by the innervating motor neuron.
Figure 3.

Extracellular DLM recordings reveal abnormal activity at restrictive temperatures that is dependent upon neuronal activity. Recordings were done on male flies of genotypes (A) CantonS, (B) Mhc S1/+, (C) para TS1;MhcS1/+, (D) Mhc S2/+, (E) para TS1;MhcS2/+, (F) Mhc 5/+, (G) para TS1;Mhc5/+, (H) Swg X118/+, (I) para TS1;SwgX118/+, (J) Brkd J29/+, and (K) para TS1;BrkdJ29/+. Representative traces are shown from a single male fly of the indicated genotype at room temperature, 38°C, and subsequent recovery to room temperature. Bar, 10 mV, 500 ms.
Samba mutations lead to hypercontraction
To further define how the Samba mutants affect Mhc function, we analyzed genetic interactions with known muscle mutants that increase or decrease the contractile state of the muscle. We characterized genetic interactions with mutations in Troponin I, Troponin T, and Tropomyosin 2 (Tm2). Troponin I is encoded by the wupA locus. A mutation in troponin I, heldup 2 (wupA hdp2), has been identified previously as a hypercontraction mutation (Deak et al., 1982; Beall and Fyrberg, 1991). A mutation in Troponin T, upheld 101 (up 101), is similar to wupA hdp2, also causing hypercontraction (Fekete and Szidonya, 1979; Homyk et al., 1980). Hemizygous flies for either wupA hdp2 or up 101 and heterozygous for Mhc S1 or Mhc S2 are synthetic lethal, whereas double heterozygous females have femur hypercontraction defects (unpublished results). A mutation in Tm2 (Tm2 D53) suppresses the hypercontraction of both up 101 and wupA hdp2 (Naimi et al., 2001). Similarly, Tm2 D53 suppresses the recessive lethality of Mhc S1, increasing viability of the homozygotes from 2.38% to 80.92% (Mhc S1 n = 435, Mhc S1;Tm2D53 n = 505). These results indicate that the Samba alleles of Mhc cause hypercontraction defects similar to wupA hdp2 and up 101. Interestingly, both up 101 and wupA hdp2 exhibit abnormal TS behavior similar to Mhc S1 and Mhc S2, suggesting that the behavioral defects are most likely a TS susceptibility resulting from an altered state of the muscle secondary to hypercontraction, as opposed to a specific TS dysfunction of the Mhc protein (Fig. 1 A). In contrast to the TS seizure behavior in hypercontraction mutants, Tm2 D53 flies do not show abnormal behavior at 38°C, nor do heterozygotes of the Mhc null (Mhc 1/+) and heterozygotes of a hypercontraction suppressor mutant (Mhc D45/+; Fig. 1 A).
To confirm that the Samba phenotype results from hypercontraction, we analyzed the structure of the IFM in Mhc S1/+ flies. The ordered array of filaments in muscles leads to birefringent properties, allowing muscle visualization under polarized light microscopy. In hypercontraction mutants characterized previously, the IFM exhibit one of two defects. Some show loss of birefringence in the middle of the muscles due to breakage or degradation, with the bulk of the muscle fiber at either one or both of the attachment sites. Others show separation from the attachment sites, with birefringence found only in the middle of the fiber (Nongthomba et al., 2003). Mhc S1/+ flies exhibited the former defect, showing birefringence at the attachment sites, with loss of birefringence in the middle of the IFM. This defect was partially suppressed in the background of Tm2 D53, as the IFM of double mutants displayed less degradation despite the presence of indented thoraces compared with similarly aged Samba flies (Fig. 1, E–G). These data confirm that the Samba mutants lead to hypercontraction defects in the muscle.
Samba mutant muscles do not alter synaptic function but move independently of neuronal input
Hypercontraction in Drosophila muscles induced by the Samba mutants leads to progressive degradation of fibers, similar to degeneration observed in muscular dystrophies. In some animal models of muscular dystrophy, loss of acetylcholine receptor clustering results in the functional denervation of diseased fibers (Rafael et al., 2000). Because Mhc S1 and Mhc S2 were isolated by TS behavioral defects and abnormal extracellular DLM activity, we hypothesized that these mutations may cause functional or structural changes at the neuromuscular junction (NMJ).
Bouton number at the NMJ is tightly regulated and is sensitive to disruptions in both presynaptic and postsynaptic function. Though poorly understood, postsynaptic defects can alter presynaptic structural and functional properties through homeostatic regulatory pathways (Petersen et al., 1997; Davis et al., 1998). To analyze the morphology of the NMJ in Samba mutants, we stained third instar larvae with α-synaptotagmin I antisera, a marker for presynaptic terminals, and analyzed muscle fibers 6 and 7 (Canton-S n = 18 larvae, 97 muscles; Mhc S1/+ n = 27, 157). Type I innervation from glutamatergic motor neurons was not altered in Mhc S1/+ animals, suggesting little effect of dysfunctional muscles on excitatory innervation (Fig. 4 J). The number of muscles showing ectopic innervation, however, was found to be more frequent than wild type, increasing from 4.1% in control animals to 15.9% in Mhc S1/+ animals (Fig. 4, A–F and K). These were determined to be type II synapses due to their morphology and the absence of postsynaptic DLG staining (Fig. 4, G–I). Type II synapses are neuromodulatory, influencing the state of excitation in body wall muscles through release of octopamine (Gramates and Budnik, 1999). Increases in type II innervation have also been reported in mutants such as tipE and nap, which reduce nerve excitability (Jarecki and Keshishian, 1995). The increase in type II innervation suggests that alterations in muscle function can lead to altered neuromodulation. Similar data was obtained with Mhc S2/+ larvae (unpublished data).
Figure 4.

Structural properties of the neuromuscular junction in MhcS1/+ mutants. Third instar larvae neuromuscular junctions from (A–C) CantonS and (D–I) Mhc S1/+ labeled with the indicated markers. Arrows indicate an axonal branch containing type II synapses as evidenced by the absence of postsynaptic DLG staining. Bar, 50 μm. (J) Excitatory type I glutamatergic innervation on muscles 6/7 does not change in mutant muscles despite altered muscular function. (K) Ectopic type II innervation increases in mutant muscles.
To determine whether the altered innervation pattern correlated with abnormal synaptic transmission, we characterized miniature excitatory junctional potential amplitude (mEJP), excitatory junctional potential amplitude (EJP), and mEJP frequency from the muscle 6 NMJ in 0.4 mM Ca2+ using intracellular recording techniques (Fig. 5 A; Table I). Wild-type fibers were found to have a mEJP of 0.95 ± 0.04 mV and EJP of 39.6 ± 1.8 mV (n = 8, 23). In mutant fibers, these values were 0.79 ± 0.02 mV and 34.2 ± 1.7 mV, respectively (n = 11, 29). Although these differences are statistically significant and may reflect subtle changes in synaptic function, the change in release is small and the differences more likely reflect the complication caused by an apparent oscillation of the resting membrane potential due to spontaneous muscle movement in Samba mutants (Fig. 5 B). Movements such as these can lead to apparent voltage changes due to electrode motion. Spontaneous muscle movements did not resemble action potential–induced contraction events. Instead, a slow, cyclic activity that did not use the full contractile potential of the muscle cell was continuously observed in the Samba mutants. Because contraction usually depends upon Ca2+ influx through L-type calcium channels in the sarcolemma, we hypothesized that alterations of Ca2+ influx through these channels may be responsible for the spontaneous contractions in Samba mutants. To test this, we recorded from mutant muscles in Ca2+-free saline. Spontaneous contractions were still observed in Ca2+-free saline similar to those in high extracellular Ca2+ (Fig. 5 B). An alternative possibility is that spontaneous contractions may reflect altered signaling from the innervating neuron. Although these recordings are normally done in preparations in which the axon has been severed from the cell body, we further tested this possibility in brain-intact preparations in the presence or absence of 3 μM TTX. Spontaneous movements were seen regardless of neuronal activity (Fig. 5 B). These results indicate that hypercontraction in Samba mutants is not due to an increase in neuronal activity or neurotransmitter release, nor to multiple release events per axonal action potential, but can be explained by autonomous movement in the absence of neuronal input. This movement is not dependent upon extracellular Ca2+, suggesting a defect in either intracellular Ca2+ homeostasis or the Ca2+ requirements of muscle contraction.
Figure 5.
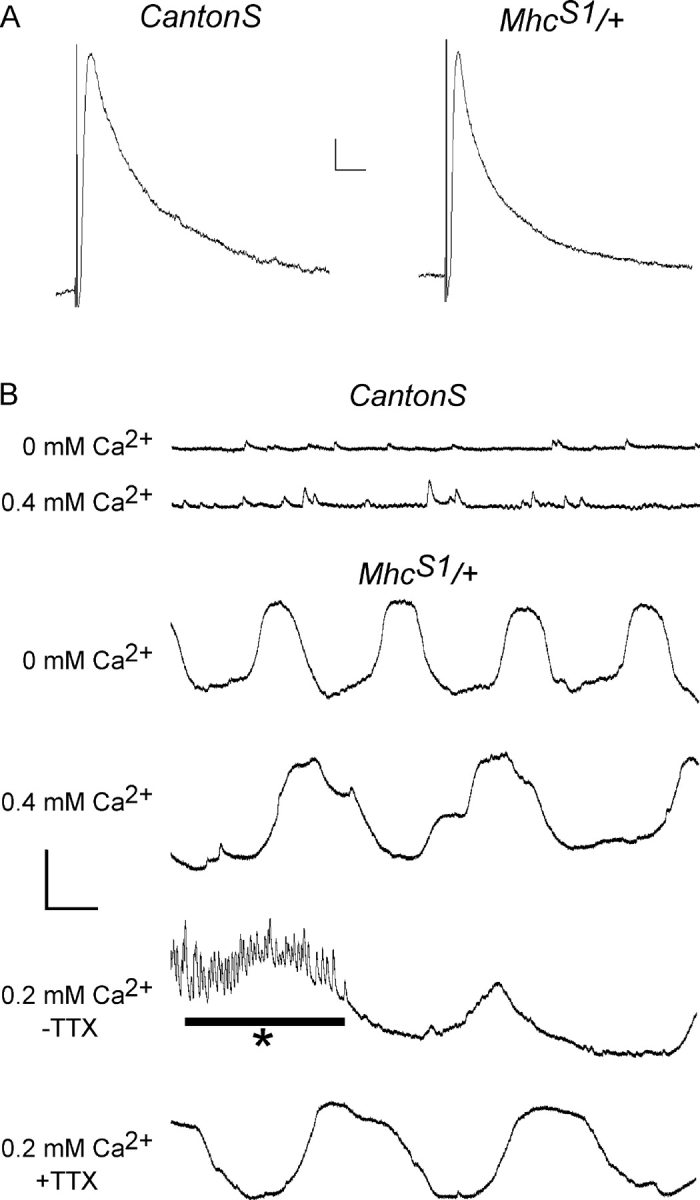
Electrophysiological properties of the MhcS1/+ third instar neuromuscular junction. Despite the increased presence of ectopic innervation, physiological properties are not altered in Mhc S1/+. (A) Representative samples of EJPs in CantonS and Mhc S1/+. Bar, 5 mV, 200 ms. (B) Resting potential oscillations found in Mhc S1/+ are present in both 0.4 and 0 mM Ca2+, as well as in the presence (−TTX) and absence (+TTX) of neuronal activity. Asterisk marks activity from the central pattern generator. Bar, 10 mV, 500 ms.
Table I.
Physiological properties of the NMJa
| Genotype | Resting potential | Miniature frequency | mEJP amplitude | EJP amplitude |
|---|---|---|---|---|
| mV | mEJP/s | mV | mV | |
| CantonSb | −69.4 ± 1.6 | 2.10 ± 0.21 | 0.95 ± 0.04 | 39.6 ± 1.8 |
| MhcS1/+ c | −62.6 ± 1.0e | 1.53 ± 0.14d | 0.79 ± 0.02e | 34.2 ± 1.7d |
Represented as value ± SEM.
n = 8,23.
n = 11,29.
P < 0.05.
P < 0.001.
Discussion
Muscle contraction requires the coordinated action of many proteins under the regulation of defined stimuli. Dysregulation of this system leads to myopathy and dystrophy syndromes in humans. Drosophila provides an efficient model to dissect the molecular and cellular functions of the individual components and their roles in the coordinated machine that underlies this process. To this end, we have begun a molecular and cellular characterization of muscle dysfunction in Drosophila using the hypercontractive Samba mutants, Mhc S1 and Mhc S2.
Hypercontraction and Mhc
Genetic perturbations can lead to hypercontraction in the Drosophila IFM by increasing actomyosin force through either a decrease in structural integrity of the sarcomere or an alteration in thin filament regulation of the cross-bridge cycle (Kronert et al., 1995; Reedy et al., 2000). Hypercontraction by the latter mechanism can be suppressed by mutations in Mhc, though it remains unclear whether suppression is obtained through an overall decrease in actomyosin force or by a direct role of Mhc on regulating thin filament dynamics (Kronert et al., 1999; Nongthomba et al., 2003). Here, we characterized two new alleles of Mhc. These alleles enhance the defects of up 101 and wupA hdp2 and are partially suppressed by Tm2 D53. These genetic interactions suggest that although Mhc S1and Mhc S2 lead to hypercontraction defects that are similar to up 101 and wupA hdp2, they do so through a different molecular mechanism, as up 101 and wupA hdp2 are fully suppressed by Tm2 D53. Using a simplified five state model of contraction based upon the allosteric/cooperative model described previously, we can hypothesize the molecular mechanisms behind hypercontraction (Fig. 6; Lehrer and Geeves, 1998).
Figure 6.

A working model for hypercontraction. A simplified five-stage model of contraction is depicted. The resting state of muscle (A) contains myosin unbound to the thin filament. During activation of the muscle (B), intracellular Ca2+ increases, allowing a limited number of troponin complexes to shift into the open state. The open state allows noncooperative binding of myosin (C) to the thin filament. When enough myosin heads bind, cooperative mechanisms facilitate binding of multiple heads (D). Release of Pi then allows contraction to occur (E), followed by ADP–ATP exchange to complete the cycle. Drosophila mutants are listed next to proposed steps at which they alter this simplified cycle.
In wild-type sarcomeres, the troponin complex remains in a closed state during rest (state A). If myosin–nucleotide complex binds this state, it is referred to as “blocked,” as it is not conducive to contraction. Neural stimulation shifts the equilibrium between state A and state B (open state) by allowing Ca2+ to enter the sarcomere, which acts as an allosteric regulator of the troponin complex via troponin C. The equilibrium shift increases the probability of myosin binding the open state in a noncooperative manner (state C). If a sufficient number of myosins bind, cooperative mechanisms facilitate binding of multiple myosin heads (state D), a low-force state of the sarcomere. Release of Pi allows myosin to enter rigor, the high-force state of the sarcomere (state E), followed by ADP–ATP exchange to complete the cycle. During this transition, an estimated 20–30% of myosins remain bound to facilitate multiple cycles during a single contraction event, as this transition state is similar to state C (Lehrer and Geeves, 1998).
Hypercontraction in Mhc mutants can be caused by two different mechanisms. One mechanism leads to hypercontraction by decreasing structural integrity of the sarcomere (Mhc 6, Mhc13). The other mechanism involves the mutations Mhc S1, Mhc S2, and Mhc 5. Hypercontraction by Mhc S1, Mhc S2, and Mhc 5 may be caused by stabilizing actin–myosin interactions during the state E–state A transition, probably by preventing proper ADP–ATP exchange. This is consistent with the localization of the amino acid substitutions in the ATPase domain of the protein. Further stabilization during this transition could lead to contraction oscillations long after the nerve-stimulated Ca2+ transient, increasing actomyosin force during relaxation periods. The spontaneous contraction oscillations observed in third instar larvae support this model. However, mutants such as up 101, wupA hdp2, and Tm2 D53 have defects which affect the state A–state B equilibrium. Therefore, in double mutants of wupA hdp2 or up 101 and Mhc S1, Mhc S2, or Mhc 5, lethality may reflect the additive effects of two distinct molecular mechanisms of hypercontraction. The differential suppressive effects of Tm2 D53 on these hypercontraction mutants support distinct molecular mechanisms as well. Alternatively, hypercontraction by Mhc S1, Mhc S2, and Mhc 5 could occur through direct Mhc–Troponin I interactions, as proposed previously (Kronert et al., 1999). However, this seems unlikely as these mutants are single amino acid substitutions which map to the ATPase domain as opposed to surfaces more accessible to protein–protein interactions.
Excitability and hypercontraction mutants
The TS seizure activity in Mhc 5, Mhc S1, and Mhc S2 as well as up 101 and wupA hdp2 is likely to reflect a temperature-dependent defect caused by an alteration in the cellular state of a hypercontractive muscle, rather than direct temperature-dependent defects of mutant proteins. The model proposed for hypercontraction may account for the activity through a dysregulation of calcium homeostasis. In normal muscles, calcium levels dramatically increase in the sarcomere in order to increase the fraction of troponin complexes in state B during regulated contraction. However, in mutants such as up 101 and wupA hdp2, rather than calcium returning to intracellular stores, the calcium remains buffered in the sarcomere. This may be due to two possibilities. One possibility is that these mutations respond to lower calcium concentrations, where calcium ions are continually binding a mutant complex, transitioning to state B, released upon return to state A, and then repeating this binding cycle long after the large calcium transient has passed for regulated contraction. Although the [Ca2+]free remains relatively low, there is an overall buffering of a significant amounts of calcium in the sarcomere by the troponin complex. The other possibility, though not mutually exclusive, is that these mutations lead to a lower activation energy for the A→B transition in the absence of calcium. State B, having a higher affinity for calcium, allows binding of calcium away from endogenous muscle calcium buffers. This can also lead to an overall aberrant buffering of calcium. Likewise, Mhc 5, Mhc S1, and Mhc S2 lead to buffering because the sarcomere is continually cycling through states C→D→E→(C). After a single cycle of unregulated contraction, calcium may unbind the troponin complexes. However, a significant number of myosins remain bound in Mhc S1, MhcS2, Mhc5, stimulating a second cycle through cooperative mechanisms, allowing state B troponin complexes to bind calcium. Buffered calcium in both mutant groups is thus continually binding and unbinding troponin complexes. During an increase in temperature, diffusion rates increase, allowing Ca2+ to diffuse farther from the contractile machinery when unbound. At sufficiently high temperatures, calcium may reach the membrane, effectively depolarizing the membrane and leading to a hyperexcitable state. This state would allow for multiple muscle action potentials once threshold is reached, but neuronal input would be required to stimulate the spike train. The suppression of seizure activity by para ts1 occurs by preventing threshold through loss of presynaptic release.
Alternatively, though not mutually exclusive, excitability defects may be caused by further increases in ectopic innervation at the adult IFM. Although the innervation defects at the third instar neuromuscular junction are modest, defects may be exacerbated at adult muscles. However, at the adult flight muscles, type II innervation seems to represent a more molecularly diverse set of synapses, and it is unknown whether muscle hypercontraction would induce increased innervation of any, a subset, or all type II–like synapses in the adult (Rivlin et al., 2004). Moreover, it is unclear how increases in type II innervation may alter excitability of muscles in a temperature-dependent fashion.
Although more experimentation will be required to discern between these possibilities as well as other potential mechanisms, it is clear that hypercontraction creates a distinct muscle state that is different from hypocontracted and normal muscles. In support of this, hypercontraction mutants have TS behavioral defects that are not evident in hypocontraction mutants or in wild-type flies. In addition, TS behavioral defects are not likely due to mixtures of differentially active myosins being expressed in the IFM, as Mhc D45/+ heterozygotes do not display TS behavioral defects such as those found in Mhc S1/+, Mhc S2/+, and Mhc 5/+. Future studies in determining the components that contribute to this altered state will be critical in understanding the underlying causes of excitability defects in hypercontraction mutants. Characterization of genetic interactors of Mhc S1 and Mhc S2 mutants such as the Swing and Breakdance loci described here may also provide insights into the molecular pathways underlying hypercontraction myopathies, as well as contribute to understanding of the mechanisms underlying human muscle diseases such as hypertrophic cardiomyopathy.
Materials and methods
Fly strains and crosses
Flies were cultured on standard medium at 22°C. All crosses using appropriate genotypes were cultured at 25°C. Mhc S1 and Mhc S2 were generated in an F1 EMS screen for X-linked and autosomal dominant TS behavioral defects. Samba mutants were recombination mapped to 2–52 cM on the second chromosome with Sp J L P marker chromosomes, deficiency mapped to 36A8–36C4, and tested for noncomplementation with P{w + mC = lacW}Mhc k10423 and Mhc 1 (Mogami and Hotta, 1981; Spradling et al., 1999). The Swg X188 and Brkd J29 mutations were also generated in screens for TS behavioral defects, and these have been mapped to 42A and 88F, respectively. In addition, revertants of Mhc S1 TS dysfunction were isolated by γ-irradiation. Mhc S1/CyO males were exposed to 6,000 rads, crossed to Gla/CyO, and F1 progeny were tested at 38°C for loss of TS behavior. Three revertants (Mhc rv1, Mhc rv2, and Mhc rv3) were isolated. All three were embryonic lethal with normal morphological development, but showed complete loss of muscle wave propagation in late stage embryos. In addition, all three revertant alleles show noncomplementation to Mhc 1. Suppression of lethality was scored as live flies that were able to survive eclosion and feed with enough motor coordination to prevent becoming trapped in the media.
Adult behavior analysis
10 flies were placed into a preheated glass vial at 38°C. Flies showing TS behavioral defects were scored in 15-s intervals. The analysis was done with 10 repetitions for each genotype and each repetition contained an independent set of 10 flies.
Polarized light miocrographs
Polarized light micrographs of the adult flight muscles were analyzed as described previously with the modification of using Xylenes as a clearing agent (Fyrberg et al., 1994). Thoraces were mounted using Permount (Fisher Scientific) and analyzed under Nomarski optics.
Mutation and crystal structure analysis
Mutations were determined by PCR and sequencing. Genomic DNA was isolated from Canton-S and Mhc S1/Df(2L)H20 flies (Simpson, 1983). Exons 4–6 were amplified by PCR and the product was sequenced at the MIT Cancer Center sequencing facility. Genomic DNA from homozygous Mhc S2 embryos was isolated and similarly processed. Amino acids are numbered according to the Mhc-P11 sequence. Crystal structure analysis of the mutations was done using the Swiss PDB Viewer software available at http://us.expasy.org/spdbv/. Crystal structures 2MYS and 1MMG were downloaded from the National Center for Biotechnology Information (NCBI) and mutations mapped according to BLAST alignments done through the NCBI BLAST website (Fisher et al., 1995; Rayment et al., 1993, 1995).
Antibodies and immunohistochemistry of third instar larvae
Wandering third instar larvae were raised at 25°C, and then dissected and fixed by standard procedures. Affinity-purified rabbit α-sytI antibodies (Littleton et al., 1993) were used at 1:1,000 and Cy2-conjugated goat α–rabbit secondary antibodies at 1:200 (Jackson ImmunoResearch Laboratories). Texas red–conjugated phalloidin was incubated simultaneously with the secondary antibody at 1:500 (Molecular Probes). Visualization and quantification was performed under light microscopy using a 40×/1.3NA oil-immersion lens. Images were taken using confocal microscopy under similar conditions and processed with Zeiss PASCAL software.
Electrophysiology
Extracellular DLM recordings.
Extracellular DLM recordings were done in male flies raised at 25°C. 1–5-MΩ electrodes were filled with 3 M KCl. The recording electrode was inserted into the lateral thorax with the ground electrode inserted into the eye. Basal activity was recorded for 2 min at 22°C. Temperature was then shifted to 38°C for 1 min, and returned to 22°C. Recordings were done using an Axoclamp-2B amplifier (Axon Instruments, Inc.) and digitized with an digitizer (model 100; Instrunet) at 10 kHz and analyzed with Superscope 3.0 software (GW Instruments). To attenuate extracellular signals from the eye, experiments were done in constant light.
Intracellular recordings.
Intracellular recordings were done at room temperature in wandering third instar larve raised at 25°C. Dissections and recordings were done in 0.4 mM Ca2+ HL3 (Stewart et al., 1994) with 4 mM MgCl2. Recordings were done from muscle 6 at segments A3–A5. 50–100-MΩ electrodes were filled with 3 M KCl. Muscles were analyzed if the resting membrane potential was below −50 mV. Data was digitized with a Digidata 1322, filtered at 10 kHz online, and analyzed using pCLAMP v8.0 software (Axon Instruments, Inc.). mEJP amplitude and frequency was determined by manual analysis, analyzing representative samples from each muscle recording. EJP amplitude was similarly analyzed, using the maximal response to suprathreshold stimulation (determined for each individual muscle). Ca2+-free recordings were done in a similar manner. Failure to evoke release was used to verify that Ca2+ was minimal in the external solution. Recordings with an intact central nervous system were done in 0.2 mM Ca2+ to prevent substantial depolarization during central pattern activity in the presence or absence of 3 μM TTX.
Acknowledgments
We thank Ilaria Rebay, Avital Rodal, Motojiro Yoshihara, and Bill Adolfsen for helpful discussions about the manuscript. We would also like to thank the Bloomington Stock Center, S.I. Bernstein, and J.C. Sparrow for Drosophila strains. The α-DLG antibody developed by C.S. Goodman was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa (Iowa City, IA).
This work was supported by grants from the National Institutes of Health, the Human Frontiers Science Program, the Searle Scholars Program, and the Packard Foundation. J. Troy Littleton is an Alfred P. Sloan Research Fellow.
Abbreviations used in this paper: DLM, dorsal longitudinal muscle; EJP, excitatory junctional potential; IFM, indirect flight muscles; mEJP, miniature excitatory junctional potential; Mhc, myosin heavy chain; NMJ, neuromuscular junction; Tm2, tropomyosin 2; TS, temperature-sensitive; wupA, wings up A.
References
- Beall, C.J., and E. Fyrberg. 1991. Muscle abnormalities in Drosophila melanogaster heldup mutants are caused by missing or aberrant troponin-I isoforms. J. Cell Biol. 114:941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, S.I., K. Mogami, J.J. Donady, and C.P. Emerson, Jr. 1983. Drosophila muscle myosin heavy chain encoded by a single gene in a cluster of muscle mutations. Nature. 302:393–397. [DOI] [PubMed] [Google Scholar]
- Collier, V.L., W.A. Kronert, P.T. O'Donnell, K.A. Edwards, and S.I. Bernstein. 1990. Alternative myosin hinge regions are utilized in a tissue-specific fashion that correlates with muscle contraction speed. Genes Dev. 4:885–895. [DOI] [PubMed] [Google Scholar]
- Davis, G.W., A. DiAntonio, S.A. Petersen, and C.S. Goodman. 1998. Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron. 20:305–315. [DOI] [PubMed] [Google Scholar]
- Deak, I.I., P.R. Bellamy, M. Bienz, Y. Dubuis, E. Fenner, M. Gollin, A. Rahmi, T. Ramp, C.A. Reinhardt, and B. Cotton. 1982. Mutations affecting the indirect flight muscles of Drosophila melanogaster. J. Embryol. Exp. Morphol. 69:61–81. [PubMed] [Google Scholar]
- Engel, J.E., and C.F. Wu. 1992. Interactions of membrane excitability mutations affecting potassium and sodium currents in the flight and giant fiber escape systems of Drosophila J. Comp. Physiol. [A]. 171:93–104. [DOI] [PubMed] [Google Scholar]
- Fekete, E., and J. Szidonya. 1979. Abnormalities of ultrastructure and calcium distribution in the flight muscle of a flightless mutant of Drosophila melanogaster Acta Biol. 30:47–57. [PubMed] [Google Scholar]
- Ferrus, A., A. Acebes, M.C. Marin, and A. Hernandez-Hernandez. 2000. A genetic approach to detect muscle protein interactions in vivo. Trends Cardiovasc. Med. 10:293–298. [DOI] [PubMed] [Google Scholar]
- Fisher, A.J., C.A. Smith, J.B. Thoden, R. Smith, K. Sutoh, H.M. Holden, and I. Rayment. 1995. X-ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP.BeFx and MgADP.AlF4. Biochemistry. 34:8960–8972. [DOI] [PubMed] [Google Scholar]
- Fyrberg, E.A., S.I. Bernstein, and K. Vijay Raghavan. 1994. Basic methods for Drosophila muscle biology. Methods Cell Biol. 44:237–258. [DOI] [PubMed] [Google Scholar]
- Ganetzky, B., and C.F. Wu. 1983. Neurogenetic analysis of potassium currents in Drosophila: synergistic effects on neuromuscular transmission in double mutants. J. Neurogenet. 1:17–28. [DOI] [PubMed] [Google Scholar]
- George, E.L., M.B. Ober, and C.P. Emerson, Jr. 1989. Functional domains of the Drosophila melanogaster muscle myosin heavy-chain gene are encoded by alternatively spliced exons. Mol. Cell. Biol. 9:2957–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramates, L.S., and V. Budnik. 1999. Assembly and maturation of the Drosophila larval neuromuscular junction. Int. Rev. Neurobiol. 43:93–117. [DOI] [PubMed] [Google Scholar]
- Hess, N.K., and S.I. Bernstein. 1991. Developmentally regulated alternative splicing of Drosophila myosin heavy chain transcripts: in vivo analysis of an unusual 3′ splice site. Dev. Biol. 146:339–344. [DOI] [PubMed] [Google Scholar]
- Homyk, T., Jr., and C.P. Emerson, Jr. 1988. Functional interactions between unlinked muscle genes within haploinsufficient regions of the Drosophila genome. Genetics. 119:105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homyk, T., Jr., J. Szidonya, and D.T. Suzuki. 1980. Behavioral mutants of Drosophila melanogaster. III. Isolation and mapping of mutations by direct visual observations of behavioral phenotypes. Mol. Gen. Genet. 177:553–565. [DOI] [PubMed] [Google Scholar]
- Hong, C.S., and B. Ganetzky. 1994. Spatial and temporal expression patterns of two sodium channel genes in Drosophila. J. Neurosci. 14:5160–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley, A.F. 2000. Cross-bridge action: present views, prospects, and unknowns. J. Biomech. 33:1189–1195. [DOI] [PubMed] [Google Scholar]
- Jarecki, J., and H. Keshishian. 1995. Role of neural activity during synaptogenesis in Drosophila. J. Neurosci. 15:8177–8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronert, W.A., P.T. O'Donnell, A. Fieck, A. Lawn, J.O. Vigoreaux, J.C. Sparrow, and S.I. Bernstein. 1995. Defects in the Drosophila myosin rod permit sarcomere assembly but cause flight muscle degeneration. J. Mol. Biol. 249:111–125. [DOI] [PubMed] [Google Scholar]
- Kronert, W.A., A. Acebes, A. Ferrus, and S.I. Bernstein. 1999. Specific myosin heavy chain mutations suppress troponin I defects in Drosophila muscles. J. Cell Biol. 144:989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb, G.D. 2000. Excitation-contraction coupling in skeletal muscle: comparisons with cardiac muscle. Clin. Exp. Pharmacol. Physiol. 27:216–224. [DOI] [PubMed] [Google Scholar]
- Lehrer, S.S., and M.A. Geeves. 1998. The muscle thin filament as a classical cooperative/allosteric regulatory system. J. Mol. Biol. 277:1081–1089. [DOI] [PubMed] [Google Scholar]
- Littleton, J.T., H.J. Bellen, and M.S. Perin. 1993. Expression of synaptotagmin in Drosophila reveals transport and localization of synaptic vesicles to the synapse. Development. 118:1077–1088. [DOI] [PubMed] [Google Scholar]
- Littleton, J.T., E.R. Chapman, R. Kreber, M.B. Garment, S.D. Carlson, and B. Ganetzky. 1998. Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron. 21:401–413. [DOI] [PubMed] [Google Scholar]
- Mogami, K., and Y. Hotta. 1981. Isolation of Drosophila flightless mutants which affect myofibrillar proteins of indirect flight muscle. Mol. Gen. Genet. 183:409–417. [DOI] [PubMed] [Google Scholar]
- Naimi, B., A. Harrison, M. Cummins, U. Nongthomba, S. Clark, I. Canal, A. Ferrus, and J.C. Sparrow. 2001. A tropomyosin-2 mutation suppresses a troponin I myopathy in Drosophila. Mol. Biol. Cell. 12:1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nongthomba, U., M. Cummins, S. Clark, J.O. Vigoreaux, and J.C. Sparrow. 2003. Suppression of muscle hypercontraction by mutations in the myosin heavy chain gene of Drosophila melanogaster. Genetics. 164:209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, S.A., R.D. Fetter, J.N. Noordermeer, C.S. Goodman, and A. DiAntonio. 1997. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 19:1237–1248. [DOI] [PubMed] [Google Scholar]
- Poage, R.E., and S.D. Meriney. 2002. Presynaptic calcium influx, neurotransmitter release, and neuromuscular disease. Physiol. Behav. 77:507–512. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D. 2000. Reflections on a quarter century of research on contractile systems. Trends Biochem. Sci. 25:607–611. [DOI] [PubMed] [Google Scholar]
- Rafael, J.A., E.R. Townsend, S.E. Squire, A.C. Potter, J.S. Chamberlain, and K.E. Davies. 2000. Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. Hum. Mol. Genet. 9:1357–1367. [DOI] [PubMed] [Google Scholar]
- Rayment, I., W.R. Rypniewski, K. Schmidt-Base, R. Smith, D.R. Tomchick, M.M. Benning, D.A. Winkelmann, G. Wesenberg, and H.M. Holden. 1993. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 261:50–58. [DOI] [PubMed] [Google Scholar]
- Rayment, I., H.M. Holden, J.R. Sellers, L. Fananapazir, and N.D. Epstein. 1995. Structural interpretation of the mutations in the beta-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA. 92:3864–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedy, M.C., B. Bullard, and J.O. Vigoreaux. 2000. Flightin is essential for thick filament assembly and sarcomere stability in Drosophila flight muscles. J. Cell Biol. 151:1483–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivlin, P.K., R.M. St Clair, I. Vilinsky, and D.L. Deitcher. 2004. Morphology and molecular organization of the adult neuromuscular junction of Drosophila. J. Comp. Neurol. 468:596–613. [DOI] [PubMed] [Google Scholar]
- Rozek, C.E., and N. Davidson. 1983. Drosophila has one myosin heavy-chain gene with three developmentally regulated transcripts. Cell. 32:23–34. [DOI] [PubMed] [Google Scholar]
- Ruff, R.L. 2003. Neurophysiology of the neuromuscular junction: overview. Ann. NY Acad. Sci. 998:1–10. [PubMed] [Google Scholar]
- Seidman, J.G., and C. Seidman. 2001. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 104:557–567. [DOI] [PubMed] [Google Scholar]
- Simpson, P. 1983. Maternal-zygotic gene interactions during formation of the dorsoventral pattern in Drosophila embryos. Genetics. 105:615–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling, A.C., D. Stern, A. Beaton, E.J. Rhem, T. Laverty, N. Mozden, S. Misra, and G.M. Rubin. 1999. The Berkeley Drosophila Genome Project gene disruption project: single P-element insertions mutating 25% of vital Drosophila genes. Genetics. 153:135–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, B.A., H.L. Atwood, J.J. Renger, J. Wang, and C.F. Wu. 1994. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J. Comp. Physiol. [A]. 175:179–191. [DOI] [PubMed] [Google Scholar]
- Suzuki, D.T., T. Grigliatti, and R. Williamson. 1971. Temperature-sensitive mutations in Drosophila melanogaster. VII. A mutation (para-ts) causing reversible adult paralysis. Proc. Natl. Acad. Sci. USA. 68:890–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigoreaux, J.O. 2001. Genetics of the Drosophila flight muscle myofibril: a window into the biology of complex systems. Bioessays. 23:1047–1063. [DOI] [PubMed] [Google Scholar]
- Wassenberg, D.R., II, W.A. Kronert, P.T. O'Donnell, and S.I. Bernstein. 1987. Analysis of the 5′ end of the Drosophila muscle myosin heavy chain gene. Alternatively spliced transcripts initiate at a single site and intron locations are conserved compared to myosin genes of other organisms. J. Biol. Chem. 262:10741–10747. [PubMed] [Google Scholar]
- Wu, C.F., B. Ganetzky, F.N. Haugland, and A.X. Liu. 1983. Potassium currents in Drosophila: different components affected by mutations of two genes. Science. 220:1076–1078. [DOI] [PubMed] [Google Scholar]
- Zhang, S., and S.I. Bernstein. 2001. Spatially and temporally regulated expression of myosin heavy chain alternative exons during Drosophila embryogenesis. Mech. Dev. 101:35–45. [DOI] [PubMed] [Google Scholar]