

Abstract
Protein translocons of the mammalian endoplasmic reticulum are composed of numerous functional components whose organization during different stages of the transport cycle in vivo remains poorly understood. We have developed generally applicable methods based on fluorescence resonance energy transfer (FRET) to probe the relative proximities of endogenously expressed translocon components in cells. Examination of substrate-engaged translocons revealed oligomeric assemblies of the Sec61 complex that were associated to varying degrees with other essential components including the signal recognition particle receptor TRAM and the TRAP complex. Remarkably, these components not only remained assembled but also had a similar, yet distinguishable, organization both during and after nascent chain translocation. The persistence of preassembled and complete translocons between successive rounds of transport may facilitate highly efficient translocation in vivo despite temporal constraints imposed by ongoing translation and a crowded cellular environment.
Keywords: FRET; TRAP; protein translocation; Sec61; TRAM
Introduction
The biogenesis of secretory and membrane proteins in mammalian cells involves several discrete steps that include targeting of nascent polypeptides to the ER, their cotranslational transport across or integration into the ER membrane, and various modification, folding, and maturation events (for review see Rapoport et al., 1996; Johnson and van Waes, 1999). The ability to reconstitute this entire process in a cell-free system amenable to biochemical fractionation has allowed each stage to be studied in isolation and has facilitated the identification of the respective factors involved. Collectively, the multiple components that define the machinery for cotranslational protein translocation constitute the translocon.
Central among the translocon components is the heterotrimeric Sec61 complex (composed of α, β, and γ subunits), multiple copies of which are thought to be assembled to form the core of a protein-conducting translocation channel (Hanein et al., 1996; Beckmann et al., 1997, 2001; Menetret et al., 2000; Morgan et al., 2002). Signal sequence–containing nascent polypeptides, recognized first by the cytoplasmic signal recognition particle (SRP), are targeted to the Sec61 channel by another translocon component, the heterodimeric SRP receptor (SR; for review see Rapoport et al., 1996). Although some nascent chains can subsequently insert directly into the Sec61 channel (Jungnickel and Rapoport, 1995), most substrates require the aid of at least one of two additional translocon components, the TRAM protein (Gorlich and Rapoport, 1993; Voigt et al., 1996) and/or the heterotetrameric TRAP complex (Fons et al., 2003). Once these critical first steps result in a commitment to initiate substrate translocation, other translocon components can interact with the nascent chain during its transport to catalyze reactions such as signal sequence cleavage and glycosylation. Thus, a biochemical approach has provided the general framework for ordering a succession of individual steps whose mechanistic basis can be further dissected.
In spite of this level of mechanistic insight, basic aspects of translocon composition and organization during its functional cycle in vivo remain poorly or not at all understood. One of the most elemental yet unresolved issues in this regard is the fate of a translocon, and in particular the translocation channel, between rounds of substrate transport. The first of two general possibilities is that the constituents of the channel, whose assembly would be directed by a nascent substrate, are disassembled upon completion of transport. Alternatively, the channel may remain assembled when not in use and closed on one or both ends to prevent compromising the permeability barrier of the ER.
The currently available data from biochemical studies have yielded evidence for both situations. Despite the lack of copurification of various components of the translocon, they can nonetheless be core-constituted into proteoliposomes that support functional protein transport (Gorlich and Rapoport, 1993). In addition, purified Sec61 complex incorporated into lipid vesicles was not observed to oligomerize into channel-like structures until the addition of ribosomes (Hanein et al., 1996). Both of these observations show that translocation channels can be assembled de novo from their isolated constituents, lending credence to a model involving use-dependent assembly and disassembly of the translocon.
In contrast, vacating the translocation channel of its nascent chain in vitro with puromycin did not result in a gross structural change or a reduction in the number of translocons that were observed by freeze-fracture EM (Hanein et al., 1996). In addition, puromycin was observed to stimulate the flux of both ions (Simon and Blobel, 1991) and a noncharged small molecule (Roy and Wonderlin, 2003) through channels that had presumably been occluded by translocating substrates. Thus, translocons may not disassemble or even close upon completion of substrate transport.
However, it is presumed that a pore large enough to transport a protein could not remain open while not in use because the permeability barrier of the ER would be compromised (Crowley et al., 1994). Indeed, ER-derived microsomal vesicles after removal of their lumenal contents with alkaline extraction were permeable to ions through channels that could be sterically blocked with either ribosomes or antibodies against Sec61α or TRAM (Hamman et al., 1998). These channels, speculated to reflect the configuration of translocons between rounds of translocation, were not only smaller than an active translocon (pore size of ∼0.9–1.7 nm vs. ∼4–6 nm) but also capable of being sealed to ions by the ER lumenal chaperone BiP (Hamman et al., 1998). These observations suggest a model in which completion of translocation results in a substantial change in the organization of translocons that remain assembled between rounds of transport. However, such large changes in pore size have yet to be observed in cryo-EM images (at ∼1.5–2 nm resolution) comparing translocons assembled onto either empty or nascent chain–containing ribosomes (Menetret et al., 2000; Beckmann et al., 2001). Recently, an X-ray structure of the archaeal Sec61 homologue (termed SecYEβ) suggested a reversibly occludable pore within a single heterotrimer (Van den Berg et al., 2004), raising the possibility that the functional state of a translocon may change without necessarily requiring changes in the overall assembly or organization of translocon components. Thus, the nature of organizational changes during the transport cycle of the core Sec61 translocation channel remains uncertain, with different conclusions being reached with different methods.
Although considerable functional data are available for SR and to a lesser extent TRAM and the TRAP complex, little is known regarding the contributions of these components to either the composition or the architecture of a native translocon. The functional activities of each protein appear to be required at specified times during substrate translocation. Whether or not these proteins are stably assembled into native translocons or recruited transiently in a use-dependent manner is not known. Analysis by blue native PAGE did not reveal clear evidence for oligomeric complexes containing any two of the essential translocon components Sec61, SR, TRAM, or TRAP (Wang and Dobberstein, 1999). Similarly, no two of these components have been observed to copurify in studies of their functional purification and reconstitution (Gorlich et al., 1992; Gorlich and Rapoport, 1993; Fons et al., 2003). Thus, biochemical analyses to this point have failed to clearly reveal stable interactions between various translocon components that must nonetheless be near each other, at least transiently, during specific stages of translocation.
Together, these various observations show that not only is the issue of translocation channel assembly and disassembly uncertain but also the nature of different structural states representing active and inactive translocons remains obscure. To begin addressing these unanswered questions regarding translocon organization, we have developed a method based on fluorescence resonance energy transfer (FRET) to probe the relative proximities of endogenously expressed translocon components in cells at low nanometer resolution. This approach was subsequently exploited to provide new insights into both the composition and organizational state of the translocon during its functional cycle in cells.
Results
Experimental rationale and strategy
An investigation of translocon composition and organization in cells requires methodology capable of providing information on the relative proximities of the constituent proteins at low nanometer resolution. With such a tool, an analysis of the positions of different combinations of translocon components can be used to infer their direct associations, and hence general features of their overall organization. By examining changes in this organization under different cellular conditions, one can potentially gain insight into the critical transition between inactive and active functional states of translocons. In this manner, we sought to distinguish between different viable models of translocon dynamics based on in vitro studies.
FRET between two fluorescently labeled proteins provides a highly sensitive and specific probe of their proximity in the low- to subnanometer range (see online supplemental material and references therein, available at http://www.jcb.org/cgi/content/full/jcb.200312079/DC1). To apply this methodology to the endogenously expressed native translocons of cultured cells, we decided to label individual components using epitope-specific antibodies directly conjugated to fluorescent dyes. We reasoned that despite their large size, FRET between labeled antibodies could potentially provide useful information regarding the proximities and organization of their bound antigens. In the first part of this work (presented largely in online supplemental material), we combined modeling and simulations to consider the feasibility of this approach to address the anticipated questions. This methodology was applied to address unanswered questions regarding the composition and organization of translocons at different stages of the transport cycle in cells.
Modeling and simulations of FRET between dye-conjugated antibodies bound to antigens in various configurations and densities were used to estimate the resolving power of antibody-mediated FRET (see online supplemental material). From these analyses, we conclude that antibody-mediated FRET should be capable of the following activities: (a) distinguishing differences in antigen proximity at the 0.5–1-nm scale; (b) distinguishing between assembled and unassembled states of oligomeric structures; and (c) discriminating even small differences in relative distances that might accompany a structural change of a multiprotein complex. These conclusions suggested that, in principle, the organization of multiprotein complexes could be assessed with antibody probes. Thus, if endogenous translocon components could be labeled with dye-conjugated antibodies while maintaining their overall organization, FRET between the dyes should provide insight into translocon composition and organization.
To apply these ideas, we used variations of previous methodology to label cellular proteins while preserving their relative proximities (Fig. 1 A). Intact live cells are rapidly fixed in formaldehyde to stabilize the in situ organization of cellular proteins with covalent cross-links between closely juxtaposed components. Because cross-links mediated by formaldehyde are extremely short (∼0.2 nm) and rapidly induced, proteins are effectively immobilized in place without an opportunity to substantially change their relative proximities or organization (Jackson, 1999). Because the preservation is covalent and, under the conditions applied here, effectively irreversible, the samples can subsequently be subjected to nonphysiological conditions that would have otherwise disrupted labile or transient interactions.
Figure 1.

Detection of FRET between fluorescently conjugated antibodies in situ. (A) Scheme for preparation of cells for FRET analysis. Insets show an individual ribosome–translocon complex. Red hatches in the inset indicate formaldehyde cross-links between interacting proteins. The dotted lines indicate permeabilization (of the cellular membranes) or partial digestion (in the case of the ribosome). In this and all subsequent diagrams, the Cy3-labeled donor and Cy5-labeled acceptor antibodies are green and red, respectively. See text for complete details. (B) Topology maps of ER proteins analyzed in this study. The positions of peptide epitopes for antibodies are indicated with white boxes. (C) Western blot analysis of canine pancreatic ER microsomes with preimmune (P) or immune (I) antisera against the indicated antigens. (D) Indirect immunofluorescence microscopy of MDCK cells with anti-Sec61β. (E) MDCK cells were stained with Sec61βCy5 followed by 2°Cy3. Shown are the fluorescence images for Cy3 (donor), Cy5 (acceptor), and merged channels (prebleach images). The Cy5 dye in the region indicated by the box was selectively photobleached, and the donor and acceptor channels imaged a second time (postbleach images). The percent change in donor intensity between the pre- and postbleach images was calculated and represented as a pseudocolor map. Note the >40% increase in donor fluorescence intensity within the boxed area upon acceptor photobleaching. (F) MDCK cells were stained with 2°Cy3 bound to unlabeled anti-CNX followed by Sec61βCy5, and the images were collected and analyzed as in E. Note the near absence of change in donor intensity upon acceptor photobleaching. The color scale represents the percentage of energy transfer (%E) in the pseudocolored map. Bars: (D–F) 5 μm.
This condition allows the fixed cells to be permeabilized and labeled with fluorescent antibodies without changing the organization of the translocon components or the multiprotein complexes in which they are assembled (Fig. 1 A). Due to the fixed nature of the antigens, the bivalent antibodies cannot induce gross changes in the existing organization of their antigens. Instead, the antibodies serve to “mark” the positions of their antigens with fluorescent probes whose relative proximities can subsequently be assessed by FRET (e.g., as modeled in online supplemental material). Thus, the ability to assess what the state of the translocon had been in intact cells under different conditions should permit us to ask if, in cells, translocon organization changes at different stages of the translocation cycle.
The sensitivity and specificity of FRET between dye-conjugated antibodies
Antibodies raised against small peptide epitopes in the cytoplasmic domains of the translocon components Sec61α, Sec61β, TRAM, TRAPα, and SRα (Fig. 1 B) were characterized by immunoblotting against ER-derived microsomes and cell lysates and indirect immunofluorescence of cultured cells (Fig. 1, C and D; and Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200312079/DC1; unpublished data). Importantly, maximal antibody binding to certain epitopes required pretreatment of the fixed and permeabilized cells with RNase (see online supplemental material; Fig. S1, B and C). Although RNase may not significantly disrupt overall ribosome structure, particularly in fixed samples, it would still digest surface-exposed loops of rRNA. The removal of these loops, together with the fact that even undigested ribosomes appear to be separated from translocons by a substantial gap (Hanein et al., 1996; Beckmann et al., 1997, 2001; Menetret et al., 2000; Morgan et al., 2002), appears to be sufficient to provide antibody access to translocon component epitopes that are otherwise sterically occluded. Whatever the precise explanation, the unmasking of epitopes is an important step that facilitates a uniform level of labeling in cells regardless of whether their translocons were actively engaged by translating ribosomes or not (Fig. S1, E and F). Therefore, we included this RNase-mediated unmasking step in all of the experiments.
For FRET analyses, antibodies were conjugated to the fluorescent dyes Cy3 or Cy5, which serve as the donor and acceptor, respectively (see online supplemental material). Then, we established conditions that allow the detection of FRET between closely juxtaposed but not more widely separated labeled antibodies. In this experiment, Cy5-conjugated anti-Sec61β (Sec61βCy5) bound to its antigen in MDCK cells served as the acceptor. The donor antibody, a Cy3-conjugated anti–rabbit secondary antibody (2°Cy3), was positioned in one of two places (Fig. 1, E and F, diagrams). In Fig. 1 E, the 2°Cy3 was bound directly to the Sec61βCy5 antibody, ensuring that all donor antibodies are adjacent to an acceptor. Alternatively, the 2°Cy3 antibody was bound to an unlabeled antibody against the COOH terminus of calnexin (CNX; Fig. 1 F), a resident ER membrane protein involved in the posttranslational quality control of proteins (Bergeron et al., 1994). Because in either case both the acceptor and donor antibodies are bound (either directly or indirectly) to ER antigens, their fluorescent signals colocalize at the resolution of light microscopy (Fig. 1, E and F). In striking contrast, the different relative positions of the donor could be readily discriminated by assessing the FRET between the Cy3 and Cy5 dyes (Fig. 1, E and F).
To quantitatively reveal and measure FRET, the Cy3 donor fluorescence is monitored before and after the Cy5 acceptor dyes are selectively destroyed by high-intensity laser photobleaching. Upon destruction of Cy5, the Cy3 fluorescence should increase by an amount corresponding to the proportion of energy that had been transferred to Cy5 (Kenworthy, 2001). It should be noted that in addition to FRET, the apparent increase in Cy3 fluorescence intensity can, under some circumstances, be caused by the photoconversion of Cy5 to a different fluorescent species that partially overlaps with the Cy3 spectra. However, in our work, this possibility was not found to be a significant source of error in the Cy3 measurements (see online supplemental material for a detailed discussion). Thus, the increase in Cy3 intensity seen with Cy5 photobleaching is largely reflective of FRET between the two fluorophores.
This increase in fluorescence intensity can be qualitatively appreciated by comparing the pre- and postbleach images of the donor (Fig. 1 E). The extent of donor dequenching can be measured in each 8 × 8 pixel region (equal to ∼0.3 μm2) and represented as a pseudocolored map that quantitatively displays the percentage of energy transfer within the photobleached region. A comparison of the percentage of energy transfer maps for Fig. 1 (E and F) reveals that the overall efficiency of FRET from the 2°Cy3 to Sec61βCy5 can vary from >40% (Fig. 1 E) to <2% (Fig. 1 F) depending on whether or not the two antibodies are directly juxtaposed or simply in the same subcellular compartment. Thus, sublight resolution differences in the relative positions of donor- and acceptor-labeled antibodies can readily be distinguished and quantified by FRET measured with acceptor photobleaching methodology.
Detection of protein proximities within translocons
Distinguishing between different models of translocon dynamics requires the reliable discrimination of assembled from disassembled multiprotein structures. To determine if antibody-mediated FRET can provide this resolution, we assessed the proximities between two resident ER membrane proteins that should be largely assembled versus two that are primarily disassociated at steady state. For the assembled case, we chose Sec61α and Sec61β, constituents of a stable Sec61 complex whose subunits do not appear to exist in a significant free pool (Gorlich and Rapoport, 1993). For comparison, we probed FRET between Sec61β and another resident ER membrane protein expressed at similar levels, CNX. Although CNX is not generally considered a core component of the translocon, a small population of CNX has been demonstrated to interact with a subset of proteins still undergoing translocation (Molinari and Helenius, 2000). Thus, the transient nature of the interactions between CNX and nascent translocating proteins results in the close proximity between Sec61β and CNX for only a small percentage of the total CNX, thereby representing a largely unassembled state between these two ER proteins.
FRET, comparable to that seen for directly interacting donor and acceptor antibodies, was observed between Sec61αCy3 and Sec61βCy5 (Fig. 2, A and C). For the same Sec61βCy5 acceptor, a CNXCy3 donor antibody directed against the cytoplasmic COOH terminus of CNX exhibited severalfold lower FRET efficiency compared with Sec61αCy3 (Fig. 2, B and C). In addition to the markedly lower FRET for the CNXCy3 donor, several other observations confirmed the specificity of the Sec61αCy3/Sec61βCy5 FRET. First, omission of the Sec61βCy5 resulted in no observable FRET (i.e., no change in Sec61αCy3 intensity on photobleaching in the Cy5 channel; unpublished data). Second, inclusion of a competitor Sec61β peptide resulted in reduced labeling by Sec61βCy5 and correspondingly lower FRET (unpublished data). Third, reducing the concentration of Sec61βCy5 also resulted in lower FRET (Fig. S9, available at http://www.jcb.org/cgi/content/full/jcb.200312079/DC1), as would be expected for lower occupancy levels of acceptor antigens with antibody (Fig. S4 E, available at http://www.jcb.org/cgi/content/full/jcb.200312079/DC1). In contrast, changing the levels of Sec61αCy3, although changing the overall brightness of the donor labeling, did not affect FRET efficiency significantly (Fig. S8, available at http://www.jcb.org/cgi/content/full/jcb.200312079/DC1), also as predicted from theoretical considerations (Fig. S4 F). Together, these results demonstrate that interactions between membrane proteins can be detected with antibody-mediated FRET in situ and establish that differences in the assembly status of abundantly expressed resident ER membrane proteins can be easily discriminated.
Figure 2.
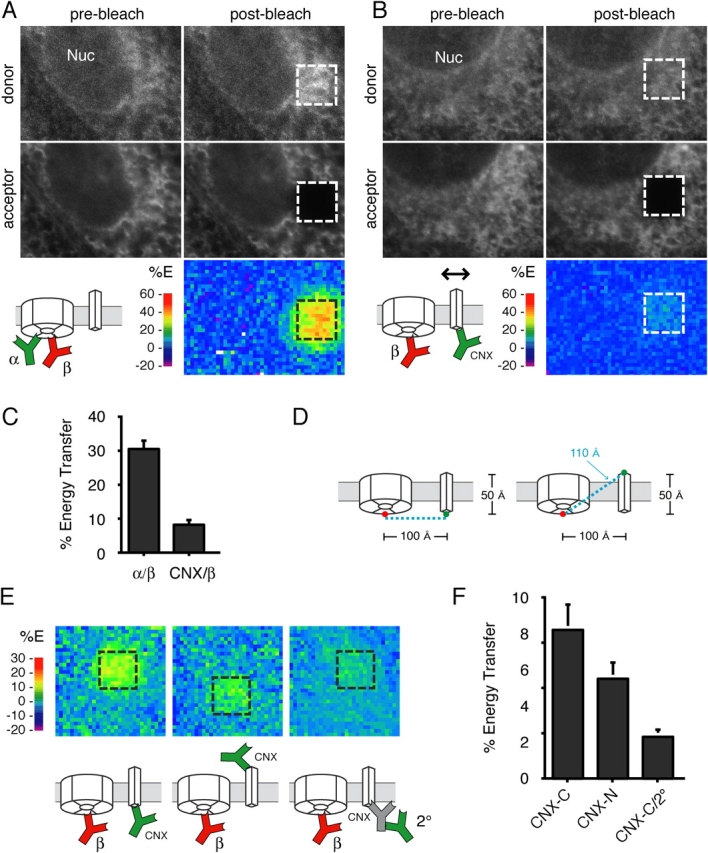
Discrimination of protein proximities with low nanometer resolution. (A and B) MDCK cells were double labeled with Sec61αCy3 and Sec61βCy5 (A) or CNXCy3 and Sec61βCy5 (B) and analyzed for FRET as in Fig. 1 E. (C) Quantitative analysis of FRET between the antibody configurations shown in A and B. The antibody pair is indicated on the x-axis, with Sec61βCy5 serving as the acceptor in each case. Each bar represents the mean ± SD (n = 10), with statistically significant differences (P < 0.01 using t test) being observed between the two conditions. (D) Illustration of the relative difference in distance for antibodies on the cytoplasmic and lumenal sides of CNX. Note that moving the donor antibody to the other side of an ∼5-nm-thick membrane changes the distance to the acceptor antibody by only ∼1 nm in this example. (E) Representative energy transfer maps of FRET to Sec61βCy5 from Cy3-labeled antibodies in three different positions. The left and middle panels used directly conjugated anti-CNX antibodies against the COOH and NH2 terminus, respectively. The right panel used 2°Cy3 bound to an unlabeled anti-CNX COOH terminus antibody. The color scale is different than in A and B to enhance visualization of differences between the energy transfer maps. (F) Quantitative analysis of FRET between the antibody configurations shown in E. The donor antibody is indicated on the x-axis, while Sec61βCy5 served as the acceptor in each case. Each bar represents the mean ± SD (n = 10), with statistically significant differences (P < 0.01 using t test) being observed between any two conditions.
Because some models of translocon dynamics posit small, conformational changes rather than gross assembly and disassembly, we also sought to experimentally determine if small changes in antigen proximity at the low nanometer scale could be reliably discriminated. Because FRET efficiency for a single donor and acceptor dye pair is inversely related to the sixth power of the distance between them (see online supplemental material and references therein), small increases in separation distance markedly reduce the energy transfer. Our simulations suggested that, even for highly flexible antibodies stochastically labeled with multiple dyes, changes in antibody proximities corresponding to even 1 nm should be readily detectable (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200312079/DC1). To assess this experimentally, we exploited an antibody against a different, NH2-terminal epitope on the lumenal domain of CNX. Although the proximity of CNX to Sec61β yields a relatively low overall FRET signal (consistent with a separation distance of ∼10 nm), we asked if changing the donor antibody position from the COOH to the NH2 terminus on CNX could be detected as a change in FRET. Remarkably, this relatively subtle repositioning of the donor antibody with respect to Sec61βCy5 (Fig. 2 D) decreased the FRET efficiency by nearly half of that seen with the COOH-terminal antibody (Fig. 2, E and F).
Positioning the donor antibody still further away from the COOH terminus of CNX (by using the 2°Cy3 donor bound to an unlabeled CNX COOH-terminal antibody) decreased the FRET efficiency to near background levels (Fig. 2, E and F). Based on the dimensions of the unlabeled spacer IgG, we infer that the 2°Cy3 donor antibody could be at most 15 nm further from the acceptor than a directly labeled CNX Cy3 COOH-terminal donor. Thus, antibody-based FRET is sufficiently sensitive to detect changes in distance in the size range of individual proteins, roughly 1–15 nm. These distances are particularly relevant to the objectives of this paper because the translocon dimensions fall within this range. In addition, two ribosome-bound translocons are sterically blocked from coming closer than 25 nm to each other (Fig. S3 B, available at http://www.jcb.org/cgi/content/full/jcb.200312079/DC1), indicating that intertranslocon FRET should not be detectable by antibody-based FRET. Thus, both theoretical predictions (Kubitscheck et al., 1993; Kenworthy and Edidin, 1998; online supplemental material) and the donor moving experiments (Fig. 2, E and F) provided confidence that antibody-based FRET was sensitive enough to measure both gross and subtle changes in the organization of components within translocons.
The Sec61 complex assembles into oligomers in cells
Purified Sec61 complex, both in solution (Hanein et al., 1996) and when bound to a eukaryotic ribosome (Menetret et al., 2000; Beckmann et al., 2001), is capable of forming ∼10-nm-diam toroidal structures estimated to contain at least three copies of the Sec61 heterotrimer. However, demonstrating the stoichiometry of the Sec61 complex in native translocons has proven more difficult to address. By cryo-EM, native channel complexes were larger, had a differently shaped and larger central pore, and contained a substantial lumenal protrusion not seen with Sec61p channels (Menetret et al., 2000; Morgan et al., 2002). Because a ribosome engaged in translocation tightly associates with several other membrane proteins comparable in abundance with the Sec61 complex (Gorlich et al., 1992; Matlack and Walter, 1995; Menetret et al., 2000), the composition and stoichiometry of the components that define the native translocation channel remain uncertain.
To determine if native translocons contain Sec61 oligomers, we asked whether or not FRET could be observed between two copies of a Sec61 complex subunit by labeling cells with a mixture of Sec61βCy3 and Sec61βCy5 antibodies at a relative ratio of 1:8. If homooligomers exist, each copy of the donor-labeled Sec61β is likely to be adjacent (within 10–15 nm) to a copy of the far more numerous acceptor-labeled antibodies. In contrast, a single Sec61 complex per translocon should result in an inter-Sec61β distance, even within polysomes, of at least 25 nm, a distance beyond which little or no FRET is observed. Thus, the ∼17% FRET efficiency for Sec61βCy3/Sec61βCy5 pair (Fig. 3 A) provides direct evidence that native translocons in cells are composed of at least two Sec61 heterotrimers.
Figure 3.

Detection and characterization of Sec61 complex oligomerization in situ. (A–C) Direct comparisons of FRET efficiencies to Sec61βCy5 from three different donor antibodies: Sec61βCy3 (at a ratio of 1:8; A), Sec61αCy3 (B), and 2°Cy3 bound to unlabeled anti-Sec61α (C). (D) Quantitation (mean ± SD; n = 10) of the comparisons from A–C are shown.
Direct comparisons of the FRET efficiency for Sec61βCy3/Sec61βCy5 with that observed between Sec61α and Sec61βCy5 provided additional support for this conclusion. Here, directly conjugated Sec61α donor antibodies displayed a FRET efficiency to Sec61βCy5 higher than that seen with Sec61βCy3/Sec61βCy5 (∼30% vs. ∼17%; Fig. 3, A and B). In contrast, spacing the donor antibody using an unlabeled Sec61α antibody showed a lower FRET efficiency (∼7%; Fig. 3 C). Because the spacer antibody dimensions are at most 15 nm and the Sec61βCy3/Sec61βCy5 FRET efficiency is bracketed between the FRET values for directly conjugated Sec61α and that seen with the spacer antibody, we conclude that one copy of Sec61β must be within ∼15 nm of another copy. In other experiments, Sec61αCy3/Sec61αCy5 also displayed comparably high FRET (see the following section; Table I), providing additional evidence for oligomeric Sec61 heterotrimers. These values are consistent with the estimated dimensions of native translocons (Menetret et al., 2000) and theoretical simulations of antibodies separated by roughly 8–10 nm (see online supplemental material). Together, these results strongly argue that the observed FRET for Sec61βCy3/Sec61βCy5 and Sec61αCy3/Sec61αCy5 is detecting oligomerized Sec61 complexes within native translocons. Thus, the data in Figs. 2 and 3 not only demonstrate that protein–protein interactions within the translocon can be detected with high resolution in cells but that the proximities of copies of a subunit of the Sec61 complex can be used to directly probe its oligomeric status.
Table I.
Changes in Sec61 complex configuration during the translocation cycle
| Donor | Acceptor | Untreated | Puromycin | Pactamycin |
|---|---|---|---|---|
| Sec61α | Sec61α | 38.3 ± 4.4 | 33.6 ± 7.2a | 35.5 ± 5.1a |
| Sec61α | Sec61β | 26.4 ± 2.1 | 27.9 ± 2.7a | 28.0 ± 2.1a |
| Sec61β | Sec61α | 41.8 ± 3.1 | 46.1 ± 2.8a | 46.8 ± 3.0a |
| Sec61β | Sec61β | 21.4 ± 3.6 | 24.8 ± 6.1a | 23.9 ± 2.5a |
FRET efficiencies (mean ± SD; n = 40) for the indicated donor–acceptor antibody pairs were measured on cells that were either left untreated or pre-treated for 15 min with 1 mM puromycin or 0.2 μM pactamycin. Each value is the mean of measurements collected from four separate experiments performed on multiple days.
Treatment is significant (P ≤ 0.01) compared with untreated cells using t test.
Core translocon organization during the transport cycle
These results permitted us to simply and directly address a long-standing unresolved issue regarding the dynamics of the translocon: what happens to the translocation channel when it is not actively engaged in substrate transport? To resolve this question, we first established conditions to complete translocation by terminating protein synthesis either prematurely with puromycin, which releases nascent polypeptides from the ribosome, or naturally with pactamycin, an inhibitor of translational initiation. Pulse labeling of cells treated with 1 mM puromycin demonstrated that within 5 min, the synthesis of radiolabeled proteins longer than ∼50–70 residues was effectively inhibited due to premature termination. Pactamycin also inhibits new protein synthesis within 5 min, but as expected for an inhibitor of initiation, an additional 10 min was required to complete translation of already engaged mRNAs (Fig. 4 A). Importantly, >95% of the translocons were bound to ribosomes in a salt-resistant manner, whereas <5% remained bound if cells were pretreated with puromycin (Fig. 4 B). Because the high salt containing solubilization conditions used in the fractionation studies maintain ribosome–translocon interactions only in the presence of a nascent chain (Gorlich et al., 1992; Jungnickel and Rapoport, 1995), this result demonstrates that the vast majority of translocons are engaged in translocation in untreated cells, whereas they are nearly quantitatively vacated upon treatment with puromycin (or pactamycin; unpublished data).
Figure 4.

Translocon component organization during the translocation cycle. (A) MDCK cells were either left untreated (unt.) or pretreated with 1 mM puromycin (puro.) or 0.2 μM pactamycin (pact.) for the indicated times before pulse labeling for 10 min with [35S]methionine in the presence of the drug. Shown are autoradiographs of total cell lysates. Coomassie blue staining of the gel revealed equal amounts of total protein in each lane (not depicted). (B) Untreated or puromycin-treated (1 mM for 15 min) cells were lysed in detergent (0.8% deoxyBigCHAP) and high salt (500 mM KAc) and analyzed by sucrose density gradient sedimentation for Sec61 complex association with ribosomes. Shown are immunoblots for Sec61β and CNX of individual fractions of the gradient. The position of ribosomes on the gradient is indicated. (C–F) Untreated, puromycin-treated, and pactamycin-treated cells were analyzed for FRET between Sec61βCy3/Sec61βCy5, Sec61βCy3/Sec61αCy5, Sec61αCy3/Sec61βCy5, and Sec61αCy3/Sec61αCy5 as indicated. Each point on the graph represents a single FRET measurement performed as in Fig. 1 D. n = 10 for each condition tested.
Next, we investigated whether or not the oligomeric state of the Sec61 complex in cells was maintained or lost upon the completion of substrate translocation by measuring FRET between all combinations of Sec61α and Sec61β under the two extremes of translocon usage and quiescence. Ten independent FRET measurements were performed on separate cells that were either treated with puromycin or pactamycin or left untreated (Fig. 4, C–F). For each antibody pair, the FRET efficiencies were comparable for actively engaged versus nontranslocating translocons. Importantly, our observation that in untreated cells, the majority of Sec61α or Sec61β labeling is dependent on RNase digestion (Fig. S1) suggests that we are indeed visualizing and making measurements on engaged translocons. Thus, the finding of comparable FRET values after puromycin and pactamycin treatments suggests that the oligomeric structure of active translocons is maintained even after completion of substrate translocation. Therefore, we conclude that the core of a native translocation channel does not disassemble into its individual components upon completion of substrate transport in vivo.
What then distinguishes an engaged from a quiescent translocon? As discussed in the Introduction, the translocon could be structurally identical in both cases (Menetret et al., 2000) or could undergo a conformational change (Hamman et al., 1997; Beckmann et al., 2001). Because antibody-mediated FRET, in principle, can discriminate even small differences in the distances separating antigens, we used this method to determine if a change in organization distinguishes translocating from quiescent translocons in cells. Careful examination of the data in Fig. 4 hinted at small, systematic differences in FRET efficiencies for some of the antibody pairs upon completion of translocation. This possibility was verified with more thorough analyses compiling 40 independent FRET measurements of engaged versus inactive translocons for each antibody pair. Statistical comparisons of untreated versus treated cells revealed small but significant differences for each of the donor/acceptor pairs (Table I).
Several observations suggest that the changes in FRET are specific and reflect the changes in organization that distinguish engaged and inactive translocons. First, the level of antibody labeling was not different for either Sec61α or Sec61β in untreated versus puromycin or pactamycin treated cells (Fig. S1, E and F). This finding argues against changes in FRET due simply to differential labeling or antibody accessibility. Second, two qualitatively different methods of vacating the translocon shift the FRET value in the same direction (either up or down) for each of the four antibody pairs tested. In addition, the lack of statistically significant differences between puromycin and pactamycin treatments for any antibody pair argues that both methods of completing translocation are equivalent from the standpoint of the structural state achieved by the Sec61 components. Third, the FRET values for the reciprocal antibody pairs Sec61αCy3/Sec61βCy5 and Sec61βCy3/Sec61αCy5 similarly increase upon completion of translocation, as would be expected for simply switching the donor and acceptor fluorophores on the same antibodies. The difference in the absolute FRET values observed for Sec61αCy3/Sec61βCy5 versus Sec61βCy3/Sec61αCy5 is likely due to small differences in dyes per antibody for the different antibody preparations and the efficiency of antigen occupancy for the two antibodies (Fig. S4). Fourth, the increase in FRET for Sec61βCy3/Sec61βCy5 with a concomitant decrease for Sec61αCy3/Sec61αCy5 argues against additional or fewer Sec61 heterotrimers being assembled into oligomers. Instead, this result, coupled with the increased FRET for both Sec61αCy3/Sec61βCy5 and Sec61βCy3/Sec61αCy5, is most consistent with the preexisting oligomerized Sec61 complexes undergoing a small but detectable change in configuration.
TRAM, TRAP, and SR associations with translocons
If the core translocon remains intact throughout the translocation cycle, what happens to proteins that do not necessarily stably associate with the translocon, and yet are essential for discrete steps of translocation? To address this question, we focused on three proteins, SRα, TRAM, and TRAPα, each of which are thought to be needed during early stages of translocation. Whether or not they are needed at stages after the initiation of substrate translocation remains unclear. Similarly, it is not known if they are recruited to translocons transiently in a use-dependent manner (e.g., as CNX is thought to be) or if they instead are more stably part of the translocon structure (e.g., as are subunits of the Sec61 complex).
We reasoned that these possibilities should be discernible with FRET in two ways. First, FRET between each component and the Sec61 complex under conditions of ongoing translocation may reveal whether, at steady-state, a significant proportion of the proteins are associated with the core translocon or not. Second, if their associations are use- or substrate-dependent, relieving translocons of their substrates with puromycin or pactamycin should result in a loss of FRET signal. Analysis of FRET between SRα, TRAM, and TRAPα donor antibodies with Sec61αCy5, and to a lesser extent Sec61βCy5, revealed FRET efficiencies higher than that seen with a CNX donor (Table II; compare for example with Fig. 2, B–D). Instead, many of the FRET efficiencies were more comparable to that seen between subunits of the Sec61 complex (Table I). It should be noted that the systematically lower FRET values seen with the Sec61β acceptor (Table II) appear to be due to small differences in dyes per IgG for the different antibody preparations and do not necessarily indicate a higher distance separating the antigens. That notwithstanding, the data suggest that at steady state, at least some proportion of SRα, TRAM, and TRAPα is likely to be part of Sec61-containing translocons.
Table II.
Maintenance of translocon component interactions during the translocation cycle
| Donor | Acceptor | Untreated | Puromycin | Pactamycin |
|---|---|---|---|---|
| SRα | Sec61α | 34.1 ± 5.2 | 31.6 ± 7.3 | 32.2 ± 4.2 |
| SRα | Sec61β | 16.0 ± 2.2 | 17.2 ± 3.3 | 18.8 ± 2.8a |
| TRAPα | Sec61α | 18.4 ± 6.4 | 13.6 ± 5.6a | 15.2 ± 4.9 |
| TRAPα | Sec61β | 13.6 ± 3.5 | 11.1 ± 3.1a | 11.8 ± 2.7 |
| TRAM | Sec61α | 17.6 ± 4.2 | 19.8 ± 2.9a | 19.7 ± 4.4 |
| TRAM | Sec61β | 12.8 ± 2.7 | 11.9 ± 2.0 | 10.4 ± 1.1a |
FRET efficiencies for the indicated donor–acceptor antibody pairs were measured and tabulated as in Table I.
Treatment is significant (P ≤ 0.01) compared with untreated cells using t test.
However, the absolute amounts of SR, TRAM, and TRAP that are incorporated into translocons are difficult to judge based on the FRET results alone. For the TRAP complex, previous biochemical analyses have demonstrated that the vast majority is tightly associated with membrane-bound ribosomes (Gorlich and Rapoport, 1993; Matlack and Walter, 1995), which is consistent with a high degree of incorporation into translocons. Similar observations for TRAM and SR are lacking because under the conditions analyzed any potential association that might have existed in intact membranes was not maintained upon detergent solubilization. However, our observation that SRα and TRAM gave quantitatively similar FRET values as TRAPα for both the Sec61α and Sec61β acceptors may suggest that, in cells, a comparably high percentage of all three components is incorporated into active translocons.
Upon puromycin or pactamycin treatment of cells, many FRET pairs in Table II underwent a small but statistically significant change, with no FRET pair decreasing to <70% of that seen in untreated cells. In some cases, the FRET efficiencies even increased slightly. In every case, the FRET values changed (either increased or decreased) in the same direction for both puromycin- and pactamycin-treated cells, although not all achieved statistical significance presumably due to an insufficiently large sample size. That notwithstanding, the relative lack of change in FRET among the components analyzed in Table II suggests that a significant degree of disassembly does not occur on completion of substrate translocation. If the majority of these components is incorporated into translocons in untreated cells (as is argued in the previous two paragraphs), these results would suggest that the close proximity of most SRα, TRAM, and TRAPα to the Sec61 complex is an inherent feature of translocon structure and not a consequence of a translocating substrate.
Discussion
In this work, we have used FRET between directly labeled antibodies to address several basic issues of protein translocon organization and composition in mammalian cells. These experiments have allowed us to draw three principal conclusions regarding translocons in vivo that had either not been addressed or had yielded conflicting results with previous approaches. First, in cells fixed at steady state, while growing under metabolically active conditions, the vast majority of Sec61 complexes are assembled into oligomeric structures. Second, these structures, while remaining oligomeric, undergo a change in configuration between rounds of protein translocation. Third, SR, TRAM, and TRAP do not appear to significantly change their associations with the Sec61 complex after completion of substrate translocation. These findings have several implications for translocation, each of which is briefly discussed in the following paragraphs.
The decisive step in cotranslational protein translocation is the achievement of a stable ribosome–translocon complex in which the nascent chain has access to the ER lumen, is shielded from the cytosol, and has committed to forward translocation (Crowley et al., 1994; Jungnickel and Rapoport, 1995; Voigt et al., 1996; Fons et al., 2003). Because completion of protein synthesis or initiation of nascent chain folding before achieving this committed stage results in a cytoplasmically localized substrate (Perara et al., 1986), the precommitment steps in translocation face severe temporal constraints. Additional constraints in vivo include a crowded cellular environment (Luby-Phelps et al., 1986), the large size and low diffusional mobility of ribosome–nascent chain complexes (Rolls et al., 2002), and a potentially limited availability of unengaged components in metabolically active cells (Fig. 4 B). How these constraints are efficiently overcome despite requiring the coordinated actions of up to five components (SRP, SR, TRAM, and the Sec61 and TRAP complexes) has been unclear.
The targeting of nascent chains to the ER includes a transient SRP-mediated slowdown in translation, which provides one mechanism for overcoming the temporal constraints on initiating translocation (Walter and Johnson, 1994). However, later steps, including transfer to the translocon, insertion of nascent chains into the channel, and initiation of translocation, do not appear to involve such translational pauses. We now provide evidence suggesting that these posttargeting steps may be spatially coupled by preassembled translocon structures that are present even before their engagement by a substrate. We suggest that this permits sequential interactions to occur in rapid succession due to the close and constant proximity of the necessary components. Thus, an initial temporal checkpoint (SRP-mediated pausing), together with spatial coupling of the remaining steps, can ensure high fidelity and efficiency of translocation despite the various obstacles posed in vivo.
Currently, it is not known how the various translocon components remain associated in vivo given that biochemical studies have yet to reveal stable associations between them. One possibility is that the conditions required to solubilize the translocon components disrupt interactions that are more stable in the context of an intact lipid bilayer. A more intriguing possibility is that a nontranslating ribosome, which can bind stably to the Sec61 complex under physiologic salt conditions (Kalies et al., 1994), provides a platform for maintaining translocon complexes in an assembled state. For example, it was recently suggested that SR may be close to the translocon (Wittke et al., 2002) and may interact directly with the ribosome (Mandon et al., 2003), which may remain translocon-bound even in the absence of ongoing translocation (Potter and Nicchitta, 2002; Nikonov et al., 2002). If ribosomes indeed do not detach from translocons after substrate translocation, then the lack of significant disassembly of translocon components between rounds of transport (Tables I and II) may be due to an organizing function imparted by the bound ribosome. Thus, the more complete disassembly observed on ER solubilization in vitro requiring high salt-mediated removal of ribosomes from translocons may not normally occur between rounds of transport in vivo. Further studies will be needed to determine the molecular basis of the maintenance of close proximities among the translocon components revealed in this work.
Materials and methods
Antibodies
Antipeptide rabbit antisera against Sec61β, SRα, and TRAPα have been described previously (Fons et al., 2003). Antipeptide antiserum against the COOH termini of canine Sec61α and TRAM were gifts from K. Kellaris and R. Gilmore (University of Massachusetts School of Medicine, Worcester, MA) and K. Matlack and P. Walter (University of California, San Francisco, San Francisco, CA). Rabbit antisera against residues 575–593 (in the COOH-terminal tail of canine CNX) and against residues 50–68 (near the NH2 terminus of canine CNX) were obtained from StressGen Biotechnologies. The IgG fraction from each of these antibodies was purified by protein A chromatography and labeled using Cy3 or Cy5 monoreactive dye packs (Amersham Biosciences) with slight modifications to the manufacturer's instructions. In brief, 3 mg IgG (in 0.9 ml PBS) was mixed with 0.1 ml 1 M NaHCO3 (pH 9.0) and added to one vial of the dye pack. After 20 min with occasional mixing, the reaction was terminated by separation of the IgG from unreacted dye by Sephadex G-25 chromatography in PBS. This procedure results in the reliable conjugation of ∼3.5–4.5 dyes/IgG. Conjugated antibodies falling outside this range were not used. Cy3-conjugated anti–rabbit IgG was obtained from Jackson ImmunoResearch Laboratories and contained an average of ∼2.5 dyes/IgG. HRP-conjugated secondary antibodies were obtained from Amersham Biosciences.
Cell culture and sample preparation
MDCK cells were grown in DME containing 10% FBS and glutamine. For imaging, cells were grown in fibronectin-coated 8-chambered Lab-Tek glass coverslips (Nunc) to 40–70% confluence. Translational inhibition (1 mM puromycin; Calbiochem) or 0.2 mM pactamycin (a gift from E. Steinbrecher, Pharmacia Corp., Peapack, NJ) was performed for 15 min at 37°C before fixation. Cells were fixed with 3.7% formaldehyde in PBS for 15 min at RT, washed with PSS (PBS with 10% FBS, 0.1% saponin), and blocked 1 h at RT with PSS containing 50 μg/ml RNase A. Three different labeling protocols were used depending on the configuration of antibodies desired. In experiments where two directly labeled primary antibodies were used, the Cy3- and Cy5-conjugated antibodies were premixed before incubating with cells. In experiments where the 2°Cy3 donor antibody was bound directly to the acceptor antibody, labeling was performed sequentially. After binding the Cy5-labeled antibody as aforementioned, the cells were washed before binding the Cy3-labeled secondary antibody. In experiments using an unlabeled “spacer” antibody to which the 2°Cy3 donor antibody was bound, labeling was performed in multiple steps. First, the unlabeled primary antibody was bound, after which the cells were washed, before binding 2°Cy3. After washing, the samples were fixed with 3.7% formaldehyde. Any remaining antigen binding sites on the secondary antibody were blocked with PBS containing 5% rabbit serum and 0.1% saponin, and subsequently incubated with the Cy5-conjugated antibodies. After completion of each of the labeling protocols, samples were washed and the cells were either viewed immediately or postfixed in 3.7% formaldehyde (which had no effect on the FRET observed) before rinsing into PSS for imaging.
Microscopy and image analysis
Images were acquired with a confocal microscope (model LSM-510; Carl Zeiss MicroImaging, Inc.) using a 1.4 NA 63× oil objective using 543 and 633 nm HeNe laser lines with 560–615 and 650 filters with open pinholes. Complete photobleaching of Cy5 was accomplished by 125 iterative scans with 5 mW illumination at zoom 4. Images were collected sequentially in the Cy3 and Cy5 channels immediately before and after photobleaching a region. FRET quantitation and generation of the energy transfer map were automated using custom macros (available upon request) written for NIH Image 1.62. Two experiments confirmed the specificity of donor dequenching. Acceptor-only labeled samples ensured no bleed-through into the donor channel. A donor-only labeled sample was shown to not change in intensity upon bleaching in the acceptor channel under the bleach conditions used.
Biochemical analyses
Lysates from MDCK cells for immunoblots (Fig. S1 A) were prepared in 100 mM Tris, pH 8, 500 mM KAc, 5 mM MgCl2, and 1% Triton X-100. After removing any insoluble material (10 min at maximum speed in a microcentrifuge), the proteins were precipitated with 15% TCA, washed in acetone, and analyzed by SDS-PAGE and immunoblotting. Canine pancreatic rough microsomal membranes were solubilized directly in SDS-PAGE sample buffer and analyzed by immunoblotting. For metabolic labeling, cells growing in 12-well dishes at ∼70% confluence were treated with translational inhibitors in methionine-free media for between 5–30 min before the addition of [35S]methionine/cysteine Translabel (ICN Biochemicals) to 100 μCi/ml. After 10 min of labeling at 37°C, cells were rinsed once in PBS, solubilized in 100 μl 1% SDS/0.1 M Tris (pH 8.0), and heated in a boiling water bath, and a 5-μl aliquot was analyzed by SDS-PAGE and autoradiography. For sucrose gradient analysis, a 6-well dish containing cells at ∼70% confluence was transferred to ice, rinsed in ice-cold PBS, and scraped into 500 μl of ice-cold 50 mM Hepes, pH 7.4, 500 mM KAc, 5 mM MgAc2, 0.8% deoxyBigCHAP (Calbiochem), and 1 mM DTT. Cells were solubilized by repeated passage through a small-bore pipette tip and sedimented at 10,000 g for 10 min in a refrigerated microcentrifuge, and 200 μl of the supernatant was applied to a 2-ml 10–50% (wt/vol) sucrose gradient containing the solubilization buffer. After centrifugation for 1 h at 55,000 rpm in a TLS-55 rotor (Beckman Coulter), 11 fractions were removed and the proteins precipitated with TCA and analyzed by immunoblotting. Fractions containing ribosomes were identified in separate gradients by monitoring absorbance at 260 nm or Coomassie blue staining of the fractions.
Miscellaneous
SDS-PAGE was performed on 12% Tris-tricine gels. Immunoblotting was performed after transfer to nitrocellulose and development was with Supersignal chemiluminescence reagents (Pierce Chemical Co.). Figures were assembled using Photoshop and Illustrator software (Adobe).
Online supplemental material
Nine supplemental figures, accompanying text, and figure legends are available online. These provide additional experimental characterization of the FRET methodology (Figs. S1 and S7–S9) and theoretical analyses of ensembles of fluorophores conjugated to antibodies (Figs. S2–S6). Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200312079/DC1.
Acknowledgments
We thank A. Kenworthy, G. Patterson, P. Randazzo, and D. Lowy for helpful discussions; K. Kellaris and R. Gilmore for antisera against Sec61α; K. Matlack and P. Walter for antisera against TRAM; and C. Nicchitta for advice on the acquisition and use of pactamycin.
E.L. Snapp was a Pharmacology Research Associate Program fellow. This work was supported by the intramural programs of the National Cancer Institute and National Institute of Child Health and Human Development.
The online version of this article includes supplemental material.
Abbreviations used in this paper: CNX, calnexin; FRET, fluorescence resonance energy transfer; SR, SRP receptor; SRP, signal recognition particle.
References
- Beckmann, R., D. Bubeck, R. Grassucci, P. Penczek, A. Verschoor, G. Blobel, and J. Frank. 1997. Alignment of conduits for the nascent polypeptide chain in the ribosome-Sec61 complex. Science. 278:2123–2126. [DOI] [PubMed] [Google Scholar]
- Beckmann, R., C.M.T. Spahn, N. Eswar, J. Helmers, P.A. Penczek, A. Sali, J. Frank, and G. Blobel. 2001. Architecture of the protein-conducting channel associated with the translating 80S ribosome. Cell. 107:361–372. [DOI] [PubMed] [Google Scholar]
- Bergeron, J.J., M.B. Brenner, D.Y. Thomas, and D.B. Williams. 1994. Calnexin: a membrane-bound chaperone of the endoplasmic reticulum. Trends Biochem. Sci. 19:124–128. [DOI] [PubMed] [Google Scholar]
- Crowley, K.S., S. Liao, V.E. Worrell, G.D. Reinhart, and A.E. Johnson. 1994. Secretory proteins move through the endoplasmic reticulum membrane via an aqueous, gated pore. Cell. 78:461–471. [DOI] [PubMed] [Google Scholar]
- Fons, R.D., B.A. Bogert, and R.S. Hegde. 2003. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J. Cell Biol. 160:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich, D., and T.A. Rapoport. 1993. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell. 75:615–630. [DOI] [PubMed] [Google Scholar]
- Gorlich, D., S. Prehn, E. Hartmann, K.U. Kalies, and T.A. Rapoport. 1992. A mammalian homolog of Sec61p and SecYp is associated with ribosomes and nascent polypeptides during translocation. Cell. 71:489–503. [DOI] [PubMed] [Google Scholar]
- Hamman, B.D., J.C. Chen, E.E. Johnson, and A.E. Johnson. 1997. The aqueous pore through the translocon has a diameter of 40-60 Å during cotranslational protein translocation at the ER membrane. Cell. 89:535–544. [DOI] [PubMed] [Google Scholar]
- Hamman, B.D., L.M. Hendershot, and A.E. Johnson. 1998. BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell. 92:747–758. [DOI] [PubMed] [Google Scholar]
- Hanein, D., K.E. Matlack, B. Jungnickel, K. Plath, K. Kalies, K.R. Miller, T.A. Rapoport, and C.W. Akey. 1996. Oligomeric rings of the Sec61p complex induced by ligands required for protein translocation. Cell. 87:721–732. [DOI] [PubMed] [Google Scholar]
- Jackson, V. 1999. Formaldehyde cross-linking for studying nucleosomal dynamics. Methods. 17:125–139. [DOI] [PubMed] [Google Scholar]
- Johnson, A.E., and M.A. van Waes. 1999. The translocon: a dynamic gateway at the ER membrane. Annu. Rev. Cell Dev. Biol. 15:799–842. [DOI] [PubMed] [Google Scholar]
- Jungnickel, B., and T.A. Rapoport. 1995. A posttargeting signal sequence recognition event in the endoplasmic reticulum membrane. Cell. 82:261–270. [DOI] [PubMed] [Google Scholar]
- Kalies, K.U., D. Gorlich, and T.A. Rapoport. 1994. Binding of ribosomes to the rough endoplasmic reticulum mediated by the Sec61p-complex. J. Cell Biol. 126:925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy, A.K. 2001. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods. 24:289–296. [DOI] [PubMed] [Google Scholar]
- Kenworthy, A.K., and M. Edidin. 1998. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of <100 Å using imaging fluorescence resonance energy transfer. J. Cell Biol. 142:69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitscheck, U., R. Schweitzer-Stenner, D.J. Arndt-Jovin, T.M. Jovin, and I. Pecht. 1993. Distribution of type I Fc epsilon-receptors on the surface of mast cells probed by fluorescence resonance energy transfer. Biophys. J. 64:110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby-Phelps, K., D.L. Taylor, and F. Lanni. 1986. Probing the structure of cytoplasm. J. Cell Biol. 102:2015–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandon, E.C., Y. Jiang, and R. Gilmore. 2003. Dual recognition of the ribosome and the signal recognition particle by the SRP receptor during protein targeting to the endoplasmic reticulum. J. Cell Biol. 162:575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlack, K.E., and P. Walter. 1995. The 70 carboxyl-terminal amino acids of nascent secretory proteins are protected from proteolysis by the ribosome and the protein translocation apparatus of the endoplasmic reticulum membrane. J. Biol. Chem. 270:6170–6180. [DOI] [PubMed] [Google Scholar]
- Menetret, J.F., A. Neuhof, D.G. Morgan, K. Plath, M. Radermarcher, T.A. Rapoport, and C.W. Akey. 2000. The structure of ribosome-channel complexes engaged in protein translocation. Mol. Cell. 6:1219–1232. [DOI] [PubMed] [Google Scholar]
- Molinari, M., and A. Helenius. 2000. Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science. 288:331–333. [DOI] [PubMed] [Google Scholar]
- Morgan, D.G., J. Menetret, A. Neuhof, T.A. Rapoport, and C.W. Akey. 2002. Structure of the mammalian ribosome-channel complex at 17 Å resolution. J. Mol. Biol. 324:871–886. [DOI] [PubMed] [Google Scholar]
- Nikonov, A.V., E. Snapp, J. Lippincott-Schwartz, and G. Kreibich. 2002. Active translocon complexes labeled with GFP-Dad1 diffuse slowly as large polysome arrays in the endoplasmic reticulum. J. Cell Biol. 158:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perara, E., R.E. Rothman, and V.R. Lingappa. 1986. Uncoupling translocation from translation: implications for transport of proteins across membranes. Science. 232:348–352. [DOI] [PubMed] [Google Scholar]
- Potter, M.D., and C.V. Nicchitta. 2002. Endoplasmic reticulum-bound ribosomes reside in stable association with the translocon following termination of protein synthesis. J. Biol. Chem. 277:23314–23320. [DOI] [PubMed] [Google Scholar]
- Rapoport, T.A., B. Jungnickel, and U. Kutay. 1996. Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes. Annu. Rev. Biochem. 65:271–303. [DOI] [PubMed] [Google Scholar]
- Rolls, M.M., D.H. Hall, M. Victor, E.H. Stelzer, and T.A. Rapoport. 2002. Targeting of rough endoplasmic reticulum membrane proteins and ribosomes in invertebrate neurons. Mol. Biol. Cell. 13:1778–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, A., and W.F. Wonderlin. 2003. The permeability of the endoplasmic reticulum is dynamically coupled to protein synthesis. J. Biol. Chem. 278:4397–4403. [DOI] [PubMed] [Google Scholar]
- Simon, S.M., and G. Blobel. 1991. A protein-conducting channel in the endoplasmic reticulum. Cell. 65:371–380. [DOI] [PubMed] [Google Scholar]
- Van den Berg, B., W.M. Clemons, Jr., I. Collinson, Y. Modis, E. Hartmann, S.C. Harrison, and T.A. Rapoport. 2004. X-ray structure of a protein-conducting channel. Nature. 427:36–44. [DOI] [PubMed] [Google Scholar]
- Voigt, S., B. Jungnickel, E. Hartmann, and T.A. Rapoport. 1996. Signal sequence–dependent function of the TRAM protein during early phases of protein transport across the endoplasmic reticulum membrane. J. Cell Biol. 134:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, P., and A.E. Johnson. 1994. Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane. Annu. Rev. Cell Biol. 10:87–119. [DOI] [PubMed] [Google Scholar]
- Wang, L., and B. Dobberstein. 1999. Oligomeric complexes involved in translocation of proteins across the membrane of the endoplasmic reticulum. FEBS Lett. 457:316–322. [DOI] [PubMed] [Google Scholar]
- Wittke, S., M. Dünnwald, M. Albertsen, and N. Johnsson. 2002. Recognition of a subset of signal sequences by Ssh1p, a Sec61p-related protein in the membrane of endoplasmic reticulum of yeast Saccharomyces cerevisiae. Mol. Biol. Cell. 13:2223–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]