

Abstract
One of the great mysteries of the nucleolus surrounds its disappearance during mitosis and subsequent reassembly at late mitosis. Here, the relative dynamics of nucleolar disassembly and reformation were dissected using quantitative 4D microscopy with fluorescent protein-tagged proteins in human stable cell lines. The data provide a novel insight into the fates of the three distinct nucleolar subcompartments and their associated protein machineries in a single dividing cell. Before the onset of nuclear envelope (NE) breakdown, nucleolar disassembly started with the loss of RNA polymerase I subunits from the fibrillar centers. Dissociation of proteins from the other subcompartments occurred with faster kinetics but commenced later, coincident with the process of NE breakdown. The reformation pathway also follows a reproducible and defined temporal sequence but the order of reassembly is shown not to be dictated by the order in which individual nucleolar components reaccumulate within the nucleus after mitosis.
Keywords: nucleolus; nucleus; mitosis; fluorescent protein; 4D imaging
Introduction
The nucleolus is a dynamic nuclear structure that assembles and disassembles during each mitotic cell division (Carmo-Fonseca et al., 2000; Olson et al., 2000; Hernandez-Verdun et al., 2002; Gerbi et al., 2003; Leung and Lamond, 2003). Nucleoli form around clusters of tandemly repeated ribosomal DNA (rDNA) genes, the so-called nucleolar organizing regions (NORs), where RNA polymerase I transcribes the rDNA repeats and generates the 45S rRNA precursor. The precursor is then cleaved and modified within nucleoli to generate mature 18S, 5.8S, and 28S rRNAs that are subsequently assembled with ribosomal proteins to form ribosome subunits. These processes occur at distinct nucleolar subcompartments, namely the fibrillar centres (FCs), dense fibrillar components (DFCs), and granular components (GCs). The rDNA clusters are located either within, or at the periphery of, FCs and rRNA transcripts appear at the boundary between the FC and DFC (Huang, 2002). The maturing rRNA transcripts progress through the DFC and then GC regions, before export of the ribosome subunits to the cytoplasm. The structural integrity of nucleoli may also play a general role in coordinating cellular stress responses. For example, disintegration of the nucleolus has been proposed as a common feature in cellular responses that activate the p53 pathway (Rubbi and Milner, 2003).
During mitosis, mammalian nucleoli disassemble and their components disperse. When cells exit mitosis nucleolar components reassemble around the respective NORs, which can later coalesce to form either one or multiple functional nucleoli. Partially processed rRNA transcripts, together with associated processing factors, form structures during mitosis termed “prenucleolar bodies” (PNBs; Jimenez-Garcia et al., 1994; Dundr et al., 2000; Savino et al., 2001). Components are subsequently transferred from the PNBs into the reforming nucleoli at NORs (Dundr et al., 2000; Savino et al., 2001). In contrast, the process of nucleolar disassembly when cells enter mitosis is not well characterized. Both the timing and mechanism of breakdown is unclear although it has been reported that a subset of nucleolar factors, including RNA polymerase I (Scheer and Rose, 1984; Gilbert et al., 1995) and the RNA Pol I upstream binding transcription factor (UBF) and transcription factor SL1 (Roussel et al., 1993; Jordan et al., 1996), remain associated with chromosomes at NORs, although no nascent rRNA transcripts are synthesized during mitosis (Weisenberger and Scheer, 1995).
Fluorescent protein (FP)-tagged nuclear factors provide markers in live cells for the location of specific nuclear bodies, such as nucleoli, or even subcompartments within nucleoli (Gerlich and Ellenberg, 2003a; Janicki and Spector, 2003). Proteins tagged with different spectral variants of either GFP, or RFP, can be coexpressed to detect two or more subnuclear structures in a single cell. The 3D localization of these markers can be recorded and quantitated over time, i.e., “4D” imaging, to provide detailed information about their dynamic movement and kinetic behavior (Gerlich et al., 2001; Dundr et al., 2002b; Gerlich and Ellenberg, 2003a). Here, we have applied quantitative 4D imaging using cell lines expressing different combinations of two or three FP-tagged markers to analyze the processes of nucleolar breakdown and reassembly during mitosis in live cells and to correlate this with parallel events affecting other nuclear structures.
Results
In vivo analysis of RNA polymerase I localization within nucleoli
A HeLa cell line termed HeLaYFP-RPA39 was established stably expressing YFP-tagged RNA polymerase I subunit RPA39 (RPA39), which is a subunit of RNA polymerase I and also RNA polymerase III (see Materials and methods). The YFP-RPA39 fusion protein concentrated in bright foci within nucleoli corresponding to the FC and also in a diffuse nucleoplasmic pool (Fig. 1 A). Both YFP-RPA39 and a transiently expressed CFP fusion of the transcription factor RNA polymerase I transcription factor RRN3 (RRN3) that is essential for initiation of transcription by RNA polymerase I (Miller et al., 2001) colocalize in the nucleolar foci (Fig. 1 A, arrowheads). These data indicate that YFP-RPA39 colocalizes inside nucleoli with RRN3 at sites including active RNA polymerase I. Analysis of HeLaYFP-RPA39 cells by FACS showed that the expression of YFP-RPA39 does not delay cell cycle progression (unpublished data).
Figure 1.

Characterization of YFP-RPA39 cell lines. (A) HeLaYFP-RPA39 cells were fixed 10 h after transient transfection with CFP-RRN3. Arrowheads indicate the colocalization of punctate structures labeled by YFP-RPA39 and CFP-RRN3 within nucleoli. The nuclear boundaries are denoted by dotted ovals. Bar, 5 μm. (B) In vitro transcription assay. Nuclear extracts prepared from HeLaYFP-RPA39 and parental HeLa cells were tested for specific transcription initiation activity on the rDNA promoter, which is shown by the presence of nascent rDNA transcript (lanes 8 and 9). Lanes 1–7 assay transcription from RNA Polymerase I complexes immunoprecipitated with anti-GFP. Anti-GFP-immunoprecipitated complexes from HeLaYFP-RPA39 nuclear extracts were transcriptionally active upon the addition of rRNA gene promoter template DNA and ribonucleoside triphosphates (lane 3). Addition of SL1 alone (lanes 4 and 6) or SL1 and UBF (lanes 5 and 7) further stimulated transcription. In the absence of anti-GFP antibody no transcription was observed (lane 1) and in the presence of anti-GFP-protein G beads but absence of a GFP tag (parental HeLa nuclear extract) no transcription was observed (lane 2). (C) Pulse-labeled rRNA transcripts transiently colocalize within the DFC after a 6-min chase. The left panel shows the localization of DFC labeled by YFP-FIB in stable HeLaYFP-FIB cell lines and the middle panel illustrates the localization of accumulated BrUTP at 6 min and the pattern of incorporation within nucleoli are shown in detail in the inserts. Bar, 5 μm.
We next addressed whether the YFP-RPA39 subunit is incorporated into functional RNA polymerase I complexes. In vitro RNA polymerase I transcription assays were performed using nuclear extracts prepared from both HeLaYFP-RPA39 and parental HeLa cell lines (Fig. 1 B). Extracts from both cell lines show equivalent levels of RNA polymerase I activity (Fig. 1 B, lanes 8 and 9). Active RNA polymerase I transcription complexes can be immunoprecipitated using anti-GFP antibodies specifically from the HeLaYFP-RPA39 nuclear extract (Fig. 1 B, compare lane 2 with lanes 3–7). The in vitro activity is stimulated by addition of purified UBF and transcription factor SL1 to the assays (Fig. 1 B, compare lane 3 with lanes 4–7). We analyzed further the relationship between YFP-RPA39 and transcription sites in vivo by performing pulse-chase incorporation of BrUTP in both HeLaYFP-RPA39 and HeLaYFP-FIB cells, the latter being a cell line stably expressing YFP-fibrillarin (FIB) as a marker for the DFC (Fig. 1 C; see Online supplemental material). The localization of BrUTP-labeled RNA within the nucleolus was analyzed by immunofluorescence, after a 6-min chase after removing BrUTP from the culture medium (see Materials and methods). These data show an accumulation of labeled RNA at the periphery of the foci containing YFP-RPA39 (i.e., FC), within the boundary region between the FC and DFC, as detected by YFP-FIB (Fig. 1 C). At longer chase times, after removal of BrUTP, we observe the labeled RNA colocalizing with the GC (within ∼20 min) and then appearing in the cytoplasm (within ∼60 min; unpublished data). The appearance of labeled RNA is inhibited by Actinomycin D at low levels that inhibit selectively RNA polymerase I activity (unpublished data). These data are consistent with previous studies on the sites of rRNA transcription in nucleoli (Huang, 2002). In summary, we conclude that YFP-RPA39 is incorporated into active RNA polymerase I transcription complexes and serves in vivo as a valid marker for detecting sites within nucleoli containing active RNA polymerase I.
We next used the HeLaYFP-RPA39 cells to monitor the localization of FP-RPA39 throughout an entire mitosis, using both confocal and deconvolution fluorescence microscopy. Time-lapse microscopy showed that RPA39 remained concentrated in chromatin-associated foci for most of mitosis, consistent with previous data (Scheer and Rose, 1984; Gilbert et al., 1995). However, detailed time-lapse analysis of single live cells consistently showed a window during metaphase, lasting ∼30 min, when RPA39 is no longer detected in chromatin foci (Fig. 2, Metaphase, arrowheads; Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200405013/DC1). Loss of RNA polymerase I subunits from chromatin, specifically during metaphase, was also observed by immunolocalization, using monoclonal antibodies specific for the RPA20 subunit (see Online supplemental material). Although RPA20 has been reported as a core subunit of RNA polymerases I, II, and III, during interphase the immunofluorescence signal of RPA20 detected using this antibody predominantly colocalizes in bright foci within nucleoli that contain RRN3 (Jones et al., 2000; see Online supplemental material). An independent study has also found that RNA polymerase I subunits RPA194, RPA39, and RPA16, but not RPA43, are transiently lost from chromatin-associated foci during metaphase (Dundr, N., and T. Misteli, personal communication). We suggest that RNA polymerase I either transiently leaves chromosomes during metaphase, or else that multiple subunits dissociate transiently from the polymerase ternary complex. However, immunolabeling with antibodies specific for the RNA polymerase I UBF, which binds to the rRNA gene repeats, showed that UBF associated with chromatin foci throughout mitosis (Fig. 2, UBF). Thus, although UBF colocalizes in foci with RPA39 during prophase, prometaphase, anaphase, and telophase, it remains in similar foci throughout metaphase when the RNA polymerase I subunits are no longer detected (Fig. 2). The loss of RNA polymerase I subunits from chromatin foci is therefore not a detection problem at this stage of metaphase. In summary, the data indicate that the previous view that RNA polymerase I remains associated with chromatin throughout the entire period of mitosis may need to be revised.
Figure 2.

The localization of UBF and RPA39 in different stages of mitosis. Unsynchronized HeLaYFP-RPA39 cells were fixed and immunolabeled with an anti-UBF antibody to denote the NORs and stained with DAPI to show the condensed chromosomes at different stages of mitosis. Arrows indicate colocalization of YFP-RPA39 and UBF, whereas arrowheads indicate the presence of UBF but absence of YFP-RPA39. The cells were also immunolabeled with an anti-RPA20 antibody, showing the exact same pattern as YFP-RPA39 (Fig. S1 D and not depicted, available at http://www.jcb.org/cgi/content/full/jcb.200405013/DC1). Bars, 5 μm.
Disassembly of nucleolar subcompartments during mitosis
To study nucleolar disassembly, we performed 4D imaging on cell lines expressing combinations of three separate marker proteins labeled with either CFP, YFP, HcRed or DsRed (see Materials and methods). Cells coexpressing YFP-RPA39 (FC), CFP-FIB (DFC), and HcRed histone H2B, showed that RNA polymerase I was consistently lost from FCs before the dissociation of FIB from the DFC in all cells examined (Fig. 3 A). This is most clear in the xz side projections for the YFP-RPA39 and CFP-FIB expressing cells (Fig. 3 A, bottom). Interestingly, the punctate distribution of FIB within the DFC changed to a more diffuse pattern, though still confined to the DFC, coincident with the loss of RNA polymerase I from nucleoli (Fig. 3 A, top, insets).
Figure 3.
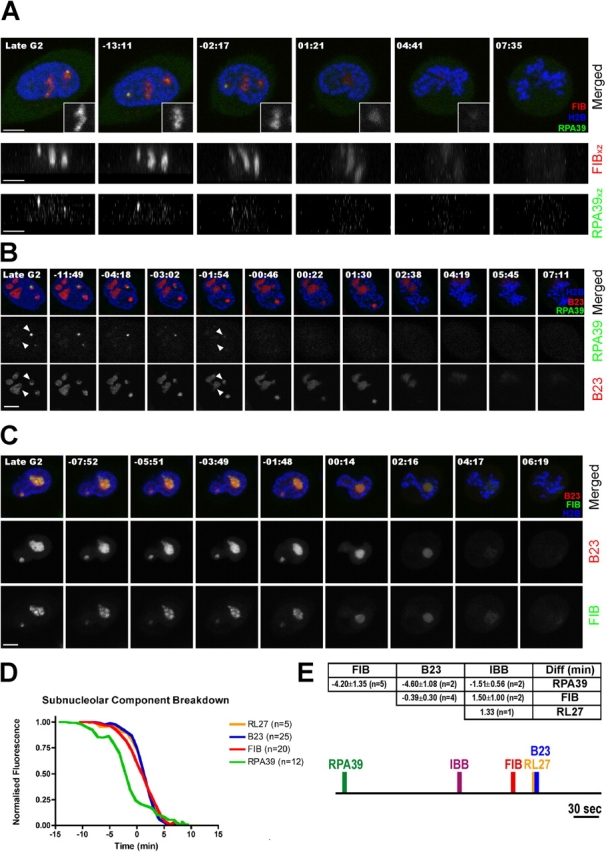
Disassembly of subnucleolar components when cells enter mitosis. (A) HeLaYFP-RPA39+CFP-FIB were transiently transfected with H2B-diHcRed and followed through mitosis. The inserts indicate the change in punctate structure labeled by CFP-FIB from the middle nucleolus. The top panels show the xy projections and the bottom panels show the xz projections of the fluorescence corresponding to FIB and RPA39 to indicate the loss of DFC and FC, respectively. (B) HeLaYFP-RPA39+CFP-H2B were transiently transfected with DsRed2-B23 and followed through mitosis. Arrowheads indicate the change in structure labeled by DsRed2-B23. (C) HeLaYFP-FIB+CFP-B23 were transiently transfected with H2B-diHcRed and followed through mitosis. For A–C, the time shown on the top of each panel indicates the time for the cell progressing through mitosis and 00:00 indicates the time at which the nuclear intensity of IBB domain dropped to 50% of its initial value (midpoint). Bars, 5 μm. (D) The mean fluorescence intensities of nucleolar components RPA39, FIB, B23, and RL27 within nucleoli were averaged, normalized and plotted against time, where time equals 00:00 is the midpoint for IBB and n = number of cells examined. The distribution of the original data is reported in Fig. S2 (available at http://www.jcb.org/cgi/content/full/jcb.200405013/DC1). (E) Pairwise midpoint-midpoint comparison between markers expressed within the same cell. For example, a negative value of −4.20 ± 1.35 on the column FIB and the row RPA39 indicates that FIB reaches its midpoint 4.20 min later than RPA39, averaging from five experiments. A possible time line to summarize the relationship between all the nucleolar markers examined and IBB is represented in the bottom panel. If the midpoint for IBB is defined at time equals 00:00, then RPA, FIB, RL27, and B23 would be −02:04, 00:56, 01:20, and 01:20, respectively. Bar, 30 s. Note that although IBB is not present in this experiment, the time line correlation is made according to a set of multiple experiments.
A similar 4D analysis was performed using cells coexpressing YFP-RPA39 (FC), DsRed-nucleophosmin (B23;GC) and CFP-H2B (Fig. 3 B). DsRed-B23 in the GC persists for ∼4–5 min after loss of RNA polymerase I from nucleoli. A subtle change in the pattern of B23 distribution in the GC is visible coincident with the loss of RNA polymerase I from nucleoli (Fig. 3 B, arrowheads, compare −01:54 with late G2). Similar kinetics were observed using FP-tagged ribosomal protein L27 (RL27) as an alternative marker for the GC (Fig. 3 D; not depicted). To compare the timing of loss of markers from the DFC and GC subcompartments, a 4D analysis was performed on cells coexpressing CFP-B23 (GC), YFP-FIB (DFC), and HcRed-H2B (Fig. 3 C and Video 2, available at http://www.jcb.org/cgi/content/full/jcb.200405013/DC1). Both CFP-B23 and YFP-FIB leave the nucleolus at approximately the same time. We conclude that nucleolar disassembly is initiated at the FC, whereas concurrent loss of both DFC and GC components occurs later.
To quantitate the pathway of nucleolar disassembly, we measured the levels of FP-fluorescence in defined cellular structures during mitotic progression (Fig. 3 D; see Online supplemental material). Data obtained from 5 to 25 separate experiments for each fluorescent marker show that the rate of loss of RPA39 from nucleoli is slower than either RL27 or B23, though comparable to FIB (Fig. 3 D). However, the loss of RPA39 is initiated earlier than any of the other markers tested. A comparison of the times at which the levels of fluorescence for each marker dropped to 50% of their initial values in nucleoli indicates that loss of RPA39 precedes the other markers by ∼4 min (Fig. 3, D and E). The loss of RNA polymerase I subunits is therefore the earliest event we have detected at the onset of nucleolar disassembly.
Comparison of nucleolar and nuclear envelope (NE) disassembly
We next compared the relative timing of nucleolar and NE disassembly using stable cell lines expressing either YFP-Lamin B receptor (HeLaYFP-LBR), or YFP-lamin B1 (HeLaYFP-LB1). These are the first and last components, respectively, to disassemble from NE during mitosis (Beaudouin et al., 2002). We performed 4D analyses using both these cell lines with DsRed-B23 expressed transiently (Fig. 4). Nucleoli containing B23 are still detected when lamin B receptor (LBR) starts to dissociate from NE (Fig. 4 A). This is most clear on the xz projection (Fig. 4 A, bottom). Loss of B23 from nucleoli occurs ∼1.5 min after the decrease in LBR signal from NE (Fig. 5 D). In contrast, analysis of HeLaYFP-LB1 cells shows that the loss of B23 from nucleoli occurs ∼2 min before dissociation of lamin B1 (LB1) from NE (Fig. 4 B; Fig. 5 D). Therefore, nucleolar disassembly occurs predominantly within the window during which NE components dissociate.
Figure 4.

Comparison of nucleolar disassembly and the structural disintegration of NE upon entering into mitosis. (A) HeLaYFP-LBR and (B) HeLaYFP-LB1 cells were transiently transfected with DsRed2-B23 for 16 h before imaging through mitosis. The location of nucleoli was denoted by DsRed2-B23 (green and in the bottom panel for xz projection) and condensing chromosomes by Hoechst 33234. The time shown on the top of each panel indicates the time for the cell progressing through mitosis and 00:00 indicates the time at which the nuclear intensity of IBB dropped to 50% of its initial value (midpoint). Bars, 5 μm.
Next, we compared the loss of NE function, as judged by leakage of HcRed-importin β binding (IBB) domain into the cytoplasm, with the timing of loss of RPA39 (Fig. 5 A) and FIB from nucleoli (Fig. 5 B). Cytoplasmic IBB is actively recruited to the nucleus by importin β in the presence of a GTP/GDP gradient maintained by an intact NE and therefore is a good reporter of NE integrity. These experiments were performed using HeLa cells stably expressing both CFP-H2B and either YFP-RPA39 (FC), or YFP-FIB (DFC), with HcRed-IBB transiently expressed in both cell lines (Fig. 5, A and B). Loss of RPA39 from nucleoli precedes the loss of IBB from the nucleoplasm (Fig. 5 A, nuclear boundary shown by dotted yellow line). In contrast, loss of IBB from the nucleus precedes the loss of FIB within nucleoli (Fig. 5 B). Loss of NE function, as judged by leakage of IBB into cytoplasm, precedes NE breakdown (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200405013/DC1). These data indicate that loss of RPA39 is one of the earliest nuclear events when cells enter mitosis.
Figure 5.
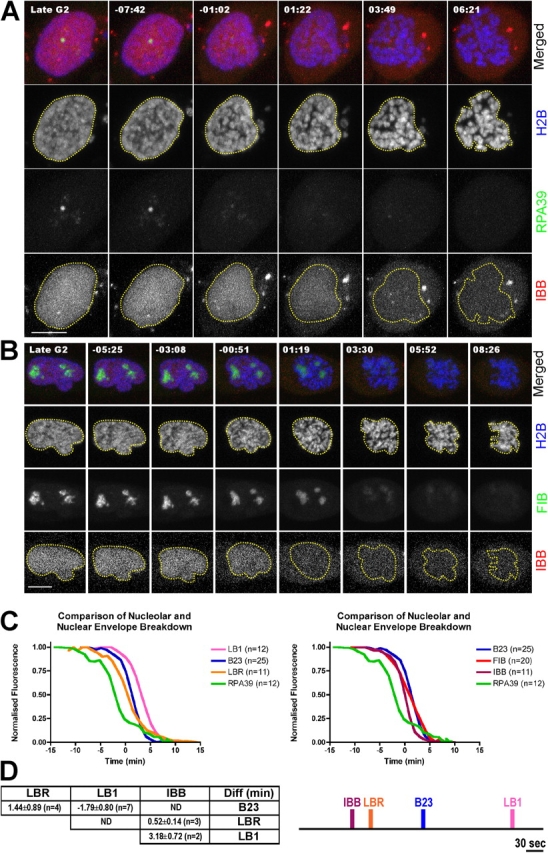
The timing of nucleolar subcompartment disassembly and the loss of functionality of NE. (A) HeLaYFP-RPA39+CFP-H2B and (B) HeLaYFP-FIB+CFP-H2B cells were transiently transfected with diHcRed-IBB for 16 h before imaging through mitosis and Hoescht 33234 was added to the medium 30 min before the experiment to label the condensing chromosomes in live cells. The time shown on the top of each panel indicates the time for the cell progressing through mitosis and 00:00 indicates the time at which the nuclear intensity of IBB dropped to 50% of its initial value (midpoint). Bar, 5 μm. (C) The mean fluorescence intensities of nucleolar components RPA39, FIB, and B23 within nucleoli and that of NE components LBR and LB1 (left) and nuclear import substrate IBB (right) within nuclei were averaged, normalized, and plotted against time, where time equals 00:00 is the midpoint for IBB and n = number of cells examined. The distribution of the original data is reported in Fig. S2. (D) The time at which the nucleolar, NE and nuclear import markers dropped to 50% of their respective initial values were compared and tabulated as in Fig. 3 E. A possible time line to summarize the relationship between nucleolar, nuclear import and NE markers is proposed in the right panel. If the midpoint for IBB is defined at time equals 00:00, then LBR, B23, and LB1 would be at 00:23, 01:20, and 03:09, respectively. Bar, 30 s.
A quantitative analysis of the rates of loss of both nucleolar and NE markers showed that initiation of RPA39 disassembly occurs before the loss of all other proteins tested from either the nucleolus or NE (Fig. 5, C and D; see Online supplemental material). However, dissociation of the GC and DFC nucleolar markers was only observed after the functional integrity of the NE is lost, as judged by loss of IBB from the nucleus. Note that the rate of loss of IBB is faster than other markers. For example, although IBB begins to decrease at least 5 min after loss of RPA39 is detected, it has fallen to 50% of its initial value within 1–2 min and is completely (>95%) lost ∼3 min before RPA39. IBB was not assembled into a subnuclear body and may leave the nucleus faster than proteins that must undergo disassembly processes. This quantitative analysis demonstrates that complete nucleolar breakdown occurs while a substantial fraction of the nuclear lamina remains intact, but after the NE no longer prevents loss of soluble proteins from the nucleus.
Reassembly of NORs
We next analyzed nuclear and nucleolar reassembly after mitosis in cells stably expressing YFP-FIB, and CFP-H2B and transiently expressing HcRed-IBB (Fig. 6). FIB is associated with chromatin in the daughter nuclei at an early stage, before the re-import of IBB (Fig. 6 A). This chromatin association is at least 5 min before the detection of any foci corresponding to the reformation of nucleoli (Fig. 6 A and Fig. 7 A). RPA39 also associates early with daughter nuclei, before the nuclear accumulation of IBB (Fig. 6 B). However, RPA39 is immediately detected in chromatin-associated foci, which only accumulate FIB ∼5 min later (Fig. 6 C, foci, arrowheads). Interestingly, although FIB is already present in the nuclei, it only accumulates at the RPA39 foci when IBB accumulates in the daughter nuclei (Fig. 6 B and C). At the same time we observe a clear increase in the number of foci containing both FIB and RPA39 (Fig. 6 B, 05:26–12:59). This suggests that reestablishment of nuclear protein import is important for stepwise reformation of nucleoli.
Figure 6.
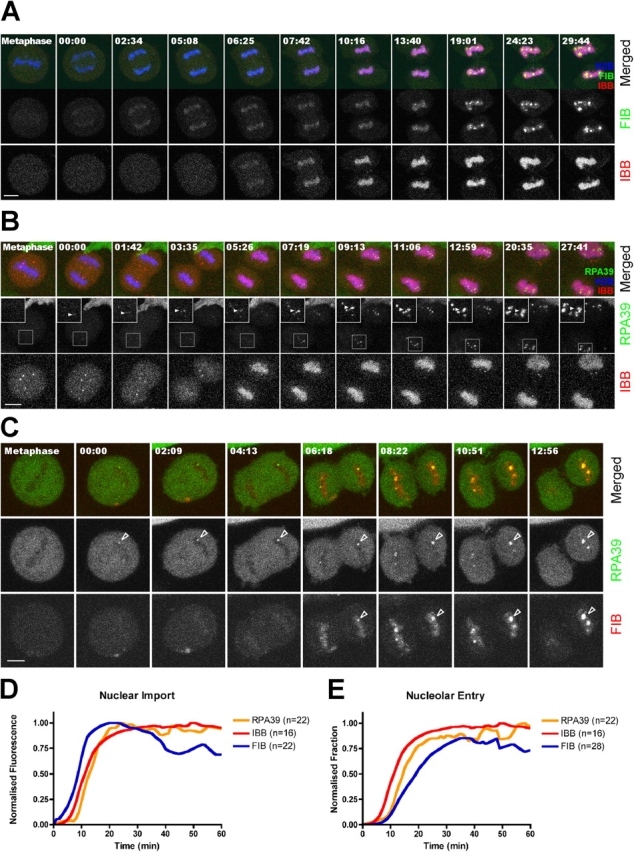
The reassembly of NOR. (A) HeLaYFP-FIB+CFP-H2B and (B) HeLaYFP-RPA39+CFP-H2B cells were transiently transfected with nuclear import substrate marker IBB-diHcRed for 16 h before imaging. The inserts in B represent the distribution of RPA39 signal within reforming NORs. Arrowheads indicate the development of the same FC foci over time. (C) HeLaYFP-RPA39+CFP-FIB cells were followed through mitosis. Arrowheads indicate the location of a reforming NOR over time. For A–C, the time shown on the top of each panel indicates the time for the cell progressing through mitosis and 00:00 indicates the beginning of chromosome migration toward its respective pole. Bars, 5 μm. (D) The mean fluorescence intensities of nuclear import substrate IBB and nucleolar subcompartment marker RPA39 and FIB within the reforming nuclei were averaged, normalized and plotted against time, where time equals 00:00 when the chromosomes start to migrate toward respective poles and n = the number of cells examined. (E) The total fluorescence intensities of nucleolar components RPA39 and FIB entering the reforming nucleoli as a fraction of the total nuclear fluorescence were averaged, normalized, and plotted against time in the same manner as of panel D and the curve for nuclear accumulation of IBB was used as a reference. The distribution of the original data is reported in Fig. S2.
Quantitative measurements of these nuclear protein concentrations confirm that FIB associates with nuclei before either RPA39 or IBB (Fig. 6 D; see Online supplemental material). However, IBB accumulation in the nucleus is detected 1–2 min earlier than RPA39 by this quantitative analysis, even though by visual inspection of the corresponding micrographs the concentrated RPA39 in nuclear foci is more striking than the diffuse nuclear accumulation of IBB. For comparison, we analyzed the rate of fluorescence increase over time within reforming nucleoli for both RPA39 and FIB, as a fraction of total nuclear fluorescence (Fig. 6 E; see Online supplemental material). This shows that FIB assembles into nucleoli later than RPA39 (∼4 min later for 50% accumulation), although it had accumulated earlier within the total nuclear volume (Fig. 6, compare D with E). Moreover, FIB is only incorporated into foci containing RPA39 (i.e., the reforming NORs) when the increase in nuclear RPA39 level is maximal. This suggests that the recruitment of FIB into the reforming nucleoli may require RNA polymerase I.
Interestingly, there is a close connection between the timing of FIB accumulation in the DFC and the time at which a functional NE is reestablished. Similarly, during nucleolar disassembly, loss of FIB from the DFC closely correlates with the time at which the functional integrity of NE is compromised (compare Fig. 5 with Fig. 6). This raises the interesting possibility that one or more factors normally excluded from the nucleus by an intact NE can contribute to signaling the dissociation of FIB from the nucleolus.
Reassembly of functional nucleoli
We define the reformation of functional nucleoli as requiring the presence of all the major interphase markers for the FC, DFC and GC subcompartments. To monitor this in live cells we analyzed cell lines stably expressing both CFP-B23 and YFP-FIB and transiently expressing DsRed-RL27 (Fig. 7). Together with the data from Fig. 6 showing the stepwise assembly of FC and DFC, these data showed for both the B23 and RL27 markers that the GC forms later than either the FC or DFC, consistent with previous immunofluorescence data (Dundr et al., 2000; Savino et al., 2001). Quantitative analysis showed that the GC forms ∼18 min after the DFC and ∼27 min after the FC. The appearance of the GC after mitosis correlates closely with the time at which BrUTP-labeled RNA appears in the GC in interphase pulse-chase experiments (unpublished data). Next, we addressed quantitatively the concentration kinetics of nucleolar factors in cells exiting mitosis (Fig. 7 B). These experiments indicate that FIB accumulates before both GC markers B23 and RL27, which reassemble with similar initial kinetics. In summary, these data support the view that the reassembly of nucleoli is normally coupled to the activation of ribosome subunit synthesis. The relationship between different nucleolar subcompartment markers in terms of their timing in nucleolar reassembly is summarized in Fig. 7 C.
Figure 7.
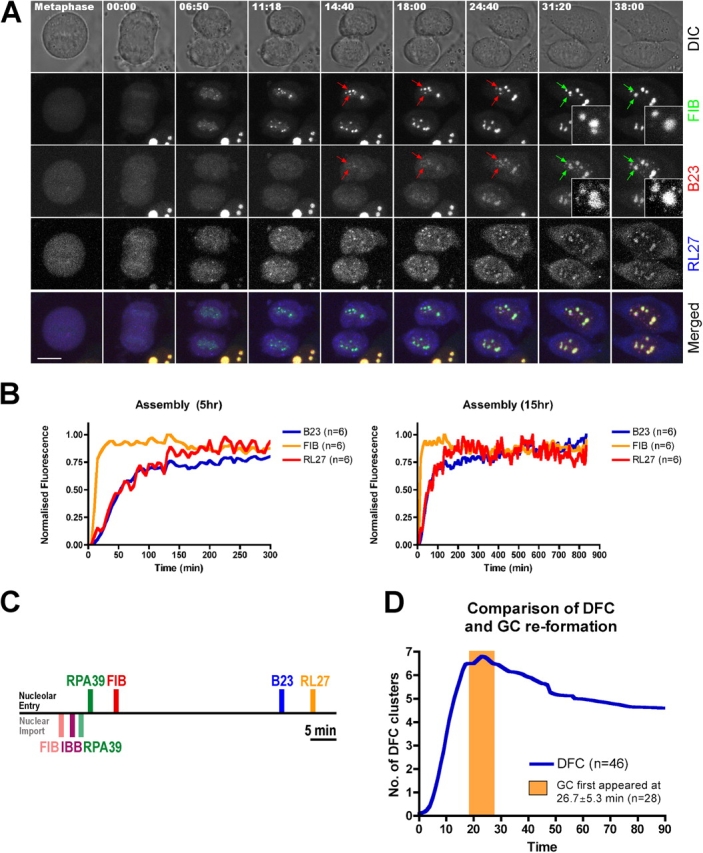
The reassembly of functional nucleoli. (A) HeLaYFP-FIB+CFP-B23 cells were transiently transfected with DsRed2-RL27 for 16 h before imaging. The time shown on the top of the panel indicates the time for the cell progressing through mitosis and 00:00 indicates the beginning of chromosome migration toward its respective pole. Bar, 5 μm. Pairs of arrows were used to follow two reforming NORs to form a single nucleolus and the color changes from red to green to represent the fusion events. (B) The mean fluorescence intensities of the nucleolar components FIB, B23 and RL27 within nucleoli were averaged, normalized and plotted over 5 (left) and 15 h (right) in the same manner as in Fig. 6 D. The distribution of the original data is reported in Fig. S2. (C) Collating data from Figs. 6 and 7, a possible time line to summarize the relationship between different nucleolar subcompartments and nuclear import markers, is proposed. The top side indicates the time for different nucleolar components to reassemble into nucleoli, whereas the bottom side indicates the time required for nucleolar markers RPA39 and FIB as well as nuclear import markers IBB to enter nucleoli after mitosis. The time line begins at 00:00 when the chromosomes start to migrate toward its respective pole. For nucleolar entry, RPA39, FIB, B23, and RL27 reach 50% of its saturated value at 14:42, 19:00, 53:00, and 60:00, respectively. For nuclear import, FIB, IBB, and RPA39 reach 50% of its saturated value at 09:06, 11:18, and 13:48, respectively. Bar, 5 min. (D) The number of clusters represented by the DFC marker FIB was counted and plotted against time, where time equals 00:00 when the chromosomes start to migrate toward its respective pole and n = the number of cells examined. An orange window was added in the same time scale to denote the first appearance of GC within the reforming nucleoli. The distribution of the original data counting the number of DFC clusters is reported in Fig. S3 A (available at http://www.jcb.org/cgi/content/full/jcb.200405013/DC1).
To investigate spatial aspects of nucleolar formation, we quantitated the number of separate DFC clusters over time as cells exit mitosis (Fig. 7 D; see Online supplemental material). We observed an increase in the mean number of DFC clusters, up to ∼7 per nucleus, during the first 20 min after mitosis (zero time defined as the time of chromosome separation during anaphase; Fig. 7 D). However, briefly after the onset of GC formation, the mean number of DFC clusters decreased on average to ∼4.5, due to fusion events between neighboring clusters (Fig. 7 D; Fig. 7 A, green arrows). In the time-lapse micrographs it is apparent that the GC markers (e.g., B23) fuse before the fusion of the DFC marker FIB (Fig. 7 A, compare inset 31:20 with inset 38:00). Therefore, the timing of the nucleolar fusion events may be driven, at least in part, by the formation of the GC around the DFCs. Intriguingly, we observed a statistically significant variation in the number of FC foci and nucleoli formed in the two daughter nuclei, although they are more similar to each other than to unrelated nuclei exiting mitosis (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200405013/DC1). This raises the possibility that stochastic events can influence nucleolar reassembly.
Discussion
Here, we used a quantitative 4D imaging approach to analyze the processes of nucleolar breakdown and reassembly during mitosis in single live cells. HeLa cell lines were constructed that stably express either one or two FP-tagged markers for either different nucleolar subcompartments, nuclear lamina components, nuclear transport reporters and/or chromatin. By establishing double-transformed stable cell lines and parallel transient transfection, we could perform multi-wavelength 3D microscopy over time to correlate changes in the relative distributions and concentrations of multiple nuclear marker proteins in the same live cell during mitosis. The data for nucleolar disassembly and reassembly during mitosis (Fig. 8) show that nucleolar breakdown begins with the loss of RNA polymerase I subunits from FCs, before the onset of NE breakdown. The subsequent disassembly of the DFC and GC subcompartments coincides with NE disassembly.
Figure 8.

The dynamics of nucleolus during mitosis. The nucleolar disassembly and reassembly pathway are shown in a–c and d–i, respectively. During disassembly, (a and b) the RPA39 (blue) dissociates from the nucleoli ∼2 min before the loss of functional integrity of the NE as illustrated by the leakage of IBB (gray). (b and c) Within ∼4 min, the DFC (yellow) and GC (magenta) subcompartments disintegrate, within the period of NE breakdown and (c) subtle changes in the distribution of proteins localized to these two subcompartments are observed (Fig. 3 C). (d) A brief period during metaphase follows, in which the RPA39 is not associated with the NORs, whereas the UBF remains bound. (e) Before anaphase, the RNA polymerase I subunits reassociate with the UBF-containing NORs and at the same time FIB reassociates with the chromosomes (shown by a change of color on the chromosome). At this stage PNBs (orange dots), which consists of partially processed rRNA transcripts and associated processing factors, are formed in the nucleoplasm. (f) Within 5 min, FIB starts to reassociate with the reforming NORs and the number of reforming NORs increases upon the activation of nuclear import. (g and h) Within 27 min after step e, the GC reforms, coincident with the maximal number of DFC fusion events. (i) Variations in the number of nucleoli between daughter nuclei imply that stochastic events can influence nucleolar reassembly.
The RPA39 dissociates from the NORs during a brief period within metaphase, although the rRNA promoter binding factor, UBF, remains bound (Fig. 2). The pathway of nucleolar reformation in live cells after mitosis showed a strictly defined and reproducible temporal sequence of incorporation of markers in the order of FC, DFC, and finally GC. However, this did not correlate with the order in which the factors were imported into nuclei, as the DFC marker FIB accumulated early in nuclei but only later was incorporated into nucleolar foci, coincident with the maximal nuclear import of RNA polymerase I.
The advantages of live cell imaging have recently been applied to study several dynamic nuclear processes (Clute and Pines, 1999; Gerlich et al., 2001; Beaudouin et al., 2002; Gerlich and Ellenberg, 2003a; Prasanth et al., 2003). A feature of the live cell approach used here is that we can conduct quantitative studies on nucleolar dynamics during mitosis at the single cell level. Moreover, we have directly correlated quantitatively the temporal changes in each of the separate nucleolar, NE and chromatin components within the nucleus. This has allowed us to detect small differences in the timing of events that would not be apparent by conventional immunofluorescence approaches, where temporal information is not available. Similarly, biochemical methods sample the mean properties of cell populations, rather than the behavior or sequence of events in individual cells. For example, we could reproducibly detect a timing difference as small as ∼4 min in the loss of RPA39 from the FC, before loss of DFC or GC markers during nucleolar breakdown. Quantitative analyses of these time-lapse data obtained from multiple cells in each experiment also provided information regarding the kinetic behavior of nuclear proteins across the cell population and showed the degree of variation between nuclei.
Recent studies on the dynamics of nucleoli during mitosis have focused mainly on the process of postmitotic nucleolar reformation (Dundr et al., 2000; Savino et al., 2001). Two previous studies have included the use of live cell imaging to analyze events during the formation of nucleoli (Dundr et al., 2000; Savino et al., 2001). Both studies used a combination of immunofluorescence and cell lines expressing a single GFP-tagged marker to analyze the temporal order of formation of PNBs and nucleoli. In particular, both the composition of PNBs, which accumulate partially processed rRNA precursors and associated components, and how these components are subsequently transferred into NORs for nucleolar assembly have been studied (Jimenez-Garcia et al., 1994; Dundr et al., 2000; Savino et al., 2001). Here, we have focused on quantitating the temporal pathway of nucleologenesis as well as studying the spatial organization of reforming NORs. In each of the live cells analyzed, multiple FP-tagged markers used for different nucleolar subcompartments allows us to distinguish reforming NORs and nucleoli from PNBs. For example, PNBs do not contain RNA polymerase I, which reassociates with the NOR early during nucleologenesis. Thus, we could perform a correlative as well as quantitative analysis of components from all three nucleolar subcompartments in parallel. More importantly, we have extended these analyses by comparing the processes of nucleolar breakdown as well as reassembly in the same cells. A novel conclusion from this work is that RNA polymerase I subunits RPA39 and RPA20 transiently leave the NORs during metaphase, whereas UBF remains associated with NORs throughout mitosis. Our findings are supported by the data from a recent independent study showing that other RNA polymerase I subunits, specifically, RPA194, RPA39, and RPA16, but not RPA43, also leave the NORs during metaphase (Dundr, M., and T. Misteli, personal communication). These results differ from the current view that RNA polymerase I, as well as UBF, remains associated with NORs throughout mitosis, based on immunofluorescence data using fixed cells, where it is difficult to detect a transient absence of RNA polymerase I. Our data in contrast indicate that multiple RNA polymerase I subunits either leave the chromosomes transiently, or else decrease in concentration below our detection limit, for a brief period at metaphase. We note that this can explain the previous observation based on high resolution in situ hybridization studies that metaphase chromosomes do not contain nascent rRNA (Weisenberger and Scheer, 1995). Our data are also consistent with the findings from run-on assays that rRNA genes in metaphase chromosomes, whereas still in the same open configuration as interphase chromosomes, are less transcriptionally active (Conconi et al., 1989). Our present data therefore indicate that the mitotic behavior of RNA polymerase I may be more similar to RNA polymerase II than was previously apparent.
We observe distinct kinetic behavior of individual proteins during nucleolar disassembly (Fig. 3 D and Fig. 5 C). For example, although dissociation of B23 and RL27 from nucleoli initiates later than RPA39 or FIB, their dissociation rate is higher. Both GC components dissociate from nucleoli at similar rates, with comparable kinetics to cytoplasmic dispersal of IBB upon NE breakdown, which is most likely a diffusion-limited event. Although the molecular mechanism of nucleolar disassembly remains poorly understood, the present data raise the possibility that distinct processes could operate sequentially and/or independently during the disassembly of FC, DFC, and GC subcompartments. We observe that RPA39 leaves the nucleolus before breakdown of nuclear lamina components, whereas the DFC and GC markers are lost during the period of nuclear lamina breakdown. It is known from previous studies that lamina disassembly is triggered via cyclin B-CDK1 mediated phosphorylation of multiple components, including LB1 (Pines and Rieder, 2001; Burke and Ellenberg, 2002). Coincidently, the same cyclin complex is involved in repression of mitotic ribosomal transcription and nucleolar reformation. For example, phosphorylation of UBF and transcription factor SL1 by CDK1 causes shut-off of RNA polymerase I transcription (Heix et al., 1998; Klein and Grummt, 1999). The CDK1 inhibitor roscovitine also causes reactivation of RNA polymerase I transcription during mitosis but not the recruitment of rRNA processing factors to the rRNA gene repeats (Sirri et al., 2000, 2002). Our data indicate that the loss of RNA polymerase I and hence transcription of rRNA genes is likely to be the initial event during mitotic disassembly of nucleoli. However, loss of rRNA gene transcription alone may not be sufficient to cause subsequent disassembly of the entire nucleolus. For example, although inhibition of ribosomal transcription during interphase causes RNA polymerase I subunits to leave nucleoli in vivo (unpublished data), the inhibition of ribosomal transcription by Actinomycin D in isolated nucleoli does not cause nucleoli to disintegrate in vitro (unpublished data). Therefore, we propose that the mitotic disassembly of the DFC and GC subcompartments is a result of an active mechanism rather than an indirect effect of the loss of transcriptional activity. This is consistent with a recent study that germ cell proteins FRGY2a and FRGY2b can reversibly disassemble somatic nucleoli in Xenopus egg cytoplasm independent of rRNA transcription (Gonda et al., 2003), suggesting that transcription activity and nucleolar integrity may not be obligatorily coupled. It will thus be interesting in future to test whether molecular mechanisms such as phosphorylation by the cyclin B–CDK1 complex may play a role in either RPA39 dissociation from FCs or in other steps in the nucleolar breakdown pathway.
In contrast with the precise temporal regulation in the order of events during both disassembly and reformation of nucleoli during mitosis, higher order aspects of nucleologenesis appear to be less strictly controlled. A statistical evaluation of the number of FC foci and functional nucleoli that appear after mitosis suggested that daughter nuclei are more similar to each other than to unrelated nuclei exiting mitosis elsewhere, consistent with the expected conservation in global chromosome positioning (Gerlich and Ellenberg, 2003b; Walter et al., 2003). Nonetheless, we observe statistically significant differences between the two daughter nuclei. At least from the point at which RNA polymerase I subunits reassociate with NORs, it appears that local effects, which can differ between daughter nuclei, influence the overall pathway of nucleologenesis. Effects acting at the level of the local chromosome environment, such as variations in chromosome orientation and decondensation (Thomson et al., 2004), variable activation of rRNA genes within the repeat clusters and variation in protein concentration kinetics (Dundr et al., 2002a), may all influence both the association of RNA polymerase I with NORs and the probability of NOR fusion and hence the number of active nucleoli formed. It is interesting to compare this view with a recent FISH analysis on the process of induced RNA polymerase II gene activation at the single cell level (Levsky et al., 2002). Both this FISH analysis and our work suggest that the pattern and level of gene activation varies at the single cell level, which had not been apparent from previous biochemical studies from cell populations. Although it is increasingly appreciated that nuclear structure, including the relative 3D distribution of chromosomes (Parada and Misteli, 2002; Bickmore and Chubb, 2003), can influence gene expression by RNA polymerase II, we infer from this work that nuclear structure may also have an important effect on events connected with RNA polymerase I transcription.
Materials and methods
Cell culture, transfection, and establishment of stable cell line
HeLa cell lines were grown in DME supplemented with 10% FCS and 100 U/ml penicillin and streptomycin (Invitrogen). Single and double stably-transformed cell lines were established using selection with G418 and/or blasticidin, after transfection with ∼2 μg of each respective plasmid construct (Table SI, available at http://www.jcb.org/cgi/content/full/jcb.200405013/DC1) per 100-mm dish. 24 independent clones were isolated for each cell line and three selected for analysis after further characterization. A description of each cell line is provided in Table SII. Plasmid transfection was performed using Effectene reagent (QIAGEN) according to the manufacturer's instructions.
Immunostaining
To prepare the fixation buffer, 2× PHEM buffer (18.14 g Pipes, 6.5 g Hepes, 0.99 g MgSO4, 3.8 g EGTA in 500 ml H2O, pH 7.0) were added to freshly prepared 37% PFA in 1:5 ratio. The fixation buffer was then warmed to 37°C and added directly to the cells slowly after pouring the media out of the dish. The cells were fixed for 10 min before being washed thrice with 1× PBS very gently. The cells were then permeabilized, stained and mounted as previously published (Leung and Lamond, 2002). Primary antibodies used were anti-UBF (1:5; Santa Cruz Biotechnology, Inc.) and anti-RPA20 (1:1; B6-2; Jones et al., 2000).
Immunoprecipitations and in vitro transcription assays
50 μg of HeLaYFP-RPA39 or HeLa nuclear extract were precleared for 30 min with 5 μl of protein G–Sepharose beads and used in immunoprecipitations with 4 μg of anti-GFP antibodies bound to 7.5 μl of protein G–Sepharose beads in 0.25 M KCl/TM10 (50 mM Tris–HCl, pH 7.9, 12.5 mM MgCl2, 1 mM EDTA, 10% glycerol, 1 mM sodium metabisulfite and 1 mM DTT) buffer. The beads were incubated with nuclear extract for 1 h, with shaking, at 4°C. After immunoprecipitation the beads were washed in TM10/0.25 M KCl buffer, then equilibrated in TM10/0.05 M KCl buffer and used in in vitro transcription assays. In vitro transcription reactions were performed as described previously (Miller et al., 2001) at a final salt concentration of 50–70 mM KCl. Supercoiled prHu3 plasmid DNA, which contains the human rRNA gene promoter from −515 to +1548, were used as templates in the transcription reaction. The resulting transcripts were analyzed in an S1 nuclease protection assay after annealing the RNA to a 5′-end–labeled oligonucleotide, which was identical to the region between −20 and +40 of the promoter template strand.
BrUTP incorporation
Coverslips seeded with HeLa cells were rinsed with hypotonic KH buffer (30 mM KCl, 10 mM Hepes, pH 7.4) briefly and incubated with 50 μl KH buffer containing 10 mM BrUTP (Sigma-Aldrich) for 5 min in a 5% CO2 incubator at 37°C. The cells on coverslips were “chased” with DME containing 20% FCS and 200 μg/ml (final concentration) G418 for a defined time in the incubator to chase the transcripts before fixation. Before methanol fixation for 20 min at −20°C, the coverslips were rinsed with PBS. The cells were then permeabilized with acetone for 30 s and air-dried for 10 min, followed by rehydration with PBS for 5 min and immunostaining using the anti-BrdU (1:5) antibody.
Mitotic studies of living cells
At each time point a 3D image stack spanning the entire nuclear volume was recorded for each of the three wavelengths. Low light imaging conditions were chosen to ensure that cells were able to progress through the entire mitosis. For these experiments a series of “double stable” cell lines were established that express combinations of two FP-tagged markers (Table SII). The third marker in each experiment was expressed by transient transfection (Table SI). Cells were seeded in a Labtek II chambered coverglasses on the previous night before imaging and an imaging medium containing 20% FCS and 0.5 mg/ml L-ascorbic acid replaced the growth medium for 3–4 h before imaging. 4D imaging was performed on a customized confocal laser scanning fluorescence microscope (model LSM510; Carl Zeiss MicroImaging, Inc.) kept at 37°C and equipped with a z-scanning stage (HRZ 200) for fast 4D acquisition using a Plan Apochromat 63× DIC oil immersion objective. Triple-color imaging of CFP, YFP and DsRed/HcRed was achieved by alternating the 413 nm Kr, 514 nm Ar, and 543 nm HeNe laser for selective excitation. Bidirectional scanning and detection were performed as published previously (Gerlich et al., 2001).
Quantitation and statistical analysis
For object identification, a reference channel was chosen that represented the structure of interest. For example, FP-H2B and FP-RPA39 are used to define the nuclear volume and reforming NORs, respectively. The images in this channel were processed by an anisotropic diffusion filtering and subsequent thresholding (Gerlich et al., 2001) to obtain a binary mask representing areas of interest. Mean fluorescence intensities were then measured in all channels of the original unfiltered images within these areas. Automated analysis of 4D data was achieved by implementation of a computer macro, which was executed in the image processing toolbox Heurisko 4.05 (Aeon). Importantly, a single threshold was selected for the analysis of entire 4D images. Although the specific threshold chosen by the user affects absolute values, the kinetics of protein concentrations measured during mitotic progression were largely unaffected (Gerlich et al., 2001). The data collected were then normalized and analyzed using GraphPad Prism 4.0 software.
Online supplemental material
Fig. S1 includes (A) representative Western blots showing expression levels of FP-fusion proteins in cell lines used, (B) FP-RPA39 localization at EM level, the localization of (C) pulsed labeled rRNA transcripts and (D) RPA20 in HeLaYFP-RPA39 cells. Distributions of quantitative measurements in Figs. 3, 5–7 are shown in Fig. S2. The statistical analysis for the differential behavior of daughter cells after mitosis is shown in Fig. S3. Fig. S4 shows that the loss of functionality of the NE precedes its structural disintegration. The plasmid and stable cell lines used in this work are tabulated in Table S1. The localization of RPA39, FIB and B23 are shown in Videos 1 and 2. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200405013/DC1.
Acknowledgments
We thank Drs. H. Kimura and P.R. Cook (University of Oxford, Oxford, UK) for the gift of mAbs B6-2, Mr. G. Rabut (EMBL) for providing macros to ensure fast acquisition after multi-point visit and multi-wavelength time series scanning as well as autofocussing during imaging of multi-point time series, and Ms. E. Zanin (EMBL) for making the IBB-FP fusion construct.
A.K.L. Leung was supported by a Croucher Foundation Scholarship, an EMBL Advanced Light Microscopy Fellowship and a Company of Biologists Ltd. travelling Fellowship. D. Gerlich is supported by an EMBO long-term fellowship. G. Miller was a Wellcome Trust Prize Student. A.I. Lamond is a Wellcome Trust Principal Research Fellow. J. Zomerdijk is a Wellcome Trust Senior Research Fellow. The Human Frontier Science Program is acknowledged for a research grant entitled “Functional organization of the cell nucleus investigated through proteomics and molecular dynamics”.
A.K.L. Leung's present address is Center for Cancer Research, Dept. of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139.
G. Miller's present address is Fred Hutchinson Cancer Research Center, MS A1-162, 1100 Fairview Ave. N., Seattle, WA 9810.
C. Lyon's present address is Cyclacel, James Lindsay Place, Dundee Technopole, Dundee DD1 5JJ, UK.
Abbreviations used in this paper: B23, nucleophosmin/nucleolar phosphoprotein B23/numatrin; DFC, dense fibrillar component; FC, fibrillar centre; FIB, fibrillarin; FP, fluorescent protein; GC, granular component; IBB, importin-β binding; LB1, lamin B1; LBR, lamin B receptor; NE, nuclear envelope; NOR, nucleolar organizing region; PNB, prenucleolar body; rDNA, ribosomal DNA; RL27, ribosomal protein L27; RPA39, RNA polymerase I subunit RPA39; RRN3, RNA polymerase I transcription factor RRN3; UBF, upstream binding transcription factor.
References
- Beaudouin, J., D. Gerlich, N. Daigle, R. Eils, and J. Ellenberg. 2002. Nuclear envelope breakdown proceeds by microtubule-induced tearing of the lamina. Cell. 108:83–96. [DOI] [PubMed] [Google Scholar]
- Bickmore, W.A., and J.R. Chubb. 2003. Dispatch. Chromosome position: now, where was I? Curr. Biol. 13:R357–R359. [DOI] [PubMed] [Google Scholar]
- Burke, B., and J. Ellenberg. 2002. Remodelling the walls of the nucleus. Nat. Rev. Mol. Cell Biol. 3:487–497. [DOI] [PubMed] [Google Scholar]
- Carmo-Fonseca, M., L. Mendes-Soares, and I. Campos. 2000. To be or not to be in the nucleolus. Nat. Cell Biol. 2:E107–E112. [DOI] [PubMed] [Google Scholar]
- Clute, P., and J. Pines. 1999. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol. 1:82–87. [DOI] [PubMed] [Google Scholar]
- Conconi, A., R.M. Widmer, T. Koller, and J.M. Sogo. 1989. Two different chromatin structures coexist in ribosomal RNA genes throughout the cell cycle. Cell. 57:753–761. [DOI] [PubMed] [Google Scholar]
- Dundr, M., T. Misteli, and M.O. Olson. 2000. The dynamics of postmitotic reassembly of the nucleolus. J. Cell Biol. 150:433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr, M., U. Hoffmann-Rohrer, Q. Hu, I. Grummt, L.I. Rothblum, R.D. Phair, and T. Misteli. 2002. a. A kinetic framework for a mammalian RNA polymerase in vivo. Science. 298:1623–1626. [DOI] [PubMed] [Google Scholar]
- Dundr, M., J.G. McNally, J. Cohen, and T. Misteli. 2002. b. Quantitation of GFP-fusion proteins in single living cells. J. Struct. Biol. 140:92–99. [DOI] [PubMed] [Google Scholar]
- Gerbi, S.A., A.V. Borovjagin, and T.S. Lange. 2003. The nucleolus: a site of ribonucleoprotein maturation. Curr. Opin. Cell Biol. 15:318–325. [DOI] [PubMed] [Google Scholar]
- Gerlich, D., and J. Ellenberg. 2003. a. 4D imaging to assay complex dynamics in live specimens. Nat. Cell Biol. 5:S14–S19. [PubMed] [Google Scholar]
- Gerlich, D., and J. Ellenberg. 2003. b. Dynamics of chromosome positioning during the cell cycle. Curr. Opin. Cell Biol. 15:664–671. [DOI] [PubMed] [Google Scholar]
- Gerlich, D., J. Beaudouin, M. Gebhard, J. Ellenberg, and R. Eils. 2001. Four-dimensional imaging and quantitative reconstruction to analyse complex spatiotemporal processes in live cells. Nat. Cell Biol. 3:852–855. [DOI] [PubMed] [Google Scholar]
- Gilbert, N., L. Lucas, C. Klein, M. Menager, N. Bonnet, and D. Ploton. 1995. Three-dimensional co-location of RNA polymerase I and DNA during interphase and mitosis by confocal microscopy. J. Cell Sci. 108:115–125. [DOI] [PubMed] [Google Scholar]
- Gonda, K., J. Fowler, N. Katoku-Kikyo, J. Haroldson, J. Wudel, and N. Kikyo. 2003. Reversible disassembly of somatic nucleoli by the germ cell proteins FRGY2a and FRGY2b. Nat. Cell Biol. 5:205–210. [DOI] [PubMed] [Google Scholar]
- Heix, J., A. Vente, R. Voit, A. Budde, T.M. Michaelidis, and I. Grummt. 1998. Mitotic silencing of human rRNA synthesis: inactivation of the promoter selectivity factor SL1 by cdc2/cyclin B-mediated phosphorylation. EMBO J. 17:7373–7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Verdun, D., P. Roussel, and J. Gebrane-Younes. 2002. Emerging concepts of nucleolar assembly. J. Cell Sci. 115:2265–2270. [DOI] [PubMed] [Google Scholar]
- Huang, S. 2002. Building an efficient factory: where is pre-rRNA synthesized in the nucleolus? J. Cell Biol. 157:739–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki, S.M., and D.L. Spector. 2003. Nuclear choreography: interpretations from living cells. Curr. Opin. Cell Biol. 15:149–157. [DOI] [PubMed] [Google Scholar]
- Jimenez-Garcia, L.F., M.L. Segura-Valdez, R.L. Ochs, L.I. Rothblum, R. Hannan, and D.L. Spector. 1994. Nucleologenesis: U3 snRNA-containing prenucleolar bodies move to sites of active pre-rRNA transcription after mitosis. Mol. Biol. Cell. 5:955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, E., H. Kimura, M. Vigneron, Z. Wang, R.G. Roeder, and P.R. Cook. 2000. Isolation and characterization of monoclonal antibodies directed against subunits of human RNA polymerases I, II and III. Exp. Cell Res. 254:163–172. [DOI] [PubMed] [Google Scholar]
- Jordan, P., M. Mannervik, L. Tora, and M. Carmo-Fonseca. 1996. In vivo evidence that TATA-binding protein/SL1 colocalizes with UBF and RNA polymerase I when rRNA synthesis is either active or inactive. J. Cell Biol. 133:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, J., and I. Grummt. 1999. Cell cycle-dependent regulation of RNA polymerase I transcription: the nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc. Natl. Acad. Sci. USA. 96:6096–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, A.K., and A.I. Lamond. 2002. In vivo analysis of NHPX reveals a novel nucleolar localization pathway involving a transient accumulation in splicing speckles. J. Cell Biol. 157:615–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, A.K., and A.I. Lamond. 2003. The dynamics of the nucleolus. Crit. Rev. Eukaryot. Gene Expr. 13:39–54. [DOI] [PubMed] [Google Scholar]
- Levsky, J.M., S.M. Shenoy, R.C. Pezo, and R.H. Singer. 2002. Single-cell gene expression profiling. Science. 297:836–840. [DOI] [PubMed] [Google Scholar]
- Miller, G., K.I. Panov, J.K. Friedrich, L. Trinkle-Mulcahy, A.I. Lamond, and J.C. Zomerdijk. 2001. hRRN3 is essential in the SL1-mediated recruitment of RNA Polymerase I to rRNA gene promoters. EMBO J. 20:1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, M.O., M. Dundr, and A. Szebeni. 2000. The nucleolus: an old factory with unexpected capabilities. Trends Cell Biol. 10:189–196. [DOI] [PubMed] [Google Scholar]
- Parada, L., and T. Misteli. 2002. Chromosome positioning in the interphase nucleus. Trends Cell Biol. 12:425–432. [DOI] [PubMed] [Google Scholar]
- Pines, J., and C.L. Rieder. 2001. Re-staging mitosis: a contemporary view of mitotic progression. Nat. Cell Biol. 3:E3–E6. [DOI] [PubMed] [Google Scholar]
- Prasanth, K.V., P.A. Sacco-Bubulya, S.G. Prasanth, and D.L. Spector. 2003. Sequential entry of components of the gene expression machinery into daughter nuclei. Mol. Biol. Cell. 14:1043–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel, P., C. Andre, C. Masson, G. Geraud, and D. Hernandez-Verdun. 1993. Localization of the RNA polymerase I transcription factor hUBF during the cell cycle. J. Cell Sci. 104:327–337. [DOI] [PubMed] [Google Scholar]
- Rubbi, C.P., and J. Milner. 2003. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 22:6068–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savino, T.M., J. Gebrane-Younes, J. De Mey, J.B. Sibarita, and D. Hernandez-Verdun. 2001. Nucleolar assembly of the rRNA processing machinery in living cells. J. Cell Biol. 153:1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer, U., and K.M. Rose. 1984. Localization of RNA polymerase I in interphase cells and mitotic chromosomes by light and electron microscopic immunocytochemistry. Proc. Natl. Acad. Sci. USA. 81:1431–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirri, V., P. Roussel, and D. Hernandez-Verdun. 2000. In vivo release of mitotic silencing of ribosomal gene transcription does not give rise to precursor ribosomal RNA processing. J. Cell Biol. 148:259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirri, V., D. Hernandez-Verdun, and P. Roussel. 2002. Cyclin-dependent kinases govern formation and maintenance of the nucleolus. J. Cell Biol. 156:969–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, I., S. Gilchrist, W.A. Bickmore, and J.R. Chubb. 2004. The radial positioning of chromatin is not inherited through mitosis but is established de novo in early G1. Curr. Biol. 14:166–172. [DOI] [PubMed] [Google Scholar]
- Walter, J., L. Schermelleh, M. Cremer, S. Tashiro, and T. Cremer. 2003. Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J. Cell Biol. 160:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberger, D., and U. Scheer. 1995. A possible mechanism for the inhibition of ribosomal RNA gene transcription during mitosis. J. Cell Biol. 129:561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]