

Abstract
Actopaxin is an actin and paxillin binding protein that localizes to focal adhesions. It regulates cell spreading and is phosphorylated during mitosis. Herein, we identify a role for actopaxin phosphorylation in cell spreading and migration. Stable clones of U2OS cells expressing actopaxin wild-type (WT), nonphosphorylatable, and phosphomimetic mutants were developed to evaluate actopaxin function. All proteins targeted to focal adhesions, however the nonphosphorylatable mutant inhibited spreading whereas the phosphomimetic mutant cells spread more efficiently than WT cells. Endogenous and WT actopaxin, but not the nonphosphorylatable mutant, were phosphorylated in vivo during cell adhesion/spreading. Expression of the nonphosphorylatable actopaxin mutant significantly reduced cell migration, whereas expression of the phosphomimetic increased cell migration in scrape wound and Boyden chamber migration assays. In vitro kinase assays demonstrate that extracellular signal-regulated protein kinase phosphorylates actopaxin, and treatment of U2OS cells with the MEK1 inhibitor UO126 inhibited adhesion-induced phosphorylation of actopaxin and also inhibited cell migration.
Keywords: phosphorylation; migration; adhesion; focal adhesions; Erk
Introduction
Sites of cell adhesion to the ECM, commonly referred to as focal adhesions or focal complexes, are important centers of signal transduction. Through the recruitment of numerous structural and regulatory proteins to the cytoplasmic tails of clustered integrin receptors, focal adhesions contribute to creating physical and functional links between the ECM and the actin cytoskeleton thereby regulating many important cellular processes including migration, proliferation, and survival (Clark and Brugge, 1995; Giancotti and Ruoslahti, 1999; Hynes, 2002).
Cell migration is a multistep process (Lauffenburger and Horwitz, 1996). A major driving force of motility is the formation of a leading edge protrusion and extension of a lamellipodium, followed by the establishment of new focal adhesion sites at the front of the cell and detachment of adhesions at the rear. These events require the dynamic reorganization of the actin cytoskeleton and the turnover of focal adhesions (Lauffenburger and Horwitz, 1996; Horwitz and Parsons, 1999; Totsukawa et al., 2004; Webb et al., 2004). A diverse range of signaling molecules have been implicated in coordinating these events including the Rho family of GTPases and their numerous effectors such as the p21-activated kinase (PAK), Rho kinase, and N-WASP (Takenawa and Miki, 2001; Bokoch, 2003; Raftopoulou and Hall, 2004); that, in turn, target key actin regulators to control actin filament assembly and actomyosin contractility (Brahmbhatt and Klemke, 2003; Totsukawa et al., 2004). Focal adhesion proteins such as FAK and paxillin also perform an important adaptor function and are essential for normal focal adhesion turnover (Turner, 2000; West et al., 2001; Webb et al., 2004).
The MAPK extracellular signal-regulated protein kinase (Erk) also localizes to focal adhesions, potentially through an interaction with paxillin (Ishibe et al., 2003). Erk contributes to the regulation of cell migration through activation of myosin light chain kinase (MLCK) and subsequent phosphorylation of myosin light chain (Klemke et al., 1997) to stimulate localized myosin-based contractility, membrane ruffling, and focal adhesion turnover (Klemke et al., 1997; Brahmbhatt and Klemke, 2003; Totsukawa et al., 2004; Webb et al., 2004). Erk also facilitates focal adhesion disassembly and thus cell retraction at the rear, in part through phosphorylation and activation of the protease Calpain (Bhatt et al., 2002; Carragher et al., 2003).
Actopaxin, also known as α-parvin/CH-ILKBP, is a focal adhesion protein that interacts with actin, paxillin, and the integrin-linked kinase (ILK; Nikolopoulos and Turner, 2000, 2002; Olski et al., 2001; Tu et al., 2001; Yamaji et al., 2001). Actopaxin is composed of an amino terminus of 95 aa followed by two calponin homology (CH) domains. A functional role for the tandem CH domains in F-actin binding has been confirmed (Nikolopoulos and Turner, 2000; Olski et al., 2001). The CH2 domain of actopaxin also contains the ILK and paxillin binding subdomain (PBS; Nikolopoulos and Turner, 2000, 2002; Tu et al., 2001). Consistent with a role for actopaxin in actin-mediated cellular processes, overexpression of actopaxin mutants defective for either paxillin- or actin-binding results in impaired cell adhesion and cell spreading (Nikolopoulos and Turner, 2000, 2002; Tu et al., 2001; Yamaji et al., 2001).
Although lacking any obvious tertiary structure, the amino terminus of actopaxin is phosphorylated by the cyclinB/cdc2 kinase during entry into mitosis (Curtis et al., 2002). Phosphorylation of actopaxin, like other focal adhesion and cytoskeletal proteins such as FAK, paxillin, caldesmon, and PAK, likely contributes to the pre-mitotic disassembly of focal adhesions associated with cell rounding and/or cell re-spreading after cytokinesis (Yamaguchi et al., 1997; Yamashiro et al., 2001; Curtis et al., 2002; Thiel et al., 2002). However, a functional role for actopaxin phosphorylation in the regulation of other cellular processes requiring cytoskeletal remodeling, such as cell migration, has not been previously evaluated.
Here, we have determined that the actopaxin amino terminus is phosphorylated in response to cell adhesion. Using nonphosphorylatable and phosphomimetic mutants of actopaxin we demonstrate that actopaxin phosphorylation is required for cell spreading, lamellipodia formation, and cell migration. In addition, the MAPK Erk phosphorylates the amino terminus of actopaxin and pharmacologic inhibition of MEK1, the upstream regulator of Erk, inhibited both actopaxin phosphorylation and cell migration. Together, these results identify actopaxin phosphorylation as a new and critical component of the cell migration machinery.
Results
Actopaxin phosphorylation is stimulated during cell spreading
Actopaxin is composed of an amino terminus of 95 aa followed by two CH domains. The CH domains function as binding sites for F-actin, paxillin, and ILK. The amino terminus lacks any obvious tertiary structure. However, it contains several phosphorylation consensus sites for cyclinB/cdc2 that are targeted during mitosis (Fig. 1), suggesting that this region of actopaxin may serve a regulatory function associated with the rearrangement of the actin cytoskeleton and/or the disassembly of focal adhesions that accompany progression through mitosis. A similar role for actopaxin phosphorylation in regulating other cellular processes involving dynamic reorganization of the cytoskeleton such as cell adhesion/spreading and motility has not previously been investigated.
Figure 1.

Actopaxin domain structure and phosphorylation sites. The domain schematic illustrates the relative positions of the two calponin homology (CH) domains, ILK, and PBS, and the principal cyclin B/cdc2 phosphorylation sites. The amino-terminal amino acid sequence indicates the location of the five potential phosphorylation sites (S4, 8, 14, 19, and T16) targeted in the current work.
Actopaxin phosphorylation was examined during cell adhesion and spreading of human osteosarcoma (U2OS) cells. Cells were in vivo labeled with inorganic 32P, harvested, and either held in suspension or re-spread on a collagen matrix for 2 h. Endogenous actopaxin was immunoprecipitated and visualized by autoradiography. Actopaxin phosphorylation was significantly enhanced in the adherent/spreading cell population (Fig. 2, lane 2) as compared with cells retained in suspension (Fig. 2, lane 1). Analysis of the cell lysates indicated identical levels of 32P-incorporation (Fig. 2, lanes 3 and 4). Paxillin phosphorylation was also stimulated during cell spreading, consistent with previous reports (Fig. 2, lanes 1 and 2; Bellis et al., 1997). These data demonstrate that actopaxin phosphorylation is not restricted to mitotic events, and suggested a role in cell adhesion, spreading, and migration.
Figure 2.

Actopaxin is phosphorylated in vivo after cell adhesion. Cells were labeled with inorganic 32P and either held in suspension (lanes 1 and 3) or spread on collagen I–coated dishes for 2 h (lanes 2 and 4). Actopaxin (top) and paxillin (bottom) proteins were immunoprecipitated, resolved on SDS-PAGE, and visualized by autoradiography. WCL, whole cell lysate.
Adhesion-mediated phosphorylation of actopaxin is restricted to the amino terminus
To evaluate the role of actopaxin phosphorylation in regulating cell adhesion and spreading, clonal U2OS cell lines expressing Xpress-tagged full-length actopaxin wild-type (WT) actopaxin and an actopaxin phosphorylation mutant were generated. The amino terminus of actopaxin contains four serine/proline phosphorylation sites and one threonine/proline site (Fig. 1), previously shown to be phosphorylated by cyclin B/cdc2 (Curtis et al., 2002). Taking these data into consideration, we generated a quintuple (Quint) nonphosphorylatable mutant of actopaxin in which each of the serine phosphorylation sites were changed to glycine and the threonine was changed to alanine (S(4,8,14,19)G/T16A). Sites were not mutated individually because previous studies indicated that this potentially contributes to phosphorylation of cryptic sites (Curtis et al., 2002). The Quint and WT proteins were then evaluated for their ability to be phosphorylated during cell adhesion using in vivo 32P-labeling assays as described above.
As was observed with endogenous actopaxin, Xpress-WT actopaxin was phosphorylated in response to cell adhesion/spreading (Fig. 3 A, lanes 1 and 2), whereas the Quint actopaxin mutant was not phosphorylated (Fig. 3 A, lanes 3 and 4). Immunoprecipitation of the WT and Quint proteins demonstrated that both are expressed and immunoprecipitated at similar levels (Fig. 3 B). These data confirmed that actopaxin is phosphorylated in response to cell adhesion/spreading in asynchronously growing cell populations, and that the sites targeted for the phosphorylation are confined to the amino terminus of actopaxin and are eliminated in the Quint actopaxin mutant.
Figure 3.

Quint mutant of actopaxin is not phosphorylated upon adhesion/spreading. (A) WT actopaxin (WT) and a nonphosphorylatable mutant (Quint) of actopaxin were stably expressed in U2OS cells. WT and Quint clone cells were labeled with inorganic 32P and either held in suspension (lanes 1 and 3) or spread on collagen-coated dishes for 2 h (lanes 2 and 4). Xpress tagged WT and Quint actopaxin proteins were immunoprecipitated, resolved on SDS-PAGE, and visualized by autoradiography. WCL are shown to indicate equivalent labeling. (B) Parental U2OS, WT, and Quint cells were spread on collagen and Xpress-tagged WT and Quint actopaxin proteins were immunoprecipitated and visualized by Western blotting. The same blot was probed with α-actinin antibodies to indicate equivalent loading.
Actopaxin phosphorylation mutants localize to focal adhesions and affect cell morphology
The first two amino-terminal (4/8) serine/proline sites are the primary sites of mitotic phosphorylation, contributing to the electrophoretic mobility shift of actopaxin in that system (Curtis et al., 2002). We hypothesized that phosphorylation of the S4/8 sites may also be a significant component of the adhesion-induced phosphorylation of actopaxin. Thus, we constructed a phosphomimetic, S4/8D, mutant and generated stable U2OS cell lines expressing this mutant. Western blot analysis confirmed equal levels of expression of each actopaxin construct in the three clonal cell lines used in the functional analyses detailed below (Fig. 4 A, top). We estimate that the exogenous actopaxin is expressed at ∼2–3 times that of the endogenous protein.
Figure 4.

Expression of actopaxin phosphorylation mutants does not affect localization to focal adhesions. (A) Western blot of U2OS parental, WT, Quint, and S4/8D cells probed with an anti-Xpress–tagged antibody to confirm their equal expression. The same blot was stripped and reprobed with actopaxin and α-actinin antibodies to visualize endogenous actopaxin and to indicate equivalent loading. (B) Immunofluorescence staining of parental (a), WT (b), Quint (c), and S4/8D (d) cells with anti-Xpress antibody to detect transfected actopaxin and rhodamine phalloidin (e–h) to visualize actin stress fibers. Micrographs were merged to demonstrate localization of actopaxin WT, Quint, and S4/8D to focal adhesions at the ends of actin stress fibers (j–l). Bar, 2 μm.
Cell spreading is accompanied by the formation of focal adhesions and reorganization of the actin cytoskeleton, thus the organization of these structures was evaluated in the actopaxin-expressing cells by indirect immunofluorescence. Parental, WT, Quint, and S4/8D cells were harvested and allowed to spread overnight on glass coverslips in complete growth medium. Xpress WT, Quint, and S4/8D actopaxin mutants all localized effectively to focal adhesions as determined by labeling with anti-Xpress antibody (Fig. 4 B, b–d). Cells were simultaneously stained with rhodamine phalloidin to visualize F-actin, and to assess possible changes in the organization of the actin cytoskeleton resulting from the overexpression of actopaxin phosphorylation mutants (Fig. 4 B, e–h). Parental and WT cells exhibited similar actin organization (Fig. 4 B, e and f). In contrast, the Quint cells exhibited more robust actin staining (Fig. 4 B, g), whereas the S4/8D cells developed very fine actin stress fibers (Fig. 4 B, h). Merged images demonstrate localization of Xpress-tagged actopaxin to the ends of actin stress fibers (Fig. 4 B, i–l). To ensure that the results were not due to a clonal phenomenon, WT, Quint, and S4/8D were transiently expressed in U2OS cells. Additional stable clones were also evaluated. Analysis of these cells under the conditions detailed above yielded similar results (unpublished data).
Actopaxin phosphorylation is required for normal cell spreading
In view of the correlation between actopaxin phosphorylation and spreading we evaluated the kinetics of spreading for the WT and mutant actopaxin-expressing cells. Parental, WT, Quint, and S4/8D cells were harvested and replated on collagen type I–coated culture dishes in serum-free conditions. Cells were evaluated over a 4-h time course by time-lapse video microscopy. Hoffman modulation contrast images of WT, Quint, and S4/8D cells spreading at 45, 90, and 150 min are presented in Fig. 5 A. Parental U2OS cells demonstrated a similar phenotype to the WT cells (unpublished data). In contrast, the Quint cells were severely impaired in their ability to spread compared with WT at each of the time points (Fig. 5 A; compare a–c with d–f), whereas the S4/8D cells exhibited greatly enhanced spreading (Fig. 5 A, g–i). In addition, as cell spreading progressed the WT and S4/8D cells matured to form a polarized phenotype, exhibiting a dominant lamellipodium. However, the Quint cells extended multiple randomly oriented protrusions and failed to develop a dominant lamellipodium at any of the time points examined (Fig. 5 A; compare d–f with a–c; Videos 1–3, available at http://www.jcb.org/cgi/content/full/jcb.200404024/DC1).
Figure 5.

Actopaxin phosphorylation mutants affect cell spreading. Actopaxin WT, Quint, and S4/8D cells were plated on collagen type 1–coated dishes and evaluated by time-lapse video microscopy. (A) Representative Hoffman illumination images were acquired at 45, 90, and 150 min after plating. Quint cells are poorly spread and lack lamellipodia at all time points (d-f) compared with WT (a–c), whereas the S4/8D cells demonstrate enhanced spreading (g–i). Bar, 5 μm. (B) Quantitation of cell area during spreading. Parental, WT, Quint, and S4/8D cells were plated on collagen coated slips for 30, 60, 120, and 240 min. For each time point, slips were fixed and processed for indirect immunofluorescence. Total cell area was then calculated from the images. Data represent the mean ± SD of at least 100 cells from three separate experiments for each time point.
In a parallel analysis, quantitation of cell area during the spreading time course on a collagen matrix confirmed the differences in spreading, with WT equivalent to parental, Quint at 50% of parental and the S4/8D cells at ∼175% of the WT/parental populations at each time point examined (Fig. 5 B). Interestingly, analysis of the spreading time-lapse also suggested a significant defect in cell migration of the Quint cells compared with the WT and S4/8D (compare Video 2 with Videos 1 and 3). Together, these data suggest that actopaxin phosphorylation plays a critical role in adhesion-mediated cell signaling events.
Actopaxin phosphorylation mimics Rac activation
The Rho GTPases play important roles in the dynamics of cell spreading. Rho regulates cell contractility and stress fiber formation, whereas Rac promotes lamellipodia formation (Raftopoulou and Hall, 2004). The results presented above suggest a close connection between actopaxin phosphorylation and Rac signaling with the Quint cells potentially inhibiting Rac activity, whereas the morphology of the S4/8D cells suggests actopaxin phosphorylation is downstream of Rac. To examine this further, WT, Quint, and S4/8D cells were transiently transfected with dominant negative N17 Rac and GFP. Cells were fixed, costained with rhodamine phalloidin to visualize F-actin and GFP positive cells were evaluated for potential changes in morphology. WT actopaxin cells expressing the N17 Rac exhibited a reduced capacity to form lamellipodia (Fig. 6, A and B), whereas the morphology of the Quint cells was unaffected. Interestingly, the S4/8D cells were significantly more resistant than the WT cells to N17 Rac and were able to maintain their lamellipodia (Fig. 6, A and B) suggesting that the phospho-mimetic S4/8D is indeed functioning as a Rac effector. GST-PBD experiments were also performed to evaluate overall Rac activity in each of the cell lines. However, the results were inconclusive since total Rac activity was unchanged suggesting that localized regulation of Rac activity/function is critical in U2OS cells (unpublished data).
Figure 6.

Actopaxin phosphorylation mimics Rac activity. WT, Quint, and S4/8D clones plated on glass coverslips were cotransfected with dominant negative N17 Rac (DN Rac) and GFP for 24 h. (A) After transfection, slips were fixed and processed for indirect immunofluorescence. Actin stress fibers were visualized by staining with rhodamine phalloidin. Bar, 5 μm. (B) Quantitation of lamellipodia formation in WT, Quint, and S4/8D transfected cells cotransfected with DN Rac was accomplished by counting GFP positive cells and compared with control nontransfected cells. The presence of at least one broad lamellipodia on a cell was counted as positive. Note that cells expressing the S4/8D actopaxin mutant were more resistant to inhibition of lamellipodia formation than the WT cells suggesting phosphorylation of actopaxin is downstream of Rac. Percentages are representative of three separate experiments and a total of 300 cells.
Actopaxin phosphorylation regulates cell migration
Dynamic phosphorylation of cytoskeletal proteins is essential for effective cell motility. To evaluate the role of actopaxin and its phosphorylation in cell migration, wound assays were performed using parental, WT, Quint, and S4/8D cells. The rate of wound closure was assessed over a 12-h time period. The parental and WT cells closed the wound at similar rates (19.5 and 19.7 μm/h, respectively; Fig. 7 A, a, e, i, and b, f, j). The Quint cells migrated at a rate of 11.5 μm/hr, ∼40% slower than both WT and parental (Fig. 7 A, c, g, k). In contrast, the S4/8D cells demonstrated an increased ability to close the wound with a rate of 26.0 μm/h, approaching 133% of parental (Fig. 7 A, d, h, l).
Figure 7.

Expression of actopaxin phosphorylation mutants interferes with wound closure. Scrape wound assays were performed using U2OS parental and actopaxin WT, Quint, and S4/8D cells. Cells were plated in complete media at a confluent density and scored with a micropipette tip. (A) Hoffman images were taken at 0, 6, and 12 h after wounding, and average wound closure rates are shown. Quint cells have reduced closure rates compared with parental and WT cells, whereas S4/8D cells demonstrated an enhanced closure rate. Average rates of closure were calculated from three separate experiments. (B) Analysis of cell morphology at the wound edge. Parental, WT, Quint, and S4/8D cells were plated on coverslips and wounded as above. Slips were processed for indirect immunofluorescence at 1 and 4 h after wounding. Cells were stained with rhodamine phalloidin to visualize actin stress fibers. Quint cells at the wound edge lacked significant lamellipodia (e and f) as compared with parental and WT cells (a–d). S4/8D cells demonstrated enhanced lamellipodia formation (g and h). Bar, 5 μm.
The wound edge was evaluated by immunofluorescence microscopy to characterize alterations in cell morphology and actin cytoskeleton during wound closure (Fig. 7 B). In contrast to the other cell lines, the S4/8D cells rapidly (within 1 h) extended broad lamellipodia into the open wound (Fig. 7 B, a–d). 4 h after wounding, the parental and WT cells also developed a leading edge similar to the S4/8D cells; however, the Quint cells exhibited only minimal protrusions at the wound edge even after 4 h (Fig. 7 B, e–h). These results correlate with the morphology exhibited during cell spreading (Fig. 5) and demonstrate an important role for actopaxin phosphorylation in lamellipodial extension and cell migration.
To extend the wound closure analysis, modified Boyden chamber migration assays were performed to evaluate additional aspects of motility including haptotaxis, chemokinesis, and chemotaxis. To evaluate haptotaxis, cells migration toward immobilized collagen or fibronectin was measured (Fig. 8 A). Chemokinesis and chemotaxis were evaluated in response to insulin-like growth factor (IGF) II (Fig. 8 B). Consistent with the wound data, the S4/8D cells migrated more efficiently than either the parental or WT cells, whereas the Quint cells exhibited significantly reduced migration rates under all conditions tested (Fig. 8, A and B). Together, these data indicate inhibition of actopaxin phosphorylation inhibits cell migration, whereas a phosphomimetic promotes motility.
Figure 8.
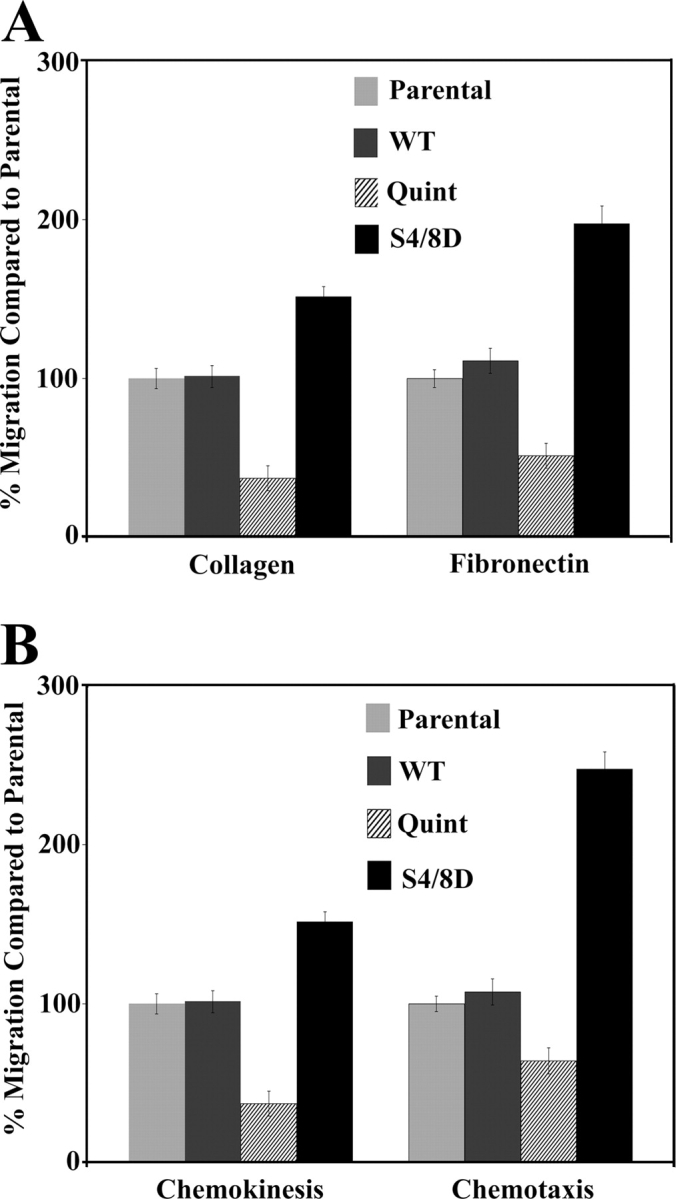
Actopaxin phosphorylation mutants affect Boyden chamber migration. Motility of parental, WT, Quint, and S4/8D cells was evaluated by modified Boyden chamber migration assays. (A) Quantification of random (haptotaxis) motility to collagen (10 µg/ml) or fibronectin (10 µg/ml) demonstrates significant reduction in Quint cell migration as compared with parental and WT cells, whereas S4/8D cells show an increase in migration. (B) Quantification of chemokinesis and chemotaxis in the presence of IGF-II (25 ng/ml) demonstrates a similar trend for each cell line to that seen in the haptotaxis assays. Values are the average of assays performed in triplicate.
Erk phosphorylates the actopaxin amino terminus
The five actopaxin phosphorylation sites conform to the consensus for Erk in addition to cdc2. Interestingly, Erk can localize to focal adhesions possibly through an association with paxillin (Ishibe et al., 2003) and has been directly linked to regulating cell motility (Klemke et al., 1997). Thus, Erk mediated phosphorylation of actopaxin provides a potential mechanism to explain our spreading and motility observations. To evaluate actopaxin as a potential target for Erk kinase activity, GST-fusion proteins of full-length actopaxin, its amino terminus, and its carboxyl terminus were incubated with active Erk2. Erk phosphorylated full-length actopaxin (amino acids 1-372), and fusion proteins encompassing amino-terminal amino acids 1-222 and 1-95 (Fig. 9 A, lanes 1, 2, and 4). However, Erk did not phosphorylate the carboxyl terminus (222-372) of actopaxin (Fig. 9 A, lane 3).
Figure 9.

Erk phosphorylates the actopaxin amino terminus in vitro. (A) GST actopaxin fusion proteins were incubated with active ERK2. Fusion proteins were resolved by SDS-PAGE and phosphorylated proteins were visualized by autoradiography. Phosphorylation is apparent on WT (full length) and the amino terminus (1-222 and 1-95) of actopaxin, but not on the carboxyl terminus (223-372). Coomassie blue-stained gel is shown to demonstrate expression of the fusion proteins. (B) In vivo inhibition of actopaxin phosphorylation in the presence of the Erk inhibitor UO126. Xpress-actopaxin WT cells labeled with 32P were held in suspension, or re-spread on collagen-coated dishes for 120 min in the presence or absence of UO126 (25 μM) followed by immunoprecipitation, SDS-PAGE, and autoradiography. Adhesion induced actopaxin phosphorylation (lane 2) is reduced in the presence of UO126 (lane 3). (C) Actopaxin WT cells were transfected with active MEK1 or DN MEK1. Cells labeled with 32P were re-spread on collagen-coated dishes for 120 min in the presence or absence of UO126 (25 μM). Xpress-tagged WT actopaxin was immunoprecipitated followed by analysis by SDS-PAGE and autoradiography.
To evaluate a potential role for Erk in actopaxin phosphorylation in vivo, adhesion-induced phosphorylation of actopaxin was evaluated in the absence or presence of the MEK1 inhibitor UO126. The addition of the MEK1 inhibitor resulted in a substantial reduction in the level of adhesion induced actopaxin phosphorylation of both the endogenous and exogenous protein, whereas incorporation of 32P into total cellular proteins was unaffected by the addition of the MEK1 inhibitor (Fig. 9 B). To compliment the inhibitor studies, WT actopaxin and either active MEK1 (MEK1) or dominant-negative MEK1 (DN MEK1) were cotransfected into parental U2OS cells followed by in vivo 32P-labeling in the absence or presence of UO126. Importantly, DN MEK1 inhibited the adhesion-induced phosphorylation of actopaxin when compared with cells expressing the active MEK1 (Fig. 9 C). Together, these results demonstrate that actopaxin is a target for Erk phosphorylation and suggest that Erk may contribute to actopaxin phosphorylation during cell spreading.
Erk inhibition impedes U2OS cell migration and actopaxin phosphorylation mutants are differentially affected
Wound healing and Boyden chamber assays were used to characterize the role of Erk in actopaxin-mediated cell migration. The inclusion of the MEK1 inhibitor UO126 during wound healing attenuated the closure rates of each of the cell populations (parental, WT, Quint, and S4/8D) indicating an important role for Erk in promoting cell motility in U2OS osteosarcoma cells, but raising the formal possibility that Erk phosphorylation of actopaxin may not play a major role in cell migration during wound healing. Interestingly however, the S4/8D cells still exhibited the highest motility rate (Fig. 10, A and B) suggesting phosphorylation of these sites contributes to Erk-dependent migration. Similar results were also observed with the addition of another MEK1 inhibitor PD98059 (unpublished data). Of note, careful evaluation of the wound edge revealed that at the later time points the S4/8D cells were still able to extend broad lamellipodia into the wound in the presence of the inhibitor, whereas the development of a leading edge in the other cell lines was impaired (unpublished data). Importantly, although actopaxin can be phosphorylated by cyclinB/cdc2 during mitosis and a recent report identified a role for cyclinB/cdc2 in αvβ3 integrin-dependent migration (Manes et al., 2003), wound closure was not inhibited in any of the cell lines in the presence of the cdc2 inhibitor alsterpaullone (Fig. 10 B) or another cdc2 inhibitor purvalanol-A (not depicted).
Figure 10.
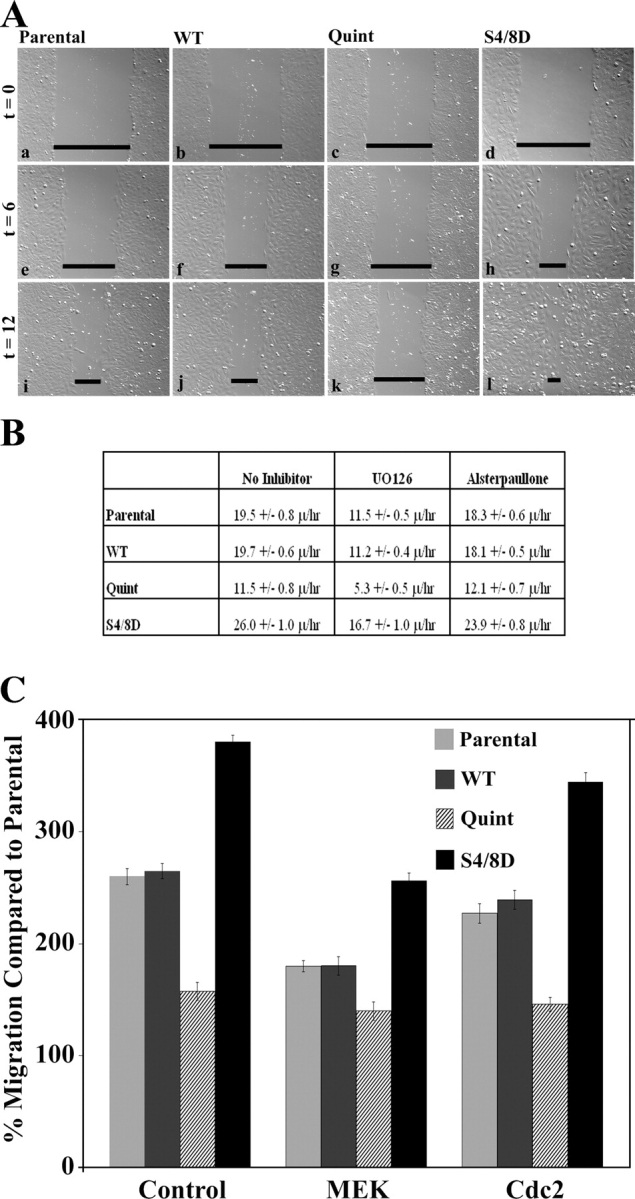
Inhibition of Erk but not cyclin B/cdc2 decreases cell migration in U2OS cells. Scrape wound assays were performed using parental, actopaxin WT, Quint, and S4/8D cells in the presence of Erk or cyclin B/cdc2 inhibitors. Cultures were incubated for 2 h with MEK1 inhibitor UO126 (25 μM) to inhibit Erk or alsterpaullone (1 μM) to inhibit cdc2, scored with a micropipette tip, and provided with fresh complete media plus inhibitor. (A) Hoffman illumination images taken at 0, 6, and 12 h after wounding in the presence of U0126 are depicted. (B) Quantitation of wound closure rates of parental, WT, Quint, and S4/8D cells in the presence of UO126 or alsterpaullone. (C) To evaluate the effect of MEK1 or cdc2 inhibition on random motility (haptotaxis), modified Boyden chamber migration assays to collagen I (10 μg/ml) were performed with or without UO126 (25 μM) or alsterpaullone (1 μM). Values are the average of three experiments.
We also evaluated the relative role of cdc2 versus Erk activity in regulating U2OS cell haptotaxis by performing Boyden chamber assays in the presence of the cdc2 inhibitor alsterpaullone or the MEK1 inhibitor UO126. As was the case with the wound assay, no significant inhibition of collagen I haptotaxis was recorded in the presence of the cdc2 inhibitor (Fig. 10 C). In contrast, U0126 inhibited migration of the parental, WT, and S4/8D actopaxin cell lines. Interestingly, in this assay the Quint cells, although exhibiting the slowest rate of migration, were essentially unaffected by the MEK1 inhibitor, suggesting that this basal motility rate is Erk-independent and that actopaxin phosphorylation is important for Erk-mediated motility in this context.
Discussion
Actopaxin was first identified as a paxillin and actin binding protein (Nikolopoulos and Turner, 2000; Olski et al., 2001). The interaction with paxillin is essential for targeting actopaxin to focal adhesions (Nikolopoulos and Turner, 2000). Consistent with a role for actopaxin in actin-mediated cellular processes, introduction of actopaxin mutants defective for either paxillin- or actin-binding results in impaired cell adhesion and cell spreading (Nikolopoulos and Turner, 2000; Tu et al., 2001; Yamaji et al., 2001). Although actopaxin is a major target for phosphorylation by the cyclinB/cdc2 kinase during mitosis (Curtis et al., 2002), the potential for actopaxin to serve as a kinase substrate in other cellular processes has not been previously evaluated. Here, we have determined that actopaxin phosphorylation is stimulated during cell adhesion and, using nonphosphorylatable and phosphomimetic mutants of actopaxin, we have determined that actopaxin phosphorylation plays a critical role in the regulation of cell spreading and cell migration, potentially as an effector of Rac activity. Furthermore, we present evidence that actopaxin is directly phosphorylated by the MAPK Erk in vitro and actopaxin phosphorylation is dependent on Erk activity in vivo, thereby identifying actopaxin as a key substrate for this important regulator of cell migration (Klemke et al., 1997; Brahmbhatt and Klemke, 2003; Webb et al., 2004).
Cell adhesion to the ECM initiates a series of integrin-mediated intracellular signaling events that promotes cell spreading via the formation of filopodia and broad lamellipodia. This process, which involves the initial activation of Cdc42 and Rac GTPases (Price et al., 1998) followed subsequently by an elevation in Rho GTPase activity, closely resembles the membrane and actin cytoskeletal remodeling events that occur in a motile cell as it extends and then stabilizes its leading edge (Nobes and Hall, 1999; Ridley, 2001). Using this cell spreading assay we were able to demonstrate for the first time that actopaxin phosphorylation is stimulated during cell adhesion. Furthermore, similar experiments with cells stably expressing a nonphosphorylatable (Quint) mutant of actopaxin revealed a significant role for actopaxin phosphorylation in promoting cell spreading. Importantly, these cells were deficient in their ability to extend and stabilize lamellipodia. This defect was also evident when analyzing cell membrane dynamics at a newly formed wound edge. Although the parental and WT actopaxin expressing cells rapidly extended a broad lamellipodium into the area of the wound denuded of cells, the actopaxin Quint mutant cells were unable to do so. Conversely, a putative phosphomimetic mutant of actopaxin, S4/8D, stimulated cell spreading and also enhanced lamellipodia formation at the wound edge. Additionally, overexpression of dominant negative N17 Rac failed to effectively block lamellipodia formation in the majority of the S4/8D mutant cells, whereas a significant reduction of lamellipodia was evident in the WT cells. Together, these results suggest that actopaxin phosphorylation functions either downstream or parallel to Rac activation, whereas unphosphorylated actopaxin may inhibit Rac activity and promote Rho signaling and the formation of actin stress fibers. Future studies will be required to fully delineate the impact of actopaxin phosphorylation on Rho GTPase family signaling.
Although the mechanism is currently unknown, it is interesting to speculate on how phosphorylation of actopaxin could impact Rho GTPase signaling. One potential link is through PIX. The PIX/COOL proteins have been shown to exhibit guanine nucleotide exchange factor activity toward Cdc42 and Rac1 (Cerione, 2004). Recent studies have demonstrated that the actopaxin family member β-parvin/affixin (Yamaji et al., 2002), interacts with and localizes with α-PIX at the leading edge of spreading cells (Olski et al., 2001; Rosenberger et al., 2003). In addition to this direct interaction, PIX isoforms also could be brought into close proximity to actopaxin in focal adhesions through their shared ability to interact with paxillin; actopaxin via a direct association (Nikolopoulos and Turner, 2000; Tumbarello et al., 2002) and PIX via association with the ARFGAP, PKL/GIT2, which in turn binds the LD4 motif of paxillin (Turner et al., 1999; Zhao et al., 2000; Brown et al., 2002). Whether actopaxin phosphorylation imparts allosteric changes that directly affect the actopaxin-PIX association and/or PIX binding to PAK, to regulate the targeting of PIX; or instead influences PIX guanine nucleotide exchange factor activity to activate PAK stimulation of membrane ruffling through PAK phosphorylation of MLCK/MLC and filamin (Kiosses et al., 1999; Vadlamudi et al., 2002) will require further study.
Alternatively, because actopaxin can bind F-actin directly via its tandem CH domains (Nikolopoulos and Turner, 2000), actopaxin phosphorylation may perform a more direct role in influencing actin filament assembly at the leading edge of motile cells. A key component in this process is the Arp2/3 complex, which binds to the side of a preexisting actin filament and nucleates new filament assembly resulting in the formation of branched networks of actin filaments at the cell cortex (Svitkina and Borisy, 1999); a process that has been shown to be essential for lamellipodial extension (Bailly et al., 2001). Recently, the Arp2/3 complex was found to bind vinculin in focal adhesions and proposed as a mechanism for delivering Arp2/3 into close proximity with its activators at the leading edge such as N-WASP, which in turn could be activated via PIX stimulated cdc42 activation (DeMali and Burridge, 2003; Cerione, 2004; Etienne-Manneville, 2004). Because vinculin and actopaxin both bind paxillin (Nikolopoulos and Turner, 2000; Turner, 2000) it will be of interest to determine if actin binding to actopaxin is regulated by phosphorylation as has been shown for other actin binding proteins such as caldesmon, filamin and tropomyosin (Vadlamudi et al., 2002; Houle et al., 2003; Huang et al., 2003; Yamakita et al., 2003) and if actopaxin is directly involved in Arp2/3-associated actin remodeling.
Another intriguing connection with the actin cytoskeleton that may be regulated through actopaxin phosphorylation involves the interaction between actopaxin and the integrin-linked kinase (ILK). First identified as an integrin binding protein, ILK has been implicated in a plethora of cell signaling events associated with cell transformation, migration, metastasis, matrix assembly, and cell survival (Wu and Dedhar, 2001). Importantly, a cassette of proteins comprising ILK-actopaxin-PINCH and in some cases paxillin, has emerged as an evolutionarily conserved integrin-actin linkage in organisms as diverse as humans, worms, and flies. Elimination of individual components of this cassette results in defective integrin-actin attachment in muscle tissue in C. elegans and D. melanogaster, whereas gene knockout of ILK in mice causes embryonic lethality and severe adhesion/spreading defects in cells derived from these embryos (Zervas et al., 2001; Mackinnon et al., 2002; Brakebusch and Fassler, 2003; Lin et al., 2003; Sakai et al., 2003). Thus, actopaxin phosphorylation could prove important in regulating these interactions and stabilizing integrin linkage to the cytoskeleton. In this regard, the actopaxin family member affixin, can be phosphorylated by ILK in vitro (Yamaji et al., 2001), although the site of phosphorylation resides in the CH2 domain, rather than the amino terminus (Mishima et al., 2004).
The Erk MAPK cascade has been implicated in the regulation of cell migration on several levels. First its activity can be stimulated via integrin mediated activation of FAK/Src (Hauck et al., 2002) and also after association with paxillin, which likely also contributes to the recruitment of Erk to focal adhesions (Fincham et al., 2000; Ishibe et al., 2003). The paxillin binding protein PAK also contributes to Erk activation via phosphorylation of its upstream regulators Raf and MEK1 (Slack-Davis et al., 2003). Paxillin-null mice are defective in Erk activation and exhibit motility defects (Hagel et al., 2002) that are consistent with a recent report detailing the importance of Erk activation and paxillin in focal adhesion turnover (Webb et al., 2004). Erk mediates cell migration via regulation of MLCK at the leading edge to promote myosin II-based contractility and membrane ruffling (Klemke et al., 1997; Brahmbhatt and Klemke, 2003; Totsukawa et al., 2004). It also facilitates focal adhesion disassembly and thus cell retraction at the rear through phosphorylation and activation of the protease Calpain (Bhatt et al., 2002; Carragher et al., 2003; Cuevas et al., 2003). Our in vitro kinase data indicate that the actopaxin amino terminus represents a bona fide target for Erk kinase activity. Clearly, actopaxin, through binding paxillin in focal adhesions is optimally positioned adjacent to active Erk to serve as a substrate. A physiologic role for Erk-mediated phosphorylation of actopaxin in regulating cell migration is supported by our results showing that MEK1 inhibitors and dominant negative MEK1 block adhesion-induced phosphorylation of actopaxin and also effectively block cell migration in the U2OS cells used here. Furthermore, the actopaxin Quint mutant lacking the Erk phosphorylation consensus sites cannot be phosphorylated in vivo and substantially inhibits cell migration, whereas the S4/8D actopaxin phosphomimetic mutant demonstrates enhanced migration compared with WT expressing cells and continues to extend lamellipodia even in the presence of the Erk inhibitor. The failure of the Quint cells to completely inhibit cell migration is consistent with the existence of multiple Erk targets and also to the continued presence of endogenous actopaxin in these cells. In contrast, although the same phosphorylation sites on actopaxin are targets for cyclinB/cdc2 kinase in mitotic cells (Curtis et al., 2002), and cyclinB/cdc2 has been shown to regulate cell migration under certain conditions (Manes et al., 2003), pharmacologic inhibitors of cdc2 activity failed to suppress cell migration in our studies suggesting that cdc2 phosphorylation of actopaxin is not playing a significant role in U2OS cells under the conditions examined.
In summary we have identified actopaxin as an Erk substrate and determined that inhibition of actopaxin phosphorylation, through site-directed mutagenesis, profoundly inhibits cell adhesion, spreading and cell migration thereby positioning actopaxin as a potentially important regulator of cell motility. Future studies will be directed toward determining actopaxin's precise role in focal adhesion turnover and actin dynamics as well as examining whether actopaxin is hyperphosphorylated in certain cancer cell populations where it may contribute to enhanced motility and metastatic potential.
Materials and methods
Reagents and antibodies
Active Erk2 was obtained from New England Biolabs, Inc. Inhibitors for MEK1 (UO126, PD98059) and cdc2 (alsterpaullone and purvalanol A) were purchased from Calbiochem. The active MEK1 construct was purchased from Stratagene and the DN MEK1 construct was a gift from M. Cobb (University of Texas, Southwestern, TX). The N17 Rac construct was a gift from M. Symons (North Shore Long Island Jewish Research Institute, Manhasset, NY). Primary antibodies to the Xpress epitope were purchased from Invitrogen and paxillin (clone 165) were purchased from BD Biosciences. The actopaxin antibody was a gift from Sigma-Aldrich. The Omniprobe antibody was purchased from Santa Cruz Biotechnology, Inc. Human plasma fibronectin was purchased from Sigma-Aldrich, and collagen type I was purchased from Cohesin.
Cell culture and transfection
Human osteosarcoma (U2OS) cells were a gift from H. Levinson (Brookdale University Hospital, Brooklyn, NY). Cells were transfected using FuGene 6 (Roche). Stable transfectants were obtained by growing in media containing 1 mg/ml G418. Stable transfectants were maintained in DME plus 0.4 mg/ml G418, 10% serum.
DNA constructs and mutagenesis
Xpress-tagged actopaxin WT was described previously (Nikolopoulos and Turner, 2000). Actopaxin double point mutant (S4/8D) and Quint mutant S(4,8,14,19)G/T16A (Quint) were generated using the QuikChange site-directed mutagenesis kit (Stratagene). The sequences of all mutated constructs were verified by sequencing on both strands.
In vivo 32P-labeling
Actopaxin-expressing cells were washed twice with PBS and then preincubated with 5 ml of phosphate-free DME containing 1% dialyzed FBS for 1 h. A total of 1.5 × 106 cells were used for each experimental condition. The cells were resuspended in 4 ml of media and 500 μCi of 32Pi (2 mCi/ml in H2O), and incubated for 1 h at 37°C. Cell suspensions plated on collagen coated dishes for 2 h. Cells were washed two times with PBS and extracted in 500 μl of lysis buffer (1% NP-40, 50 mM Tris, HCl, pH 7.6, 10% glycerol, 50 mM NaCl, 0.1% β-mercaptoethanol, 1 mM PMSF, 10 μg/ml leupeptin, 5 mM NaF, and 1 mM NaVO4).
To evaluate actopaxin and paxillin phosphorylation, 3 μl of Xpress (or Omniprobe) or Pax 165 antibody was added to each cell lysate. After 2 h, protein A/G beads were then added for 2 h at 4°C. Immunocomplexes were washed four times in lysis buffer before SDS-PAGE. Gels were stained, dried, and exposed to film.
In vitro phosphorylation
GST proteins were prepared as described previously (Nikolopoulos and Turner, 2000). Fusion proteins were washed two times in kinase buffer (New England Biolabs, Inc.) and resuspended in the same buffer. 10 μCi of γ-[32P]ATP and 100 U of recombinant ERK2 were added to each reaction. After 30 min, the kinase reactions were boiled in sample buffer and run on a 12.5% gel, dried, and exposed to film.
Spreading and motility assays
Cells were suspended in 1 ml PBS/EDTA, harvested and washed twice with serum-free DME,10 μg/ml soybean trypsin inhibitor, 1% BSA. After 1 h in suspension, cells were replated on 10 μg/ml collagen- or 10 μg/ml fibronectin-coated coverslips in serum-free media for the indicated time period, fixed, and processed for indirect immunofluorescence as described previously (Brown et al., 1996).
Modified Boyden chamber migration assays were performed essentially as described previously (Riedy et al., 1999). For haptotaxis, an 8-μm pore membrane was coated on the bottom with 10 μg/ml collagen I. Chemotaxis was evaluated by including 25 ng/ml IGF-II in the bottom well. Stimulation of random motility (chemokinesis) was tested by adding 25 ng/ml IGF-II to the upper well. Scrape wound assays were performed as described previously (West et al., 2001). Images were captured every 6 h, starting at 0 h (t = 0) after wounding using a microscope (model Eclipse TE-300; Nikon).
Time-lapse video microscopy for spreading and wound healing assays was performed as follows. For spreading assays, cells were plated in serum-, phenol-, and bicarbonate-free DME supplemented with 25 mM Hepes, pH 7.35, on 10 μg/ml collagen-coated plates for 30 min at 37°C. Images were acquired using a microscope (model Eclipse TE-300; Nikon), a SPOT camera, and Compix Simple PCI software Imaging. The images were taken at 1-min intervals for 6–8 h and organized into videos using Compix Simple PCI 5.0.
Online supplemental material
Video 1 shows WT actopaxin expressing U2OS cells spreading on a collagen matrix. Video 2 shows Quint actopaxin expressing U2OS cells spreading on a collagen matrix. Video 3 shows S4/8D actopaxin expressing U2OS cells spreading on a collagen matrix. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200404024/DC1.
Acknowledgments
We are grateful to Abby Racette for excellent technical assistance and members of the Turner lab for helpful comments. We also thank Dr. Sotiris Nikolopoulos (Memorial Sloan-Kettering Cancer Center, New York, NY) for the generation of some of the actopaxin constructs used in this work. Additionally, we would like to thank Melanie Cobb for the DN MEK1 construct and Marc Symons for the N17 Rac construct.
This work was supported by a National Institutes of Health grant RO1 HL070244 to C.E. Turner.
Abbreviations used in this paper: CH, calponin homology; DN MEK1; dominant-negative MEK1; Erk, extracellular signal-regulated protein kinase; IGF, insulin-like growth factor; ILK, integrin-linked kinase; MLCK, myosin light chain kinase; PAK, p21-activated kinase; PBS, paxillin binding subdomain; Quint, quintuple; WT, wild-type.
References
- Bailly, M., I. Ichetovkin, W. Grant, N. Zebda, L.M. Machesky, J.E. Segall, and J. Condeelis. 2001. The F-actin side binding activity of the Arp2/3 complex is essential for actin nucleation and lamellipod extension. Curr. Biol. 11:620–625. [DOI] [PubMed] [Google Scholar]
- Bellis, S.L., J.A. Perrotta, M.S. Curtis, and C.E. Turner. 1997. Adhesion of fibroblasts to fibronectin stimulates both serine and tyrosine phosphorylation of paxillin. Biochem. J. 325:375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt, A., I. Kaverina, C. Otey, and A. Huttenlocher. 2002. Regulation of focal complex composition and disassembly by the calcium-dependent protease calpain. J. Cell Sci. 115:3415–3425. [DOI] [PubMed] [Google Scholar]
- Bokoch, G.M. 2003. Biology of the p21-activated kinases. Annu Rev Biochem. 72:743–781. [DOI] [PubMed] [Google Scholar]
- Brahmbhatt, A.A., and R.L. Klemke. 2003. ERK and RhoA differentially regulate pseudopodia growth and retraction during chemotaxis. J. Biol. Chem. 278:13016–13025. [DOI] [PubMed] [Google Scholar]
- Brakebusch, C., and R. Fassler. 2003. The integrin-actin connection, an eternal love affair. EMBO J. 22:2324–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M.C., J.A. Perrotta, and C.E. Turner. 1996. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J. Cell Biol. 135:1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M.C., K.A. West, and C.E. Turner. 2002. Paxillin-dependent paxillin kinase linker and p21-activated kinase localization to focal adhesions involves a multistep activation pathway. Mol. Biol. Cell. 13:1550–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher, N.O., M.A. Westhoff, V.J. Fincham, M.D. Schaller, and M.C. Frame. 2003. A novel role for FAK as a protease-targeting adaptor protein: regulation by p42 ERK and Src. Curr. Biol. 13:1442–1450. [DOI] [PubMed] [Google Scholar]
- Cerione, R.A. 2004. Cdc42: new roads to travel.Trends Cell Biol. 14:127–132. [DOI] [PubMed] [Google Scholar]
- Clark, E.A., and J.S. Brugge. 1995. Integrins and signal transduction pathways: the road taken. Science. 268:233–239. [DOI] [PubMed] [Google Scholar]
- Cuevas, B.D., A.N. Abell, J.A. Witowsky, T. Yujiri, N.L. Johnson, K. Kesavan, M. Ware, P.L. Jones, S.A. Weed, R.L. DeBiasi, et al. 2003. MEKK1 regulates calpain-dependent proteolysis of focal adhesion proteins for rear-end detachment of migrating fibroblasts. EMBO J. 22:3346–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M., S.N. Nikolopoulos, and C.E. Turner. 2002. Actopaxin is phosphorylated during mitosis and is a substrate for cyclin B1/cdc2 kinase. Biochem. J. 363:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMali, K.A., and K. Burridge. 2003. Coupling membrane protrusion and cell adhesion. J. Cell Sci. 116:2389–2397. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S. 2004. Cdc42 - the centre of polarity. J. Cell Sci. 117:1291–1300. [DOI] [PubMed] [Google Scholar]
- Fincham, V.J., M. James, M.C. Frame, and S.J. Winder. 2000. Active ERK/MAP kinase is targeted to newly forming cell-matrix adhesions by integrin engagement and v-Src. EMBO J. 19:2911–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1999. Integrin signaling. Science. 285:1028–1032. [DOI] [PubMed] [Google Scholar]
- Hagel, M., E.L. George, A. Kim, R. Tamimi, S.L. Opitz, C.E. Turner, A. Imamoto, and S.M. Thomas. 2002. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol. Cell. Biol. 22:901–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck, C.R., D.A. Hsia, and D.D. Schlaepfer. 2002. The focal adhesion kinase–a regulator of cell migration and invasion. IUBMB Life. 53:115–119. [DOI] [PubMed] [Google Scholar]
- Horwitz, A.R., and J.T. Parsons. 1999. Cell migration–movin' on. Science. 286:1102–1103. [DOI] [PubMed] [Google Scholar]
- Houle, F., S. Rousseau, N. Morrice, M. Luc, S. Mongrain, C.E. Turner, S. Tanaka, P. Moreau, and J. Huot. 2003. Extracellular signal-regulated kinase mediates phosphorylation of tropomyosin-1 to promote cytoskeleton remodeling in response to oxidative stress: impact on membrane blebbing. Mol. Biol. Cell. 14:1418–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, R., L. Li, H. Guo, and C.L. Wang. 2003. Caldesmon binding to actin is regulated by calmodulin and phosphorylation via different mechanisms. Biochemistry. 42:2513–2523. [DOI] [PubMed] [Google Scholar]
- Hynes, R.O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell. 110:673–687. [DOI] [PubMed] [Google Scholar]
- Ishibe, S., D. Joly, X. Zhu, and L.G. Cantley. 2003. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol. Cell. 12:1275–1285. [DOI] [PubMed] [Google Scholar]
- Kiosses, W.B., R.H. Daniels, C. Otey, G.M. Bokoch, and M.A. Schwartz. 1999. A role for p21-activated kinase in endothelial cell migration. J. Cell Biol. 147:831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke, R.L., S. Cai, A.L. Giannini, P.J. Gallagher, P. de Lanerolle, and D.A. Cheresh. 1997. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 137:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger, D.A., and A.F. Horwitz. 1996. Cell migration: a physically integrated molecular process. Cell. 84:359–369. [DOI] [PubMed] [Google Scholar]
- Lin, X., H. Qadota, D.G. Moerman, and B.D. Williams. 2003. C. elegans PAT-6/Actopaxin plays a critical role in the assembly of integrin adhesion complexes in vivo. Curr. Biol. 13:922–932. [DOI] [PubMed] [Google Scholar]
- Mackinnon, A.C., H. Qadota, K.R. Norman, D.G. Moerman, and B.D. Williams. 2002. C. elegans PAT-4/ILK functions as an adaptor protein within integrin adhesion complexes. Curr. Biol. 12:787–797. [DOI] [PubMed] [Google Scholar]
- Manes, T., D.Q. Zheng, S. Tognin, A.S. Woodard, P.C. Marchisio, and L.R. Languino. 2003. αvβ3 integrin expression up-regulates cdc2, which modulates cell migration. J. Cell Biol. 161:817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima, W., A. Suzuki, S. Yamaji, R. Yoshimi, A. Ueda, T. Kaneko, J. Tanaka, Y. Miwa, S. Ohno, and Y. Ishigatsubo. 2004. The first CH domain of affixin activates Cdc42 and Rac1 through alphaPIX, a Cdc42/Rac1-specific guanine nucleotide exchanging factor. Genes Cells. 9:193–204. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos, S.N., and C.E. Turner. 2000. Actopaxin, a new focal adhesion protein that binds paxillin LD motifs and actin and regulates cell adhesion. J. Cell Biol. 151:1435–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolopoulos, S.N., and C.E. Turner. 2002. Molecular dissection of actopaxin-integrin-linked kinase-Paxillin interactions and their role in subcellular localization. J. Biol. Chem. 277:1568–1575. [DOI] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1999. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144:1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olski, T.M., A.A. Noegel, and E. Korenbaum. 2001. Parvin, a 42 kDa focal adhesion protein, related to the alpha-actinin superfamily. J. Cell Sci. 114:525–538. [DOI] [PubMed] [Google Scholar]
- Price, L.S., J. Leng, M.A. Schwartz, and G.M. Bokoch. 1998. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell. 9:1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou, M., and A. Hall. 2004. Cell migration: Rho GTPases lead the way. Dev. Biol. 265:23–32. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J. 2001. Rho GTPases and cell migration. J. Cell Sci. 114:2713–2722. [DOI] [PubMed] [Google Scholar]
- Riedy, M.C., M.C. Brown, C.J. Molloy, and C.E. Turner. 1999. Activin A and TGF-beta stimulate phosphorylation of focal adhesion proteins and cytoskeletal reorganization in rat aortic smooth muscle cells. Exp. Cell Res. 251:194–202. [DOI] [PubMed] [Google Scholar]
- Rosenberger, G., I. Jantke, A. Gal, and K. Kutsche. 2003. Interaction of alphaPIX (ARHGEF6) with beta-parvin (PARVB) suggests an involvement of alphaPIX in integrin-mediated signaling. Hum. Mol. Genet. 12:155–167. [DOI] [PubMed] [Google Scholar]
- Sakai, T., S. Li, D. Docheva, C. Grashoff, K. Sakai, G. Kostka, A. Braun, A. Pfeifer, P.D. Yurchenco, and R. Fassler. 2003. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 17:926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack-Davis, J.K., S.T. Eblen, M. Zecevic, S.A. Boerner, A. Tarcsafalvi, H.B. Diaz, M.S. Marshall, M.J. Weber, J.T. Parsons, and A.D. Catling. 2003. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J. Cell Biol. 162:281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1999. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenawa, T., and H. Miki. 2001. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci. 114:1801–1809. [DOI] [PubMed] [Google Scholar]
- Thiel, D.A., M.K. Reeder, A. Pfaff, T.R. Coleman, M.A. Sells, and J. Chernoff. 2002. Cell cycle-regulated phosphorylation of p21-activated kinase 1. Curr. Biol. 12:1227–1232. [DOI] [PubMed] [Google Scholar]
- Totsukawa, G., Y. Wu, Y. Sasaki, D.J. Hartshorne, Y. Yamakita, S. Yamashiro, and F. Matsumura. 2004. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 164:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, Y., Y. Huang, Y. Zhang, Y. Hua, and C. Wu. 2001. A new focal adhesion protein that interacts with integrin-linked kinase and regulates cell adhesion and spreading. J. Cell Biol. 153:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello, D.A., M.C. Brown, and C.E. Turner. 2002. The paxillin LD motifs. FEBS Lett. 513:114–118. [DOI] [PubMed] [Google Scholar]
- Turner, C.E. 2000. Paxillin interactions. J. Cell Sci. 113:4139–4140. [DOI] [PubMed] [Google Scholar]
- Turner, C.E., M.C. Brown, J.A. Perrotta, M.C. Riedy, S.N. Nikolopoulos, A.R. McDonald, S. Bagrodia, S. Thomas, and P.S. Leventhal. 1999. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J. Cell Biol. 145:851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadlamudi, R.K., F. Li, L. Adam, D. Nguyen, Y. Ohta, T.P. Stossel, and R. Kumar. 2002. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat. Cell Biol. 4:681–690. [DOI] [PubMed] [Google Scholar]
- Webb, D.J., K. Donais, L.A. Whitmore, S.M. Thomas, C.E. Turner, J.T. Parsons, and A.F. Horwitz. 2004. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6:154–161. [DOI] [PubMed] [Google Scholar]
- West, K.A., H. Zhang, M.C. Brown, S.N. Nikolopoulos, M.C. Riedy, A.F. Horwitz, and C.E. Turner. 2001. The LD4 motif of paxillin regulates cell spreading and motility through an interaction with paxillin kinase linker (PKL). J. Cell Biol. 154:161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C., and S. Dedhar. 2001. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J. Cell Biol. 155:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, R., Y. Mazaki, K. Hirota, S. Hashimoto, and H. Sabe. 1997. Mitosis specific serine phosphorylation and downregulation of one of the focal adhesion protein, paxillin. Oncogene. 15:1753–1761. [DOI] [PubMed] [Google Scholar]
- Yamaji, S., A. Suzuki, Y. Sugiyama, Y. Koide, M. Yoshida, H. Kanamori, H. Mohri, S. Ohno, and Y. Ishigatsubo. 2001. A novel integrin-linked kinase-binding protein, affixin, is involved in the early stage of cell–substrate interaction. J. Cell Biol. 153:1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji, S., A. Suzuki, H. Kanamori, W. Mishima, M. Takabayashi, K. Fujimaki, N. Tomita, S. Fujisawa, S. Ohno, and Y. Ishigatsubo. 2002. Possible role of ILK-affixin complex in integrin-cytoskeleton linkage during platelet aggregation. Biochem. Biophys. Res. Commun. 297:1324–1331. [DOI] [PubMed] [Google Scholar]
- Yamakita, Y., F. Oosawa, S. Yamashiro, and F. Matsumura. 2003. Caldesmon inhibits Arp2/3-mediated actin nucleation. J. Biol. Chem. 278:17937–17944. [DOI] [PubMed] [Google Scholar]
- Yamashiro, S., H. Chern, Y. Yamakita, and F. Matsumura. 2001. Mutant Caldesmon lacking cdc2 phosphorylation sites delays M-phase entry and inhibits cytokinesis. Mol. Biol. Cell. 12:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervas, C.G., S.L. Gregory, and N.H. Brown. 2001. Drosophila integrin-linked kinase is required at sites of integrin adhesion to link the cytoskeleton to the plasma membrane. J. Cell Biol. 152:1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z.S., E. Manser, T.H. Loo, and L. Lim. 2000. Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol. Cell. Biol. 20:6354–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]