

Abstract
Yeast cells can initiate bud formation at the G1/S transition in a cue-independent manner. Here, we investigate the dynamic nature of the polar cap and the regulation of the GTPase Cdc42 in the establishment of cell polarity. Using analysis of fluorescence recovery after photobleaching, we found that Cdc42 exchanged rapidly between the polar caps and cytosol and that this rapid exchange required its GTPase cycle. A previously proposed positive feedback loop involving actomyosin-based transport of the Cdc42 GTPase is required for the generation of robust cell polarity during bud formation in yeast. Inhibition of actin-based transport resulted in unstable Cdc42 polar caps. Unstable polarity was also observed in mutants lacking Bem1, a protein previously implicated in a feedback loop for Cdc42 activation through a signaling pathway. When Bem1 and actin were both inhibited, polarization completely failed. These results suggest that cell polarity is established through coupling of transport and signaling pathways and maintained actively by balance of flux.
Keywords: feedback; cell polarity; actin; Cdc42; dynamic
Introduction
The establishment of cell polarity is important for such processes as cell motility, embryogenesis, and stem cell differentiation. In most physiological processes, the axis of polarity is dictated by preexisting asymmetric cues; however, without these cues, many systems can still polarize in random orientations (Wedlich-Soldner and Li, 2003). For example, the cortical rotation and polarization of Xenopus laevis eggs can occur in random directions upon egg activation, independent of a sperm entry site that normally directs this process (Gerhart et al., 1989). Dictyostelium discoideum and leukocytes, which polarize in response to gradients of chemoattractants, still polarize and move in random directions when exposed to a uniform concentration of chemoattractant (Devreotes and Zigmond, 1988). The intrinsic abilities of cells to break symmetry and polarize reflect a self-organization capacity that is likely to be fundamental to many morphogenetic processes (Misteli, 2001).
During the cell cycle of the budding yeast Saccharomyces cerevisiae, cells polarize at the G1/S transition in order to orient cell growth for bud formation. Initiation of polarization is triggered by the Cdk1 (Cdc28) when complexed with G1 cyclins. The key mediator of cell polarization is Cdc42, a highly conserved member of the Rho family GTPases. Cdc42 is active when bound to GTP, and the exchange of GDP for GTP is catalyzed by the Dbl family guanine nucleotide exchange factor (GEF) Cdc24. Upon Cdk1 activation at G1/S, Cdc24 is released from the nucleus and is activated by pathways that are not yet clearly delineated (Gulli et al., 2000; Bose et al., 2001; Moffat and Andrews, 2003). This chain of activation from Cdk1 via Cdc24 to Cdc42 temporally links polarization with the START phase of the cell cycle. Through multiple downstream effectors, Cdc42 controls the nucleation of actin cables and actin patches (Johnson, 1999). The actin cables are nucleated by the formin family proteins, which are activated by Rho family GTPases (Dong et al., 2003). Orientation of actin cables toward the bud site provides a vectorial pathway for the delivery of membrane and protein components necessary for polarized growth and morphogenesis of the bud (Pruyne and Bretscher, 2000).
A critical event that marks the success of symmetry breaking and bud site establishment is the localization of GTP-bound Cdc42 to a single discrete site on the plasma membrane. In haploid yeast cells, the site of Cdc42 accumulation is usually adjacent to the bud scars, which are remnants from previous cell divisions (Casamayor and Snyder, 2002). This spatial specification is controlled by the bud site selection pathway, the central player being the Bud1 GTPase, which is thought to be activated at or near the newest bud scar (Chant and Herskowitz, 1991; Park et al., 1997). Activated Bud1 binds directly to Cdc24, which activates Cdc42 at the bud site (Park et al., 1997, 2002). However, when Bud1 or its upstream regulators are abrogated, cells still polarize and bud efficiently, albeit in random directions (Chant and Herskowitz, 1991). This observation suggests that the spatial cue from the bud scar is not necessary for polarization per se but is only required to orient the bud with respect to the previous cell division. Understanding how yeast cells polarize independent of the bud scar could provide fundamental insights into how cell polarity can be achieved through self-organization.
We previously developed an artificial system to study cell polarization that occurs through purely intrinsic mechanisms (Wedlich-Soldner et al., 2003). Natural spatial or temporal cues for polarization were circumvented by arresting cells in G1 and expressing a GFP-myc6 (MG)–tagged, constitutively active form of Cdc42 (MG-Cdc42Q61L), which is locked in the GTP-bound state. These cells formed randomly positioned polar caps of MG-Cdc42Q61L. Polarization in this assay did not require any preexisting asymmetry but was completely dependent on actin polymerization and actin-based transport. These results, combined with mathematical modeling, led us to conclude that spontaneous polarization can be achieved through a positive feedback loop involving actin-based transport of Cdc42GTP and Cdc42GTP-stimulated actin polymerization. This feedback loop has the ability to amplify and stabilize a stochastic accumulation of Cdc42GTP on the plasma membrane.
It was unclear whether and how the mechanism uncovered by the above work might be relevant to the physiological process of bud formation. The above system clearly deviates from normal conditions for polarization: MG-Cdc42Q61L was overexpressed and locked in the GTP-bound form, and the GEF Cdc24 was inactive due to the G1 arrest. More importantly, a number of previous works found that Cdc42 localization during bud formation was not affected by latrunculin A (LatA), an actin polymerization inhibitor (Ayscough et al., 1997; Jaquenoud and Peter, 2000; Irazoqui et al., 2003). These observations reinforced the idea that polarization is normally achieved through a hierarchical pathway from Cdc42 to actin. Recently, a signaling-based mechanism for bud scar-independent polarization was proposed, involving the adaptor protein Bem1 (Butty et al., 2002; Irazoqui et al., 2003). Bem1 interacts directly with Cdc42GTP and Cdc24, and these interactions are thought to enable a positive feedback loop that could potentially result in symmetry breaking through amplification of stochastic accumulations of Cdc24 or Cdc42GTP on the plasma membrane. An additional feature of this hypothesis is that Bem1 forms a polymeric scaffold that stabilizes Cdc42 or Cdc24 polar caps by restricting diffusion (Irazoqui et al., 2003).
Although the actin-based and signaling-based hypotheses for the establishment of cell polarity represent somewhat contrasting views, it is possible that both mechanisms could coexist. Several crucial pieces of data are needed to properly evaluate the role of scaffolding and actin during physiological polarization. First, there has been no molecular dynamics analysis of the polarized state, and thus it is unclear whether this state is truly dynamic or static. Second, there has been no careful kinetics analysis of Cdc42 cap formation in the presence or absence of LatA, using an assay where bud formation occurs with high synchrony and efficiency. In this work, we address these questions through analysis of protein dynamics by FRAP, and characterization of the polarization process using a highly synchronized polarization assay and live cell imaging.
Results
The polarized state is dynamic
The goal of this work is to understand how cells establish a polarized distribution of the Cdc42 GTPase during the physiological process of bud formation. We first performed FRAP experiments to investigate whether the polarized state, as marked by Cdc42, is dynamic or static. The physical nature of the polarized state can provide crucial insights for understanding both the establishment and maintenance of cell polarity. For this and, more importantly, for subsequent experiments, we used strains bearing deletions of CLN1, 2, and 3, and expressing CLN2 under the control of the Met3 promoter (Amon et al., 1994). The advantage of using this strain is the ability to arrest cells in G1 by turning off Cln2 expression in the presence of methionine and to release from arrest by methionine wash-out to allow highly synchronized polarization (this assay will be referred to as the release assay). This allowed us to obtain cell populations enriched for cells with a polar cap of Cdc42 before bud emergence. To visualize Cdc42, MG-Cdc42 was expressed under either the CDC42 promoter or the inducible Gal1 promoter (the latter was particularly important for expressing alleles of Cdc42 that prohibit cell proliferation if constitutively expressed). No differences in the experimental results described in this work were observed between MG-Cdc42 expressed using the CDC42 promoter or the Gal1 promoter (see Fig. 3 A and not depicted).
Figure 3.

Polarization of Cdc42 after release from G1 arrest. (A) Polarization of MG-Cdc42 expressed from the Gal1 promoter (pGal, RLY1948) or the CDC42 promoter (p42, RLY1951) upon release from G1 arrest in the presence or absence of LatA. The percentage of cells with polarized MG-Cdc42 was determined at different time points (given in min) after release. Error bars correspond to SD. (B) Kinetics of MG-Cdc42 cap formation in two representative wild-type (RLY1950) cells determined by time-lapse imaging started at 10 min after release from G1 arrest. Fluorescence intensity in the cap was measured every 20 s. (C) MG-Cdc42 caps monitored in the presence or absence of LatA by time-lapse imaging 20 min after release from G1 arrest. Normalized average intensities in the cap region were measured every 15 s and plotted against time. (D) Chymographs showing the stability of two caps monitored in C.
FRAP experiments were performed on cells with polar caps of MG-Cdc42. These cells were arrested as described above and released for 30–40 min, at which time >80% of cells had a polar cap of Cdc42. After photobleaching, MG-Cdc42 fluorescence recovered rapidly to near prebleach levels (Fig. 1 A, half-time for recovery, t 1/2 = 4.29 ± 1.63 s, n = 10), suggesting that the Cdc42 polar cap is highly dynamic. To test if the rapid recovery of Cdc42 was due to the GTPase cycle or due to actin-based membrane trafficking, FRAP was performed on caps of MG-Cdc42, MG-Cdc42Q61L (GTP bound), or MG-Cdc42D57Y (GDP bound). Both MG-Cdc42Q61L and MG-Cdc42D57Y formed polar caps in the release assay (the untagged wild-type Cdc42 was also present in these strains). The recovery time of MG-Cdc42Q61L (Fig. 1 B, t 1/2 = 57.95 ± 13.07 s, n = 9) and MG-Cdc42D57Y (Fig. 1 B, t 1/2 = 37.79 ± 6.46 s, n = 7) in untreated cells was much slower than that of MG-Cdc42, suggesting that the GTPase cycle plays a major role in the high rate of exchange of Cdc42 in the polar caps. The slow recovery of MG-Cdc42Q61L was unlikely to be due to scaffolding, because recovery of the bleached gap to the surrounding fluorescence level occurred within <20 s indicating that lateral diffusion was not notably restricted (Fig. 1, C and D). In cells treated with LatA during release, MG-Cdc42 recovery was delayed, but still rapid (Fig. 1 A, t 1/2 = 7.21 ± 1.20 s, n = 9, P = 0.0004), which is consistent with the idea that actin-based transport contributes to Cdc42 delivery to the polar caps.
Figure 1.

FRAP analysis of MG-Cdc42 polar caps. In all graphs, the time points of bleaching are indicated by arrows and the area of bleaching is indicated by circles. Average intensities are given relative to the prebleach state. Bars, 5 μm. (A) FRAP of MG-Cdc42 caps formed in RLY1948 cells 30 min after release from G1 arrest. Caps formed in the presence (wt+LatA, gray line) or absence (wt, black line) of LatA were bleached multiple times and recovery monitored by time-lapse microscopy at 1 frame/s. Images from representative examples show caps before bleaching and at the indicated times after bleaching. (B) FRAP of MG-Cdc42Q61L (RLY1703) and MG-Cdc42D57Y (RLY1991) caps 35 min after release from G1 arrest. Recovery was monitored as in A with frames taken every 5 s. (C) Chymograph showing recovery of the MG-Cdc42Q61L cap shown in B. The initial recovery by lateral diffusion is indicated by dotted lines. (D) Line scans of the MG-Cdc42Q61L cap analyzed in B and C at the indicated times after bleaching (0 s represents the prebleach state). Arrows at the 5-s time point indicate the equalizing of fluorescence by diffusion seen during the initial recovery period.
Next, we performed FRAP on MG-Cdc42 caps in Δbem1 cells. The prediction would be that if Bem1 forms a polymeric scaffold, then without Bem1, Cdc42 becomes more dynamic. However, Cdc42 recovery did not drastically change and was even slightly slower in Δbem1 cells (t 1/2 = 5.99 ± 2.44 s, n = 9, P = 0.09). These results argue against the idea that Bem1 immobilizes Cdc42 at the polar cap. We also examined the dynamics of other polar cap resident proteins such as Cdc24 and Bem1 itself. Cdc24-GFP showed similar recovery dynamics to Cdc42 (Fig. 2 A, t 1/2 = 4.99 ± 2.05 s, n = 11). Interestingly, Bem1-GFP fluorescence recovered even more rapidly (t 1/2 = 2.37 ± 1.39 s, n = 10) but only to ∼60% of the prebleach level (Fig. 2 B). To test whether this was due to the presence of a nonexchanging pool of Bem1 in the polar cap or a limited pool in the cytosol, we repeatedly bleached a spot in the cytosol and monitored fluorescence loss in photobleaching in the cap. As shown in Fig. 2 C, fluorescence of the Bem1 cap decreased rapidly during bleaching and was completely lost after repeated bleaching, suggesting that all Bem1-GFP molecules in the polar cap exchange rapidly with the cytosolic pool.
Figure 2.
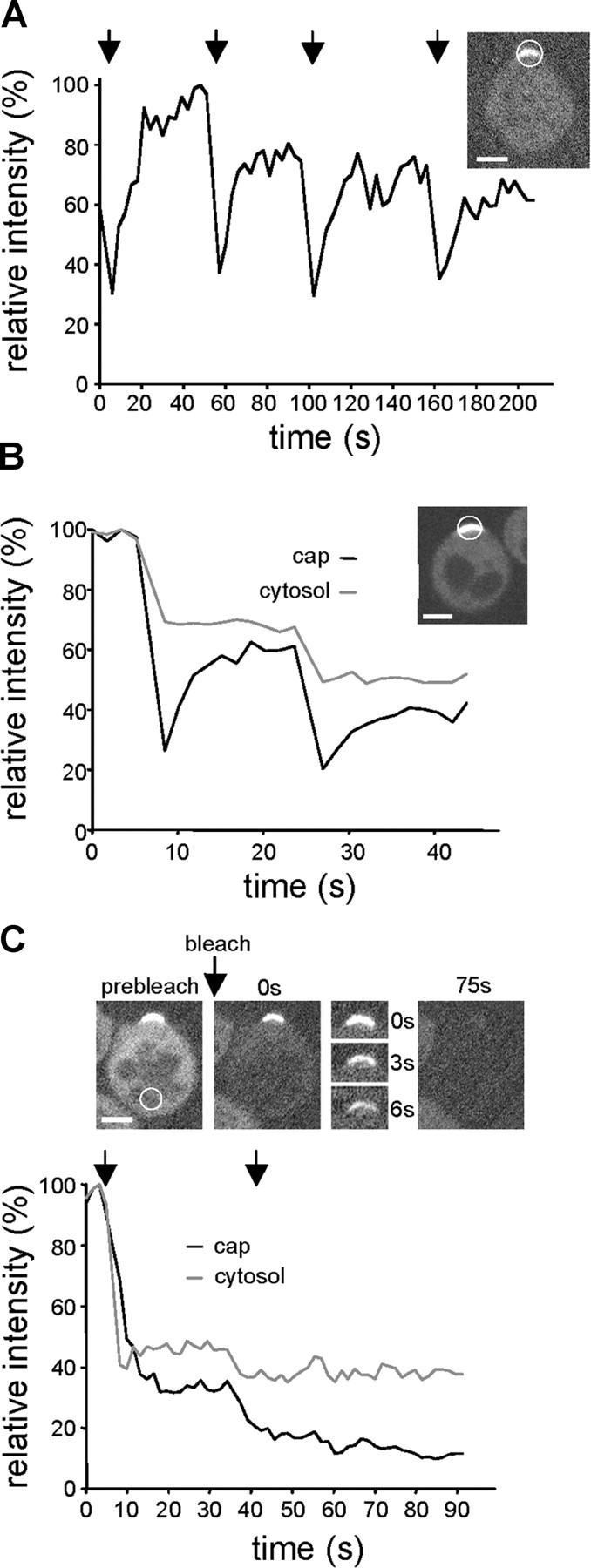
FRAP analysis of Cdc24-GFP and Bem1-GFP polar caps. The time points of bleaching are indicated by arrows and the area of bleaching is indicated by circles. Average intensities are given relative to the prebleach state. Images were taken every second. Bars, 5 μm. (A) FRAP of a Cdc24-GFP cap in an RLY1891 cell formed 25 min after release of cells from G1 arrest. One representative example of multiple bleaching and recovery is shown. (B) FRAP of a Bem1-GFP cap in an RLY1963 cell formed 25 min after release from G1 arrest. Fluorescence was monitored both in the cap (black line) and the cytosol (gray line). (C) Fluorescence loss in photobleaching of Bem1-GFP in an RLY1963 cell in the cytosol 25 min after release from G1 arrest. After bleaching the cytosol at the opposite side of the Bem1-GFP cap, fluorescence was measured in the cytosol (gray line) and the cap (black line). Images show the whole cell or the cap area at the indicated times after bleaching.
F-actin plays an important role in the establishment of Cdc42 polar caps
The FRAP experiments suggest that Cdc42 in the polar cap is dynamic and that its rapid exchange requires both the GTPase cycle and, to a lesser extent, actin. We next investigated the role of actin in the establishment of Cdc42 polar caps. First, we tested whether LatA affects Cdc42 polarization by analyzing the kinetics of polar cap formation in the release assay. In the absence of LatA, cells polarized with high synchrony after release from G1. At 40 min, >80% of cells had formed an MG-Cdc42 polar cap (Fig. 3 A). When cells were released into media containing 100 μM LatA, the rate of polarization was reduced by roughly twofold, and even at 70 min, only ∼60% of LatA-treated cells formed an MG-Cdc42 cap (Fig. 3 A). Rhodamine-phalloidin staining of LatA-treated cells showed no visible actin structures (not depicted).
To better understand the polarization defect caused by actin disruption, time-lapse videos were made of Cdc42 polar cap formation before bud emergence. Cells polarized rapidly and caps of MG-Cdc42 formed within a 3-min time window after an initial lag of 15–25 min, suggesting that polarization is controlled in a switch-like manner (Fig. 3 B and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200405061/DC1). The caps were stable and maintained similar intensity during observation (>9 min, Fig. 3, C and D, n = 20 and Video 2). In about half of the LatA-treated cells that formed an MG-Cdc42 cap (44%, n = 32), the cap disappeared within 10 min of observation. Most of the unstable caps (79%) flickered once or twice with intermittent stability of 2–3 min before complete disappearance (Fig. 3, C and D; Video 3). This instability of MG-Cdc42 cap in the presence of LatA could account for the reduced polarization measured at the level of cell populations (Fig. 3 A). At later time points (>40 min), all observed caps (n = 15) appeared to be stable. Interestingly, the unstable caps at earlier time points were significantly weaker than the stable caps (comparing maximum intensity in the initially present cap). The fraction of Cdc42 in the cap (ratio of total fluorescence intensity in the cap/total intensity in the cell) was 0.021 ± 0.009 (n = 13) for the stable caps, and 0.013 ± 0.004 (n = 11) for the unstable caps (t test, P = 0.011), suggesting that a certain threshold concentration of MG-Cdc42 in the forming polar cap has to be surpassed to achieve stable polarization.
Membrane-bound Cdc42 is polarized through actin-based transport
We investigated whether or not the LatA effect reflected an involvement of actin-based membrane transport. First, we examined Cdc42 polar cap formation in strains with temperature-sensitive mutations of either Myo2 (Johnston et al., 1991) or tropomyosin (Pruyne et al., 1998). Upon release at the permissive temperature, both strains polarized with kinetics comparable to wild-type cells (not depicted). At the restrictive temperature of 35°C, polarization of MG-Cdc42 in both strains was delayed to a similar extent as with LatA treatment (Fig. 4 A). These observations suggest that actomyosin-dependent membrane transport is required for optimal efficiency of Cdc42 polarization.
Figure 4.

A role for actin-based membrane transport in the polarization of wild-type and mutant Cdc42. (A) Polarization of MG-Cdc42 in strains temperature sensitive for actin-based transport upon release from G1 arrest at 35°C. Polarization was assayed as in Fig. 3 A for strains wt (RLY1948), myo2-66 (RLY1954), and tpm ts (RLY1897). (B) Polarization of MG-Cdc42Q61L (RLY1703) and MG-Cdc42D57Y (RLY1991) upon release from G1 arrest in the presence or absence of LatA. Note that the graphs for the LatA-treated cells run along the x axis. (A and B) Error bars correspond to SD. (C) Membrane fractionation of MG-Cdc42 in wild-type (RLY1948) and sec6-4 (RLY1894) mutant backgrounds. Cell extracts were prepared and separated by differential centrifugation as described in Materials and methods. In brief, cleared cell lysates (S1) were separated into S2 and P2 in a 10,000 g centrifugation step and S2 was further separated into S3 and P3 in a 100,000 g step. A representative example is shown for each strain. Quantification was performed on an Odyssey imager and is represented as an average of three independent experiments. All amounts are normalized to the total present in the lysate. Because loss of material occurred in the pellet fractions P values were calculated from the respective S values. (D) Localization of MG-Cdc42, MG-Cdc42C188S (RLY1952), MG-Cdc42Q61L, and MG-Cdc42D57Y 25 min after release from G1 arrest. Whereas MG-Cdc42C188S was mostly in the cytosol, all prenylated forms of Cdc42 were present on membranes to varying degrees. MG-Cdc42 showed a clear cytosolic pool (note cytosolic fluorescence in top right panel). MG-Cdc42Q61L and MG-Cdc42D57Y were mostly found on internal membranes and the plasma membrane. Bar, 5 μm. (E) Membrane fractionation of different Cdc42 forms. Cell extracts for strains RLY1948, RLY1703, and RLY1991 were prepared and separated by differential centrifugation as described in C. A representative example is shown for each Cdc42 form. Quantification is represented as average of two independent experiments.
Next, we asked which nucleotide-bound form of Cdc42 might be polarized through actin-based transport. Our previous experiments with Cdc42Q61L suggest that the GTP-bound form of Cdc42 is mostly associated with the membrane fractions and that actin is absolutely required for its polarized localization in G1-arrested cells (Wedlich-Soldner et al., 2003). To test if actin is required for polarization of GTP-bound Cdc42 during bud formation, G1-arrested cells expressing MG-Cdc42Q61L (in the presence of the endogenous Cdc42) were initially treated with LatA or shifted to the restrictive temperature for 15 min to eliminate any preformed polar caps, then washed and released from arrest. In the absence of LatA, MG-Cdc42Q61L polarized with similar kinetics to MG-Cdc42 and the cells budded with normal kinetics (Fig. 4 B and not depicted). However, when released in the presence of LatA (Fig. 4 B), or at the restrictive temperature in the myo2-66 and tpm ts backgrounds (not depicted), MG-Cdc42Q61L completely failed to polarize and remained evenly distributed on the cell surface. To test if the role of actin in Cdc42 polarization is restricted to the GTP-bound form, we examined the polarization of MG-Cdc42D57Y. Surprisingly, the formation of MG-Cdc42D57Y cap after release from G1 arrest was also completely blocked by LatA (Fig. 4 B), suggesting that both nucleotide-bound forms of Cdc42 could be polarized via actin-based transport.
To verify whether the observed requirement for actin-based transport could be due to secretion of Cdc42 bound to secretory vesicles, we analyzed the membrane fractionation of MG-Cdc42 in arrested cells of wild-type and sec6-4 mutant backgrounds. At the restrictive temperature, sec6-4 mutants accumulate secretory vesicles because vesicle fusion to the plasma membrane is blocked. As previously reported for MG-Cdc42Q61L and vesicle marker Sec4 (Wedlich-Soldner et al., 2003), MG-Cdc42 was highly enriched (∼5-fold) in the P3 fraction, which contains the secretory vesicles (Fig. 4 C). These results indicate that MG-Cdc42 can associate with secretory vesicles and is able to polarize via actomyosin dependent secretion.
We wanted to understand why polarization of MG-Cdc42Q61L and MG-Cdc42D57Y, but not wild-type MG-Cdc42, was completely dependent on actin-based transport. We noticed that both MG-Cdc42Q61L and MG-Cdc42D57Y were abundantly associated with various membrane compartments with relatively low cytosolic signals, whereas wild-type MG-Cdc42 appeared to be more evenly distributed between the cytosol and membranes. Mutation of the COOH-terminal prenylation site (C188 to S) caused Cdc42 to completely distribute to the cytosol (Fig. 4 D). To more quantitatively compare the distributions of wild-type Cdc42 and the distribution of the Cdc42 mutants locked in either nucleotide-bound form, we performed cell fractionation using cultures enriched for polarized cells. We found that whereas 52% of MG-Cdc42 was cytosolic (S3), only 7.7% of MG-Cdc42Q61L and 9.7% of MG-Cdc42D57Y were present in the soluble pool (Fig. 4 E). These results suggest that the ability to cycle between the GTP- and GDP-bound states correlates with a significant cytosolic pool of Cdc42 as well as an ability to establish a Cdc42 polar cap through an actin-independent mechanism. If the GTPase cycle is blocked, Cdc42 cannot dissociate from the membrane and can only polarize via the actin-based pathway.
Actin-independent polarization does not require preexisting spatial cues
The results described above confirmed the existence of an actin-independent mechanism for polarization that requires the Cdc42 GTPase cycle. A crucial component of the Cdc42 GTPase cycle is the GEF Cdc24. In haploid yeast strains that bud axially, Cdc24 is recruited to the presumptive bud site adjacent to the newest bud scar by the Bud1 GTPase (Park et al., 1997). However, bud site selection cannot account for the actin-independent polarization in our assays because the W303a strain background (used in this work) is naturally deficient in bud site selection (Fig. 5 A). Additionally, disruption of BUD1 caused only a slight reduction in Cdc42 polarization kinetics, compared with the kinetics in wild-type cells, with or without LatA (Fig. 5 B). Nocodazole, a microtubule polymerization inhibitor, did not affect polarization kinetics either in the presence or absence of LatA, which is consistent with the fact that microtubules are not required for polarity establishment (not depicted). These results, together with the previous demonstration that PI(4,5)P2 and cholesterol-rich lipids (Bagnat and Simons, 2002; Takenawa and Itoh, 2001) were symmetrically distributed in G1 cells (Wedlich-Soldner et al., 2003), suggest that an intrinsic, cytoskeleton-independent mechanism can generate cell polarity in random orientations.
Figure 5.

Cdc42 polarization is independent of bud site selection. (A) Budding pattern of RLY1683 cells. The position of the first bud emerging after release from G1 arrest was scored in relation to the closest bud scar as proximal, distal or to the side (see diagram). (B) Effect of BUD1 deletion on MG-Cdc42 polarization. Wild-type (RLY1948) or Δbud1 (RLY1959) cells were released from G1 arrest and polarization of MG-Cdc42 was scored in the presence or absence of LatA. Error bars correspond to SD.
Actin- and Bem1-dependent feedback loops work in parallel to generate cell polarity
Previous works have led to a hypothesis that a positive feedback loop involving activation of Cdc42 by Cdc24 and Cdc42GTP-facilitated recruitment of Cdc24, can lead to spontaneous symmetry breaking (Bose et al., 2001; Irazoqui et al., 2003). It is suggested that recruitment of Cdc24 by Cdc42GTP is mediated through Bem1, which can form a ternary complex with Cdc42GTP and Cdc24. We tested whether Cdc24 can be recruited to ectopic Cdc42Q61L polar caps. First, polar caps were induced by overexpression of HA-Cdc42Q61L in G1-arrested cells that also expressed Cdc24-GFP. We looked at cells that had formed two polar caps of HA-Cdc42Q61L. The polar caps in G1-arrested cells, as visualized by either actin staining or immunofluorescence staining against HA, did not recruit Cdc24 (unpublished data); however, this is not surprising as Cdc24 is both sequestered in the nucleus and inactive in G1 (Shimada et al., 2000). When released from the G1 arrest, both of the Cdc42Q61L caps in bipolar G1 cells develop into buds (Wedlich-Soldner et al., 2003). Cdc24-GFP was recruited to both Cdc42Q61L caps after the release and remained associated with both buds (Fig. 6 A), suggesting that the ectopic Cdc42Q61L caps can recruit Cdc24. When BEM1 was disrupted in that strain, most buds contained HA-Cdc42Q61L in the bud tip (not depicted), but completely failed to recruit Cdc24-GFP (Fig. 6 B). These results suggest that recruitment of Cdc24 to sites of Cdc42GTP accumulation requires Bem1, which is consistent with the idea of a feedback loop based on interactions of those three proteins.
Figure 6.

Actin and Bem1 function in parallel during cell polarization. (A) Recruitment of Cdc24-GFP by Cdc42Q61L. HA-tagged Cdc42Q61L was expressed from the Gal1-promoter in G1-arrested cells that also expressed Cdc24-GFP (RLY1989). Representative DIC and fluorescence images of cells with two buds, one of which must be ectopic. (B) Cdc24-GFP is not recruited to buds of Δbem1 cells expressing HA-Cdc42Q61L. Representative DIC and fluorescence images of cells with small and medium buds are shown. (A and B) Buds are indicated with arrows. (C) Kinetics of polarization of Cdc24-GFP expressed from the CDC24 promoter in wild-type (RLY1891) or Δbem1 (RLY1961) cells released from G1 arrest. Images show typical caps formed at 35 min after release. Note that the graph for the LatA-treated Δbem1 cells runs along the x axis. (D) Kinetics of polarization of MG-Cdc42 expressed from the CDC42 promoter in wild-type (RLY1950) or Δbem1 (RLY1965) cells released from G1 arrest. Images show typical caps formed at 35 min after release. Note that the graph for the LatA-treated Δbem1 cells runs along the x axis. Error bars correspond to SD. (E) Localization of Cdc24-GFP to multiple caps in Δbem1 cells 25 min after release from G1 arrest. Caps are indicated with arrows. Bars, 5 μm.
We also examined the effect of LatA on the polarization of Cdc24-GFP and the effect of Δbem1 on the polarization of Cdc24-GFP and MG-Cdc42 in the release assay. Similar to MG-Cdc42, Cdc24-GFP polarized rapidly upon release, and by 40 min, >90% of the cells had a polar cap of Cdc24-GFP (Fig. 6 C). Cdc24 polarization was consistently delayed by LatA, although the extent of the delay was much less than that on Cdc42 polar cap formation (compare Fig. 3 A with Fig. 6 D). In Δbem1 cells, both Cdc24-GFP and MG-Cdc42 polarized with kinetics slower than those in wild-type cells (Fig. 6, C and D). Consistent with a previous finding (Butty et al., 2002), Cdc24-GFP caps in Δbem1 cells were unstable and disappeared upon bud emergence (Fig. 6 C). Furthermore, ∼5% of the Δbem1 formed two caps of Cdc24, whereas wild-type cells always had unique caps (Fig. 6, C and E), suggesting that loss of Bem1 also diminished the cell's ability to restrict polarity to a unique axis. Strikingly, in Δbem1 cells treated with LatA, both Cdc24-GFP and MG-Cdc42 completely failed to polarize (Fig. 6, C and D). These results suggest that the actin- and Bem1-dependent pathways function in parallel to generate cell polarity.
Discussion
We have described here three findings regarding the mechanism of cell polarization in budding yeast. First, through FRAP analysis, we found that the initial polarized state, as marked by a polar cap of Cdc42, is dynamic, and this dynamicity is largely dependent on the Cdc42 GTPase cycle. Second, through careful kinetic analysis and single cell imaging, we showed that actin polymerization and actin-based transport are important for polarization of Cdc42 during bud formation. When actin function was inhibited, polarization occurred with significantly reduced efficiency and the resulting Cdc42 polar caps were unstable. Finally, we showed that a Bem1- and GTPase cycle dependent pathway acts in parallel with the actin-dependent feedback loop in the generation of robust cell polarity (Fig. 7).
Figure 7.

A schematic diagram depicting the two positive feedback loops (in dark lines) that control the generation of cue- independent cell polarity in yeast. The actin-dependent feedback loop results in accumulation of Cdc42 in both nucleotide-bound forms on the plasma membrane, whereas the Bem1-dependent feedback loop results in recruitment and/or activation of the GEF Cdc24 to the site of Cdc42 accumulation. Coupling of these feedback loops is required for the generation of robust cell polarity.
The polarized state: scaffold versus dynamic flux
The original theoretical formulation of spontaneous pattern formation by Meinhardt and Gierer (1974) is based on local positive feedback loops and does not require the formation of scaffolds, a term that is frequently used but poorly defined. The term “scaffold”, used with a mechanistic implication in the context of cell polarization, implies the assembly of a polymer of proteins or other cellular materials, which itself is stable and restricts the diffusion of other bound molecules (Blumer and Cooper, 2003). Components of such scaffolds are expected to exchange slowly with the soluble pool, as exemplified by the subunits of the septin ring, which do not undergo FRAP (Dobbelaere et al., 2003). Even at their most dynamic state, septin fluorescence recovery occurs with a t 1/2 > 175 s (Dobbelaere et al., 2003). The multivalent adaptor protein Bem1 has been speculated to form a polymeric scaffold; however, it exchanges with a t 1/2 = 2.37 ± 1.39 s, even faster than Cdc42, suggesting that Bem1 is unlikely to play the role of restricting Cdc42 from diffusing out of the polar cap.
Our result does not rule out the existence of other scaffolding factors that could slow down the diffusion of certain components of the polar cap. Intuitively, the polar cap structures must be maintained in a dynamic fashion because the plasma membrane in the growth region is constantly being modified through exocytic and endocytic events. Regardless of whether scaffolding agents indeed exist, as long as diffusion is not completely restricted, continuous targeting and recycling events must occur in order to maintain the steady-state flux balance of polar cap components. The implication of such a polarized state is that cell polarity can be altered by changes in rate constants of any of the processes that contribute to steady-state flux balance, such as diffusion (influenced directly by scaffolding), transport, and recycling through cytosolic and membrane compartments. Such a dynamic polarized system offers many possibilities for fine tuning and differential regulation by a variety of internal or environmental signals.
Dynamics of Cdc42: the role of GTPase cycle and actin cytoskeleton
We found that Cdc42 mutants locked in either the GTP- or GDP-bound form exhibit drastically reduced mobility in the FRAP experiments. The half times of recovery were >10-fold that of the cycling Cdc42 and comparable to the values measured for yeast membrane proteins like Snc1 or SsoI (Valdez-Taubas and Pelham, 2003). In contrast, loss of actin slowed down fluorescence recovery by only less than twofold. Furthermore, polarization of MG-Cdc42Q61L and MG-Cdc42D57Y was completely abolished by LatA treatment. These observations suggest that there are two pathways for localization of Cdc42: a fast pathway that requires the GTPase cycle and a slower pathway that requires actin. Because >90% of Cdc42Q61L and Cdc42D57Y are associated with membranes, whereas ∼50% of the wild-type Cdc42 is in the cytosol, we suspect that the fast recovery of wild-type Cdc42 is mainly achieved through rapid exchange between the polar cap and the cytosolic pool, whereas slow recovery is mediated through actin-based membrane transport. How the GTPase cycle facilitates the rapid dissociation/association of Cdc42 with the membrane should be of much interest for future study. We hypothesize that the energy of GTP hydrolysis drives the extraction of the prenyl moiety of Cdc42 from the membrane, an event that is thought to involve the guanine nucleotide dissociation factor.
The role of the actin-based positive feedback loop in the generation of cell polarity
Our previous work using an artificial assay showed that simply elevating the level of Cdc42GTP can drive cell polarization through a positive feedback loop, composed of actin-dependent Cdc42 transport and Cdc42-stimulated actin nucleation, is sufficient to achieve stable polarity, and the model predicted properties of the system that could be demonstrated experimentally (Wedlich-Soldner et al., 2003). This positive feedback loop should be present in normal cells and contribute to cell polarization during the physiological process of bud formation. Several previous works had reported that LatA did not prevent polarization of Cdc42 (Ayscough et al., 1997; Jaquenoud and Peter, 2000; Irazoqui et al., 2003). None of these works, however, could rule out partial effects because kinetic analysis was either lacking or done using cell synchronization protocols where polarization occurs on the order of hours instead of minutes.
The assay that we have used involved release of cells from an arrest point where they were poised to polarize. Polarization occurred in a highly synchronized fashion and reached the plateau with 80–90% of the cells polarized within 30–40 min, which is a more accurate time scale for polarization, given that the cell cycle normally takes 90 min. This assay, together with live cell imaging, clearly revealed a role for actin in cell polarization. The reduced population kinetics and final percentage of Cdc42 polarization in the presence of LatA can be accounted for by the fraction of cells that underwent flickering and eventually reverted back to the nonpolarized state, as revealed by live cell imaging. Therefore, without actin, cells have a diminished ability to establish polarity with temporal precision and high stability.
The role of actin in the establishment of Cdc42 polar caps is likely to involve actin-based transport of Cdc42-containing secretory vesicles. Mutations that inactivated actin cable–based transport resulted in a similar reduction in polarization to that observed with LatA, and blocking vesicle-plasma membrane fusion resulted in an increase of Cdc42 in the membrane fraction containing secretory vesicles. Consistently, Cdc42 mutants locked in the GTP or GDP-bound forms, which partition mostly in the membrane-bound fractions, polarize in a completely actin-dependent manner. Therefore, the actin-based transport indistinguishably targets both nucleotide-bound forms of Cdc42 to the polar caps. It is worth noting that G1-released cells overexpressing Cdc42Q61L could polarize with kinetics similar to those observed in the wild type. This indicates that lethality (assayed as an inability to eventually form colonies after days of incubation) induced by the expression of this allele (Irazoqui et al., 2003) was unlikely to be due to impaired cell polarization, but more likely due to defects in processes later in the cell cycle (e.g., cytokinesis; Gladfelter et al., 2002) or a de-coupling of the nuclear and morphogenetic cycles.
A Bem1-dependent but actin-independent pathway for cell polarization
The observation that only wild-type Cdc42, but not Cdc42 mutants locked in the GTP or GDP-bound form, could polarize in the absence of F-actin, suggests that the actin-independent pathway requires the Cdc42 GTPase cycle. Additionally, this intrinsic, cytoskeleton-independent cell polarization requires the adaptor protein Bem1. A previous work also arrived at a similar conclusion; however, that work concluded that Bem1 is absolutely required for Cdc42 polarization in the absence of bud site selection, based on the observation that bem1 and bud1 mutants are synthetically lethal (Irazoqui et al., 2003). In our work, Δbem1 cells clearly can polarize and undergo random budding, despite relatively short-lived Cdc24 polar caps and delayed Cdc42 polar cap formation. We suggest that the synthetic lethality of bem1 and bud1 is due to their shared biochemical function in Cdc24 activation, as shown by a recent work (Shimada et al., 2004), instead of a failure of bem1 mutant cells to polarize without the spatial cue provided by the bud scar.
The molecular details of the Bem1-mediated polarization pathway still remain unclear. The FRAP data suggest that it is unlikely that Bem1 functions through the formation of a polymeric scaffold. Our work supports the idea that Bem1 mediates a positive feedback loop connecting Cdc24 and Cdc42 (Butty et al., 2002; Irazoqui et al., 2003). Such a feedback loop could effectively amplify stochastic variations of Cdc24 or Cdc42 on the plasma membrane, leading to formation of unique polar caps. We observed that polar caps of Cdc42GTP (due to Cdc42Q61L expression) could indeed recruit Cdc24 in a Bem1-dependent manner. Bem1 contains multiple protein-interaction domains such as the SH3 domain and PB1 domain (Ito et al., 2001; van Drogen-Petit et al., 2004). We speculate that Bem1 is a functional analogue of the metazoan protein Par6, which binds activated Cdc42 and mediates the cooperative assembly of a multi-functional polarity complex (Joberty et al., 2000; Fukata et al., 2003; Garrard et al., 2003). Understanding how cell polarity can be generated through these multivalent adaptor proteins should benefit from works in both systems.
Bi-stable control of cell polarization through coupled positive feedback loops
Controls of cell cycle events are switch-like, and this property allows cell cycle transitions to occur with temporal precision and bi-stability and ensures the fidelity of cell division (Ferrell, 2002; Qu et al., 2003). Cell polarization during the yeast cell cycle appears to be no exception. Single cell imaging revealed that the actual process of Cdc42 polar cap formation occurs within 3 min after a much longer lag phase, and once established, the polarity is stably maintained throughout bud growth. The latter phenomenon may reflect hysteresis, as the burst of G1 Cdk1 activity that triggers polarization declines soon after START (Moffat and Andrews, 2003). When the actin-based feedback loop was inhibited, the polarization “switch” flickered and eventually resulted in a bimodal distribution of polarized and nonpolarized cells. This is reminiscent of a synthetic switch controlling gene expression by a single positive feedback loop (Becskei et al., 2001). Cell polarization during bud formation must also occur with spatial precision, that is, the polar axis must be unique and once formed must maintain spatial stability. When the actin-dependent mechanism is operating alone (i.e., in the Δbem1 background), we observed bipolar cells, which never occur in the wild-type population. This is consistent with our previous results, in which bipolarity was observed in a small population of the G1 cells that polarized due to Cdc42Q61L overexpression (Wedlich-Soldner et al., 2003). On the other hand, the polar axis drifted in some of the LatA-treated cells that exhibited “flickering” polar caps (Fig. 3 D). Therefore, coupling the actin-dependent feedback loop with the Bem1-dependent one is required for achieving robust temporal and spatial stability as well as uniqueness of cell polarity.
Recent work on neutrophil and Dictyostelium chemotaxis has also implicated a positive feedback loop, involving the phospholipid PIP3 and PI3 kinase (PI3K), which is required for amplification of the asymmetry that originates from gradients of chemoattractant, as well as spontaneous cell polarization in the absence of a gradient (Li et al., 2003; Merlot and Firtel, 2003; Xu et al., 2003). It is thought that PIP3 could stimulate, through the action of a GEF, the local production of an activated Rho-type GTPase, which could in turn activate PI3K to generate more PIP3 (Weiner et al., 2002). Actin also seems to play a role in the amplification of gradient signals and spontaneous polarization in neutrophils (Wang et al., 2002), but it is unclear whether actin participates in the same feedback loop as PIP3 or represents a parallel pathway as we observed in this work. Because chemotactic cells must be able to switch directions rapidly in order to track down agents such as a moving bacterium, their polarization pathways may not have evolved the same strong bi-stability as that observed in the budding yeast. The similarities as well as differences of these physiological systems could illuminate our understanding of nature's design principles underlying the control of cell polarity.
Materials and methods
Yeast strain construction
Techniques for yeast cell culture and genetics were essentially as described previously (Sherman et al., 1981). All strains used in this work are described in Table I.
Table I. Yeast strains used in this work.
| Strain | Genotype a | Source |
|---|---|---|
| RLY261 | MATa ura3 leu2-112 his3 trp1 ade2 Δsst1 | E. Elion |
| RLY1683 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 can1-100 | Amon et al., 1994 |
| RLY1703 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Gal-myc-GFP-CDC42L61::URA3 (pRL367) |
Wedlich-Soldner et al., 2003 |
| RLY1733 | MATα cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Gal1-myc-GFP-CDC42Q61L::URA3 (pRL367) |
This work |
| RLY1788 | MATα cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Δtpm2::HIS3 tpm1-2::LEU2 |
Wedlich-Soldner et al., 2003 |
| RLY1891 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 pCDC24-CDC24-GFP::URA3 (pRL370) |
This work |
| RLY1894 | MATα cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 sec6-4 Gal1-myc-GFP-CDC42::URA3 (pRL368) |
This work |
| RLY1897 | MATα cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Δtpm2::HIS3 tpm1-2::LEU2 Gal1-myc-GFP-CDC42::URA3 (pRL368) |
This work |
| RLY1941 | MATa cln1::hisG cln2-Δ cln3::LEU2::hisG yipLac204-MET-CLN2::TRP1 ura3 leu2 his3 | Butty et al., 2002 |
| RLY1948 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Gal1-myc-GFP-CDC42 (pRL368) |
This work |
| RLY1949–51 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 pCDC42-myc-GFP-CDC42::URA3 (pRL369) |
This work |
| RLY1952 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 can1-100 Gal-myc-GFP-CDC42188S::URA3 (pSTIL8) |
This work |
| RLY1953 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 myo2-66 | This work |
| RLY1954–56 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 myo2-66 Gal1-myc-GFP-CDC42::URA3 (pRL368) |
This work |
| RLY1957 | MATa cln1::hisG cln2-Δ cln3::LEU2 MET3-CLN2::TRP1 ura3 his3-11,15 Δbud1::G418 | This work |
| RLY1958 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Δbud1::G418 CDC24-GFP::URA3 (pRL370) |
This work |
| RLY1959 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Δbud1::G418 Gal-myc-GFP-CDC42::URA3 (pRL368) |
This work |
| RLY1960 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Δbem1::G418 | This work |
| RLY1960/61 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Δbem1::G418 pCDC24-CDC24-GFP::URA3 (pRL370) |
This work |
| RLY1963/64 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Δbem1::G418 pBEM1-BEM1-GFP (ACB514) |
This work |
| RLY1965/66 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 ura3 his3-11,15 ade2-1 Δbem1::G418 pCDC42-myc-GFP-CDC42::URA3 (pRL369) |
This work |
| RLY1988 | MATa cln1::hisG cln2-Δ cln3::LEU2::hisG yipLac204-MET-CLN2::TRP1 pGAL-HA-CDC42Q61L::URA3 (pRL227) leu2 his3 | This work |
| RLY1989 | MATa cln1::hisG cln2-Δ cln3::LEU2::hisG yipLac204-MET-CLN2::TRP1 pGAL-HA-CDC42Q61L::URA3 (pRL227) p24-Cdc24-GFP::LEU2 (pRL270LEU2) his3 | This work |
| RLY1990 | MATa cln1::hisG cln2-Δ cln3::LEU2::hisG yipLac204-MET-CLN2::TRP1 Δbem1::G418 pGAL-HA-CDC42Q61L::URA3 (pRL227) p24-Cdc24-GFP::LEU2 (pRL270LEU2) his3 | This work |
| RLY1991 | MATa cln1::hisG cln2-Δ cln3::LEU2 pMET3-CLN2::TRP1 pGAL-GFP-myc-CDC42D57Y::URA3 (pTS198) his3-11,15 | This work |
All strains are in the W303 background.
Triple cln deletion strains were gifts from A. Amon (Massachusetts Institute of Technology, Cambridge, MA) and M. Peter (Swiss Federal Institute of Technology Zurich [ETH], Zurich, Switzerland). Deletion of BUD1 in the triple cln deletion background (to obtain RLY1957) was done by crossing RLY1733 with a Δbud1::G418 strain from the research genetics knockout library. Deletion of BEM1 in the triple cln deletion background (to obtain RLY1960) was done similarly by crossing RLY1732 with a Δbem1::G418 strain from the same knockout library. The temperature-sensitive alleles myo2-66 and tpm ts (tpm ts strain was a gift from A. Bretscher; Cornell University, Ithaca, NY) were introduced into the triple cln deletion background by backcrosses to obtain strains RLY1953 and RLY1788, respectively.
Plasmids used for this work
pRL227, pRL367, pRL368, pTS198, pRL369, pSTIL8, and pRL370 are all based on a pRS306 (Sikorski and Hieter, 1989) backbone and are used for integration into the URA3 locus after linearization. The plasmid ACB514 (a gift from M. Peter; Butty et al., 2002) was used to integrate a BEM1-GFP fusion construct under the control of the endogenous BEM1-promoter into the URA3 locus of RLY1960. See Table II for more plasmid description.
Table II. Plasmids used in this work.
| Plasmid | Description |
|---|---|
| pRL227 | HA- CDC42Q61L under the control of the Gal1/10 promoter |
| pRL367 | GFP-myc6-CDC42Q61L under the control of the Gal1/10 promoter |
| pSTIL8 | GFP-myc6-CDC42C188S under the control of the Gal1/10 promoter (the prenylated cysteine at the COOH terminus of Cdc42 was mutated to serine) |
| pRL368 | GFP-myc6-CDC42 under the control of the Gal1/10 promoter |
| pTS198 | GFP-myc6-CDC42D57Y under the control of the Gal1/10 promoter |
| pRL369 | GFP-myc6-CDC42 under the control of the endogenous CDC42 promoter |
| pRL370 | CDC24-GFP under the control of the endogenous CDC24 promoter |
| pRL370/LEU2 | CDC24-GFP under the control of the endogenous CDC24 promoter in a pRS305 backbone for integration into the LEU2 locus after linearization |
| ACB514 | BEM1-GFP under the control of the endogenous BEM1-promoter |
Release assay
Logarithmically growing triple cln cells were arrested in G1 by growth for 3 h in YP medium supplemented with 2 mM methionine containing either 2% raffinose (for expression under the Gal1/10 promoter) or 2% glucose (for expression under the endogenous promoters). To induce expression of Cdc42 constructs under the Gal1/10 promoter, galactose was added to 2% and cells grown for another 2–3 h. To release from the G1 arrest, cells were washed three times with water and resuspended in synthetic complete (SC) medium containing 2% glucose and no methionine. LatA (a gift from P. Crews; University of California, Santa Cruz, Santa Cruz, CA) was added to 100 μM 30 min before the release or at the time of release with no visible difference in its effect. To assay for effects of the temperature-sensitive tpm1-2 and myo2-66 alleles cells were first grown and arrested at the permissive temperature and then shifted to the restrictive temperature 30 min before release. For all quantifications, means and SDs were calculated from at least three independent experiments with >40 cells per time point per experiment.
Membrane fractionation
Logarithmically growing triple cln cells containing MG-Cdc42 (RLY1948), MG-Cdc42Q61L (RLY1703), or MG-Cdc42D57Y (RLY1991) under the Gal1 promoter were arrested in G1 for 2 h and then processed for fractionation (Fig. 4 C) or induced for another 2 h, and subsequently released from arrest as described above in the release assay (Fig. 4 E). After ∼1 h of release, when 90–100% of the cells were polarized, cells were harvested and washed with cold 10 mM sodium azide. Membrane fractionation was performed as described previously (Novick et al., 1993). In brief, cleared cell lysates (S1) were centrifuged at 10,000 g to yield a supernatant (S2) and pellet (P2). S2 was further centrifuged at 100,000 g to give a supernatant (S3) and pellet (P3). Previous characterization showed that P2 contains large amounts of plasma membrane, ER, and mitochondria markers, whereas P3 is enriched for secretory vesicles. All pellets were resuspended in the same volume as before centrifugation and the same amount of each fraction was loaded on a gel. Immunoblot analysis was performed using a mouse anti-myc (9E10) antibody. Quantitative analysis was performed using the Odyssey Infrared Imaging system from LI-COR Biotechnology. Because pellets could often not be dissolved completely, leading to loss of material, P2 and P3 values were calculated from the respective S values. All values were normalized to the total amount in the lysates. Fractionations were done at least three times with the same qualitative results.
Microscopy
All wide-field fluorescence microscopy was performed on a fluorescence microscope (Nikon E800) with a 100× Plan Apo TIRF n = 1.45 lens and a cooled CCD camera (model CCD782-Y; Princeton Instruments). Image acquisition and analysis were performed with Metamorph (version 5.1; Universal Imaging Corp.). For long term imaging of cap formation pictures were taken in one focal plane and only videos with negligible z-drift were analyzed. To ensure that caps had disappeared and not just moved, cells were checked live in all planes after a video and sometimes during video acquisition.
Actin staining with rhodamine-phalloidin was done as described previously (Pringle et al., 1989). Calcofluor staining of bud scars was also performed as described previously (Wedlich-Soldner et al., 2000).
FRAP analysis was performed in the Nikon-Harvard imaging facility on a spinning disk confocal laser (PerkinElmer, Ultraview) attached to an inverted microscope (model TE2000U; Nikon). Bleaching was done with the MicroPoint Laser system from Photonic Instruments. In brief, a pulsed nitrogen laser was used to excite a dye cell and the emitted light then directed through a fiber optic connection into the back of the inverted microscope. A graded neutral density filter was used to attenuate the signal. Pulse frequency was set to 20/s and bleaching was performed for 1–4 s.
Image analysis
All measurements were performed in Metamorph. Data analysis was done with Excel (Microsoft). Chymographs were done with Metamorph. For measurements of cap intensities, average intensity values were normalized for photobleaching by dividing by the average intensity of the rest of the cell and then plotted as percent of the maximum cap intensity in a given series (video). Regression analysis to determine t 1/2 in FRAP experiments was done using a one-phase exponential association function (Y = bottom + (top − bottom)·(1-exp[−k·x]), where k is the rate constant and t 1/2 is 0.69/k) in Prism (version 4.00; GraphPad).
Online supplemental material
Video 1 shows cap formation of MG-Cdc42. The video corresponds to the black line in Fig. 3 B (RLY1950). Frames were taken every 10 s starting 10 min after release from G1 arrest and are played back at 5 frames/s (50×). Video 2 shows stable MG-Cdc42 cap formed after release of RLY1950 cells from G1 arrest in the absence of LatA. The video corresponds to wt (dotted line) in Fig. 3 (C and D). Frames were taken 25 min after release with 10 s/frame. Playback is at 5 frames/s (50×). Video 3 shows flickering MG-Cdc42 cap after release of RLY1950 from G1 arrest into LatA. The video corresponds to LatA-1 (black line) in Fig. 3 (C and D). Frames were taken 25 min after release with 10 s/frame. Playback is at 5 frames/s (50×). Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200405061/DC1.
Acknowledgments
We thank P. Crews for latrunculin A; A. Amon, A. Bretscher, and M. Peter for yeast strains and plasmid constructs; J. Water-Shuler and the Nikon-Harvard Imaging Center for help on FRAP analysis; L. Wu and S. Altschuler for stimulating discussions; and T. Mitchison and S. Wedlich for critical reading of the manuscript.
This work was supported by an EMBO fellowship to R.Wedlich-Soldner and by a National Institutes of Health grant GM057063 to R. Li.
R. Wedlich-Soldner and S.C. Wai contributed equally.
Abbreviations used in this paper: GEF, guanine nucleotide exchange factor; LatA, latrunculin A; MG, GFP-myc6.
References
- Amon, A., S. Irniger, and K. Nasmyth. 1994. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell. 77:1037–1050. [DOI] [PubMed] [Google Scholar]
- Ayscough, K.R., J. Stryker, N. Pokala, M. Sanders, P. Crews, and D.G. Drubin. 1997. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J. Cell Biol. 137:399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnat, M., and K. Simons. 2002. Cell surface polarization during yeast mating. Proc. Natl. Acad. Sci. USA. 99:14183–14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becskei, A., B. Seraphin, and L. Serrano. 2001. Positive feedback in eukaryotic gene networks: cell differentiation by graded to binary response conversion. EMBO J. 20:2528–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumer, K.J., and J.A. Cooper. 2003. Go ahead, break my symmetry! Nat. Cell Biol. 5:1048–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose, I., J.E. Irazoqui, J.J. Moskow, E.S. Bardes, T.R. Zyla, and D.J. Lew. 2001. Assembly of scaffold-mediated complexes containing Cdc42p, the exchange factor Cdc24p, and the effector Cla4p required for cell cycle-regulated phosphorylation of Cdc24p. J. Biol. Chem. 276:7176–7186. [DOI] [PubMed] [Google Scholar]
- Butty, A.C., N. Perrinjaquet, A. Petit, M. Jaquenoud, J.E. Segall, K. Hofmann, C. Zwahlen, and M. Peter. 2002. A positive feedback loop stabilizes the guanine-nucleotide exchange factor Cdc24 at sites of polarization. EMBO J. 21:1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casamayor, A., and M. Snyder. 2002. Bud-site selection and cell polarity in budding yeast. Curr. Opin. Microbiol. 5:179–186. [DOI] [PubMed] [Google Scholar]
- Chant, J., and I. Herskowitz. 1991. Genetic control of bud site selection in yeast by a set of gene products that constitute a morphogenetic pathway. Cell. 65:1203–1212. [DOI] [PubMed] [Google Scholar]
- Devreotes, P.N., and S.H. Zigmond. 1988. Chemotaxis in eukaryotic cells: a focus on leukocytes and Dictyostelium. Annu. Rev. Cell Biol. 4:649–686. [DOI] [PubMed] [Google Scholar]
- Dobbelaere, J., M.S. Gentry, R.L. Hallberg, and Y. Barral. 2003. Phosphorylation-dependent regulation of septin dynamics during the cell cycle. Dev. Cell. 4:345–357. [DOI] [PubMed] [Google Scholar]
- Dong, Y., D. Pruyne, and A. Bretscher. 2003. Formin-dependent actin assembly is regulated by distinct modes of Rho signaling in yeast. J. Cell Biol. 161:1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell, J.E., Jr. 2002. Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr. Opin. Cell Biol. 14:140–148. [DOI] [PubMed] [Google Scholar]
- Fukata, M., M. Nakagawa, and K. Kaibuchi. 2003. Roles of Rho-family GTPases in cell polarization and directional migration. Curr. Opin. Cell Biol. 15:590–597. [DOI] [PubMed] [Google Scholar]
- Garrard, S.M., C.T. Capaldo, L. Gao, M.K. Rosen, I.G. Macara, and D.R. Tomchick. 2003. Structure of Cdc42 in a complex with the GTPase-binding domain of the cell polarity protein, Par6. EMBO J. 22:1125–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart, J., M. Danilchik, T. Doniach, S. Roberts, B. Rowning, and R. Stewart. 1989. Cortical rotation of the Xenopus egg: consequences for the anteroposterior pattern of embryonic dorsal development. Development. 107:37–51. [DOI] [PubMed] [Google Scholar]
- Gladfelter, A.S., I. Bose, T.R. Zyla, E.S. Bardes, and D.J. Lew. 2002. Septin ring assembly involves cycles of GTP loading and hydrolysis by Cdc42p. J. Cell Biol. 156:315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulli, M.P., M. Jaquenoud, Y. Shimada, G. Niederhauser, P. Wiget, and M. Peter. 2000. Phosphorylation of the Cdc42 exchange factor Cdc24 by the PAK-like kinase Cla4 may regulate polarized growth in yeast. Mol. Cell. 6:1155–1167. [DOI] [PubMed] [Google Scholar]
- Irazoqui, J.E., A.S. Gladfelter, and D.J. Lew. 2003. Scaffold-mediated symmetry breaking by Cdc42p. Nat. Cell Biol. 5:1062–1070. [DOI] [PubMed] [Google Scholar]
- Ito, T., Y. Matsui, T. Ago, K. Ota, and H. Sumimoto. 2001. Novel modular domain PB1 recognizes PC motif to mediate functional protein-protein interactions. EMBO J. 20:3938–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaquenoud, M., and M. Peter. 2000. Gic2p may link activated Cdc42p to components involved in actin polarization, including Bni1p and Bud6p (Aip3p). Mol. Cell. Biol. 20:6244–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joberty, G., C. Petersen, L. Gao, and I.G. Macara. 2000. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat. Cell Biol. 2:531–539. [DOI] [PubMed] [Google Scholar]
- Johnson, D.I. 1999. Cdc42: An essential Rho-type GTPase controlling eukaryotic cell polarity. Microbiol. Mol. Biol. Rev. 63:54–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, G.C., J.A. Prendergast, and R.A. Singer. 1991. The Saccharomyces cerevisiae MYO2 gene encodes an essential myosin for vectorial transport of vesicles. J. Cell Biol. 113:539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., M. Hannigan, Z. Mo, B. Liu, W. Lu, Y. Wu, A.V. Smrcka, G. Wu, L. Li, M. Liu, et al. 2003. Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell. 114:215–227. [DOI] [PubMed] [Google Scholar]
- Meinhardt, H., and A. Gierer. 1974. Applications of a theory of biological pattern formation based on lateral inhibition. J. Cell Sci. 15:321–346. [DOI] [PubMed] [Google Scholar]
- Merlot, S., and R.A. Firtel. 2003. Leading the way: Directional sensing through phosphatidylinositol 3-kinase and other signaling pathways. J. Cell Sci. 116:3471–3478. [DOI] [PubMed] [Google Scholar]
- Misteli, T. 2001. The concept of self-organization in cellular architecture. J. Cell Biol. 155:181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat, J., and B. Andrews. 2003. Late-G1 cyclin-CDK activity is essential for control of cell morphogenesis in budding yeast. Nat. Cell Biol. 6:59–66. [DOI] [PubMed] [Google Scholar]
- Novick, P., P. Brennwald, N.C. Walworth, A.K. Kabcenell, M. Garrett, M. Moya, D. Roberts, H. Muller, B. Govindan, and R. Bowser. 1993. The cycle of SEC4 function in vesicular transport. Ciba Found. Symp. 176:218–228. [PubMed] [Google Scholar]
- Park, H.O., E. Bi, J.R. Pringle, and I. Herskowitz. 1997. Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc. Natl. Acad. Sci. USA. 94:4463–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, H.O., P.J. Kang, and A.W. Rachfal. 2002. Localization of the Rsr1/Bud1 GTPase involved in selection of a proper growth site in yeast. J. Biol. Chem. 277:26721–26724. [DOI] [PubMed] [Google Scholar]
- Pringle, J.R., R.A. Preston, A.E. Adams, T. Stearns, D.G. Drubin, B.K. Haarer, and E.W. Jones. 1989. Fluorescence microscopy methods for yeast. Methods Cell Biol. 31:357–435. [DOI] [PubMed] [Google Scholar]
- Pruyne, D., and A. Bretscher. 2000. Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. J. Cell Sci. 113:365–375. [DOI] [PubMed] [Google Scholar]
- Pruyne, D.W., D.H. Schott, and A. Bretscher. 1998. Tropomyosin-containing actin cables direct the Myo2p-dependent polarized delivery of secretory vesicles in budding yeast. J. Cell Biol. 143:1931–1945. [DOI] [PubMed] [Google Scholar]
- Qu, Z., W.R. MacLellan, and J.N. Weiss. 2003. Dynamics of the cell cycle: checkpoints, sizers, and timers. Biophys. J. 85:3600–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F., G.R. Fink, and J.B. Hicks, and Cold Spring Harbor Laboratory. 1981. Methods in Yeast Genetics: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. 119 pp.
- Shimada, Y., M.P. Gulli, and M. Peter. 2000. Nuclear sequestration of the exchange factor Cdc24 by Far1 regulates cell polarity during yeast mating. Nat. Cell Biol. 2:117–124. [DOI] [PubMed] [Google Scholar]
- Shimada, Y., P. Wiget, M.P. Gulli, E. Bi, and M. Peter. 2004. The nucleotide exchange factor Cdc24p may be regulated by auto-inhibition. EMBO J. 23:1051–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski, R.S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenawa, T., and T. Itoh. 2001. Phosphoinositides, key molecules for regulation of actin cytoskeletal organization and membrane traffic from the plasma membrane. Biochim. Biophys. Acta. 1533:190–206. [DOI] [PubMed] [Google Scholar]
- Valdez-Taubas, J., and H.R. Pelham. 2003. Slow diffusion of proteins in the yeast plasma membrane allows polarity to be maintained by endocytic cycling. Curr. Biol. 13:1636–1640. [DOI] [PubMed] [Google Scholar]
- van Drogen-Petit, A., C. Zwahlen, M. Peter, and A.M. Bonvin. 2004. Insight into molecular interactions between two PB1 domains. J. Mol. Biol. 336:1195–1210. [DOI] [PubMed] [Google Scholar]
- Wang, F., P. Herzmark, O.D. Weiner, S. Srinivasan, G. Servant, and H.R. Bourne. 2002. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell Biol. 4:513–518. [DOI] [PubMed] [Google Scholar]
- Wedlich-Soldner, R., and R. Li. 2003. Spontaneous cell polarization: undermining determinism. Nat. Cell Biol. 5:267–270. [DOI] [PubMed] [Google Scholar]
- Wedlich-Soldner, R., M. Bolker, R. Kahmann, and G. Steinberg. 2000. A putative endosomal t-SNARE links exo- and endocytosis in the phytopathogenic fungus Ustilago maydis. EMBO J. 19:1974–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedlich-Soldner, R., S. Altschuler, L. Wu, and R. Li. 2003. Spontaneous cell polarization through actomyosin-based delivery of the Cdc42 GTPase. Science. 299:1231–1235. [DOI] [PubMed] [Google Scholar]
- Weiner, O.D., P.O. Neilsen, G.D. Prestwich, M.W. Kirschner, L.C. Cantley, and H.R. Bourne. 2002. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat. Cell Biol. 4:509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J., F. Wang, A. Van Keymeulen, P. Herzmark, A. Straight, K. Kelly, Y. Takuwa, N. Sugimoto, T. Mitchison, and H.R. Bourne. 2003. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 114:201–214. [DOI] [PubMed] [Google Scholar]