

Abstract
Mammary epithelial cells cultured on a concentrated laminin-rich extracellular matrix formed 3D acinar structures that matured to polarized monolayers surrounding a lumen. In the absence of glucocorticoids mature acinus formation failed and the expression of an acinus-associated, activator protein 1 (AP1) and nuclear factor κB transcription factor DNA-binding profile was lost. Treatment with the JNK inhibitor, SP600125, caused similar effects, whereas normal organization of the mammary epithelial cells as acini caused JNK activation in a glucocorticoid-dependent manner. The forming acini expressed BRCA1, GADD45β, MEKK4, and the JNK activating complex GADD 45β−MEKK4 in a glucocorticoid-dependent fashion. JNK catalyzed phosphorylation of c-Jun was also detected in the acini. In addition, expression of β4 integrin and in situ occupation of its promoter by AP1 components, c-Jun and Fos, was glucocorticoid dependent. These results suggest that glucocortocoid signaling regulates acinar integrity through a pathway involving JNK regulation of AP1 transcription factors and β4 integrin expression.
Keywords: glucocorticoid; mammary epithelial cell; acinus; JNK; BRCA1
Introduction
Acini are spherical monolayers of epithelial cells that enclose a central lumen (O'Brien et al., 2002). Culturing mouse mammary epithelial cells on a laminin-rich Engelbreth-Holm-Swarm (EHS) tumor-derived ECM generates such acinar structures (Barcellos-Hoff et al., 1989; Blatchford et al., 1999). The spatial organization of these structures is maintained through cell–ECM and cell–cell interactions. ECM detection by the epithelial cells occurs through interactions with integrins. For instance, abrogating expression of the collagen receptor, α2β1 integrin, caused MDCK acini to apoptose because they could not detect the surrounding ECM (Saelman et al., 1995). Essential cell–cell interactions occur via the epithelial junctional complex of tight junctions, adherens junctions, and desmosomes (O'Brien et al., 2002). In 2D mammary epithelial model systems, glucocorticoids contribute to such cell–cell interactions by inducing the reorganization of tight and adherens junctions leading to enhanced cell–cell adhesion and decreased paracellular permeability (Buse et al., 1995; Wong et al., 1999).
The supporting EHS matrix contains laminin, collagen IV, entactin, and heparan sulfate proteoglycan (Kleinman et al., 1986). Even single mammary epithelial cells are able to assimilate information from this laminin-rich ECM and can be induced to synthesize β-casein (Streuli et al., 1995). Function-blocking anti–β1-integrin antibodies inhibit mammary epithelial acinus organization and function (Howlett et al., 1995). Indeed, expression of a dominant-negative β1-integrin in mouse mammary epithelium delayed the glandular maturation of late pregnancy, showed deficient milk protein expression at the onset of lactation and defects in epithelial cell polarity (Faraldo et al., 2000). In vitro, treatment of mammary tumor cells loosely organized as 3D assemblies in culture on EHS ECM with an inhibitory β1-integrin antibody lead to a morphological and functional reversion to a normal acinar phenotype (Weaver et al., 1997). These reverted acini displayed a reorganized cytoskeleton and a now functional basement membrane. Thus, the ECM through interactions with integrins can determine the phenotype of the mammary epithelial cell, regardless of its cellular genotype, i.e., normal or malignant.
During acinus formation, loss of luminal mammary epithelial cells is due to apoptosis (Blatchford et al., 1999) and autophagy (Mills et al., 2004). Debnath et al. (2002) demonstrated that this apoptosis followed apicobasal cell polarization and preceded proliferation suppression. Acinus cavitation is accompanied by a redistribution of ECM constituents to the periphery of the structure (Barcellos-Hoff et al., 1989).
Through β4-integrin mediated function, in hemidesmosomes, acini resist apoptotic insults to which monolayers readily succumb (Weaver et al., 2002). Furthermore, function-blocking antibodies against desmocollin-2 and desmoglein-2, the principal desmosomal cadherins in mammary alveolar luminal cells, disrupted the formation of 3D spheres of mammary epithelial Fsk-7.1 cells (Runswick et al., 2001). Morphogenesis was also inhibited by function-blocking antibodies against E-cadherin suggesting its critical dependence on both desmosomal adhesion and cadherin junctions (Runswick et al., 2001).
Mammary epithelial acinus maintenance and function requires lactogenic hormones (Topper and Freeman, 1980): prolactin will trigger the vectoral secretion of milk proteins into their lumena (Blatchford et al., 1999); insulin or IGF-1 may be required to support cell survival (Farrelly et al., 1999); and it seems that glucocorticoids could be required for the formation and maintenance of tight junctional complexes that would support acinus structure and function (Buse et al., 1995). The present work investigates the role of the glucocorticoids.
3D organization of mammary epithelial cells, as acini, is shown to require glucocorticoids which activate the MAPK, JNK. Acinus formation is compromised by a JNK inhibitor and JNK activation is glucocorticoid dependent. JNK activates the activator protein 1 (AP1) component, c-Jun, and glucocorticoid and JNK regulated targets of AP1, in the acini, may include β4 integrin expression. Thus, glucocorticoid and JNK signaling support the formation of mammary epithelial acini.
Results
Formation of mammary epithelial acini
Primary mouse mammary epithelial cells cultured on concentrated EHS ECM formed 3D cell clusters, as judged by phase-contrast microscopy (Fig. 1 A, i), whereas cells plated on diluted EHS ECM formed a monolayer (Fig. 1 A, ii). Maturation of these structures occurs within 4 d (Fig. 1 B): DAPI staining (top) and vital dye staining (bottom; Debnath et al., 2002) shows death of the internal lumenal cells (ethidium bromide [EtBr] staining) contrasting with eventual survival of the outer monolayer of cells (Calcein AM, green staining). Mature cell clusters consist of polarized acini, as judged by association of laminin with the basal surface, lateral distribution of E-cadherin and the apical distribution of their actin cytoskeleton (Fig. 1 C, i, ii, and iii). That clearing of the lumen involves apoptosis was suggested by cleaved caspase 3 expression in the lumenal cells of the early forming spheres (Fig. 1 C, iv). Treatment of the cultures with prolactin induced milk protein secretion into the lumen of the spheres (Fig. 1 C, v, β-casein staining).
Figure 1.
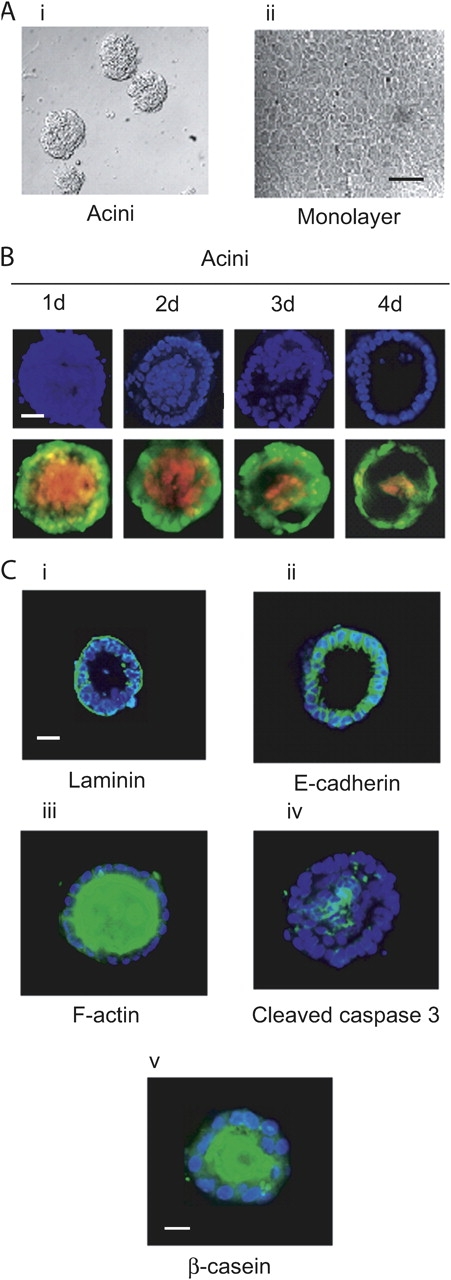
Mammary epithelial cells cultured on concentrated EHS ECM form functional acini. (A) Phase-contrast microscopy of primary mouse mammary epithelial cells cultured on concentrated EHS ECM (i) and on EHS ECM diluted (1:1.4) (ii) for 4 d (10X). Bar, 50 μm. (B) Time course of acinus formation from dispersed cells (1–4 d), fluorescence microscopic analysis (staining: DAPI; and Calcein AM and EtBr; 40X). Bar, 20 μm. (C) Fluorescence microscopic analysis of 4 d acini stained with anti–laminin V (i), anti–E-cadherin antibody (ii), phalloidin-FITC (iii), anticleaved caspase 3 (iv; all 40X; bar, 20 μm) and anti–β-casein antibody (v; 20X; bar, 10 μm).
Glucocorticoids are required for formation of mammary epithelial acini
In contrast to the norm, omitting hydrocortisone from the cultures resulted, after 4 d, in more disordered assemblies showing less defined cell–cell association with incomplete lumen formation (Fig. 2 A, compare DAPI in rows 1 and 3 with DAPI in rows 2 and 4). In addition, polarized lateral β-catenin distribution and basal laminin association was lost (Fig. 2 A, compare β-catenin and Laminin in rows 1 and 3 with β-catenin and Laminin in rows 2 and 4).
Figure 2.
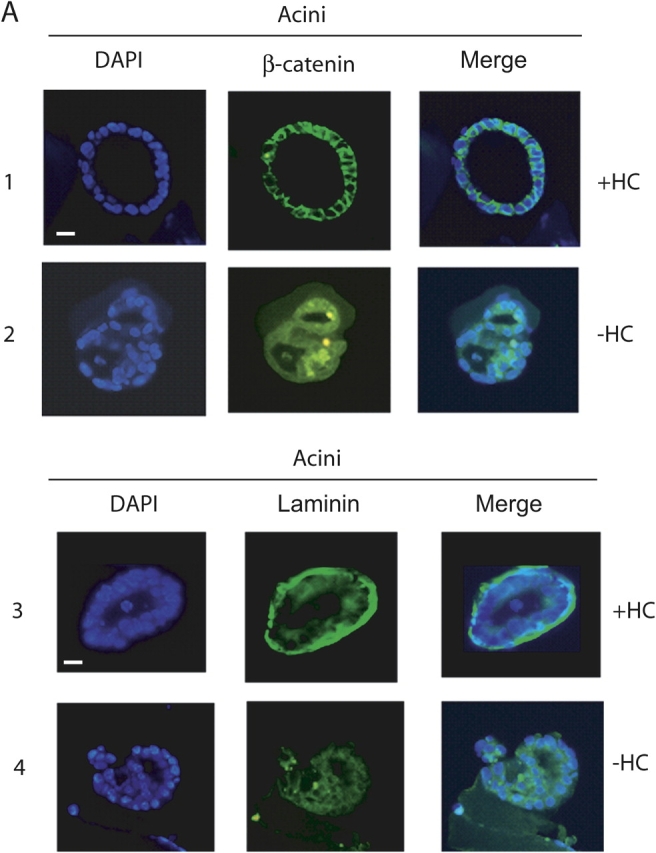

Generation of acini requires glucocorticoids. (A) Fluorescence microscopic analysis of 4 d acini stained with DAPI (left), anti–β-catenin antibody (middle) and merged image (right) cultured from time of seeding in the presence (+HC; row 1) and absence (−HC; row 2) of hydrocortisone (all 40X). Bar, 10 μm. Rows 3 and 4, same as above but stained with DAPI (left), anti–laminin V antibody (middle), and merged image (right). (B) RT-PCR analysis of total RNA isolated from 4 d acini cultured in the presence and absence of hydrocortisone for cyclin D1 and p21 transcripts (i); and 2 and 4 d acini and 2 d acini cultured in the absence of hydrocortisone (2d-HC) for occludin, ZO1, ZO2, and ZO3 transcripts (ii). GAPDH mRNA levels were measured as a control for equal input amounts of RNA. No reverse transcriptase (RT) added controls, lanes (−), show absence of DNA contamination. (iii) Western analyses of ZO1 and occludin in 2 and 4 d acini and in acini cultured in the presence of RU486 (50 μM; RU). β-Actin was measured as a loading control. (C) EMSA analysis, using a 32P-labeled NFκB-DNA binding element, performed on whole cell extracts from mammary epithelial cells at harvest (H, lane 1), cells maintained as monolayers on plastic (M, lane 2), cells cultured on concentrated EHS ECM (acini) for 2 and 4 d (lanes 3 and 4), monolayers on dilute EHS ECM matrix (lanes 5 and 6), monolayers on plastic overlaid with the EHS ECM matrix (lanes 7 and 8), and acini cultured in the presence of (+) and absence of (−HC) hydrocortisone (lanes 9 and 10). Arrowheads indicate NFκB complexes. PNFκB indicates binding element. In this and subsequent figures all discontinuities in gel images are marked with a vertical line. (D) EMSA analysis, as C, but using 32P-labeled AP1-DNA binding element. Light colored arrowheads indicate acinus-associated complexes. (E) Western analysis of phospho(ser63)-c-Jun (i), total c-Jun (ii), IP (JunD)/Western (anti–phospho-serine) (iii), and IP (JunD)/Western (JunD) (iv), in 2 and 4 d acini and in acini cultured in the presence of RU486 (50 μM; RU).
Glucocorticoids were also required for maintenance of p21 RNA transcript expression in the acini (Fig. 2 B, i). p21 expression, which had been correlated with functional organization of mammary epithelial acini (Weaver et al., 1997), was apparent in 4 d acini, whereas cyclin D1 expression was reciprocally lost. This pattern was reversed in the absence of hydrocortisone (Fig. 2 B, i). Cyclin D1 is selectively required for successful mammary alveolar epithelial cell proliferation in pregnancy and its expression is positively regulated by nuclear factor kB (NFκB; Guttridge et al., 1999; Cao et al., 2001). Acinar expression of the tight junction protein transcripts, occludin, ZO1 and ZO3, was also glucocorticoid dependent (Fig. 2 B, ii). ZO2 expression was constitutive. In addition, ZO1 and occludin protein expression was glucocorticoid dependent (Fig. 2 B, iii), in this context.
Lastly, organization of the cells into acini had an associated transcription factor expression profile: it caused loss of NFκB-DNA binding activity which was not seen in epithelial cell monolayers formed on dilute EHS ECM, or in monolayers overlaid with matrix or monolayers cultured on plastic (Fig. 2 C, compare lanes 3 and 4 with lanes 5 and 6; compare lanes 7 and 8 with lane 2). In addition, acinar extracts contained AP1 DNA binding complexes not seen in the monolayer cultures (Fig. 2 D, compare lanes 3 and 4 with 5 and 6; compare lanes 7 and 8 with lane 2). Omission of glucocorticoids from the acinar cultures led to the loss of this profile: NFκB was abundant in the absence of glucocorticoids and different AP1 DNA complexes detected (Fig. 2, C and D, compare lane 9 with lane 10). The AP1 and NFκB complexes were shown to be DNA binding site specific as excess of unlabeled binding element DNA competed away the complexes, whereas nonspecific competitor oligonucleotides did not (unpublished data). Retention of active NFκB in glucocorticoid-minus acini correlated with loss of IκBα and IKKα activation (unpublished data).
The AP1 transcription factor complexes detected should most likely contain c-Jun. Total c-Jun levels remained constant in the acini and were unaffected by glucocorticoid receptor inhibition (Fig. 2 E, ii). However, c-Jun was phosphorylated on a JNK substrate site (ser63) in a glucocorticoid-dependent manner (Fig. 2 E, i). Because the total c-Jun band runs at ∼43 kD and the phospho(ser63)-c-Jun at 48 kD we conclude that the c-Jun phosphorylated on ser63 represents only a part of the total cellular c-Jun, a “phosphorylated” pool (Binetruy et al., 1991). In contrast, levels of phospho-JunD and JunD were not consistently affected by RU486 treatment (Fig. 2 E, iii and iv), nor were JunB and phospho-JunB (not depicted). That the AP1 complexes might contain c-Jun phosphorylated by JNK is supported by our later identifying JNK activation as being glucocorticoid dependent in the acini (see next section).
JNK activity is required for mammary epithelial acinus formation
Acini generated in the presence of the specific JNK inhibitor, SP600125 (Bennett et al., 2001), failed to clear their lumena and to exhibit polarized F-actin, E-cadherin, β-catenin, β4 integrin, and ZO-1 distribution (Fig. 3 A, compare left column with middle column). The glucocorticoid receptor antagonist, RU486, had a similar effect (Fig. 3 A, i, right column). That this effect is specific to MAPK–JNK pathway activity is suggested by the fact that 2 d acini forming in the presence of the ERK–MAPK pathway inhibitor (PD98059) or p38 inhibitor (SB230580) show normal vital dye distribution (Fig. 3 A, ii) in contrast to acini formed in the presence of SP600125 which show a disturbed EtBr staining.
Figure 3.
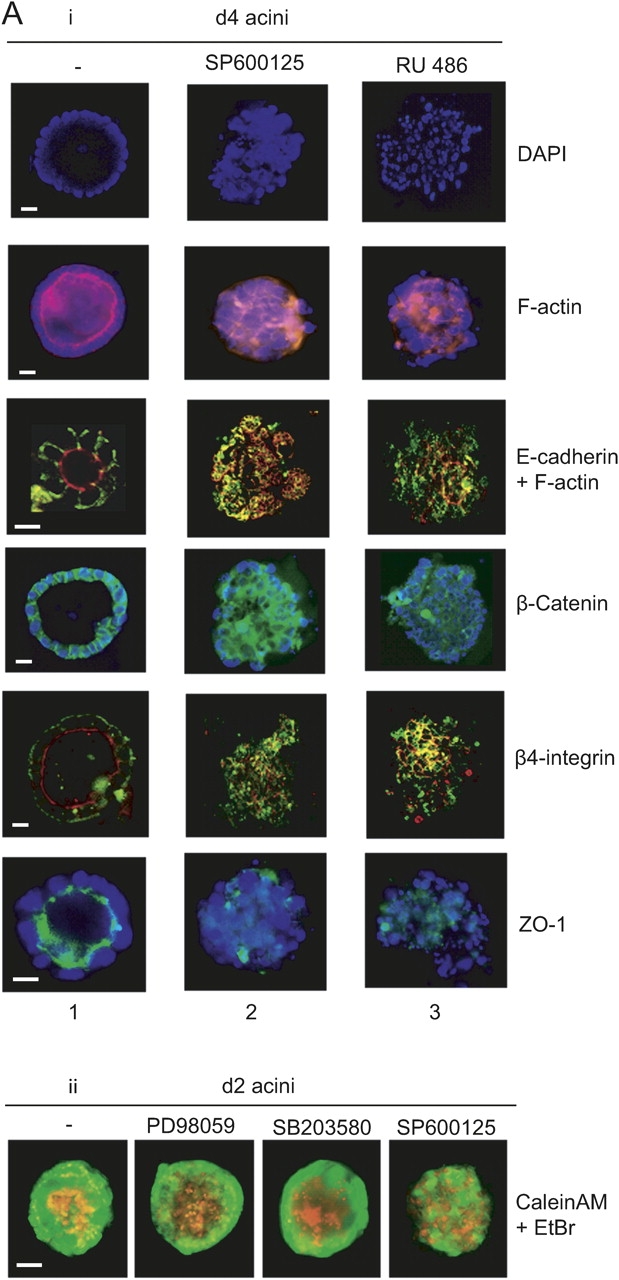

Acinus formation requires JNK activity. (A, i) Fluorescence microscopic analysis of 4 d acini cultured in the absence (−) or presence of SP600125 (50 μM) or RU486 (50 μM); staining with DAPI (top), rhodamine-phalloidin and DAPI (second from top; both 40X; bar, 10 μm), anti–E-cadherin (FITC) and rhodamine-phalloidin (third from top; 20X; bar, 20 μm), anti–β-catenin and DAPI (fourth from top), anti–β4-integrin (FITC) and rhodamine-phalloidin (second from bottom; both 40X; bar, 10 μm) and anti-ZO1 and DAPI (bottom; 20X; bar, 20 μm). (ii) 2 d acini formed in presence of PD98059 (10 μM), SB230580 (1 μM), and SP600125 (50 μM) stained with Calcein AM and EtBr. Bar, 10 μm. (B) EMSA analysis, using AP1 (left) and NFκB (right) binding elements, on extracts from acini treated with the JNK inhibitor, SP600125 (50 μM), for 1, 4, and 24 h. (C) Western analysis of phospho(thr183/tyr185)-JNK and JNK in extracts from cells at harvest (H), cells maintained as a monolayer on plastic (M), 2 and 4 d acini, and 2 d acini in the absence of hydrocortisone (2d-HC). (D) Western analysis of phospho(ser63)- c-Jun (i), c-Jun (ii), phospho(thr183/tyr185)-JNK (iii), JNK (iv), phospho(thr202/tyr204)-ERK (v), and ERK (vi) in 2 and 4 d acini and 2 d acini cultured in the presence of SP600125.
Inhibition of JNK activity with SP600125 also transiently impaired expression of the acinus-associated AP1 complex and this was accompanied by the transient reappearance of a slower migrating AP1 complex (Fig. 3 B, i). In addition, it caused a transient maintenance of the NFκB complex in acini formed in its presence (Fig. 3 B, ii). We do not presently know why these changes are only transient in the acini and not longer lasting.
While JNK inhibition impaired acinus formation, JNK phosphorylation on p-thr183 and p-tyr185 (which leads to its activation) was detected in normal acini (Fig. 3 C) but not in acini generated in the absence of hydrocortisone (or in mammary epithelial cells maintained as monolayers on plastic; Fig. 3 C, lanes 2d-HC and M, respectively). SP600125 also inhibited phosphorylation of c-Jun on the JNK substrate site (ser63; Fig. 3 D, i) but did not inhibit acinus-associated activation of JNK (phosphorylation on thr183/tyr185; Fig. 3 D, iii) nor ERK phosphorylation (Fig. 3 D, v). Thus, a degree of specificity of action of SP600125 in this context is evident.
Glucocorticoids induce BRCA1 and growth arrest and DNA damage inducible (GADD45) β expression and activate JNK in acini
Rajan et al. (1996) observed glucocorticoid-dependent BRCA1 expression in differentiated HC11 mammary epithelial cells and Harkin et al. (1999) showed that BRCA1 overexpression induced the DNA damage-associated regulator, GADD45β, and subsequent JNK activation to trigger apoptosis in a cultured mammary tumor cell line. GADD45 proteins associate with multiple cellular proteins, including proliferation cell nuclear antigen (Smith et al., 1994), p21 (Kearsey et al., 1995), and the MAPK kinase kinase, MEKK4 (Takekawa and Saito, 1998). Thus, glucocorticoid-dependent BRCA1 and GADD45β expression was examined: BRCA1 mRNA was found in 2 d acini but was not detectable in mammary epithelial cells cultured as a monolayer on plastic or in 2 d acini formed in the absence of hydrocortisone (Fig. 4 A, i). BRCA1 protein expression was found, in parallel, in 2 d acini and again its presence was hydrocortisone dependent (Fig. 4 A, ii). GADD45β (mRNA and protein) expression was similar and was also glucocorticoid dependent (Fig. 4 B).
Figure 4.

Glucocorticoids support BRCA1 and GADD45β expression and GADD45β–MEKK4 complex formation in acini. (A, i) RT-PCR analysis of BRCA1 mRNA levels in cells maintained on plastic under proliferating conditions (M), in 2 and 4 d acini and acini cultured for 2 d in the absence of hydrocortisone (2d-HC). GAPDH and no RT added controls as Fig. 2 B, i. (ii) Western analysis of BRCA1 in cells at harvest (H), cells cultured as a monolayer on plastic (M), 2 and 4 d acini, and acini cultured for 2 d in the absence of hydrocortisone (2d-HC). (B, i) RT-PCR analysis of GADD45β mRNA levels (A, i). (ii) Western analysis of GADD45β levels (A, ii). (C, i) Diagram: release of MEKK4 autoinhibition by GADD45β binding (Mita et al., 2002). (ii) Western analysis of MEKK4 levels (A, ii). (iii) Analysis of MEKK4–GADD45β complexes. Complexes were immunoprecipitated from cell extracts with anti-MEKK4 at harvest (H), from cells maintained on plastic (M), 2 and 4 d acini, and acini cultured for 2 d in the absence of hydrocortisone (2d-HC); followed by anti-GADD45β immunoblot. (D) JNK activity assay: JNK was harvested onto GST-c-Jun (amino acids 1–89) beads from extracts of cells at harvest (H), from cells maintained on plastic under proliferating conditions (M), 2 and 4 d acini, and acini cultured for 2 d in the absence of hydrocortisone (2d-HC). Shown is the anti–phospho-c-Jun immunoblot performed after incubation of the JNK–GST-c-Jun bead complexes with ATP. Cell cultures treated with (+) and without (−) pervanadate (25 mM) were used as a positive and negative control, respectively.
Activation of JNK is catalyzed by the MAPK-kinase, MKK-4/SEK1 (Sanchez et al., 1994), whose activity is in turn regulated by MEKK4 (Gerwins et al., 1997). Mita et al. (2002) have shown that MEKK4 is activated by alleviating its autoinhibition. This occurs on formation of GADD45β–MEKK4 complexes (Fig. 4 C, i). MEKK4 was expressed in 2 and 4 d acini (Fig. 4 C, ii). Formation of GADD45β–MEKK4 complexes was analyzed by immunoprecipitation with an anti-MEKK4 antibody and detection of GADD45β by immunoblotting. GADD45β–MEKK4 complexes were generated in the 2 and 4 d acini in a hydrocortisone-dependent manner (Fig. 4 C, iii). These complexes were not detectable in monolayers cultured on plastic (Fig. 4 C, lane M). This formation of MEKK4–GADD45β complexes potentially links GADD45β to MEKK4-JNK signaling, under these conditions.
Downstream activation of the MEKK4 substrate, MKK4, was detected indirectly by demonstrating the induction of JNK activity. Immobilized GST-c-Jun (amino acids 1–89) was used to bind activated JNK. After ATP addition and incubation in kinase buffer, Western blot analysis with an anti–phospho-c-Jun (ser63; JNK target) specific antibody was performed. c-Jun phosphorylation was detected in acini in a hydrocortisone-dependent manner (Fig. 4 D). Cultures of monolayers of these cells on plastic with or without pervanadate served as positive and negative controls for this assay (Fig. 4 D, lanes + and −). Thus, JNK phosphorylation at MKK4-target sites, thr183/tyr185 (Fig. 3 C), inducing JNK activation (reflected by phosphorylation of GST-c-Jun), was detected in the acini and was glucocorticoid dependent.
To confirm that BRCA1 expression was activated by glucocorticoids upstream of JNK activation, it was shown by RT-PCR analysis that RU486 treatment blocked BRCA1 transcript expression but that SP600125 had no effect (Fig. 5 A, i). BRCA1 regulates GADD45β transcription through its interaction with transcription factors of the OCT family. Disruptions in the octamer/OCT-1 binding region abolish activation of the GADD45β promoter by BRCA1 (Fan et al., 2002). To test whether BRCA1 was recruited to the GADD45β promoter in acini in a glucocorticoid-dependent manner chromatin immunoprecipitation (ChIP) analysis (Boyd and Farnham, 1999) was performed. BRCA1 was found to be associated in situ with the GADD45β promoter, in 2 d acini, by PCR detection of GADD45β promoter fragments (encompassing the OCT binding element) after anti-BRCA1 antibody ChIP (Fig. 5 A, ii). BRCA1 was not associated with the GADD45β promoter in acini generated in the presence of RU486. In addition, the association of BRCA1 with another of its established promoter targets, the p21 promoter (Somasundaram et al., 1997), was also found to be glucocorticoid dependent (Fig. 5 A, iii).
Figure 5.
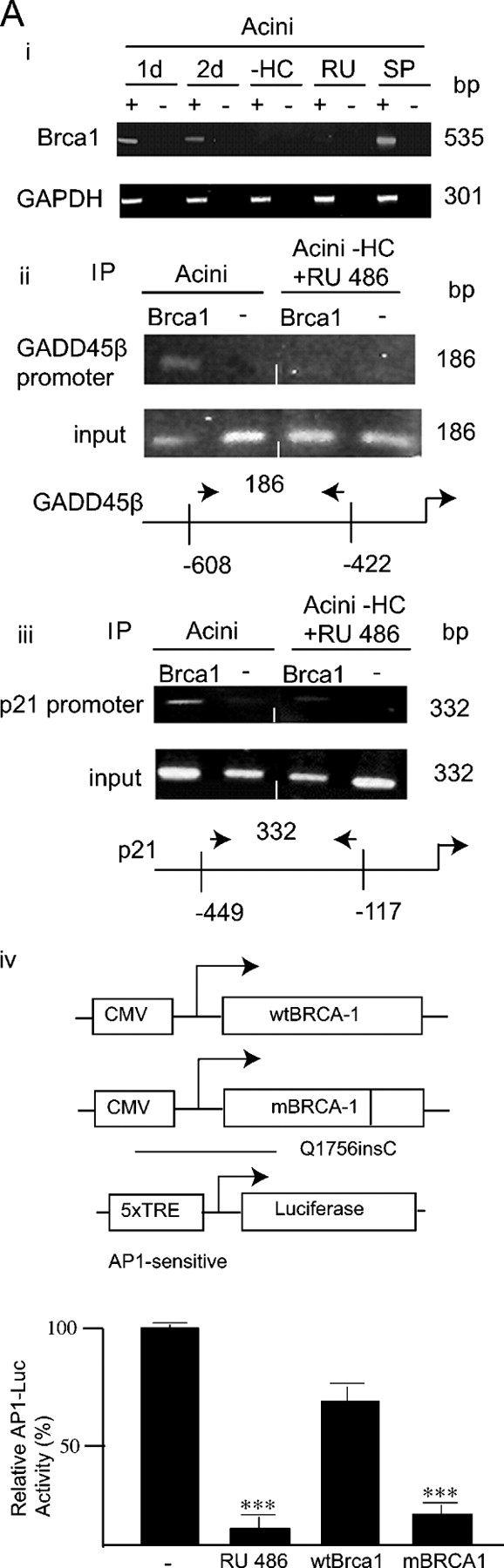
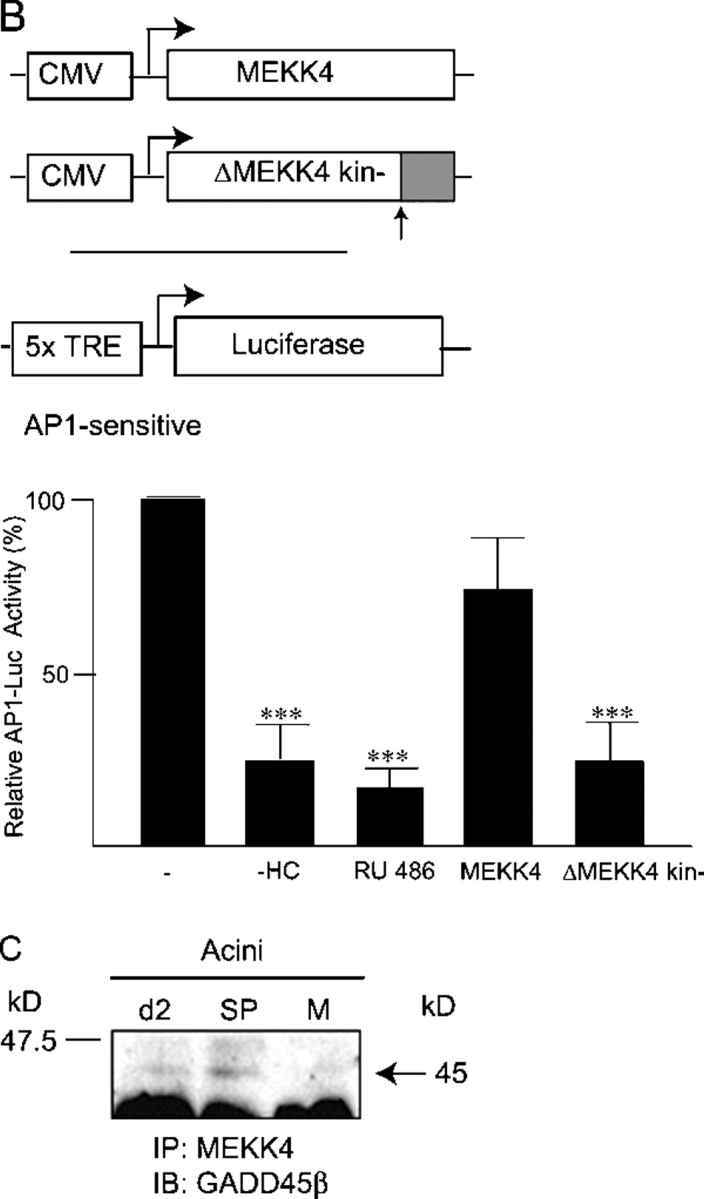
Analysis of glucocorticoid signaling in acini. (A, i) RT-PCR analysis of BRCA1 mRNA levels in acini cultured for 1 and 2 d, acini formed for 2 d in the absence of hydrocortisone (2d-HC), and 2 d acini cultured in the presence of RU486 and SP600125 (Fig. 2 B, i). (ii and iii) ChIP analysis of BRCA1 occupation of the GADD45β or p21 promoters in acini cultured for 2 d in the presence and absence of glucocorticoid receptor function (−HC + RU486). IP was with anti-BRCA1 antibody (+) or no antibody (−). After reversal of cross-linking, DNA recovered from the chromatin was analyzed by PCR using primers specific for the GADD45β (ii) and p21 (iii) promoters (see diagrams). Promoter DNA purified from sheared chromatin “input” to the IP is shown as control. (iv) Relative luciferase activity in extracts of acini formed from cells 1 d after suspension transfection with the pAP1-luciferase (1.0 μg) (−), transfected and treated with RU486, cotransfected with pCR3-BRCA1 (1.0 μg) (wtBRCA1) and cotransfected with the BRCA1 mutant expression vector, pCR3-BRCA1-Q1756insC (1.0 μg) (mBRCA1). ***, indicates value to be significantly (P < 0.05; n = 3; t test) less than lane (−). (B) Relative luciferase activity in extracts of acini formed from cells 1 d after suspension transfection with the pAP1-luciferase (1.0 μg) (−), transfected in the absence of hydrocortisone (−HC) or treated with RU486, cotransfected with pCMV5-MEKK4 (1.0 μg) and cotransfected with the MEKK4 dominant-negative mutant expression vector, pCMV5-ΔMEKK4 kin− (1.0 μg). ***, same as A iv, above. (C) Analysis of MEKK4–GADD45β complexes. Complexes were immunoprecipitated with anti-MEKK4 antibody from cell extracts from 2 d acini, 2 d acini cultured in the presence of SP600125 (+SP) and cells maintained as a monolayer on dilute EHS ECM (M) and followed by anti-GADD45β immunoblot.
We had observed that differentiation of mammary epithelial cells (HC11) with dexamethasone and prolactin triggered expression from an AP1 sensitive promoter-reporter system (unpublished data). Expression from a transiently transfected AP1-dependent promoter-reporter (luciferase) construct, in the acini, was also glucocorticoid dependent (Fig. 5 A, iv and Fig. 5 B): it was reduced by RU486 (Fig. 5 A, iv and Fig. 5 B) or in the absence of hydrocortisone (Fig. 5 B). Cotransfection with an expression vector encoding functional BRCA1 had no significant affect on AP1 reporter gene expression but cotransfection with an expression vector encoding a naturally occurring “dominant-negative” BRCA1 mutant, Q1756insC (Somasundaram et al., 1997), significantly decreased AP1-dependent luciferase activity (Fig. 5 A, iv). The Q1756insC mutation is an insertion of a cytosine in codon 1756 (in exon 20) that induces a frameshift and disables the transcriptional activating capacity of BRCA1 that resides in the COOH terminus (Monteiro et al., 1996). Thus, expressing a transactivating defective BRCA1 impeded a glucocorticoid-dependent output in the forming acini.
Expressing a dominant-negative, kinase defective MEKK4 had a similar effect (Fig. 5 B): expression of functional MEKK4 did not significantly affect reporter gene expression but cotransfection of an expression vector encoding a dominant-negative MEKK4 (Gerwins et al., 1997) significantly repressed AP1-dependent luciferase activity. This functionally links a requirement for the potential to activate the MEKK4 signaling pathway to a glucocorticoid action in the acini. Finally, it was shown that the GADD 45β–MEKK4 complex was generated in the presence of SP600125 (Fig. 5 C) demonstrating that activation of JNK occurred downstream of GADD45β association with MEKK4 in the acini.
β4 integrin expression: a downstream target of the glucocorticoid and JNK action
Potapova et al. (2002) reported that β4 integrin expression is regulated by JNK signaling in human PC3 prostate carcinoma cells. Because β4 integrin expression has been documented to be vital for acinus formation (Weaver et al., 2002), the possibility that it is a downstream target of the glucocorticoid and JNK signaling was examined. β4 integrin mRNA was not detected in RU486 treated 2 d acini and its levels were reduced on SP600125 treatment (Fig. 6 A, i). β4 integrin protein levels were also decreased in 2 d acini treated with SP600125 and undetectable with RU486 treatment (Fig. 6 A, ii). The mouse β4 integrin promoter contains multiple AP1 DNA binding elements. Using ChIP analysis, in situ occupation of the β4 integrin promoter in 2 d acini by c-Jun and c-Fos was demonstrated and this occupation was glucocorticoid receptor and JNK dependent (Fig. 6, B and C). This suggests that maintenance of β4 integrin expression is a potential target of glucocorticoid and JNK signaling in the developing acini.
Figure 6.

β4 integrin expression, a target for glucocorticoid and JNK regulation. (A, i) RT-PCR analysis of β4 integrin RNA in 2 d acini (−) and 2 d acini treated with RU486 (RU) and SP600125 (SP) (Fig. 2 B, i). (ii) Western analysis, using anti–β4 integrin antibody, of extracts from 2 and 4 d acini generated in the absence and presence of RU486 (RU) and SP600125 (SP). β-Actin expression was measured as loading control. (B and C) ChIP analysis of c-Jun and c-Fos occupation of the β4 integrin promoter (see diagram) in acini cultured for 2 d in the presence and absence of RU486 and SP600125. IP was with anti–c-Jun (B) or anti-Fos antibody (C) (Fig. 5 A, ii).
BRCA1, GADD45β and phospho-JNK expression in the pregnancy/lactation transition in the mouse mammary gland
Mouse mammary acini are in vitro alveolar-like structures that functionally mimic the differentiated epithelial alveoli of the early lactating mammary gland. It was of interest to see if selective BRCA1, GADD45β, and phospho-JNK expression was associated with the pregnancy/lactation transition in the mammary gland, in vivo. Western blot analysis of cell extracts showed that BRCA1 and GADD45β were not detectable in whole cell extracts from late pregnancy mammary gland but expression of these proteins in cell extracts was detectable on day 1 of lactation (Fig. 7 A, top and second from top). JNK phosphorylation, reflecting the activation of JNK–MAPK pathway, had a slightly broader expression range. It was detected in late pregnancy (day 18, but not on day 16), and up to 3 d into lactation (Fig. 7 A, third from top). Finally, using ChIP analysis, it could be seen that BRCA1 is recruited to the GADD45β and p21 promoter primarily in the early lactating mammary gland (Fig. 7 B). These results support the possibility that JNK signaling is activated in the transition from pregnancy to lactation in the mouse mammary gland, in vivo.
Figure 7.
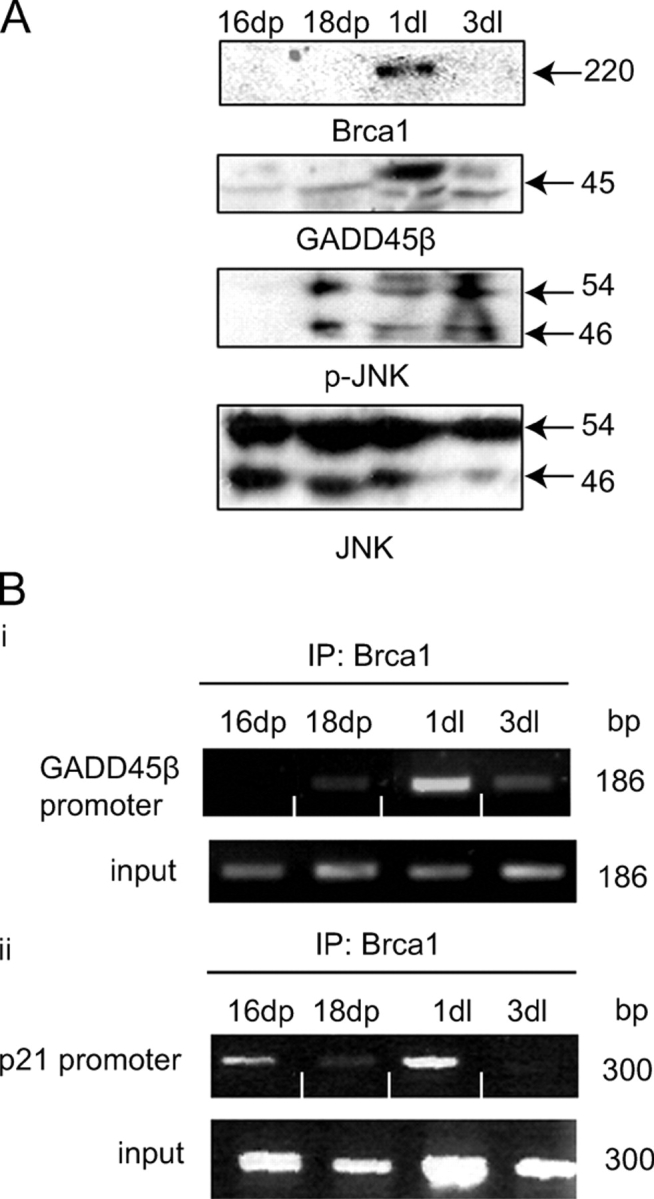
BRCA1, GADD45β, and phospho-JNK expression in pregnancy/lactation transition in the mouse mammary gland. (A) Western analysis of whole cell extracts from 16- and 18-d pregnant (16dp and 18dp) and 1- and 3-d lactating (1dl and 3dl) mammary glands. Analysis was performed using anti-BRCA1 (top), anti-GADD45β (second from top), anti–phospho-JNK (third from top) and anti-JNK antibodies (bottom). (B) ChIP analysis of BRCA1 occupation of the GADD45β or p21 promoters (Fig. 5 A, ii and iii) in 16- and 18-d pregnant and 1- and 3-d lactating mammary gland. IP was with anti-BRCA1 antibody (Fig. 5 A, ii).
Discussion
Debnath et al. (2002) suggest that a primary event in acinus formation is establishment of epithelial cell polarity and that this triggers the restriction in cell proliferation and the apoptotic activity that are required for acinus maturation and lumen formation. The establishment of polarity in epithelial cells most probably goes hand-in-hand with the development of cell–cell contacts through apical junctions (O'Brien et al., 2002). Firestone and coworkers (Buse et al., 1995; Wong et al., 1999; Woo et al., 1999; Rubenstein et al., 2003) suggest that in mammary tumor cell monolayers, glucocorticoids induce tight junction formation and cell polarity through a multi-step cascade involving early rapid stimulation of the transcriptional regulator, Id-1, and later induced recruitment of tight junction proteins, adherens junction proteins and Ras and PI3 kinase signaling proteins to the sites of cell–cell contact (Woo et al., 1999). This parallels necessary down-regulation of fascin, an adherens junction-associating actin-bundling protein (Wong et al., 1999). This work suggests that glucocorticoids act to support acinus formation and may do this by regulating the expression of proteins required for cell–cell (ZO-1 and occludin) and ECM–cell contacts (β4 integrin).
Acinus formation was accompanied by JNK phosphorylation (Fig. 3 C) and activation (Fig. 4 D) and this effect was lost when glucocorticoid receptor function was inhibited. However, there remains the possibility that loss of glucocorticoid receptor function primarily undermines morphological activities such as establishment of cell–cell junctions, signaling from which might support JNK activation. Thus, there is the possibility that the glucocorticoid-modulated activation of JNK is indirect. Inhibition of JNK activity with SP600125 had similar effects to inhibiting glucocorticoid receptor function both on acinus morphology and transcription factor binding activity. But note must be taken that SP600125 has been shown to act with similar potency on a range of other kinases in the studies of Bain et al. (2003). However, it was the only one of the three inhibitors of Erk, p38, and JNK–MAPK pathway activities, respectively, to inhibit acinus formation in this work (Fig. 3 A, ii).
BRCA1 and GADD45β expression were induced during acinus maturation (Fig. 4) and the absence of BRCA1 transcripts in acini treated with RU486 but their presence on SP600125 treatment (Fig. 5 A, i) defined the induction of BRCA1 expression as upstream of JNK. Also, using ChIP analysis, the in situ association of BRCA1 with the GADD 45β promoter in a glucocorticoid-dependent manner was demonstrated. The formation of GADD45β–MEKK4 complexes, which is reported to relieve autoinhibition of the MEKK4 kinase and permit JNK-pathway activation (Mita et al., 2002), occurred in the acini and was glucocorticoid dependent (Fig. 4 C, iii). The detection of their presence established a potential physical link between the glucocorticoid induction of BRCA1 and GADD45β expression and JNK pathway signaling in the acini. This is supported by the inhibition of glucocorticoid-dependent activation of a transfected AP1-dependent promoter-reporter system by coexpression of a dominant-negative BRCA1 protein (Fig. 5 A, iv) and a kinase-defective dominant-negative MEKK4 (Fig. 5 B), respectively. This leaves open the possibility that glucocorticoids signal to activate JNK through modulation of BRCA1 and GADD45β action during acinus formation. The late pregnancy/early lactation transition is accompanied by “closure” of epithelial cell–cell junctions to facilitate efficient transfer of milk constituents to the alveolar lumina (Nguyen et al., 2001). Lactation is associated with high circulating glucocorticoid levels (Feng et al., 1995) and it was of interest that selective BRCA1, GADD45β, and phospho-JNK expression could be detected at this transition (Fig. 7).
A target of the glucocorticoid and JNK signaling was activation of c-Jun and associated AP1 activation. c-Jun was phosphorylated on serine-63, a JNK substrate site, in the acini in a glucocorticoid-dependent manner (Fig. 2 E) and AP1 activity, as judged by reporter gene expression, was maintained in a glucocorticoid, BRCA1, and MEKK4-dependent manner (Fig. 5 A, iv; Fig. 5 B). In addition, glucocorticoid-dependent in situ occupation of the “target” β4 integrin promoter by c-Jun and Fos was seen (Fig. 6). Such indirect regulation of AP1 activity by glucocorticoids was unexpected. The direct interactions of activated glucocorticoid receptor with AP1 that have been reported are DNA-binding independent and inhibitory (for review see Karin and Chang, 2001).
The significant failure of mammary gland development during pregnancy that is seen in BRCA1 conditional knockout mice is suggested to reflect cell loss arising from failure of DNA damage repair (Xu et al., 1999). Apart from direct actions in DNA damage repair complexes (for review see Venkitaraman, 2001), BRCA1 also induces a wide range of transcriptional responses (Harkin et al., 1999; Fan et al., 2002). The induction by BRCA1 of the expression of the DNA damage repair associated regulator, GADD45β, has been linked experimentally to activation of JNK and apoptosis of breast and ovarian tumor cell lines (Harkin et al., 1999; Thangaraju et al., 2000). However, it seems unlikely that the only outcome of inducing JNK activation in these acini would be the induction of apoptosis. This work suggests that JNK signaling is required for establishment or maintenance of acinar cell function but it may also trigger apoptosis in those lumenal cells not supported by ECM association. Glucocorticoids, and perhaps BRCA1, independently of JNK activation, would also seem implicated in the cell cycle arrest that accompanies acinus maturation. An induction of p21 and suppression of cyclin D1 expression accompanies acinus formation and this is glucocorticoid dependent (Fig. 2 B). In addition, BRCA1 occupation of the p21 promoter, in situ, is glucocorticoid dependent, but it may not be JNK dependent. BRCA1 regulation of p21 promoter function has been reported previously (Somasundaram et al., 1997).
Debnath et al. (2002) emphasize the co-coordinated requirement of both antiproliferative and apoptotic activities, triggered by cell polarization, to achieve lumen formation in mammary epithelial acini. The work of Weaver et al. (2002) demonstrates how these structures are resistant to apoptotic insults to which some mammary tumor cells or mammary epithelial cells organized as monolayers readily succumb. This work suggests that glucocorticoids and JNK activity are required to support mammary epithelial cells to organize into functional acini. In the immediate future it will be of particular interest to identify further downstream targets of glucocorticoid and JNK signaling in acini. That β4 integrin, possibly through AP1, and ZO-1 and occludin expression is regulated by glucocorticoids, suggests a role in maintaining cell–ECM and cell–cell component expression that are essential for acinus viability. Typically mammary tumor cells fail to organize as acini (Petersen et al., 1992; Howlett et al., 1995). It will be of interest to assess responsiveness to glucocorticoids and JNK signaling in such cells.
Materials and methods
Cell culture
Mouse mammary epithelial cells were harvested from mid- to late-pregnant CD-1 mice as described previously (Furlong et al., 1996). Cells were seeded on tissue culture dishes coated with concentrated, growth factor depleted, EHS ECM (Matrigel®; BD Biosciences; 1.74 mg protein/6 cm2 plate; or diluted 1:1.4 with F12 medium) at a density of 2.4 × 106 cells/ml and cultured for 48 h with 5 ng/ml epidermal growth factor (Promega), 5 μg/ml insulin (Sigma-Aldrich), 1 μg/ml hydrocortisone (Sigma-Aldrich), and 50 μg/ml gentamycin (Sigma-Aldrich) in F12 medium (Life Technologies, Inc.). Cells cultured on EHS ECM were washed, and the medium was changed to: F12, 50 μg/ml gentamycin, 5 μg/ml insulin, 1 μg/ml hydrocortisone, and 3 μg/ml prolactin. These cells were harvested after 1–4 d in culture. Cells cultured on plastic were maintained for 72 h in the initial medium supplemented with 10% heat inactivated FCS (Life Technologies). Cells were recovered by scraping and were pelleted by centrifugation. The cell pellets were snap frozen and stored at −80°C before RNA or protein extract preparation. 50 μM RU486 (Sigma-Aldrich), 50 μM SP600125 (Calbiochem), 10 μM PD98059 (Calbiochem), and 1 μM SB230580 (Calbiochem) were added to cells cultured on EHS ECM at the time of plating.
Cell extract preparation, Western blot analysis, immunoprecipitation, kinase assays, and electrophoretic mobility shift analysis (EMSA)
Whole cell extracts were prepared by resuspending pelleted cells in 50–100 μl of extraction buffer (Furlong et al., 1996). The samples were kept on ice for 30 min before centrifugation at 14,000 rpm at 4°C for 20 min. The supernatant constituted the whole cell extract.
Western blot analysis was performed as described previously (Furlong et al., 1996). Antibodies against BRCA1 (Upstate Biotechnology), occludin (BD Transduction Laboratories), β4 integrin, and ZO1 (CHEMICON International, Inc.), GADD45β, MEKK4, and JunD (Santa Cruz Biotechnology, Inc.), ERK, phospho-ERK, c-Jun, phospho-c-Jun, JNK, and phospho-JNK (Cell Signaling Technology), and phospho-serine (Calbiochem), were used according to the manufacturer's recommendations.
For immunoprecipitation-Western analysis, cell pellets were resuspended in 250 μl of NP-40 lysis buffer (150 mM NaCl, 1% NP-40, 50 mM Tris-HCl, pH 8.0) and allowed settle on ice for 30 min before centrifugation at 14,000 rpm at 4°C for 20 min. The resultant supernatant was incubated with 1 μl of anti-MEKK4 or anti-JunD antibody for 1 h on ice. 100 μl of protein A bead suspension (Sigma-Aldrich) was then added to the antibody–antigen reaction and incubated at 4°C overnight, with rotation. The protein A beads were pelleted by brief centrifugation at 14,000 rpm for 15 s at 4°C and the immunocomplex was washed three times with lysis buffer. After removing the final wash, 50 μl of 2X Laemmli buffer was added and the samples boiled for 10 min. The samples were then centrifuged at 14,000 rpm for 2 min and the supernatant loaded on a 10% SDS PAGE gel and Western analysis for the appropriate antigen performed.
For the JNK activity assay, cell extracts were prepared as above and the assay was performed using a SAPK/JNK assay kit from Cell Signaling Technology.
EMSA was performed as described previously (Furlong et al., 1996) using 12 μg of protein extract and 15,000 cpm of 32P-end-labeled double stranded oligonucleotide transcription factor binding elements for AP1 (Marti et al., 1994) and NFκB (Geymayer and Doppler, 2000).
RT-PCR analysis
Cultured cells or cell assemblies were gently scraped from the plates, pelleted, washed by resuspension in PBS, pelleted, and resuspended in TRIzol® Reagent (Invitrogen). Reverse transcription and limited cycle PCR were performed as described previously (O'Connell and Martin, 2000). Primers were designed against murine β4 integrin (fwd, gctgagttggacttggaagc; rev, gtagtgtccttcgagcagcc), BRCA1 (fwd, tcggcgcttgcaagtacggatct; rev, aaggttagacagctggacaccta), cyclin D1 (fwd, agcagaagtgcgaagaggag; rev, ctggcattttggagaggaag), GADD45β (fwd, caccctgatccagtcgttct; rev, ttgcctctgctctcttcaca), GAPDH (fwd, accacagtccatgccatcac; rev, tccaccaccctgttgctgta), occludin (fwd, cctactcctccaatggcaaa; rev, aggtggatattccctgaccc), p21 (fwd, gtccaatcctggtgatgtcc; rev, ctcaggtagaccttgggcag), ZO1 (fwd, caaaacgctctacaggctcc; rev, gaagagctggacagaggtgg), ZO2 (fwd, tctcaagatcaacggcactg; rev, tgatcagaatactgctggcg), and ZO3 (fwd, accagctctcagaccaggaa; rev, ctgactgcctagcttcaccc) using Primer3 software (Rozen and Skaletsky, 2000).
Microscopy
For whole acinus microscopy, cells were cultured on concentrated EHS ECM in 2 well chamber slides (Lab Tek®). Medium was removed and the cell assemblies were fixed in situ using 4% PFA. Cells were permeabilized using 0.1% Triton X-100 for 10 min before blocking in 5% normal serum (appropriate species). Cells were incubated with primary antibodies (or phalloidin-FITC or -rhodamine; Molecular Probes) overnight at 4°C and then with FITC- or rhodamine-conjugated secondary antibodies for 20 min. The primary antibodies used were against: β4 integrin and ZO1 (CHEMICON International, Inc.) and E-cadherin (BD Transduction Laboratories). Nuclei were counterstained with DAPI (Sigma-Aldrich). For sectioning, cell assemblies were released from the ECM by gentle scraping, briefly centrifuged to pellet, and fixed in 10% formalin (Sigma-Aldrich) for at least 24 h before embedding in paraffin. 5-μm sections were cut. Sections were deparaffinized and treated for either immunoflourescence: sections were blocked in 5% normal serum and then incubated with primary antibodies against β-catenin (BD Transduction Laboratories), laminin V (Sigma-Aldrich), β-casein (a gift from E. Reichman, University of Zurich, Zurich, Switzerland), and cleaved caspase 3 (Cell Signaling Technology) overnight at 4°C. Sections were counterstained with DAPI (Sigma-Aldrich). For vital dye studies, acini were incubated with 10 μg/ml EtBr and 10 μM Calcein AM (Molecular Probes) for 10 min at 37°C. Phase contrast and fluorescence were visualized with an Axiocam HRC (Carl Zeiss MicroImaging, Inc.) mounted on a fluorescence microscope (model DMLB HBO100; Leica) and images were captured using Axiovision v3.1 software (Carl Zeiss MicroImaging, Inc.). Confocal microscopy was performed using a confocal microscope (model MRC 1024; Bio-Rad Laboratories) and images were captured using Laser Capture software.
Transfection and luciferase assays
Plasmids were transfected into freshly harvested primary mammary epithelial cells suspended in F12 medium, before plating onto EHS, using the FuGene 6 transfection system (Roche Molecular Biochemicals). Plasmids used were pAP1-luc (Stratagene), pCMV5-MEKK4, and pCMV5-ΔMEKK4 kin− (Gerwins et al., 1997; a gift from G. Johnson, University of Colorado Health Sciences Center, Denver, CO), and pCR3-BRCA1 and pCR3-BRCA1-Q1756insC (Somasundaram et al., 1997; a gift from B. Weber, University of Pennsylvania, Philadelphia, PA). The pRL-CMV, renilla luciferase reporter plasmid (Promega), was cotransfected and its activity used to correct for variation in transfection efficiency. After 24 h, cells were harvested, cell extracts prepared, and luciferase assays were performed using a Dual-Luciferase® Reporter Assay System (Promega). Statistical analysis was performed using the t test.
ChIP analysis
Cross-linking ChIP analysis (Boyd and Farnham, 1999) was performed using a ChIP assay kit purchased from Upstate Biotechnology. Mouse mammary glands were minced with a razor blade, brought up to 10.0 ml with PBS, and cross-linked for 10 min in a final volume of 1% formaldehyde. The cross-linking reaction was stopped by the addition of 0.125 M glycine, the glands pelleted by gentle centrifugation, washed with cold PBS, homogenized in an ULTRA-TURRAX® T8 disperser (IKA®), in 2.0 ml PBS, transferred to a 1.5 ml tube, centrifuged at 2,000 rpm for 10 min at 4°C, and the pellet resuspended in 200 μl SDS lysis buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris, pH 8.1, 16.7 mM NaCl, and 1 μg/ml each of aprotinin, leupeptin, and pepstatin A) on ice for 10 min. Acini were cross-linked on the cell culture plates by adding 135 μl 37% formaldehyde to the 5.0-ml medium and incubating at 37°C for 10 min. After glycine addition and washing with cold PBS, the cell assemblies were scraped from the plates in PBS into a 1.5-ml centrifuge tube and treated with lysis buffer (as above). Samples were sonicated on ice (setting 15, 3 × 10-s pulses; Ultrasonics sonicator) to reduce DNA length to 200–1,000 bp and centrifuged for 10 min at 14,000 rpm at 4°C. The supernatant was diluted 10-fold in ChIP buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris, pH 8.1, 16.7 mM NaCl, and 1 μg/ml each of aprotinin, leupeptin, and pepstatin A) and the chromatin precleared with salmon sperm DNA/protein A agarose (Upstate Biotechnology) for 30 min at 4°C with agitation. Precleared chromatin solution (1.0 ml) and 5 μl antibody (BRCA1; Upstate Biotechnology), p21 (Santa Cruz Biotechnology, Inc.), c-Fos (Oncogene), and c-Jun (Cell Signaling Technology) or no antibody (negative control) were incubated overnight at 4°C with rotation. Before immunoprecipitation, 50 μl of chromatin solution was saved (input chromatin) and was processed with the eluted immunoprecipitates beginning at the cross-link reversal step. The immunocomplexes were collected by adding 60 μl of salmon sperm DNA/protein A agarose and rotating for 1 h at 4°C. The beads were pelleted by brief centrifugation, washed with low salt complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCL, pH 8.1, 150 mM NaCl), high salt complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCL, pH 8.1, 500 mM NaCl), LiCl immunocomplex buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10mM Tris-HCl, pH 8.1) and TE, pH 8.0, with brief centrifugation between each wash to recover the beads. The immunocomplexes were eluted from the beads by the addition of 500 μl 1% SDS in 0.1 M NaHCO3 to pelleted beads, vortexing and rotating at RT for 30 min. To reverse the cross-linking process, 200 mM NaCl was added to the eluates which were incubated at 65°C for 4 h. 10 mM EDTA, 40 mM Tris-HCL, pH 6.5, and 2 μl of 10 mg/ml Proteinase K was then added and incubated at 45°C for 1 h. DNA was recovered by phenol/chloroform extraction and ethanol precipitation. Promoter sequences were detected in immunoprecipitated and input DNA by PCR using specific primers: GADD45β (fwd, gatgctagggtgccttggta; rev, cctagcctttcccaagctct), p21 (fwd, gtatgctgccacaaccacac; rev, ccatcaggccaatcaaaagt), and β4-integrin (fwd, tgaagtttgtcagcaggc; rev, ttcatgcgtatcagccat). Parallel ChIP analyses for each promoter was performed using an antiacetylated histone antibody as a positive control.
Acknowledgments
We thank Drs. G. Johnson, E. Reichman, and B. Weber for gifts of reagents.
This work was supported by the Health Research Board, Ireland and IRCSET/Enterprise Ireland.
J. Murtagh's current address is Dept. of Oncology, Montefiore Medical Center, 111 East 210th St., Bronx, NY 10467.
Abbreviations used in this paper: AP1, activator protein 1; ChIP, chromatin immunoprecipitation; EHS, Engelbreth-Holm-Swarm; EMSA, electrophoretic mobility shift analysis; EtBr, ethidium bromide; GADD45, growth arrest and DNA damage inducible; NFκB, nuclear factor κB.
References
- Bain, J., H. McLauchlan, M. Elliott, and P. Cohen. 2003. The specificities of protein kinase inhibitors: an update. Biochem. J. 371:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff, M.H., J. Aggeler, T.G. Ram, and M.J. Bissell. 1989. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development. 105:223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, B.L., D.T. Sasaki, B.W. Murray, E.C. O'Leary, S.T. Sakata, W. Xu, J.C. Leisten, A. Motiwala, S. Pierce, Y. Satoh, et al. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N- terminal kinase. Proc. Natl. Acad. Sci. USA. 98:13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binetruy, B., T. Smeal, and M. Karin. 1991. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature. 351:122–127. [DOI] [PubMed] [Google Scholar]
- Blatchford, D.R., L.H. Quarrie, E. Tonner, C. McCarthy, D.J. Flint, and C.J. Wilde. 1999. Influence of microenvironment on mammary epithelial cell survival in primary culture. J. Cell. Physiol. 181:304–311. [DOI] [PubMed] [Google Scholar]
- Boyd, K.E., and P.J. Farnham. 1999. Coexamination of site-specific transcription factor binding and promoter activity in living cells. Mol. Cell. Biol. 19:8393–8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buse, P., P.L. Woo, D.B. Alexander, A. Reza, and G.L. Firestone. 1995. Glucocorticoid-induced functional polarity of growth factor responsiveness regulates tight junction dynamics in transformed mammary epithelial tumor cells. J. Biol. Chem. 270:28223–28227. [DOI] [PubMed] [Google Scholar]
- Cao, Y., G. Bonizzi, T.N. Seagroves, F.R. Greten, R. Johnson, E.V. Schmidt, and M. Karin. 2001. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 107:763–775. [DOI] [PubMed] [Google Scholar]
- Debnath, J., K.R. Mills, N.L. Collins, M.J. Reginato, S.K. Muthuswamy, and J.S. Brugge. 2002. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 111:29–40. [DOI] [PubMed] [Google Scholar]
- Fan, W., S. Jin, T. Tong, H. Zhao, F. Fan, M.J. Antinore, B. Rajasekaran, M. Wu, and Q. Zhan. 2002. BRCA1 regulates GADD45 through its interactions with the OCT-1 and CAAT motifs. J. Biol. Chem. 277:8061–8067. [DOI] [PubMed] [Google Scholar]
- Faraldo, M.M., M.A. Deugnier, J.P. Thiery, and M.A. Glukhova. 2000. Development of mammary gland requires normal beta 1-integrin function. Adv. Exp. Med. Biol. 480:169–174. [DOI] [PubMed] [Google Scholar]
- Farrelly, N., Y.J. Lee, J. Oliver, C. Dive, and C.H. Streuli. 1999. Extracellular matrix regulates apoptosis in mammary epithelium through a control on insulin signaling. J. Cell Biol. 144:1337–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Z., A. Marti, B. Jehn, H.J. Altermatt, G. Chicaiza, and R. Jaggi. 1995. Glucocorticoid and progesterone inhibit involution and programmed cell death in the mouse mammary gland. J. Cell Biol. 131:1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong, E.E., N.K. Keon, F.D. Thornton, T. Rein, and F. Martin. 1996. Expression of a 74-kDa nuclear factor 1 (NF1) protein is induced in mouse mammary gland involution. Involution-enhanced occupation of a twin NF1 binding element in the testosterone-repressed prostate message-2/clusterin promoter. J. Biol. Chem. 271:29688–29697. [DOI] [PubMed] [Google Scholar]
- Gerwins, P., J.L. Blank, and G.L. Johnson. 1997. Cloning of a novel mitogen- activated protein kinase kinase kinase, MEKK4, that selectively regulates the c-Jun amino terminal kinase pathway. J. Biol. Chem. 272:8288–8295. [DOI] [PubMed] [Google Scholar]
- Geymayer, S., and W. Doppler. 2000. Activation of NF-kappaB p50/p65 is regulated in the developing mammary gland and inhibits STAT5-mediated beta-casein gene expression. FASEB J. 14:1159–1170. [DOI] [PubMed] [Google Scholar]
- Guttridge, D.C., C. Albanese, J.Y. Reuther, R.G. Pestell, and A.S. Baldwin Jr. 1999. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 19:5785–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkin, D.P., J.M. Bean, D. Miklos, Y.H. Song, V.B. Truong, C. Englert, F.C. Christians, L.W. Ellisen, S. Maheswaran, J.D. Oliner, and D.A. Haber. 1999. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell. 97:575–586. [DOI] [PubMed] [Google Scholar]
- Howlett, A.R., N. Bailey, C. Damsky, O.W. Petersen, and M.J. Bissell. 1995. Cellular growth and survival are mediated by beta 1 integrins in normal human breast epithelium but not in breast carcinoma. J. Cell Sci. 108:1945–1957. [DOI] [PubMed] [Google Scholar]
- Karin, M., and L. Chang. 2001. AP-1–glucocorticoid receptor crosstalk taken to a higher level. J. Endocrinol. 169:447–451. [DOI] [PubMed] [Google Scholar]
- Kearsey, J.M., P.J. Coates, A.R. Prescott, E. Warbrick, and P.A. Hall. 1995. Gadd45 is a nuclear cell cycle regulated protein which interacts with p21Cip1. Oncogene. 11:1675–1683. [PubMed] [Google Scholar]
- Kleinman, H.K., M.L. McGarvey, J.R. Hassell, V.L. Star, F.B. Cannon, G.W. Laurie, and G.R. Martin. 1986. Basement membrane complexes with biological activity. Biochemistry. 25:312–318. [DOI] [PubMed] [Google Scholar]
- Marti, A., B. Jehn, E. Costello, N. Keon, G. Ke, F. Martin, and R. Jaggi. 1994. Protein kinase A and AP-1 (c-Fos/JunD) are induced during apoptosis of mouse mammary epithelial cells. Oncogene. 9:1213–1223. [PubMed] [Google Scholar]
- Mills, K.R., M. Reginato, J. Debnath, B. Queenan, and J.S. Brugge. 2004. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc. Natl. Acad. Sci. USA. 101:3438–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita, H., J. Tsutsui, M. Takekawa, E.A. Witten, and H. Saito. 2002. Regulation of MTK1/MEKK4 kinase activity by its N-terminal autoinhibitory domain and GADD45 binding. Mol. Cell. Biol. 22:4544–4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro, A.N., A. August, and H. Hanafusa. 1996. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc. Natl. Acad. Sci. USA. 93:13595–13599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, D.A., A.F. Parlow, and M.C. Neville. 2001. Hormonal regulation of tight junction closure in the mouse mammary epithelium during the transition from pregnancy to lactation. J. Endocrinol. 170:347–356. [DOI] [PubMed] [Google Scholar]
- O'Brien, L.E., M.M. Zegers, and K.E. Mostov. 2002. Opinion: building epithelial architecture: insights from three-dimensional culture models. Nat. Rev. Mol. Cell Biol. 3:531–537. [DOI] [PubMed] [Google Scholar]
- O'Connell, F.C., and F. Martin. 2000. Laminin-rich extracellular matrix association with mammary epithelial cells suppresses BRCA1 expression. Cell Death Differ. 7:360–367. [DOI] [PubMed] [Google Scholar]
- Petersen, O.W., L. Ronnov-Jessen, A.R. Howlett, and M.J. Bissell. 1992. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA. 89:9064–9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova, O., S.V. Anisimov, M. Gorospe, R.H. Dougherty, W.A. Gaarde, K.R. Boheler, and N.J. Holbrook. 2002. Targets of c-Jun NH(2)-terminal kinase 2-mediated tumor growth regulation revealed by serial analysis of gene expression. Cancer Res. 62:3257–3263. [PubMed] [Google Scholar]
- Rajan, J.V., M. Wang, S.T. Marquis, and L.A. Chodosh. 1996. Brca2 is coordinately regulated with BRCA1 during proliferation and differentiation in mammary epithelial cells. Proc. Natl. Acad. Sci. USA. 93:13078–13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen, S., and H. Skaletsky. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:365–386. [DOI] [PubMed] [Google Scholar]
- Rubenstein, N.M., Y. Guan, P.L. Woo, and G.L. Firestone. 2003. Glucocorticoid down-regulation of RhoA is required for the steroid-induced organization of the junctional complex and tight junction formation in rat mammary epithelial tumor cells. J. Biol. Chem. 278:10353–10360. [DOI] [PubMed] [Google Scholar]
- Runswick, S.K., M.J. O'Hare, L. Jones, C.H. Streuli, and D.R. Garrod. 2001. Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nat. Cell Biol. 3:823–830. [DOI] [PubMed] [Google Scholar]
- Saelman, E.U., P.J. Keely, and S.A. Santoro. 1995. Loss of MDCK cell alpha 2 beta 1 integrin expression results in reduced cyst formation, failure of hepatocyte growth factor/scatter factor-induced branching morphogenesis, and increased apoptosis. J. Cell Sci. 108(Pt 11):3531–3540. [DOI] [PubMed] [Google Scholar]
- Sanchez, I., R.T. Hughes, B.J. Mayer, K. Yee, J.R. Woodgett, J. Avruch, J.M. Kyriakis, and L.I. Zon. 1994. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 372:794–798. [DOI] [PubMed] [Google Scholar]
- Smith, M.L., I.T. Chen, Q. Zhan, I. Bae, C.Y. Chen, T.M. Gilmer, M.B. Kastan, P.M. O'Connor, and A.J. Fornace Jr. 1994. Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science. 266:1376–1380. [DOI] [PubMed] [Google Scholar]
- Somasundaram, K., H. Zhang, Y.X. Zeng, Y. Houvras, Y. Peng, G.S. Wu, J.D. Licht, B.L. Weber, and W.S. El-Deiry. 1997. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature. 389:187–190. [DOI] [PubMed] [Google Scholar]
- Streuli, C.H., C. Schmidhauser, N. Bailey, P. Yurchenco, A.P. Skubitz, C. Roskelley, and M.J. Bissell. 1995. Laminin mediates tissue-specific gene expression in mammary epithelia. J. Cell Biol. 129:591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekawa, M., and H. Saito. 1998. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 95:521–530. [DOI] [PubMed] [Google Scholar]
- Thangaraju, M., S.H. Kaufmann, and F.J. Couch. 2000. BRCA1 facilitates stress- induced apoptosis in breast and ovarian cancer cell lines. J. Biol. Chem. 275:33487–33496. [DOI] [PubMed] [Google Scholar]
- Topper, Y.J., and C.S. Freeman. 1980. Multiple hormone interactions in the developmental biology of the mammary gland. Physiol. Rev. 60:1049–1106. [DOI] [PubMed] [Google Scholar]
- Venkitaraman, A.R. 2001. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J. Cell Sci. 114:3591–3598. [DOI] [PubMed] [Google Scholar]
- Weaver, V.M., O.W. Petersen, F. Wang, C.A. Larabell, P. Briand, C. Damsky, and M.J. Bissell. 1997. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 137:231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, V.M., S. Lelievre, J.N. Lakins, M.A. Chrenek, J.C. Jones, F. Giancotti, Z. Werb, and M.J. Bissell. 2002. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, V., D. Ching, P.D. McCrea, and G.L. Firestone. 1999. Glucocorticoid down-regulation of fascin protein expression is required for the steroid-induced formation of tight junctions and cell-cell interactions in rat mammary epithelial tumor cells. J. Biol. Chem. 274:5443–5453. [DOI] [PubMed] [Google Scholar]
- Woo, P.L., D. Ching, Y. Guan, and G.L. Firestone. 1999. Requirement for Ras and phosphatidylinositol 3-kinase signaling uncouples the glucocorticoid-induced junctional organization and transepithelial electrical resistance in mammary tumor cells. J. Biol. Chem. 274:32818–32828. [DOI] [PubMed] [Google Scholar]
- Xu, X., Z. Weaver, S.P. Linke, C. Li, J. Gotay, X.W. Wang, C.C. Harris, T. Ried, and C.X. Deng. 1999. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell. 3:389–395. [DOI] [PubMed] [Google Scholar]