

Abstract
The Cockayne syndrome B (CSB) protein is essential for transcription-coupled DNA repair (TCR), which is dependent on RNA polymerase II elongation. TCR is required to quickly remove the cytotoxic transcription-blocking DNA lesions. Functional GFP-tagged CSB, expressed at physiological levels, was homogeneously dispersed throughout the nucleoplasm in addition to bright nuclear foci and nucleolar accumulation. Photobleaching studies showed that GFP-CSB, as part of a high molecular weight complex, transiently interacts with the transcription machinery. Upon (DNA damage-induced) transcription arrest CSB binding these interactions are prolonged, most likely reflecting actual engagement of CSB in TCR. These findings are consistent with a model in which CSB monitors progression of transcription by regularly probing elongation complexes and becomes more tightly associated to these complexes when TCR is active.
Keywords: Cockayne syndrome; GFP; photobleaching studies; TCR
Introduction
Metabolic by-products such as reactive oxygen species, environmental compounds, and short-wave electromagnetic radiation (γ and UV) continuously jeopardize the DNA structure. DNA injuries directly disturb vital DNA-transacting processes such as replication, transcription, and cell cycle progression. Evidence in the literature suggests that DNA damage-induced transcriptional interference triggers apoptosis (Yamaizumi and Sugano, 1994; Ljungman and Zhang, 1996). Our previous work on repair-deficient mice provided strong support for our hypothesis that this damage-induced apoptosis leads to segmental ageing (de Boer et al., 2002). Moreover, DNA lesions may result in permanent mutations in the DNA sequence, eventually causing cancer. To prevent the severe consequences of genetic erosion a variety of distinct and partially overlapping DNA repair pathways has evolved, each specialized in the removal of specific types of damage (Friedberg et al., 1995; Hoeijmakers, 2001). Priority is given to remove the highly cytotoxic transcription-blocking injuries, allowing quick resumption of transcription. This process, referred to as transcription-coupled repair (TCR; Mellon et al., 1987), is directly triggered by lesion-induced obstruction of elongating RNA polymerase II (RNAP II). An example of transcription-blocking lesions are DNA helix distorting, UV-induced cyclo-butane pyrimidinedimers (CPDs) and 6-4 photoproducts (6-4PPs). Dependent on the type of lesion stalled RNAP II complexes are first identified by TCR-specific factors and further processed by the core nucleotide and perhaps base excision repair (NER and BER, respectively) factors (Le Page et al., 2000). Removal of lesions in nontranscribed areas of the genome is dependent on global genome repair (GGR).
Inherited defects within genes involved in the TCR pathway give rise to the rare autosomal recessive disorder Cockayne syndrome (CS; Venema et al., 1990; Van Hoffen et al., 1993). CS patients display mainly progeroid symptoms, growth failure, and severe neurological abnormalities and are not cancer-prone (Nance and Berry, 1992). Most of the salient clinical symptoms expressed among CS individuals, except sun-sensitive skin, are difficult to explain by a DNA repair defect only. Within classical Cockayne syndrome two genes are involved, CSA and CSB (Troelstra et al., 1992; Henning et al., 1995). The proteins encoded by these genes are essential for TCR, however their exact function in this process remains elusive. The 44-kD CSA protein contains five WD repeats; polypeptides with these repeats are usually involved in formation of macromolecular complexes via the WD-repeat regions (Neer et al., 1994). The 168-kD CSB protein is a member of the SWI2/SNF2 protein family of putative helicases, which includes a variety of proteins involved in transcriptional regulation, chromatin remodeling, and DNA repair (Pazin and Kadonaga, 1997). Biochemical studies showed that recombinant CSB is a DNA-dependent ATPase and is able to remodel chromatin at the expense of ATP (Citterio et al., 1998, 2000).
Because TCR only occurs in the presence of active transcription, it was suggested that the CS proteins probably interact with elongating RNA polymerase complexes. Moreover, besides a pivotal role in TCR, several lines of evidence suggest an additional function of CS proteins, particularly CSB, in the elongation phase of RNAP II transcription. Gel filtration and immunoprecipitation studies showed that CSB resides in a high molecular weight complex and that a part of these higher order assemblies contain RNAP II (van Gool et al., 1997). Gel mobility shift assays further revealed that CSB interacts with a ternary complex of DNA, RNAP II, and nascent RNA (Tantin et al., 1997) and in vitro transcription experiments showed that CSB stimulates RNAP II elongation (Selby and Sancar, 1997). In addition, Rad26 (the yeast counterpart of CSB) was found to be required for transcription in vivo (Lee et al., 2001). Recently, in a genetic screen for suppressor mutants of Rad26, Spt4 was identified (Jansen et al., 2000). Spt4 is part of a protein complex known to associate with and regulate the processivity of RNAP II, further supporting a function of CSB in transcription elongation (Hartzog et al., 1998; Wada et al., 1998).
A potential problem for the repair machinery is that lesion-stalled polymerases impede the accessibility of repair factors to these lesions by steric hindrance (Donahue et al., 1994). Several models have been suggested that describe the function of the CS proteins and the fate of stalled RNAP II complexes at lesions: (a) backtracking of the RNAP II complex, providing access to lesions (Tornaletti and Hanawalt, 1999); (b) pushing the elongating polymerase past the lesion, translesion transcription; (c) physical removal of the complex; (d) proteolytic degradation of the stalled polymerase; or (e) recruiting NER proteins that compete with RNAP II. Efforts to set up an in vitro system for TCR have met with little success. A possible explanation is that in vitro systems lack the structural elements that are required for proper TCR function (such as nuclear matrix attachment or chromatinated DNA). Particularly, the topological and ATP-dependent chromatin remodeling activity of CSB suggests a role for this protein in remodeling the chromatin structure or the interaction surface of the stalled RNAP II with DNA, to permit admission of the NER machinery to the lesion. However, an adequate model at which stage the TCR factors CSA and CSB are operational is lacking.
To address some of these issues, we investigated the involvement of CSB in TCR and RNAP II transcription elongation in the most relevant context, the living cell. We generated a cell line that stably expresses physiologically relevant levels of a biologically active fusion protein of the GFP and CSB. Spatial and temporal distribution of GFP-CSB was monitored with live cell confocal microscopy. In addition, we determined the in vivo reaction parameters of this protein when engaged in repair and transcription by using FRAP analysis to measure the mobility of GFP-CSB in untreated transcriptionally active cells and compared these with transcription-inhibited cells and cells challenged with a high dose of UV light (DNA damage induction).
Results
Expression of GFP-CSB in human fibroblasts
To study the nuclear localization and dynamics of the CSB protein in living cells, we tagged the protein with GFP. Enhanced GFP was fused to the amino terminus of CSB (Fig. 1 A), resulting in a GFP-CSB fusion protein, which was stably expressed in CSB-deficient human fibroblasts (CS1AN-Sv). Immunoblot analysis, using anti-CSB (Fig. 1 B) and anti-GFP (not depicted) antibodies, showed that GFP-CSB migrates at the expected height of full-length fusion protein (∼195 kD) in two independent clones, and was expressed at physiological levels. In addition, the GFP-CSB cDNA was able to fully correct the UV sensitivity of CS-B cells (Fig. 1 C), stressing that GFP-CSB is functional in vivo.
Figure 1.
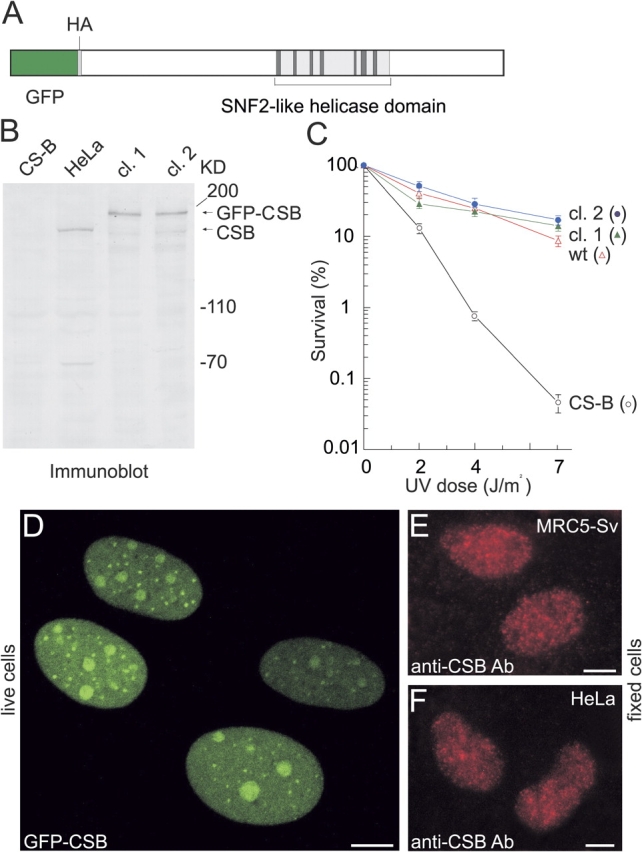
Characterization of stably expressed GFP-CSB in CS1AN-Sv human fibroblasts. (A) Schematic representation of GFP-CSB fusion protein. The SNF2-like helicase domain is indicated. In between the GFP cDNA and the CSB open reading frame an HA tag is present. (B) Immunoblot analysis of GFP-CSB expression. Equal amounts of WCE from CS1AN-Sv, HeLa, and GFP-CSB transfected CS1AN-Sv fibroblasts (two independent clones, 1 and 2 respectively) were probed with affinity-purified polyclonal anti-CSB antibodies. (C) UV survival of GFP-CSB expressing fibroblasts. The percentage of surviving cells is plotted against the applied UV dose. Survival of clones 1 and 2 and control cell lines after UV treatment was determined by pulse labeling with [3H]thymidine: CS1AN-Sv, CS-B (black open circles); VH10-Sv, wt (red open triangles), clone 1 (green triangles), clone 2 (blue circles). Both clones show a complete restoration of the CS-specific UV sensitivity, indicating that the GFP-tagged CSB protein is fully functional. (D) Subnuclear localization of GFP-CSB in living stably transfected CS1AN-Sv human fibroblasts. All cells show a strict nuclear distribution and focal and nucleolar accumulations of GFP-CSB in the nucleoplasm. (E and F) Epifluorescent images of MRC5-Sv human fibroblasts (F) and HeLa cells (G) immunostained with affinity-purified anti-CSB antibodies. Bars, 10 μm.
Localization of GFP-CSB in living cells
Confocal microscopy demonstrated that CSB-GFP predominantly resided in the nucleus (Fig. 1 D), where it accumulated in small bright foci, in addition to a homogeneous distribution in the nucleoplasm. The foci were present in a large fraction of the investigated cells in varying number and size. In addition, the fusion protein was enriched in nucleoli (Fig. 1 D). Immunofluorescence studies in MRC5-SV and HeLa cells, using affinity-purified polyclonal anti-CSB antibodies to determine endogenous nontagged CSB, revealed a more disrupted focal pattern than GFP-imaging in live cells. Immunostaining of GFP-tagged CSB with anti-HA (GFP is tagged with HA) showed the same disrupted pattern (unpublished data) as endogenously stained CSB. From these observations, we conclude that the disrupted foci are caused by the applied fixation procedure. Together, these data suggest that the observed distribution is not due to GFP-tagging or to overexpression of the protein and likely reflects the physiologically relevant nuclear organization of CSB.
Nuclear mobility of GFP-CSB in living cells
To investigate whether GFP-CSB is bound to subnuclear structures or is moving freely we measured the GFP-CSB nuclear mobility and the dynamic properties of GFP-CSB molecules by applying a FRAP protocol to the cells. Briefly, a 2-μm wide strip spanning the nucleus was bleached and fluorescence recovery in the strip was measured at 100-ms intervals (Fig. 2 A). The rate of recovery of fluorescence in the bleached strip is a measure for the diffusion rate of the tagged protein (Fig. 2 B). Note that the fluorescent intensity in the strip does not recover to prebleach levels (set to one) because a fraction of the molecules is permanently bleached. Analysis of the FRAP data revealed that the majority of GFP-CSB molecules was freely mobile in the nucleoplasm with an effective diffusion coefficient (D eff) of 7 μm2/s (Fig. 2 C). In addition, the 163-kD XPF–ERCC1-GFP complex, which is involved in the core NER reaction (Houtsmuller et al., 1999), had a D eff of 12 μm2/s, clearly higher than that of GFP-CSB. This suggests that GFP-CSB resides in a high MW complex (>800 kD), confirming our previous gelfiltration studies with HA-tagged CSB (van Gool et al., 1997).
Figure 2.
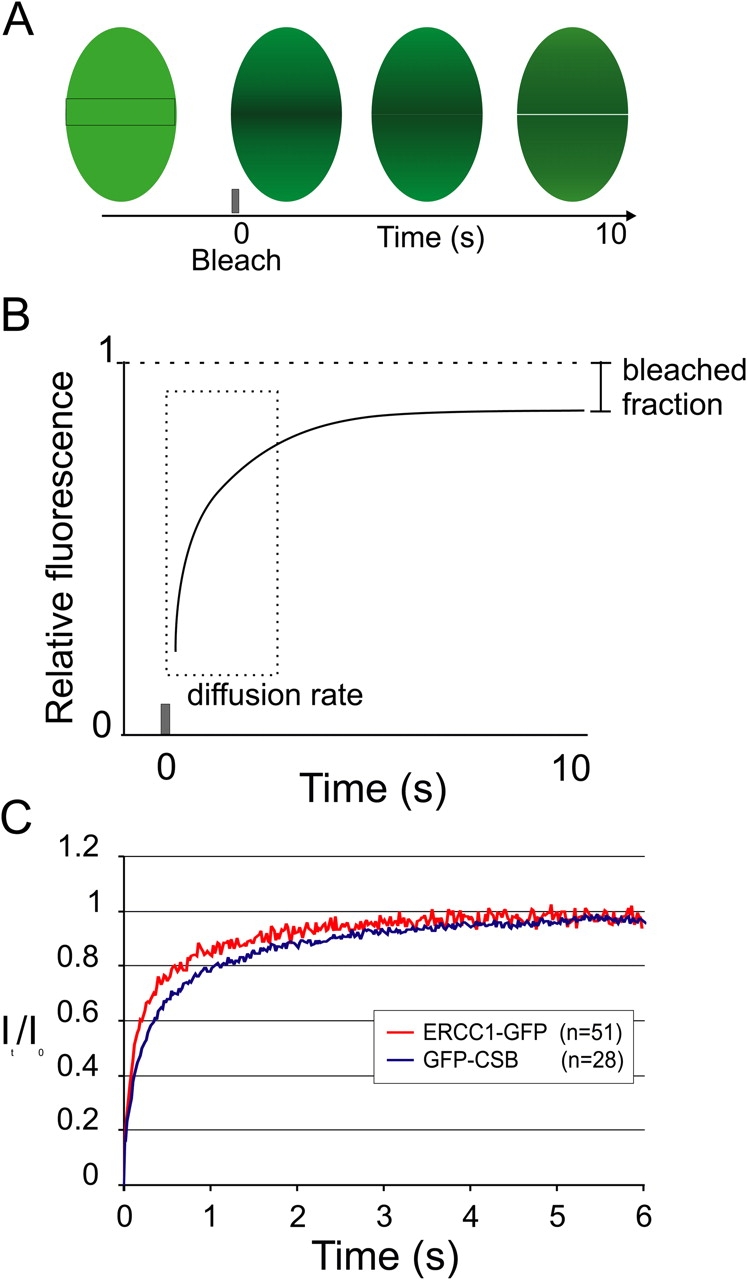
FRAP analysis of GFP-CSB nuclear mobility. (A) During a FRAP experiment all fluorescent molecules in a small defined strip spanning the nucleus are bleached and subsequent fluorescent recovery is measured. (B) Plotting the recovery relative fluorescent intensity of the strip in time shows a small permanently bleached fraction as caused by the initial bleach pulse. The rate of fluorescent recovery in the strip is a measure for the effective diffusion rate (Deff) of a protein. (C) FRAP analysis of GFP-CSB and ERCC1-GFP–expressing cells show a recovery of fluorescence in the strip.
Transient immobilization of GFP-CSB in transcription
Previous in vitro experiments suggest that CSB interacts with RNAP II (Tantin et al., 1997; van Gool et al., 1997). Therefore, we studied the dynamic behavior of GFP-CSB molecules in relation to transcription in living cells, using a combined FLIP/FRAP procedure (Fig. 3 A; Hoogstraten et al., 2002). Briefly, a region at one pole of the nucleus is bleached and the influx of fluorescence in the bleached area is monitored (FRAP) as well as the fluorescence loss in photobleaching (FLIP) at the opposite pole of the nucleus. The difference in relative fluorescent intensity between the FLIP and FRAP region in time is plotted on a logarithmic scale. The time to reach 90% redistribution of GFP-CSB fluorescence was 59 ± 8 s. For comparison with other conditions this value was set to 1 in the subsequent experiments. To investigate the relation between GFP-CSB mobility and transcription we incubated cells with various transcription inhibitors. The transcription elongation inhibitor H8 clearly induced a reduction in redistribution time (∼20%) as compared with transcriptionally active cells (Fig. 3 B), suggesting a faster overall mobility of GFP-CSB molecules when transcription is inhibited. In sharp contrast, the DNA-intercalating agent Actinomycin D resulted in a severe loss of the ability for GFP-CSB molecules to redistribute, which is indicative of a long-term immobilization of CSB proteins (Fig. 3 C). A similar differential response to transcription inhibitors was reported for RNAP II, where Actinomycin D also resulted in almost complete immobilization, whereas the H8-like transcription inhibitor DRB led to increased mobility (Kimura et al., 2002). It was argued by the authors that DRB treatment results in release of RNAP II, whereas Actinomycin D irreversibly stalls the polymerase on its template. The similar behavior in response to transcription inhibitors suggests a close relationship between elongating RNAP II and CSB in living cells. In contrast, XPF–ERCC1-GFP was not immobilized in presence of Actinomycin D, indicating that the immobilization of CSB is not caused by repair activity (unpublished data).
Figure 3.
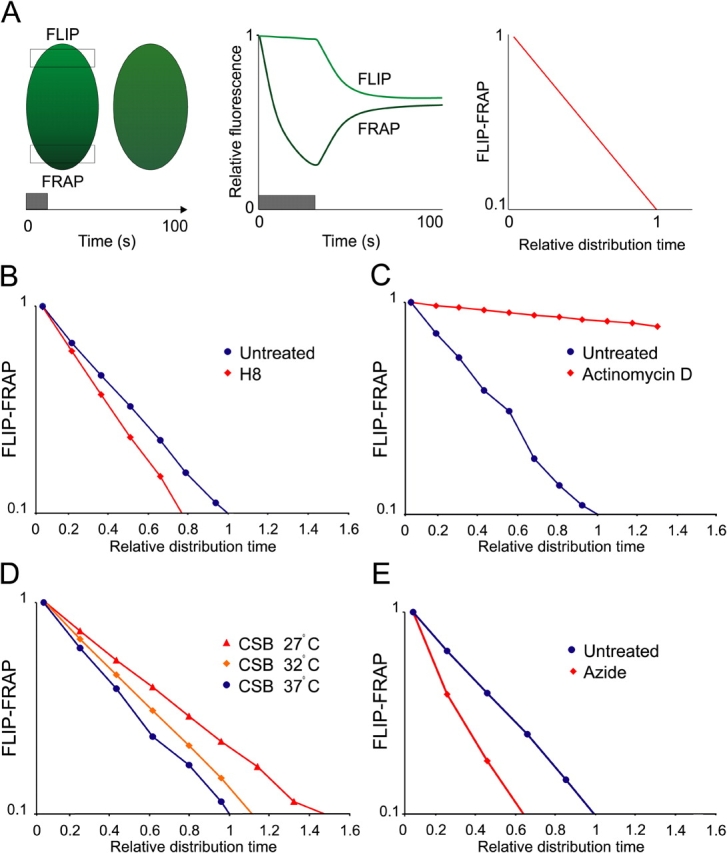
Dynamic measurements of the GFP-CSB nuclear mobility by combined FLIP/FRAP analysis. (A) Combined FLIP/FRAP analysis was performed by bleaching at one pole of the nucleus and simultaneously monitoring the fluorescent recovery at the bleached (FRAP) and opposite (FLIP) poles of the cell. After bleaching, the FRAP curve shows a drop in fluorescent intensity followed by a recovery of fluorescence and the FLIP curve shows a slow decrease of fluorescent intensity due to redistribution of the bleached molecules. The relative intensities of FLIP and FRAP were subtracted and plotted (y axis) against the relative redistribution time of untreated cells (x axis). (B) Combined FLIP/FRAP experiment of untreated cells (blue circles; n = 10) and H8-treated cells (red diamonds; n = 10). H8-treated cells display a decreased relative redistribution time as compared with untreated cells. (C) Combined FLIP/FRAP experiment of untreated cells (blue circles; n = 10) and Actinomycin D–treated cells (red diamonds; n = 10). Actinomycin D treatment results in an increased relative redistribution time as compared with untreated cells. (D) Combined FLIP/FRAP experiments at different temperatures (27°C, red triangles; 32°C, orange diamonds; and 37°C, blue circles). At low temperatures the relative redistribution time is increased. (E) Combined FLIP/FRAP experiment of untreated cells (blue circles, n = 10) and azide-treated cells (red diamonds; n = 10). Azide-treated cells display a decreased relative redistribution time as compared with untreated cells.
Next, we applied the combined FLIP-FRAP procedure to cells cultured at different temperatures. The rationale behind this is that a relatively small difference in (absolute) temperature (Kelvin) has a negligible effect on diffusion rate, but strongly affects the duration of temperature-dependent enzymatic processes such as transcription and active transport (Phair and Misteli, 2000; Hoogstraten et al., 2002). Combined FLIP-FRAP of GFP-CSB–expressing cells cultured at respectively 37, 32, and 27°C revealed a significant decrease of mobility when temperature was reduced (Fig. 3 D). In contrast, diffusion of freely mobile XPF–ERCC1-GFP (Houtsmuller et al., 1999) did not change upon lowering temperature (unpublished data). Furthermore, ATP depletion by azide induced an increased GFP-CSB mobility (Fig. 3 E).
In conclusion, these observations strongly suggest that GFP-CSB mobility is decreased in transcriptionally active cells by transient temperature-dependent immobilizations, most likely due to association with elongating transcription complexes. The fluorescence recovery plots fitted best to curves generated by computer simulation of FRAP on molecules (see Materials and methods) of which a small fraction (∼17%) is shortly immobilized (2–5 s).
CSB-associated RNAP II is transcriptionally active in vitro
To provide biochemical evidence for the suggested interaction with the transcription machinery we isolated the CSB-RNAP II complex as described before using our previously generated human cell line expressing CSB tagged with a HA epitope (HA-CSB-[His]6, referred to as 2tCSB; van Gool et al., 1997) and immunoaffinity purification by binding to an anti-HA antibody resin followed by elution with excess of HA-peptide. Immunopurified CSB was tested in an in vitro reconstituted transcription system (RTS) using an adenovirus major late promoter template (Gerard et al., 1991; Coin et al., 1999). The HA-eluate from dtCSB whole cell extracts (WCE) was able to support the synthesis of the 309 nt transcript when RNAP II was omitted from the RTS (Fig. 4, compare lanes 3 and 14 with lanes 1 and 8, respectively), indicating that the CSB-associated RNAP II is transcriptionally active. In contrast, no signal was detected by addition of HA-eluate from normal HeLa WCE expressing normal CSB without HA-tag (Fig. 4, lane 5) or in the absence of any extract (Fig. 4, lane 7), supporting the specificity of the CSB-RNAP II interaction. To determine the presence of additional basal transcription components in the 2tCSB HA-eluate, transcription was performed in the absence of TBP (Fig. 4, lanes 2, 4, 6, and 13), TFIIE, TFIIB, TFIIF, or TFIIH (Fig. 4, lanes 9 to 12, respectively). In neither of these cases complementation for the lack of any of these factors was detected (Fig. 4, compare lane 2 with lane 3 and lanes 9 to 12 with lane 14), indicating that none of the transcription initiation factors were present in the 2tCSB (HA-elution) fraction in detectable amounts.
Figure 4.

In vitro transcriptional activity of CSB-associated RNA polymerase II. RNAP II transcriptional activity is present in the 2tCSB HA-eluate. In vitro transcription was performed in a reconstituted transcription system (RTS) containing human recombinant TBP, TFIIB, TFIIE, and highly purified HeLa TFIIH, TFIIF, and RNAP II and the adenovirus major late promoter as a template (309 nt). Lanes 1 and 8 show complete reactions. To determine the presence of transcription components in the tagged-CSB fraction, individual transcription factors (indicated on top of each lane) were omitted from reactions containing HA-eluate from 2tCSB WCE (lanes 2 and 3 and lanes 9–14). As a control, HA-eluate from HeLa WCE (lanes 4 and 5), or no protein (lanes 6 and 7) were added to reactions lacking TBP or RNAP II.
Nuclear mobility of GFP-CSB in UV-irradiated cells
To investigate the behavior of GFP-CSB in TCR we determined the overall nuclear mobility in UV-irradiated cells by FRAP analysis. Fluorescence recovery plots of UV-damaged cells (16 J/m2, a repair-saturating UV dose; Fig. 5 A, red line) revealed a small but reproducible reduction of fluorescence recovery when compared with non-UV–damaged cells (blue line), indicating that a fraction of GFP-CSB molecules is immobilized for a longer period. In addition, the diffusion rate of the mobile GFP-CSB fraction in untreated and UV-irradiated cells is unaltered, indicating that the size of the CSB complex is not altered upon DNA damage induction (unpublished data).
Figure 5.

FRAP analysis of GFP-CSB after UV irradiation. (A) FRAP analysis of untreated cells (blue line; n = 148) and UV-irradiated cells (16 J/m2; red line; n = 125). GFP-CSB mobility in UV-treated cells is measured between 5 and 25 min after irradiation. UV-treated cells show an immobilization of GFP-CSB. (B) Dose dependency of GFP-CSB immobilization based on three independent experiments. (C) FRAP analysis of untreated cells (blue line; n = 148), cells treated with DRB before UV irradiation (16 J/m2; green line; n = 79) and cells treated with UV solely (16 J/m2; red line; n = 125). DRB was added to the medium 3 h before the experiment. (D) Dynamics of the immobile fraction of GFP-CSB in time after UV (16 J/m2) based on three independent experiments.
The amount of UV-induced immobilized molecules was proportional to UV dose: from ∼5% at 4 J/m2 to a plateau of ∼15% at 16 J/m2 (Fig. 5 B). A similar UV dose-dependent immobilization was observed with core NER factors, such as ERCC1-GFP–XPF (Houtsmuller et al., 1999), GFP-XPA (Rademakers et al., 2003), and TFIIH-GFP (Hoogstraten et al., 2002), although the maximum fraction of GFP-CSB immobilization (∼15%) is significantly lower than found with the other NER factors (35–40%). No UV-induced immobilization was found with non-NER factors tagged with GFP (Houtsmuller et al., 1999) stressing the notion that immobilization is related to NER. This suggests that GFP-CSB binds more stably to stalled RNAP II than to elongating RNAP II.
When prolonged immobilization is dependent on stalled polymerases, and implicitly on TCR, we predict that the UV-induced immobilization requires active transcription. To verify this hypothesis we treated the cells with the transcription inhibitor DRB before UV irradiation. As shown in the mobility plot of Fig. 5 C (green line) a significant decrease of the immobile fraction upon transcriptional inhibition was apparent when compared with transcriptional active UV-irradiated cells (DRB did not completely prevent UV-induced immobilization, likely caused by incomplete transcription inhibition). This indicates that the observed immobilization of GFP-CSB is most likely due to its engagement in TCR.
When the immobilization of CSB reflects actual participation in TCR we expect that the immobilized fraction would decrease in time depending on progression of repair. Therefore, we measured UV-dependent immobilization of GFP-CSB molecules at various time points after irradiation (Fig. 5 D, 16 J/m2). These experiments revealed that the bound fraction gradually decreased to background levels within 16 h after UV. This indicates that UV-dependent immobilization of GFP-CSB is a reversible process. Interestingly, the kinetics of this process was much slower than anticipated on the basis of the efficient damage repair by TCR measured in selected genes (Mellon et al., 1987).
GFP-CSB mobility in NER-deficient (XP-A) cells
To investigate the effect of defective DNA repair on the mobility of GFP-CSB we examined UV-induced immobilization in repair deficient cells, lacking functional XPA (XP12RO). Comparative analysis of FRAP experiments applied to cells expressing GFP-CSB in either CSB- or XPA-deficient cells showed a slightly larger immobile fraction of CSB molecules in XPA cells upon UV irradiation, using a subsaturating UV dose of 8 J/m2 (Fig. 6, A and B). This contrasts to UV treatment after transcription inhibition, where a smaller immobilized fraction was observed (Fig. 5 C). Importantly and in contrast to GFP-CSB molecules in repair proficient cells (Fig. 6 C and Fig. 5 B), the fraction of immobilization in XPA-deficient cells did not decrease (nor increase) 4 h after UV-damage induction (Fig. 6 D). The notion that the fraction of immobilized molecules is not substantially increased as compared with NER-proficient cells, suggest that CSB molecules are not permanently trapped in repair complexes, but transiently interact with blocked polymerases. This transient interaction with abortive repair complexes is significantly different from the stable association of CSB with stalled RNAP II by Actinomycin D treatment (Fig. 3 C; Kimura et al., 2002).
Figure 6.

FRAP analysis of GFP-CSB in NER-deficient (XP group A) cells. (A) FRAP curves of untreated (blue line; n = 20) and 8 J/m2 UV-irradiated (red line; n = 20) GFP-CSB–expressing CS1AN cells, measured between 5 and 15 min after UV irradiation. 8 J/m2 induced a smaller immobile fraction as 16 J/m2 as shown in Fig. 5 A. (B) Identical to A, however here GFP-CSB is expressed in XP group A cells, lacking functional NER. A slight increase in the immobile fraction is visible as compared with repair-proficient cells in A. (C and D) Identical to A and B, respectively, with the exception that here also the FRAP measurements were plotted that were performed 4 h after UV irradiation (green lines, n = 200), with an intermediate immobilization in NER-proficient cells and no recovery of the immobilized fraction in XP-A cells.
GFP-CSB accumulates at sites of local damage
DNA damage-dependent immobilization of GFP-CSB argues for a model in which CSB complexes that transiently interact with the transcription machinery remain longer bound to lesion-blocked polymerases than to elongating complexes. To obtain further evidence for this hypothesis we locally inflicted UV lesions in cells expressing GFP-CSB using a porous UV-blocking membrane (Volker et al., 2001). Shortly after UV irradiation, we detected accumulations of GFP-CSB similar to those found for other NER proteins such as XPA (Fig. 7 A). XPA is involved in GGR and TCR and is known to accumulate at locally damaged areas in the cell (Volker et al., 2001; Rademakers et al., 2003). This indicates that GFP-CSB also accumulates at sites of DNA damage, most likely active in TCR. Interestingly, the number of accumulated GFP-CSB molecules in the damaged area is relatively stable at least up to 8 h after UV (Fig. 7 A). These results are in line with our findings that upon overall UV irradiation an immobilization of GFP-CSB is measured until 16 h after UV. This is in contrast to XPA, which showed an intense concentration of proteins a few minutes after UV that in the first 2 h drops to a lower steady-state level (2 h after UV irradiation) and then slowly reduces toward background levels. In addition, immunofluorescence analysis showed that 6-4PPs are removed within 2 h after UV, whereas the vast majority of CPD lesions are still present up to 8 h after local irradiation (Volker et al., 2001; unpublished data). This suggests that the early intense accumulation of XPA mainly reflects repair of 6-4PPs via the GGR pathway, whereas the subsequent less intense accumulation of XPA represents repair of CPDs.
Figure 7.

Accumulation of GFP-CSB at local UV-damaged subnuclear areas. (A) Epifluorescent images of fixed GFP-CSB–expressing cells at various time points (15 min, 2 h, 4 h, 8 h) after local irradiation. Immunofluorescent analysis with anti-XPA antibody shows accumulation of XPA at sites of damage. GFP-CSB shows accumulation in the same areas. Top, GFP signal; bottom, Cy3 signal. The arrows all point to UV-induced local accumulations of GFP-CSB and XPA, respectively. Bar, 5 μm. (B) Fluorescence recovery plot of a local damage (red diamonds) and an undamaged control region (blue circles). The calculated average residence time is 135 ± 20 s.
Do these local accumulations reflect long-term (a few hours) immobilization of CSB molecules or are they the result of a dynamic equilibrium between binding and releasing molecules at the site of damage? To investigate this, we determined the GFP-CSB residence time in the accumulations by FRAP on damaged and nondamaged areas in the nucleus. For this purpose we bleached the fluorescence in the locally damaged region. Interestingly, a quick recovery of fluorescence within the local damaged area was observed (Fig. 7 B), indicating that repair-bound GFP-CSB molecules rapidly exchange with the mobile pool. However, the fluorescence recovery in the damaged area is slower than in an equally sized control region in a nondamaged cell (Fig. 7 B). We calculated an average residence time of GFP-CSB molecules in the local damage of 135 ± 20 s. This binding time is short relative to other core NER factors like XPA, TFIIH, and ERCC1, which are bound in a locally damaged area for 3–5 min (Houtsmuller et al., 1999; Hoogstraten et al., 2002; Rademakers et al., 2003).
Discussion
Here, we present a study on the dynamic behavior of the TCR protein CSB in living cells, using a cell line that stably expresses functional GFP-tagged CSB protein at physiological levels. Confocal microscopy and quantitative digital image analysis of different photobleaching (FRAP) procedures revealed transient interactions of CSB with the transcription machinery, which are prolonged when RNA polymerases are arrested at sites of DNA damage.
Mobility of GFP-CSB
FRAP analysis indicated that the overall CSB mobility is remarkably slow compared with its calculated molecular size and the observed effective diffusion rate (Deff) of other NER-factors (XPA, ERCC1–XPF, and TFIIH) tested in a similar fashion that all have a Deff that is in concordance with their molecular sizes, arguing against a stable preassembled NER “holo” complex (Houtsmuller et al., 1999; Hoogstraten et al., 2002; Rademakers et al., 2003). The relatively slow mobility of GFP-CSB in vivo, even in transcription inhibited cells, confirms previous biochemical evidence that CSB resides in a complex with an estimated hydrodynamic velocity of particles >700 kD (van Gool et al., 1997).
Dynamic interactions of CSB with the transcription machinery
A significant fraction (∼25%) of RNAP II in mammalian cells is bound to DNA and a single polymerase typically is bound for ∼20 min during transcription elongation (Kimura et al., 2002). The suggested role of CSB in transcription elongation predicts that CSB-containing complexes show similar dynamics as RNAP II. However, FRAP studies suggest that only a small fraction of CSB-containing complexes (∼17%) are immobilized for ∼2–5 s in a transcription-dependent fashion. The drop in mobility at 27°C as compared with 37°C suggests that CSB is immobilized in a temperature-dependent fashion, most likely due to its involvement in transcription. Interestingly, upon treatment with Actinomycin D an unexpectedly large fraction of CSB is immobilized. A similar strong immobilization, induced by Actinomycin D treatment, was reported for GFP-tagged RNAP II. The authors explain this observation by a permanent block of the elongating complex caused by the DNA intercalating agent (Kimura et al., 2002). Apparently, these “frozen” complexes permanently trap CSB molecules. In conclusion, our findings suggest a model in which there is a dynamic equilibrium between mobile CSB complexes and CSB transiently bound to elongating RNAP II.
Dynamic interactions of transcription factors with active transcription sites have been noticed before using similar procedures. Hager and colleagues (McNally et al., 2000) described a rapid exchange between chromatin-bound and freely mobile GFP-tagged glucocorticoid receptors. Moreover, our dynamic studies on GFP-tagged TFIIH and androgen receptors (Hoogstraten et al., 2002; Farla et al., 2004), revealed a similar short transcription-dependent interaction. However, both factors stimulate transcription initiation rather than elongation (Chandler et al., 1983; Lu et al., 1992). Obviously, elongating complexes, by virtue of their nature, are longer associated to DNA than we observe here for CSB. The transient interactions of elongation stimulating factors, such as CSB, may provide a flexible response to different chromatin conformations or changing conditions during elongation allowing different factors to bind on demand. This is the first in vivo example of a transcription elongation factor that is not a stably associated component of the RNAP II elongation holocomplex.
Previously, a fraction of CSB molecules was found to interact with RNAP II (Tantin et al., 1997; van Gool et al., 1997). In addition, recombinant CSB was claimed to stimulate RNAP II elongation in vitro (Selby and Sancar, 1997) and RNAP II transcription was reported to be slightly impaired in CSB-deficient cells (Balajee et al., 1997; Dianov et al., 1997). Furthermore, a specific role in transcription elongation for the Rad26 protein (the yeast homologue of CSB) was found in yeast (Lee et al., 2001) and deduced from genetic interactions between Rad26 and Spt4 (Jansen et al., 2000), which is implicated in regulation of RNAP II processivity (Hartzog et al., 1998; Wada et al., 1998). Here, we demonstrate that in a reconstituted transcription assay CSB-associated RNAP II is transcriptionally competent. No other general transcription initiation factors were detected in the immunoprecipitated fraction. Together, these studies support a role for CSB in transcription elongation.
Besides a function in RNAP II-driven transcription, a more general role for CSB in RNA polymerase I and III mediated transcription was suggested (Yu et al., 2000; Bradsher et al., 2002). The nucleolar localization of GFP-CSB in vivo presented here supports a role of CSB in processivity of other RNA polymerases. Furthermore, the absence of the CSB protein in human fibroblasts derived from CS group B patients caused fragility of metaphase chromosomes at specific loci, these include the U1 and U2 snRNA genes (RNAP II) and the 5S RNA genes (transcribed by RNAP III; Yu et al., 2000). It was suggested that this fragility is provoked by the absence of a (general) stimulating function of CSB in elongation during transcription of these highly structured RNAs.
Participation of CSB in TCR
The fraction of immobilized molecules after UV irradiation decreased to background levels (∼16 h) much slower than core NER factors appeared to decrease to background levels in 4–6 h after UV (Houtsmuller et al., 1999; Hoogstraten et al., 2002; Rademakers et al., 2003). These kinetics of core NER factors were similar to the rate of 6-4PP removal (Van Hoffen et al., 1995) and thus suggest that with the applied methods predominantly repair of 6-4PP was monitored. DNA repair of the most abundant UV lesion, CPD, appeared to occur at much slower rates. Repair of these lesions mainly happens in the transcribed strand of active genes by TCR, in contrast to 6-4PP removal that are almost as efficiently repaired by GGR as by TCR (Van Hoffen et al., 1995). Assuming that our observed immobilizations of GFP-CSB actually reflect participation in TCR, this further implies that completion of TCR is slower than completion of the bulk of GGR. However, this assumption contrasts to the general accepted model that TCR is more efficient than GGR (Bohr et al., 1985; Mellon et al., 1987). A possible explanation for this apparent contradiction is that the relatively fast removal of CPDs by TCR was measured on frequently transcribed genes (i.e., DHFR), whereas our live cell studies are performed on the total pool of transcriptional units, including long and less frequently transcribed genes. Because TCR is likely initiated by the blockage of transcription elongation on lesions, the efficiency of TCR-dependent lesion removal (such as CPDs) is likely determined both by the rate of transcription and the size of the transcriptional unit.
In addition, similar to FRAP analysis, the local accumulation of CSB was observed at least until 8 h after UV irradiation, further providing evidence for a relatively long lasting active TCR pathway after UV irradiation.
Dynamic interaction of CSB with TCR complexes
The relative small increase in immobilized GFP-CSB fraction in XPA-deficient cells as compared with NER-proficient cells and the fact that this fraction does not increase in time after UV, suggest that CSB molecules are not permanently bound to lesion-stalled RNAPII complexes that are not further processed because of defective NER. It is remarkable that the absence of repair does not lead to a complete immobilization as observed after treatment with Actinomycin D RNAPII (Fig. 3 C). The DNA intercalating drug Actinomycin D forms a permanent block for RNAPII thereby capturing the molecule in a template-bound form (Kamitori and Takusagawa, 1992). In contrast, UV-induced stalling of RNAPII in an XPA-deficient background probably induces a different type of conformation which is still permissive to removal from the DNA template by CSB. Evidence for the accessibility of RNAPII stalled on UV lesions was very recently provided by Tremeau-Bravard et al. (2004). This different behavior of CSB might be explained by differential structural consequences for the stalled RNAPII provoked by Actinomycin D adducts versus bonafide NER lesions such as CPDs. Actinomycin D adducts do have a distinct effect on the DNA structure as compared with NER lesions because this drug does not induce NER (unpublished data).
In addition, lesion-stalled polymerases are targets for ubiquitination (Bregman et al., 1996) and, at least in yeast, proteolysis of polyubiquitinated Rpb1 (largest RNAPII subunit) by Def1 (Woudstra et al., 2002) plays a role in clearing of stalled RNAPII from the lesion. This process provides an escape route when Rad26 (yeast orthologue of CSB) is exhausted or not present (van den Boom et al., 2002; Woudstra et al., 2002). If a similar system would be present in mammalian cells this would provide an explanation that UV-induced RNAPII are cleared in a Def1-dependent fashion, and do not likely form such permanent roadblocks as Actinomycin D.
Distinct kinetic pools of CSB
Here, we have shown that the equilibrium between different kinetic pools of freely diffusing CSB complexes and a “transcription bound” fraction can shift under different transcriptional conditions. The observed temperature dependence of CSB mobility might be due to its ATPase function during association in transcription. Because CSB is essential for TCR, we investigated the consequences of DNA damage on the distribution of CSB over the distinct kinetic pools. Shortly after UV exposure we observed a change in the duration of the transcription-related immobilization of CSB. The maximal immobilization of ∼15% in TCR is significantly smaller than the GGR-induced maximal immobilization of ∼40% for other (core) NER factors (Houtsmuller et al., 1999; Hoogstraten et al., 2002; Rademakers et al., 2003). This implies that the molecular equilibrium, which is established between DNA damage bound and freely diffusing molecules, is different for GGR and TCR. Probably, the immobile fraction of GGR proteins is directly dependent on the DNA damage load, whereas the number of stalled RNAP II elongation complexes in a cell determines the immobile fraction of TCR proteins.
In summary, we find that CSB complexes transiently interact with the transcription machinery during elongation. A possible explanation for this behavior of CSB is that this protein constantly monitors the elongation status of the transcribing polymerases. When a complex is stalled on a DNA lesion the transient interactions of the CSB protein are stabilized, to allow CSB to exert its function in damage removal.
Materials and methods
Generation and characterization of GFP-CSB fusion protein
To generate the GFP-CSB fusion gene, the NH2-terminal HA-tagged CSB cDNA (van Gool et al., 1997) was cloned downstream of the GFP cDNA in the SacI–SalI sites of the pEGFP-C3 expression vector (CLONTECH Laboratories, Inc.). GFP-CSB was stably expressed in CS-B–deficient human fibroblasts (CS1AN-Sv) using SuperFect transfection reagent (QIAGEN). After selection with 300 μg/ml G418, stable transfectants were isolated and selected for UV resistance by exposing cells three times to a UV dose of 4 J/m2 UV-C (254 nm) with daily intervals. Stably expressing clones were characterized for protein expression by immunoblot analysis using an affinity-purified rabbit polyclonal anti-CSB and by UV survival together with VH10-Sv (wt) and untransfected CS1AN-Sv fibroblasts as described previously (van Gool et al., 1997).
Cell culture and specific treatments
The human fibroblasts CS1AN-Sv (CS-B), XP12RO-Sv (XP-A), and wild-type VH10-Sv were grown in a 1:1 mixture of Ham's F10 and DME (GIBCO BRL) supplemented with antibiotics and 10% FCS at 37°C, 5% CO2. Transcription inhibitors were used according to the following conditions: 100 μM N-(2[methylamino]ethyl)-5-isoquinolinesulfonamide (H8; 2 h), 10 μg /ml Actinomycin D (2 h). Treatment with UV light was at 254 nm (UV-C) using a germicidal lamp at the indicated doses. DNA damage in localized areas of the nucleus was performed as described previously (Volker et al., 2001). For azide treatment cells were cultured for 15 min in glucose-free medium (GIBCO BRL) supplemented with 60 mM deoxyglucose and 0.2% Na-azide.
Light microscopy and image analysis
Cells were cultured on sterile glass coverslips. For indirect immunofluorescence, fixation was in 2% PFA in PBS for 10 min at RT. After fixation, cells were permeabilized with 0.1% Triton X-100 in PBS. Endogenous CSB in wild-type VH10-Sv cells was detected with affinity-purified, rabbit polyclonal anti-CSB. Secondary antibody staining was performed with anti–rabbit Alexa 594–conjugated antibodies (Molecular Probes). For fixed cells, fluorescent microscopy images were obtained with a Leitz Aristoplan microscope equipped with epifluorescence optics and a PLANAPO 63X/1.40 oil immersion lens. Confocal laser scanning microscopy images of live cells were recorded with a LSM 410 (Carl Zeiss MicroImaging, Inc.). GFP images were obtained after excitation with 455–490 and long pass emission filter (>510 nm). Alexa 595 images were obtained after excitation with 515–560 and long pass emission filter (580 nm).
In vitro transcription assay
Transcription was assayed in an RTS-containing human recombinant TBP, TFIIB, and TFIIE and purified HeLa TFIIA, TFIIF, TFIIH, and RNAP II as described previously (Gerard et al., 1991). In brief, HA-elution fractions containing CSB were preincubated with the indicated transcription factors and with 100 ng of the adenovirus two major late promoter (Ad2MLP)-containing template for 15 min at 25°C. After the addition of nucleotides, transcription was allowed to proceed for 45 min at 25°C. The 309 nt α-[32P]-CTP run off transcripts were resolved by electrophoresis through a 5% acrylamide/50% urea gel and analyzed by autoradiography.
FRAP
A LSM410 (Carl Zeiss MicroImaging, Inc.) was used for the FRAP experiments. Recovery curves for evaluation of protein mobility were obtained as described previously (Hoogstraten et al., 2002). For FRAP analysis, a 2-μm wide strip, spanning the entire nucleus, was bleached for 200 ms at highest intensity of the 488-nm line of a 15-mW Ar-laser focused by a 40X 1.3 NA oil immersion lens. Subsequently, the recovery of fluorescence in the strip was monitored at intervals of 100 ms with the same laser at 5% of the intensity applied for bleaching, using a dichroic beamsplitter (488/543 nm) and an additional 515–540 nm band pass filter for emission detection. Similarly, combined FLIP and FRAP analysis was performed by giving a 6-s bleach pulse to a strip at the bottom side of the cell. Next, the fluorescent images were made with low laser intensity every 6 s for a total of 3 min.
Computer simulation
For optimal interpretation of the FRAP data we developed a computer modeling environment to simulate FRAP applied to fluorescent molecules inside a finite volume. The FRAP procedures were simulated using experimentally obtained parameters describing lens (beam shape and 3-D intensity distribution, during monitoring and bleach pulse), GFP (quantum yield, susceptibility to bleaching), and nuclear properties (size and shape). Three protein mobility parameters, diffusion coefficient, bound fraction, and duration of binding of individual molecules were varied and the best fit with experimental data was obtained using least square fitting.
Acknowledgments
We thank Drs. L.H.F. Mullenders, A.A. van Zeeland, R. van Driel, and G. Mari-Giglia for helpful suggestions and discussion.
This work was supported by the Centre for Biomedical Genetics, the Dutch Scientific Organization (NWO-ALW, NWO-ZonMW and Spinoza award), the EC contract (Dnage), and the Dutch Cancer Society (Koningin Wilhelmina fonds).
V. van den Boom and E. Citterio contributed equally to this work.
E. Citterio's current address is IFOM-FIRC Institute of Molecular Oncology, via Adamello 16, 20139 Milano, Italy.
Abbreviations used in this paper: 6-4PP, 6-4 photoproduct; CPD, cyclo-butane pyrimidinedimer; CS, Cockayne syndrome; FLIP, fluorescence loss in photobleaching; GGR, global genome repair; RTS, reconstituted transcription system; TCR, transcription-coupled repair; WCE, whole cell extracts.
References
- Balajee, A.S., A. May, G.L. Dianov, E.C. Friedberg, and V.A. Bohr. 1997. Reduced RNA polymerase II transcription in intact and permeabilized Cockayne syndrome group B cells. Proc. Natl. Acad. Sci. USA. 94:4306–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr, V.A., C.A. Smith, D.S. Okumoto, and P.C. Hanawalt. 1985. DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell. 40:359–369. [DOI] [PubMed] [Google Scholar]
- Bradsher, J., J. Auriol, L. Proietti de Santis, S. Iben, J.L. Vonesch, I. Grummt, and J.M. Egly. 2002. CSB is a component of RNA pol I transcription. Mol. Cell. 10:819–829. [DOI] [PubMed] [Google Scholar]
- Bregman, D.B., R. Halaban, A.J. Van Gool, K.A. Henning, E.C. Freidberg, and S.L. Warren. 1996. UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne's syndrome cells. Proc. Natl. Acad. Sci. USA. 93:11586–11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler, V.L., B.A. Maler, and K.R. Yamamoto. 1983. DNA sequences bound specifically by glucocorticoid receptor in vitro render a heterologous promoter hormone responsive in vivo. Cell. 33:489–499. [DOI] [PubMed] [Google Scholar]
- Citterio, E., S. Rademakers, G.T. van der Horst, A.J. van Gool, J.H. Hoeijmakers, and W. Vermeulen. 1998. Biochemical and biological characterization of wild-type and ATPase-deficient Cockayne syndrome B repair protein. J. Biol. Chem. 273:11844–11851. [DOI] [PubMed] [Google Scholar]
- Citterio, E., V. Van Den Boom, G. Schnitzler, R. Kanaar, E. Bonte, R.E. Kingston, J.H. Hoeijmakers, and W. Vermeulen. 2000. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell. Biol. 20:7643–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin, F., E. Bergmann, A. Tremeau-Bravard, and J.M. Egly. 1999. Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J. 18:1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer, J., J.O. Andressoo, J. de Wit, J. Huijmans, R.B. Beems, H. van Steeg, G. Weeda, G.T. van der Horst, W. van Leeuwen, A.P. Themmen, et al. 2002. Premature aging in mice deficient in DNA repair and transcription. Science. 296:1276–1279. [DOI] [PubMed] [Google Scholar]
- Dianov, G.L., J.F. Houle, N. Iyer, V.A. Bohr, and E.C. Friedberg. 1997. Reduced RNA polymerase II transcription in extracts of cockayne syndrome and xeroderma pigmentosum/Cockayne syndrome cells. Nucleic Acids Res. 25:3636–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue, B.A., S. Yin, J.-S. Taylor, D. Reines, and P.C. Hanawalt. 1994. Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc. Natl. Acad. Sci. USA. 91:8502–8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farla, P., R. Hersmus, B. Geverts, P.O. Mari, A.L. Nigg, H.J. Dubbink, J. Trapman, and A.B. Houtsmuller. 2004. The androgen receptor ligand-binding domain stabilizes DNA binding in living cells. J. Struct. Biol. 147:50–61. [DOI] [PubMed] [Google Scholar]
- Friedberg, E.C., G.C. Walker, and W. Siede. 1995. DNA repair and mutagenesis. American Society for Microbiology, Washington D.C. 698 pp.
- Gerard, M., L. Fischer, V. Moncollin, J.-M. Chipoulet, P. Chambon, and J.-M. Egly. 1991. Purification and interaction properties of the human RNA polymerase B(II) general transcription factor BTF2. J. Biol. Chem. 266:20940–20945. [PubMed] [Google Scholar]
- Hartzog, G.A., T. Wada, H. Handa, and F. Winston. 1998. Evidence that Spt4, Spt5, and Spt6 control transcription elongation by RNA polymerase II in Saccharomyces cerevisiae. Genes Dev. 12:357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning, K.A., L. Li, N. Iyer, L. McDaniel, M.S. Reagan, R. Legerski, R.A. Schultz, M. Stefanini, A.R. Lehmann, L.V. Mayne, and E.C. Friedberg. 1995. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell. 82:555–564. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers, J.H. 2001. Genome maintenance mechanisms for preventing cancer. Nature. 411:366–374. [DOI] [PubMed] [Google Scholar]
- Hoogstraten, D., A.L. Nigg, H. Heath, L.H. Mullenders, R. van Driel, J.H. Hoeijmakers, W. Vermeulen, and A.B. Houtsmuller. 2002. Rapid switching of TFIIH between RNA polymerase I and II transcription and DNA repair in vivo. Mol. Cell. 10:1163–1174. [DOI] [PubMed] [Google Scholar]
- Houtsmuller, A.B., S. Rademakers, A.L. Nigg, D. Hoogstraten, J.H.J. Hoeijmakers, and W. Vermeulen. 1999. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science. 284:958–961. [DOI] [PubMed] [Google Scholar]
- Jansen, L.E.T., H. den Dulk, R.M. Brouns, M. de Ruijter, J.A. Brandsma, and J. Brouwer. 2000. Spt4 modulates Rad26 requirement in transcription-coupled nucleotide excision repair. EMBO J. 19:6498–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitori, S., and F. Takusagawa. 1992. Crystal structure of the 2:1 complex between d(GAAGCTTC) and the anticancer drug actinomycin D. J. Mol. Biol. 225:445–456. [DOI] [PubMed] [Google Scholar]
- Kimura, H., K. Sugaya, and P.R. Cook. 2002. The transcription cycle of RNA polymerase II in living cells. J. Cell Biol. 159:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Page, F., E.E. Kwoh, A. Avrutskaya, A. Gentil, S.A. Leadon, A. Sarasin, and P.K. Cooper. 2000. Transcription-coupled repair of 8-oxoguanine: requirement for XPG, TFIIH, and CSB and implications for Cockayne syndrome. Cell. 101:159–171. [DOI] [PubMed] [Google Scholar]
- Lee, S.K., S.L. Yu, L. Prakash, and S. Prakash. 2001. Requirement for yeast RAD26, a homolog of the human CSB gene, in elongation by RNA polymerase II. Mol. Cell. Biol. 21:8651–8656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungman, M., and F. Zhang. 1996. Blockage of RNA polymerase as a possible trigger for u.v. light-induced apoptosis. Oncogene. 13:823–831. [PubMed] [Google Scholar]
- Lu, H., L. Zawel, L. Fisher, J.-M. Egly, and D. Reinberg. 1992. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature. 358:641–645. [DOI] [PubMed] [Google Scholar]
- McNally, J.G., W.G. Müller, D. Walker, R. Wolford, and G.L. Hager. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 287:1262–1265. [DOI] [PubMed] [Google Scholar]
- Mellon, I., G. Spivak, and P.C. Hanawalt. 1987. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 51:241–249. [DOI] [PubMed] [Google Scholar]
- Nance, M.A., and S.A. Berry. 1992. Cockayne syndrome: review of 140 cases. Am. J. Med. Genet. 42:68–84. [DOI] [PubMed] [Google Scholar]
- Neer, E.J., C.J. Schmidt, R. Nambudripad, and T.F. Smith. 1994. The ancient regulatory-protein family of WD-repeat proteins. Nature. 371:297–300. [DOI] [PubMed] [Google Scholar]
- Pazin, M.J., and J.T. Kadonaga. 1997. SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein-DNA interactions? Cell. 88:737–740. [DOI] [PubMed] [Google Scholar]
- Phair, R.D., and T. Misteli. 2000. High mobility of proteins in the mammalian cell nucleus. Nature. 404:604–609. [DOI] [PubMed] [Google Scholar]
- Rademakers, S., M. Volker, D. Hoogstraten, A.L. Nigg, M.J. Mone, A.A. Van Zeeland, J.H. Hoeijmakers, A.B. Houtsmuller, and W. Vermeulen. 2003. Xeroderma pigmentosum group A protein loads as a separate factor onto DNA lesions. Mol. Cell. Biol. 23:5755–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby, C.P., and A. Sancar. 1997. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. USA. 94:11205–11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantin, D., A. Kansal, and M. Carey. 1997. Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol. Cell. Biol. 17:6803–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti, S., and P.C. Hanawalt. 1999. Effect of DNA lesions on transcription elongation. Biochimie. 81:139–146. [DOI] [PubMed] [Google Scholar]
- Tremeau-Bravard, A., T. Riedl, J.M. Egly, and M.E. Dahmus. 2004. Fate of RNA polymerase II stalled at a cisplatin lesion. J. Biol. Chem. 279:7751–7759. [DOI] [PubMed] [Google Scholar]
- Troelstra, C., A. Van Gool, J. De Wit, W. Vermeulen, D. Bootsma, and J.H.J. Hoeijmakers. 1992. ERCC6, a member of a subfamily of putative helicases, is involved in Cockaynes syndrome and preferential repair of active genes. Cell. 71:939–953. [DOI] [PubMed] [Google Scholar]
- van den Boom, V., N.G. Jaspers, and W. Vermeulen. 2002. When machines get stuck–obstructed RNA polymerase II: displacement, degradation or suicide. Bioessays. 24:780–784. [DOI] [PubMed] [Google Scholar]
- van Gool, A.J., E. Citterio, S. Rademakers, R. van Os, W. Vermeulen, A. Constantinou, J.M. Egly, D. Bootsma, and J.H.J. Hoeijmakers. 1997. The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in a RNA polymerase II containing complex. EMBO J. 16:5955–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoffen, A., A.T. Natarajan, L.V. Mayne, A.A. Van Zeeland, L.H.F. Mullenders, and J. Venema. 1993. Deficient repair of the transcribed strand of active genes in Cockaynes syndrome cells. Nucleic Acids Res. 21:5890–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoffen, A., J. Venema, R. Meschini, A.A. van Zeeland, and L.H.F. Mullenders. 1995. Transcription-coupled repair removes both cyclobutane pyrimidine dimers and 6-4 photoproducts with equal efficiency and in a sequential way from transcribed DNA in xeroderma pigmentosum group C fibroblasts. EMBO J. 14:360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema, J., L.H.F. Mullenders, A.T. Natarajan, A.A. van Zeeland, and L.V. Mayne. 1990. The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcripionally active DNA. Proc. Natl. Acad. Sci. USA. 87:4707–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volker, M., M.J. Mone, P. Karmakar, A. van Hoffen, W. Schul, W. Vermeulen, J.H. Hoeijmakers, R. van Driel, A.A. van Zeeland, and L.H. Mullenders. 2001. Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell. 8:213–224. [DOI] [PubMed] [Google Scholar]
- Wada, T., T. Takagi, Y. Yamaguchi, A. Ferdous, T. Imai, S. Hirose, S. Sugimoto, K. Yano, G.A. Hartzog, F. Winston, et al. 1998. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 12:343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woudstra, E.C., C. Gilbert, J. Fellows, L. Jansen, J. Brouwer, H. Erdjument-Bromage, P. Tempst, and J.Q. Svejstrup. 2002. A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature. 415:929–933. [DOI] [PubMed] [Google Scholar]
- Yamaizumi, M., and T. Sugano. 1994. U.v.-induced nuclear accumulation of p53 is evoked through DNA damage of actively transcribed genes independent of the cell cycle. Oncogene. 9:2775–2784. [PubMed] [Google Scholar]
- Yu, A., H.-Y. Fan, D. Liao, A.D. Bailey, and A.M. Weiner. 2000. Activation of p53 or loss of the Cockayne syndrome group B repair protein causes metaphase fragility of human U1, U2, and 5S genes. Mol. Cell. 5:801–810. [DOI] [PubMed] [Google Scholar]