

Abstract
Ezrin, a membrane–actin cytoskeleton linker, which participates in epithelial cell morphogenesis, is held inactive in the cytoplasm through an intramolecular interaction. Phosphatidylinositol 4,5-bisphosphate (PIP2) binding and the phosphorylation of threonine 567 (T567) are involved in the activation process that unmasks both membrane and actin binding sites. Here, we demonstrate that ezrin binding to PIP2, through its NH2-terminal domain, is required for T567 phosphorylation and thus for the conformational activation of ezrin in vivo. Furthermore, we found that the T567D mutation mimicking T567 phosphorylation bypasses the need for PIP2 binding for unmasking both membrane and actin binding sites. However, PIP2 binding and T567 phosphorylation are both necessary for the correct apical localization of ezrin and for its role in epithelial cell morphogenesis. These results establish that PIP2 binding and T567 phosphorylation act sequentially to allow ezrin to exert its cellular functions.
Keywords: ERM proteins; PIP2; actin cytoskeleton; epithelial cell morphogenesis
Introduction
Ezrin is a member of the ezrin/radixin/moesin (ERM) family that acts as a linker between the plasma membrane and the actin cytoskeleton. ERM proteins are involved in cell adhesion, motility, morphogenesis, and also participate in signal transduction pathways (Bretscher et al., 2002; Gautreau et al., 2002).
ERM proteins are recruited to the plasma membrane via their NH2-terminal domain (∼300 residues), which contains both protein and phosphatidylinositol 4,5-bisphosphate (PIP2) binding sites (Algrain et al., 1993; Niggli et al., 1995). They bind to F-actin through the last 34 aa of their COOH-terminal domain (Turunen et al., 1994). In the cytoplasm, ERM proteins are maintained in an inactive conformation through an intramolecular interaction between their NH2-terminal ERM association domain (N-ERMAD) and the last 100 residues of the COOH-terminal ERM association domain (C-ERMAD). This interaction masks membrane and F-actin binding sites (Gary and Bretscher, 1995; Magendantz et al., 1995).
The activation of ERM proteins, resulting in the unmasking of their functional binding sites, occurs through conformational changes triggered by events including the binding to PIP2 and the phosphorylation of a conserved threonine in the actin binding site of the C-ERMAD (T567 in ezrin). ERM proteins phosphorylated at this conserved threonine are localized in membrane extensions rich in actin (Nakamura et al., 1996; Hayashi et al., 1999). Expression of ERM mutants mimicking phosphorylated forms (threonine mutated to aspartic acid, T567D mutation in ezrin) induces the formation of actin-rich membrane projections such as lamellipodia or microvilli (Oshiro et al., 1998; Yonemura et al., 1999; Gautreau et al., 2000). In vitro, the C-ERMAD phosphorylation strongly weakens its interaction with the N-ERMAD (Matsui et al., 1998; Simons et al., 1998). By looking at the crystal structure of a complex between the N- and C-ERMADs of moesin, the threonine residue is seen located at the interface of these two domains (Pearson et al., 2000). Its phosphorylation is predicted to destabilize the N-ERMAD–C-ERMAD interaction through both steric and electrostatic effects. These observations imply that phosphorylation of this conserved threonine contributes to the conformational activation of ERM proteins. However, although this phosphorylation is required, it is not sufficient in vitro for the association of ERM proteins with F-actin indicating that this phosphorylation event is only one step of the activation process (Nakamura et al., 1999).
The binding to PIP2 has also been proposed to play an essential role in the conformational activation of ERM proteins (Nakamura et al., 1999; Yonemura et al., 2002). A PIP2 binding mutant of ezrin is not recruited to the plasma membrane, suggesting that PIP2 binding is essential for the membrane localization of ERM proteins (Barret et al., 2000). Moreover, in vitro, PIP2 regulates the binding of ERM proteins to the cytoplasmic tail of several transmembrane proteins (Hirao et al., 1996; Heiska et al., 1998) and together with the C-ERMAD threonine phosphorylation, the binding to F-actin (Nakamura et al., 1999). The crystal structure of the N-ERMAD of radixin complexed with the polar headgroup of PIP2 shows a slight change of conformation in contrast to N-ERMAD alone (Hamada et al., 2000). These observations indicate that the binding to PIP2 is an additional step required in the conformational activation of ERM proteins.
To analyze the synergy between these two events in the conformational activation of ezrin in vivo, we made use of the mutations abolishing PIP2 binding alone or in combination with the T567D mutation. We demonstrate that PIP2 binding is the primary requirement in the conformational activation of ezrin followed by the threonine phosphorylation. Moreover, we show that this sequence of events is necessary for the apical targeting of ezrin and for the morphogenesis of epithelial cells.
Results and discussion
PIP2 − ezrin is maintained in an inactive conformation in the cytoplasm
To explore the role of PIP2 in the conformational activation of ezrin, we used the LLC-PK1 epithelial cell line, which is derived from the kidney proximal tubule and develops microvilli at the apical surface. We derived stable clones producing an ezrin variant in which the binding to PIP2 is abolished (PIP2 − ezrin; Barret et al., 2000). This ezrin mutant has two lysine doublets mutated to asparagine (K253/254N and K262/263N). Wild-type (wt) ezrin was mainly localized at the apical microvilli but also observed as a weak diffuse staining corresponding to the cytoplasmic pool and as a faint signal at cell–cell contacts. In contrast, the PIP2 − ezrin was completely cytoplasmic and absent from microvilli (Fig. 1 A). To assess the ability of PIP2 − ezrin to associate with the actin cytoskeleton we performed, before immunofluorescence, an extraction with a Triton X-100 buffer preserving the cytoskeleton and cytoskeleton-associated proteins. No staining could be detected in cells expressing PIP2 − ezrin in contrast to wt ezrin, which remained associated with the microvillus actin cytoskeleton (Fig. 1 A). Quantification after Western blot analysis of soluble and insoluble fractions confirmed that PIP2 − ezrin was threefold less insoluble than wt ezrin (Fig. 1 B). This indicates that PIP2 − ezrin cannot bind to the actin cytoskeleton. Therefore, we quantified the amount of ezrin in the membrane and cytosolic pools after cell fractionation. We found a threefold reduction in the amount of PIP2 − ezrin in the membrane fraction in comparison with wt ezrin showing that PIP2 − ezrin can no longer be recruited at the membrane (Fig. 1 C). To confirm the role of PIP2 in the recruitment of ezrin to the membrane, we treated cells with ionomycin in presence of Ca2+. This pharmacological treatment leads to the hydrolysis of PIP2 (Várnai and Balla, 1998). After 30 min, wt ezrin was released from the membrane and resided in the cytoplasm where upon it became fully extractable with Triton X-100 buffer (Fig. 1 D). Thus, in these treated cells, wt ezrin behaved similarly to PIP2 − ezrin. Because the cytoplasmic localization of PIP2 − ezrin is not due to a folding defect, confirmed by both tryptophan fluorescence spectrum analysis and chymotrypsin digestion (unpublished data), we conclude that the cytoplasmic localization of PIP2 − ezrin is due to its inability to bind PIP2.
Figure 1.

PIP2 − ezrin is restricted to the cytoplasm. (A) Localization of wt and PIP2 − ezrin by double fluorescence with anti–VSV G antibody (ezrin VSVG) and phalloidin (actin) with or without preextraction with a 0.5% Triton X-100 buffer. Images of xz sections, and apical, median, basal focal xy sections are shown. wt ezrin is associated with actin of the apical microvilli. PIP2 − ezrin is present in the cytoplasm and is totally extracted with detergent. Bars, 10 μm. (B) Association of wt, PIP2 −, T/D, and PIP2 − T/D ezrin with the cytoskeleton. Similar amount of total (Tot), soluble (Sol) or insoluble material (Ins) were analyzed by Western blotting with an anti–VSV G antibody. (C) Distribution of ezrin mutants in membrane and cytosol fractions. Total (Tot), cytosolic (Cyt), and membrane-associated material concentrated eightfold (Mb 8x) were immunoblotted with anti–VSV G antibody. The amount of ezrin was quantified by densitometric analysis of serially diluted fractions obtained from three independent experiments. Average values are given ± SEM. (D) wt ezrin is released from the membrane and extracted with Triton X-100 buffer (+ext) after ionomycin treatment (iono).
The accumulation of PIP2 − ezrin in the cytoplasm suggests either that PIP2 is the major membrane binding partner of ezrin or alternatively that PIP2 is required for its conformational activation unmasking other NH2-terminal membrane binding sites. To discriminate between these two possibilities we derived stable LLC-PK1 clones expressing the NH2-terminal domain either in its wt form (Nter ezrin) or carrying the PIP2 − mutation (NterPIP2 − ezrin). We showed that both Nter ezrin and NterPIP2 − ezrin were localized at the membrane in an unpolarized manner and were not found in the cytoplasm (Fig. 2 A). Consistent with these fluorescence data, analysis of membrane and cytosolic pools indicated that both Nter ezrin and NterPIP2 − ezrin were almost exclusively in the membrane fraction (Fig. 2 B). This result highlights the importance of the N-ERMAD–C-ERMAD interaction in the regulation of the recruitment of ezrin at the membrane because, in comparison, only 10% of full-length ezrin is present at the membrane (Fig. 1 C). Most importantly, it demonstrates that PIP2 is not the major membrane partner that dictates the membrane association of ezrin but is the primary requirement in the conformational activation of the full-length molecule unmasking other membrane binding sites. Altogether, these results indicate that PIP2 − ezrin is cytoplasmic because it is maintained in an inactive conformation.
Figure 2.

Membrane localization of Nter and NterPIP 2 − ezrin. (A) Fluorescence was performed as in Fig. 1. Nter and NterPIP2 − ezrin are localized at the membrane but in a nonpolarized manner. Bar, 10 μm. (B) Distribution of ezrin mutants in membrane and cytosol fractions. Total (Tot), cytosol (Cyt), and membrane-associated material (Mb) were immunoblotted with an anti–VSV G antibody.
Ezrin phosphorylation at T567 requires the interaction of the N-ERMAD with PIP2
Because the T567 phosphorylation of ezrin has been shown to play an important role in its conformational activation, we examined the level of T567 phosphorylation of PIP2 − ezrin using a T567 phosphospecific antibody. Western blots were performed after gel electrophoresis of immunoprecipitated proteins. Although a signal was detected with wt ezrin, which corresponded to the phosphorylated protein because it was abolished after phosphatase treatment, no signal was observed with PIP2 − ezrin (Fig. 3 A). This result indicates that the binding of ezrin to PIP2 is required for and precedes T567 phosphorylation in the mechanism of ezrin activation. The interaction of ezrin with PIP2 could target the protein at the membrane at the vicinity of the kinase. To test this hypothesis, we fused wt and PIP2 − ezrin to the pleckstrin homology (PH) domain of the phospholipase C-δ1 (wtPH and PIP2 − PH ezrin), which interacts with PIP2 at the plasma membrane, and we analyzed the subcellular distribution and the phosphorylation of these chimeras after transient transfection of LLC-PK1 cells. Both wtPH and PIP2 − PH ezrin were localized at the plasma membrane and in the microvilli (Fig. 3 B). After cell fractionation, both chimeras were found almost exclusively in the membrane fraction (Fig. 3 C). Although these two chimeras had the same localization in the cells, only wtPH ezrin was phosphorylated at T567 (Fig. 3 D). Altogether, these results show that the targeting of ezrin to the membrane is not sufficient for its phosphorylation at T567. Rather, our results indicate that the interaction of the ezrin N-ERMAD with PIP2 is necessary for the subsequent phosphorylation of T567. This suggests that the interaction of the N-ERMAD with PIP2 induces a conformational rearrangement that allows the phosphorylation at T567. Such a conformational change has been observed upon analysis of the crystal structure of the N-ERMAD complexed to the polar headgroup of PIP2 (Hamada et al., 2000).
Figure 3.

Ezrin phosphorylation at T567 requires the interaction of the N-ERMAD with PIP2. (A) Immunoprecipitated wt and PIP2 − ezrin were analyzed by Western blot with an anti–VSV G antibody (VSVG) or with an antibody recognizing ezrin phosphorylated at T567 (T567P). +PPase indicates than immunoprecipitated proteins were treated with λ-phosphatase. In contrast to wt ezrin, PIP2 − ezrin is not phosphorylated at T567. (B) After transient transfection, wtPH and PIP2 − PH ezrin were localized by immunofluorescence with an anti–VSV G antibody. Both proteins are localized at the plasma membrane and in microvilli. Bar, 10 μm. (C) Distribution of wtPH and PIP2 − PH ezrin in membrane and cytosol fractions was obtained as in Fig. 2. (D) Immunoprecipitated wtPH ezrin is phosphorylated at T567, whereas PIP2 − PH ezrin is not phosphorylated.
The T567D mutation restores the membrane–cytoskeleton linker capacity of PIP2 − ezrin
Because the binding of ezrin to PIP2 is required for its subsequent phosphorylation, we determined whether the T567D mutation in the PIP2 − ezrin mutant (PIP2 − T/D ezrin) would be sufficient to restore the apical membrane localization and the binding to the actin cytoskeleton. Whereas PIP2 − ezrin was found restricted to the cytoplasm (Fig. 1 A), PIP2 − T/D ezrin was also recruited to the plasma membrane, similar to ezrin carrying the T567D mutation alone (T/D ezrin; Fig. 4 A). This suggests that the T567D mutation allows the unmasking of the PIP2-independent membrane binding sites (Fig. 1 C). In addition, both T/D ezrin and PIP2 − T/D ezrin remained associated with the cortical actin cytoskeleton after extraction of cells with the Triton X-100 buffer indicating that T567D mutation is sufficient to unmask the actin binding site independently of PIP2 binding (Fig. 1 B and Fig. 4 A). Moreover, ionomycin treatment had no effect on either the membrane localization or the association with the actin cytoskeleton of both T/D and PIP2 − T/D ezrin (Fig. 4 B). Altogether, our results show that the T567D mutation restores the capacity of PIP2 − ezrin to act as a membrane–actin cytoskeleton linker and demonstrates that binding of ezrin to PIP2 is dispensable for the unmasking of functional binding sites in vivo, in contrast to what was described in vitro (Nakamura et al., 1999).
Figure 4.
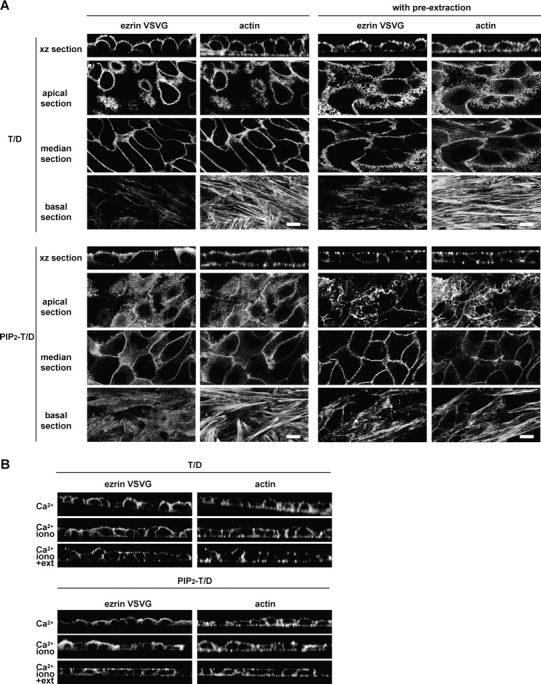
Unpolarized localization of T/D and PIP2 − T/D ezrin at the plasma membrane. Fluorescence was performed as in Fig. 1. T/D ezrin was mainly located on the dorsal surface of the cells and in cell–cell contacts. PIP2 − T/D ezrin was uniformly distributed at the plasma membrane. T/D and PIP2 − T/D ezrin remained associated with the cortical actin cytoskeleton after extraction with the Triton X-100 buffer. Bars, 10 μm. (B) Membrane localization of T567D and PIP2 − T/D ezrin and cytoskeleton association are preserved after ionomycin treatment followed by extraction with the Triton X-100 buffer before fixation.
However, neither PIP2 − T/D ezrin, nor T/D ezrin had a restricted apical localization (Fig. 4 A). The factors controlling the apical localization of ezrin are still unknown. But our results indicate that the release of the N-ERMAD–C-ERMAD interaction leading to the unmasking of PIP2 independent membrane binding sites is not sufficient, in itself, for the apical localization of ezrin. This indicates that regulated unmasking of the membrane binding sites in the NH2-terminal domain is required for this apical localization consistent with our observations using the Nter mutants (Fig. 2 A). The sequence of events consisting of binding to PIP2 followed by phosphorylation at T567 is critical for the restricted apical localization of wt ezrin.
Unlike T/D ezrin, PIP2 − T/D ezrin does not alter epithelial cell morphology
We have shown previously that production of T/D ezrin in epithelial cells induced the formation of lamellipodia, membrane ruffles, tufts of microvilli, and impaired cell–cell contacts indicating a critical role for ezrin in epithelial cell morphogenesis (Gautreau et al., 2000; Pujuguet et al., 2003). To test whether ezrin binding to PIP2 is necessary for its role in epithelial cell morphogenesis we analyzed the surface of cells expressing PIP2 − and PIP2 − T/D ezrin by scanning electron microscopy (Fig. 5). In contrast to cells producing T/D ezrin, we observed that cells producing PIP2 − T/D ezrin formed a regular monolayer with surface microvilli resembling those of cells overexpressing wt ezrin (Fig. 5). This suggests that despite the ability of PIP2 − T/D ezrin to act as a linker between the membrane and the actin cytoskeleton, its expression does not affect epithelial cell morphology as seen with T/D ezrin. This suggests that the conformational activation unmasking functional binding sites alone is not sufficient to allow ezrin to exert its signaling functions. Therefore, correct regulation of ezrin activation is dependent on the binding of ezrin to PIP2.
Figure 5.
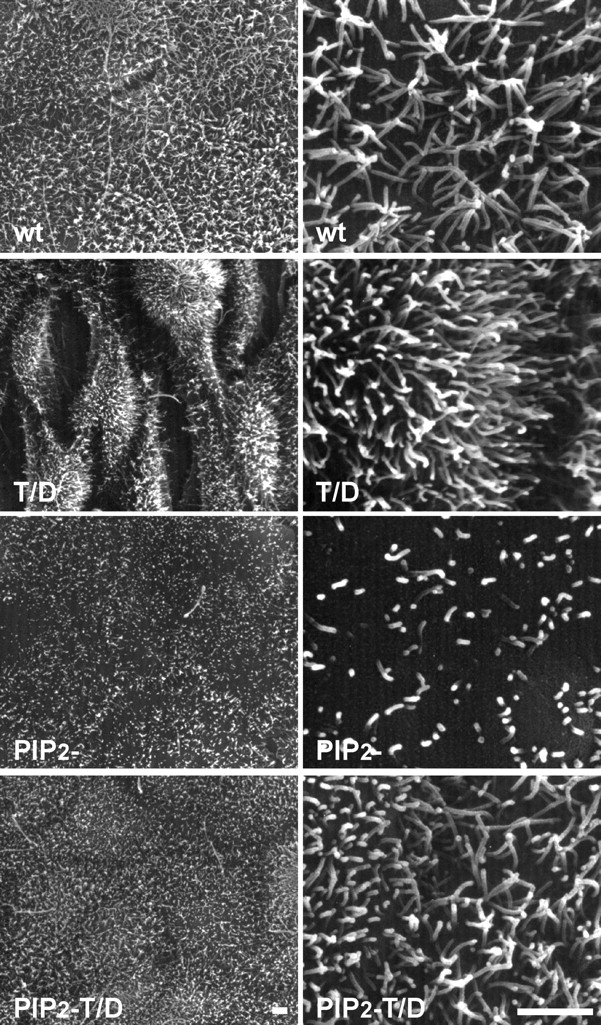
Binding of ezrin to PIP2 is required for its function in epithelial cell morphogenesis. Scanning electron microscopy of confluent cells grown on filters for 4 d producing wt, T/D, PIP2 −, and PIP2 − T/D ezrin. Clones producing PIP2 − ezrin formed a well-organized monolayer displaying shorter and more disperse microvilli on their cell surface as compared with cells producing wt ezrin. Cells producing PIP2 − T/D ezrin did not show the aberrant morphology observed with cells producing T/D ezrin. Bars, 2 μm.
We observed that although cells expressing PIP2 − ezrin formed a well-organized monolayer, they displayed, on their apical surface, fewer and shorter microvilli compared with the abundant and regular microvilli seen on cells expressing wt ezrin. Thus, production of PIP2 − ezrin had a strong dominant negative effect on microvillus formation. We did not detect any effect of PIP2 − ezrin on the amount and localization of the phosphorylated endogenous ERM proteins (unpublished data). This indicates that the absence of microvilli in cells producing PIP2 − ezrin is not due to a dominant negative effect on the activation of endogenous ERM proteins, but rather is due to an effect on a downstream effector of ezrin involved in microvillus formation.
Recently, genetic analysis of Drosophila moesin, the only ERM protein in this organism, has stressed the role of this protein in the control of epithelial cell integrity and polarity (Polesello et al., 2002; Speck et al., 2003). Our experiments have demonstrated that to exert its cellular functions at the apical pole of epithelial cells, ezrin undergoes a conformational activation, which requires primarily PIP2 binding and the subsequent phosphorylation at T567. Future works using these well-characterized mutants of ezrin should further the understanding of how the regulated conformational activation of ezrin controls its signaling functions in the development and maintenance of epithelial cell polarity.
Materials and methods
Cell culture
LLC-PK1 cells (CCL 101; American Type Culture Collection) were grown in DME (GIBCO BRL) and supplemented with 10% FCS, at 37°C in 10% CO2.
Antibodies
The mouse monoclonal anti–vesicular stomatitis virus glycoprotein (VSV G) antibody (clone P5D4) was described previously (Kreis, 1986). T567 phospho-ezrin pAb was purchased from Cell Signaling Technology.
DNA constructs and stable transfection
The pCB6 vectors containing cDNA coding for VSV G–tagged ezrin carrying the K253N, K254N, K262N, and K263N mutations (PIP2 − ezrin) or the T567D mutation (T/D ezrin) were described previously (Barret et al., 2000; Gautreau et al., 2000). The pCB6 vector containing cDNA coding for VSV G–tagged Nter ezrin (1-309) was described previously (Algrain et al., 1993). The pCB6 vectors containing cDNA coding for wtPH and PIP2 − PH ezrin were obtained by an in-frame insertion of a fragment coding for the PH domain of rat phospholipase C-δ1 (10-139) at the 3′ end of the VSV G–ezrin cDNA. All constructs were obtained by standard techniques and verified by sequencing. Stable LLC-PK1 clones were obtained as described previously (Gautreau et al., 2000). All experiments were performed with three independent clones for each construct and gave similar results.
Immunofluorescence
105 cells were seeded on 1-cm2 polyester filters (Transwell; Costar Corp.) and grown for 4 d. Cells were fixed with 3% PFA and permeabilized with 0.5% Triton X-100. Cells were subsequently incubated with anti–VSV G antibody and then with Alexa 488–conjugated goat anti–mouse secondary antibody (Jackson ImmunoResearch Laboratories) and with TRITC-labeled phalloidin (Sigma Chemical Co.). The samples were analyzed with an SP2 confocal laser scanning microscope (Leica). When indicated, living cells were incubated for 30 min with 10 mM CaCl2 and 30 μM ionomycin (Sigma Chemical Co.) in PBS. Extraction was performed before fixation by treating the cells for 1 min with a Triton X-100 buffer (50 mM MES, 3 mM EGTA, 5 mM MgCl2, 0.5% Triton X-100, pH 6.4) at 20°C.
Scanning electron microscopy
5 × 105 cells were seeded on 4.7 cm2 polycarbonate filters and grown for 4 d. Samples were fixed with 2.5% glutaraldehyde, dehydrated in a graded series of ethanol incubation, dried by the critical point method using liquid CO2, coated with gold palladium, and observed with a microscope (model JSM 840A; JEOL).
Immunoprecipitation
Immunoprecipitations were performed as described previously (Gautreau et al., 2000). When indicated, immunoprecipitated proteins were treated with λ-phosphatase (New England Biolabs, Inc.) according to the manufacturer's instructions.
Analysis of detergent-soluble and -insoluble fractions
Cellular fractions were obtained from 6-well plates confluent cultures. Total cellular fractions were collected with Laemmli buffer at 100°C. Soluble fractions were prepared by a 1-min extraction with Triton X-100 buffer at 20°C and supplemented with 3× Laemmli buffer. The insoluble fractions were extracted with Laemmli buffer at 100°C. Samples were analyzed by SDS-PAGE and immunoblotting. Densitometric analysis were performed with the Scion image program (NIH image).
Cytosol/membrane fractionation
Confluent cultures from 10-cm dishes were mechanically disrupted using a cell cracker in 10 mM Hepes, 1 mM EDTA, 150 mM NaCl, pH 7.4, buffer containing a cocktail of protease inhibitors. The homogenates were clarified by centrifugation at 600 g. An aliquot of the supernatant was supplemented with 3× Laemmli buffer and corresponded to the total fraction. The supernatant was subjected to a 30-min centrifugation at 100,000 g. The resulting supernatant was supplemented with 3× Laemmli buffer (cytosolic fraction) and the membrane pellets were solubilized in Laemmli buffer (membrane fraction). Samples were analyzed by SDS-PAGE and immunoblotting.
Acknowledgments
We thank M. Grasset for her assistance in scanning electron microscopy and Drs. J. Srivastava and P. Pujuguet for critical reading of the manuscript.
This work was supported by grants from Ligue Nationale contre le Cancer (équipe labellisée), Association pour la Recherche contre le Cancer (ARC 5599 and 4601). B.T. Fievet is a recipient of a fellowship from the Ministère de l'Education Nationale, de la Recherche et de la Technologie.
Abbreviations used in this paper: C-ERMAD, COOH-terminal ERM association domain; ERM, ezrin/radixin/moesin; N-ERMAD, NH2-terminal ERMAD; PH, pleckstrin homology; PIP2, phosphatidylinositol 4,5-bisphosphate; VSV G, vesicular stomatitis virus glycoprotein; wt, wild-type.
References
- Algrain, M., O. Turunen, A. Vaheri, D. Louvard, and M. Arpin. 1993. Ezrin contains cytoskeleton and membrane binding domains accounting for its proposed role as a membrane–cytoskeletal linker. J. Cell Biol. 120:129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barret, C., C. Roy, P. Montcourrier, P. Mangeat, and V. Niggli. 2000. Mutagenesis of the phosphatidylinositol 4,5-biphosphate (PIP2) binding site in the NH2-terminal domain of ezrin correlates with its altered cellular distribution. J. Cell Biol. 151:1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher, A., K. Edwards, and R.G. Fehon. 2002. ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3:586–599. [DOI] [PubMed] [Google Scholar]
- Gary, R., and A. Bretscher. 1995. Ezrin self-association involves binding of an N-terminal domain to a normally masked C-terminal domain that includes the F-actin binding site. Mol. Biol. Cell. 6:1061–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautreau, A., D. Louvard, and M. Arpin. 2000. Morphogenic effects of ezrin require a phosphorylation-induced transition from oligomers to monomers at the plasma membrane. J. Cell Biol. 150:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautreau, A., D. Louvard, and M. Arpin. 2002. ERM proteins and NF2 tumor suppressor: the Yin and Yang of cortical actin organization and cell growth signaling. Curr. Opin. Cell Biol. 14:104–109. [DOI] [PubMed] [Google Scholar]
- Hamada, K., T. Shimizu, T. Matsui, S. Tsukita, S. Tsukita, and T. Hakoshima. 2000. Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 19:4449–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, K., S. Yonemura, T. Matsui, and S. Tsukita. 1999. Immunofluorescence detection of ezrin/radixin/moesin (ERM) proteins with their carboxyl-terminal threonine phosphorylated in cultured cells and tissues. J. Cell Sci. 112:1149–1158. [DOI] [PubMed] [Google Scholar]
- Heiska, L., K. Alfthan, M. Grönholm, P. Vilja, A. Vaheri, and O. Carpén. 1998. Association of ezrin with intercellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2). J. Biol. Chem. 273:21893–21900. [DOI] [PubMed] [Google Scholar]
- Hirao, M., N. Sato, T. Kondo, S. Yonemura, M. Monden, T. Sasaki, Y. Takai, S. Tsukita, and S. Tsukita. 1996. Regulation mechanism of ERM (ezrin/radixin/moesin) protein/plasma membrane association: possible involvement of phosphatidylinositol turnover and rho-dependent signaling pathway. J. Cell Biol. 135:37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis, T.E. 1986. Microinjected antibodies against the cytoplasmic domain of vesicular stomatitis virus glycoprotein block its transport to the cell surface. EMBO J. 5:931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magendantz, M., M.D. Henry, A. Lander, and F. Solomon. 1995. Interdomain interactions of radixin in vitro. J. Biol. Chem. 270:25324–25327. [DOI] [PubMed] [Google Scholar]
- Matsui, T., M. Maeda, Y. Doi, S. Yonemura, M. Amano, K. Kaibuchi, S. Tsukita, and S. Tsukita. 1998. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J. Cell Biol. 140:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, F., M.R. Amieva, C. Hirota, Y. Mizuno, and H. Furthmayr. 1996. Phosphorylation of T-558 of moesin detected by site-specific antibodies in RAW264.7 macrophages. Biochem. Biophys. Res. Commun. 226:650–656. [DOI] [PubMed] [Google Scholar]
- Nakamura, F., L. Huang, K. Pestonjamasp, E.J. Luna, and H. Furthmayr. 1999. Regulation of F-actin binding to platelet moesin in vitro by both phosphorylation of threonine 558 and polyphosphatidylinositides. Mol. Biol. Cell. 10:2669–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niggli, V., C. Andréoli, C. Roy, and P. Mangeat. 1995. Identification of a phosphatidylinositol-4, 5-biphosphate-binding domain in the N-terminal region of ezrin. FEBS Lett. 376:172–176. [DOI] [PubMed] [Google Scholar]
- Oshiro, N., Y. Fukata, and K. Kaibuchi. 1998. Phosphorylation of moesin by Rho-associated kinase (Rho-kinase) plays a crucial role in the formation of microvilli-like structures. J. Biol. Chem. 273:34663–34666. [DOI] [PubMed] [Google Scholar]
- Pearson, M.A., D. Reczek, A. Bretscher, and P.A. Karplus. 2000. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell. 101:259–270. [DOI] [PubMed] [Google Scholar]
- Polesello, C., I. Delon, P. Valenti, P. Ferrer, and F. Payre. 2002. Dmoesin controls actin-based cell shape and polarity during Drosophila melanogaster oogenesis. Nat. Cell Biol. 4:782–789. [DOI] [PubMed] [Google Scholar]
- Pujuguet, P., L. Del Maestro, A. Gautreau, D. Louvard, and M. Arpin. 2003. Ezrin regulates E-cadherin-dependent adherens junction assembly through Rac1 activation. Mol. Biol. Cell. 14:2181–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, P.C., S.F. Pietromonaco, D. Reczek, A. Bretscher, and L. Elias. 1998. C-terminal threonine phosphorylation activates ERM proteins to link the cell's cortical lipid bilayer to the cytoskeleton. Biochem. Biophys. Res. Commun. 253:561–565. [DOI] [PubMed] [Google Scholar]
- Speck, O., S.C. Hughes, N.K. Noren, R.M. Kulikauskas, and R.G. Fehon. 2003. Moesin functions antagonistically to the Rho pathway to maintain epithelial integrity. Nature. 421:83–87. [DOI] [PubMed] [Google Scholar]
- Turunen, O., T. Wahlström, and A. Vaheri. 1994. Ezrin has a COOH-terminal actin-binding site that is conserved in the ezrin protein family. J. Cell Biol. 126:1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Várnai, P., and T. Balla. 1998. Visualization of phosphoinositides that bind pleckstrin homolgy domains: calcium- and agonist-induced dynamic changes and relationship to Myo-[3H]inositol–labeled phosphoinositide pools. J. Cell Biol. 143:501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura, S., S. Tsukita, and S. Tsukita. 1999. Direct involvement of ezrin/radixin/moesin (ERM)-binding membrane proteins in the organization of microvilli in collaboration with activated proteins. J. Cell Biol. 145:1497–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura, S., T. Matsui, S. Tsukita, and S. Tsukita. 2002. Rho-dependent and -independent activation mechanisms of ezrin/radixin/moesin proteins: an essential role for polyphosphoinositides in vivo. J. Cell Sci. 115:2569–2580. [DOI] [PubMed] [Google Scholar]