

Abstract
Presenilin-1 null mutation (PS1 −/−) in mice is associated with morphological alterations and defects in cleavage of transmembrane proteins. Here, we demonstrate that PS1 deficiency also leads to the formation of degradative vacuoles and to the aberrant translocation of presynaptic α- and β-synuclein proteins to these organelles in the perikarya of primary neurons, concomitant with significant increases in the levels of both synucleins. Stimulation of autophagy in control neurons produced a similar mislocalization of synucleins as genetic ablation of PS1. These effects were not the result of the loss of PS1 γ-secretase activity; however, dysregulation of calcium channels in PS1 −/− cells may be involved. Finally, colocalization of α-synuclein and degradative organelles was observed in brains from patients with the Lewy body variant of AD. Thus, aberrant accumulation of α- and β-synuclein in degradative organelles are novel features of PS1 −/− neurons, and similar events may promote the formation of α-synuclein inclusions associated with neurodegenerative diseases.
Keywords: autophagy; calcium dysregulation; neurodegenerative diseases
Introduction
Presenilin-1 (PS1) is one of several proteins linked to early onset, familial Alzheimer's disease (AD). PS1 null (−/−) mice die in utero, and PS1 −/− embryos have multiple abnormalities including smaller size, skeletal malformations, nervous system hemorrhages, and fewer neurons (Wong et al., 1997). PS1 was initially shown to play a role in the intramembrane cleavage of the amyloid precursor protein (APP; De Strooper et al., 1998) and Notch (De Strooper et al., 1999) and was subsequently documented to cleave additionally a growing list of type I transmembrane proteins including Erb-b4, E-cadherin, the low density lipoprotein-related protein, nectin-1α, and CD44, among others (Selkoe and Kopan, 2003). PS1 has additional functions unrelated to proteolytic cleavage, including effects on the wnt signaling pathway (Kang et al., 1999), cell adhesion junction assembly (Georgakopoulos et al., 1999), calcium channel regulation (Leissring et al., 2000; Yoo et al., 2000), trafficking of secretory proteins to the cell surface (Naruse et al., 1998; Kaether et al., 2002) and kinesin-dependent vesicle transport (Kamal et al., 2001; Pigino et al., 2003). Thus, the diverse pathologies seen in PS1 −/− mice likely reflect the pleiotropic activities of PS1.
Here, we have identifed a novel phenotype characterized by clusters of autophagic lysosomal organelles and the aberrant localization of synuclein proteins to perikarya in neurons from PS1 −/− mice. Synucleins are a family of small acidic proteins comprised of α-, β-, and γ-synuclein (George, 2002). Abnormal cytoplasmic aggregation of α-synuclein has been implicated as the primary pathology in a variety of neurodegenerative diseases including Parkinson's disease (PD), dementia with Lewy bodies, the Lewy body variant of AD (LBVAD), multiple system atrophy, and neurodegeneration with brain iron accumulation, which are collectively termed α-synucleinopathies (Duda et al., 2000). β- and γ-Synuclein accumulations also are seen in some α-synucleinopathies (Galvin et al., 1999). In normal brains, α- and β-synuclein are localized predominantly to central nervous system presynaptic terminals (Maroteaux et al., 1988; Jakes et al., 1994), especially near synaptic vesicles (George et al., 1995; Iwai et al., 1995).
The presynaptic location of α- and β-synuclein suggests they play a role in synaptic function (Withers et al., 1997), perhaps via interaction with synaptic vesicles (Maroteaux and Scheller, 1991). Depletion of α-synuclein in cultured hippocampal neurons or mice selectively diminishes the distal pool of synaptic vesicles in presynaptic terminals implicating α-synuclein in vesicle positioning or recycling (Murphy et al., 2000; Cabin et al., 2002). Synucleins may be additionally involved in signal transduction (Nakajo et al., 1993; Shibayama-Imazu et al., 1993; Ostrerova et al., 1999; Iwata et al., 2001) or have chaperone-like functions (Ostrerova et al., 1999; Souza et al., 2000).
Furthermore, because a hydrophobic fragment of α-synuclein was isolated from amyloid plaques in AD (Ueda et al., 1993) and α-synuclein–positive Lewy bodies frequently occur in familial AD (FAD) brains associated with PS1 mutations (Lippa et al., 1998), the formation of α-synuclein inclusions may be linked to mechanisms underlying AD. We examined this possibility and show here that degradative organelles increase and selectively accumulate α- and β-synuclein in PS1 −/− neurons, thereby implicating PS1 deficiencies in mechanisms of α- and β-synuclein trafficking and degradation which could play a role in α-synuclein lesions in neurodegenerative diseases such as FAD and LBVAD.
Results
PS1 deficiency resulted in the formation of enlarged lysosomes in fibroblasts and primary neurons
Examination by differential interference contrast microscopy revealed the presence of enlarged organelles in immortalized fibroblasts derived from PS1 −/− mice. These enlarged structures occupied a significant volume of the cytoplasm (Fig. 1 A), distending the flat surface of the fibroblasts. To determine the identity of these structures, we probed the cells with a panel of organelle-specific antibodies. Antibodies to lysosomal membrane-associated protein 2 (Lamp-2) selectively labeled these organelles, suggesting a lysosomal origin (Fig. 1, B and C). Similarly, an accumulation of Lamp-2 immunolabeled organelles also was detected in primary neurons derived from PS1 −/− mice (Fig. 1, D and E) but not from those derived from PS1 +/? mice (Fig. 1 F), suggesting that the accumulation of these organelles is a general property of PS1 deficiency. Significantly, these organelles were not present in PS2 −/− cells, suggesting that this phenotype is independent of PS2 (unpublished data).
Figure 1.
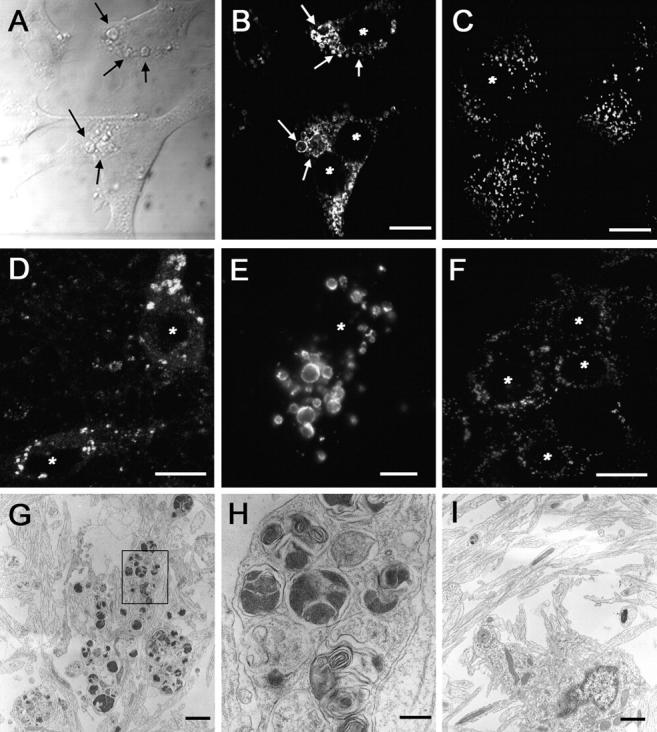
Enlarged lysosomal organelles are visible in PS1 −/− cells. (A) Differential contrast interference imaging of the cell surface of PS1 −/− fibroblasts demonstrated that a significant volume of the cells was occupied by large, spherical structures (arrows point to examples of enlarged vesicular structures). Lamp-2 immunolabeling of PS1 −/− (B) or PS1 +/? (C) fibroblasts demonstrated that these enlarged structures expressed lysosomal marker proteins and were present only within PS1 −/− cells. Similar accumulations of Lamp-2–positive organelles were detected in (D and E) PS1 −/− but not (F) PS1 +/? mouse primary neurons. Transmission EM analysis of PS1 −/− (G and H) or PS1 +/?. (I) Neurons demonstrated that PS1 −/− neurons contain large clusters of organelles. H is a higher magnification of the boxed area in G. *, denotes nucleus. Bars: (B–D and F) 10 μm; (E) 5 μm; (G and I) 1 μm; (H) 200 nm.
To further confirm that the enlarged structures represent lysosomal vacuoles, we used transmission EM to visualize large clusters of lysosomal organelles that occupied a significant portion of PS1 −/− neuronal perikarya (Fig. 1, G and H), but not control PS1 +/? neurons (Fig. 1 I). These organelles consisted of both electron-dense and multilamellar material. Thus, ultrastructural analyses suggest that these organelles are likely to represent enlarged lysosomal or autophagic vacuoles.
α- and β-synuclein are selectively mislocalized to enlarged lysosomes in PS1 −/− neurons
To determine the biological consequences of these enlarged organelles, we examined the distribution of a number of synaptic and cytoskeletal proteins in PS1 −/− neurons. α- and β-synuclein, two presynaptic proteins, were selectively redistributed to neuronal perikarya. In mouse PS1 +/+ (not depicted) or PS1 +/? neurons cultured for 10 d, both α-synuclein (Fig. 2 A) and β-synuclein (not depicted) were almost completely localized to punctate structures immunoreactive for the presynaptic marker proteins. However, when PS1 −/− neurons were immunolabeled with antibodies to either α-synuclein (Fig. 2 B) or β-synuclein (Fig. 2 C), most synuclein immunoreactivity did not colocalize with the synaptic marker proteins, but instead was localized to vesicles clustered around nuclei and extending into the proximal processes of 30–40% of PS1 −/− neurons (Fig. 2, B and C, arrows). Double labeling of PS1 −/− neurons with antibodies to α-synuclein and Lamp-2 demonstrated a clear colocalization of the perikaryal synuclein immunoreactivity with the enlarged, Lamp-positive organelles (Fig. 2, E and F), not seen in PS1 +/? neurons (Fig. 2 D). Higher magnification images revealed both Lamp-2 and synuclein immunoreactivity colocalized to the outer edges of these lysosomal structures, producing a ringlike appearance (Fig. 2 F). Similar colocalization was demonstrated with antibodies to additional lysosomal proteins (Lamp-1, cathepsin D; unpublished data).
Figure 2.
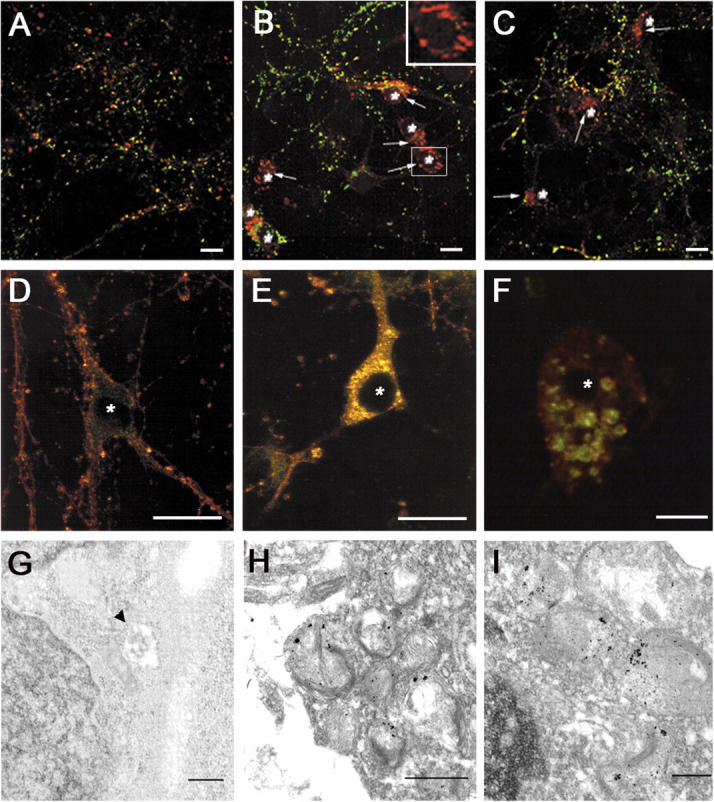
Perinuclear synuclein accumulations can be visualized in association with lysosomes in PS1 −/− neurons. (A) PS1 +/? or (B and C) PS1 −/− primary neuronal cultures were immunolabeled with antibodies to synaptophysin (green) and α-synuclein (A and B, red; Ab SNL-1) or synapsin I (green) and β-synuclein (C, red; Ab Syn 207). In PS1 −/− neurons, α- and β-synuclein are found in perinuclear locations that do not contain other presynaptic proteins (B and C, arrows). (D–F) Colocalization of lysosomal-associated membrane protein 2 (LAMP-2) with perinuclear synuclein accumulations in PS1 −/− neurons. (D) PS1 +/? or (E and F) PS1 −/− neurons cultured for 10 d were immunolabeled with antibodies to synuclein (Syn 202, red) and LAMP-2 (ABL93, green). (G–I) Immuno-EM of synuclein association with perinuclear multivesicular and autophagic organelles in PS1 −/− neurons. Synuclein is not found in association with lysosomes in PS1 +/? neurons (G, arrowhead) but is localized to enlarged lysosomal organelles in PS1 −/− neurons (H and I). *, denotes nucleus. Bars: (A–E) 10 μm; (F) 5 μm; (H) 500 nm; (G and I) 100 nm.
Immuno-EM was conducted to determine the spatial relationship of the perikaryal α- and β-synuclein proteins to these structures. Although no synuclein immunoreactivity was found near the nuclei of PS1 +/? neurons (Fig. 2 G), synuclein immunoreactivity was associated with the aberrant lysosomal organelle clusters of PS1 −/− neurons, most of which was localized along the limiting membrane of these organelles (Fig. 2, H and I), similar to the pattern seen by immunofluorescence. Although some synuclein immunoreactivity appeared to be intralysosomal, it also is plausible that α- and β-synuclein are bound to the cytoplasmic surface of these lysosomes.
Expression levels of α- and β-synuclein are increased in PS1 −/− neurons
Expression of both α- and β-synuclein was increased in 10-d-old PS1 −/− neurons relative to control neurons (Fig. 3). In contrast, levels of other synaptic or axonal proteins, including synaptophysin, synapsin I, neurofilament-L (NFL), and tau, were nearly unchanged or increased less than twofold in PS1 −/− neurons (Fig. 3).
Figure 3.
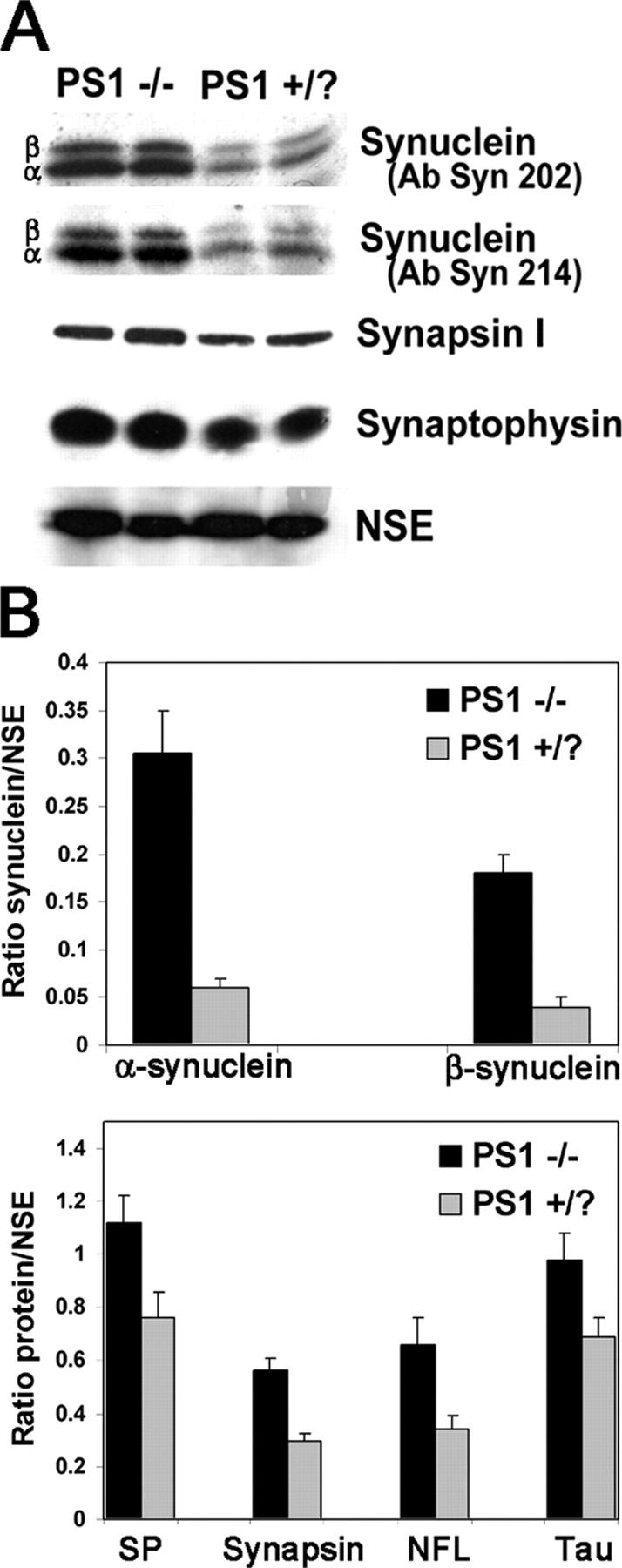
α- and β-synuclein are overexpressed in PS1 −/− neurons. (A) Increased levels of both α- and β-synuclein in PS1 −/− lysates were demonstrated with the synuclein antibodies Syn 202 and Syn 214. Other synaptic proteins including synapsin I and synaptophysin were increased only slightly in PS1 −/− neurons. NSE was used as a loading control. (B) 125I-quantification of synuclein, synaptophysin, synapsin I, NFL, and tau levels relative to NSE. Error bars represent SD.
Human PS1 rescues synuclein aberrations in PS1 −/− neurons
As shown in Fig. 4, the alterations in α- and β-synuclein were specifically due to the absence of PS1, based on analysis of primary cortical cultures from PS1 −/− mice rescued with a transgenic human WT presenilin construct (Qian et al., 1998). No somatic synuclein deposits were found in PS1-WT rescued cultures (Fig. 4 C), and synuclein protein levels were not increased in these cells (Fig. 4 E). Similarly, in neuronal cultures from PS1 −/− mice rescued with transgenic human FAD PS1-A246E, α- and β-synuclein were not found in somatic accumulations (Fig. 4 D), and levels of both α- and β-synuclein were similar to those seen in control cultures (Fig. 4 E). Thus, PS1 reexpression in PS1 null neurons is sufficient to rescue the abnormal α- and β-synuclein phenotype, and mutant PS1 appears to rescue equally well as WT PS1.
Figure 4.

Expression of human wild-type or FAD mutant PS1 eliminated synuclein perinuclear accumulations and overexpression. (A–D) Immunolabeling of neuronal cultures with antibodies to synapsin I (green) and α- and β-synuclein (red, Syn 202). Neuronal cultures were generated from mice (A) PS1 −/−, (B) PS1 +/?, (C) PS1 −/− rescued with wild-type human PS, or (D) PS1 −/− rescued with PS1 containing FAD mutant A246E. Perinuclear synuclein accumulations appeared only in PS1 −/− neurons (A, arrow). *, denotes nucleus. Bar, 10 μm. (E) Western blots of lysates from 10-d-old cultures. The mouse PS1 carboxy-terminal fragment was present only in PS1 +/? cultures, and the larger human fragment was present only in the two rescue lines. α- and β-synuclein levels were increased solely in the PS1 −/− cultures. NSE was used as a loading control.
Cause of increased synuclein expression
Because increased steady-state levels of α- and β-synuclein may be due to either increases in the synthesis of these proteins or decreases in degradation, we examined the levels of synuclein mRNA, translation of protein, and protein turnover at timed intervals in PS1 −/− and PS1 +/? neurons. Synuclein mRNA increased steadily with time in both cell types (Fig. 5 A), however, there were no differences in the levels of synuclein mRNA between PS1 −/− and PS1 +/? neurons at any time point examined. Nevertheless, despite the identical mRNA levels between the two cell types, the amount of newly synthesized α-synuclein after 1.5 h of metabolic labeling was demonstrably higher in PS1 −/− neurons at all times in culture tested, although β-synuclein protein synthesis did not differ (Fig. 5 B). Previous studies have likewise suggested that regulation of α-synuclein protein expression likely occurs at the posttranscriptional level (Petersen et al., 1999).
Figure 5.
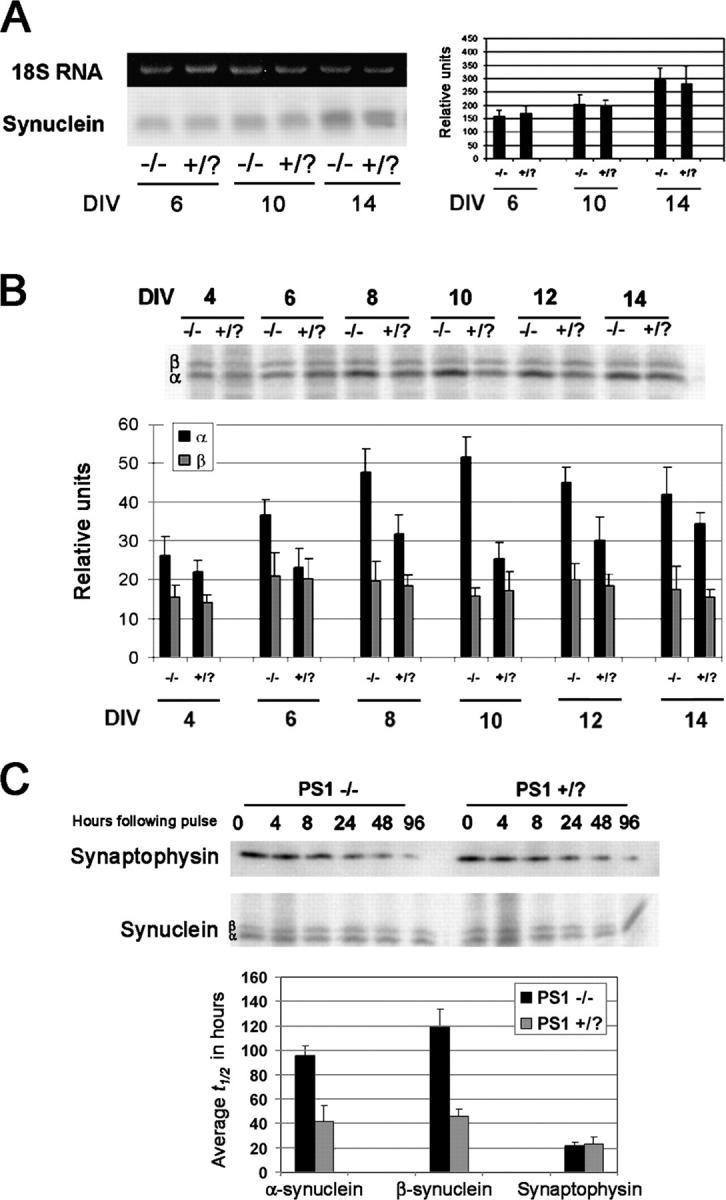
Examination of synuclein synthesis and degradation in PS1 −/− neurons. (A) Northern blot and PhosphorImager quantification of synuclein mRNA levels in PS1 −/− and PS1 +/? neurons after multiple DIV. α- and β-synuclein appeared as one band on the Northern blot. There were no differences in the levels of synuclein mRNA. (B) Immunoblot and quantification of synuclein translation. Translation of α-synuclein, but not β-synuclein, was significantly increased in PS1 −/− neurons. (C) Pulse chase and quantification of synuclein and synaptophysin turnover. The average half-life of both α- and β-synuclein was increased in PS1 −/− neurons, whereas that of synaptophysin was unchanged between the two cell types. Error bars represent SD.
Turnover of radiolabeled proteins was measured over a 4-d period to assay synuclein degradation. α-Synuclein has been shown to be very stable in transfected HEK293 and PC12 cells (Okochi et al., 2000). Although the half-life of endogenous α- and β-synuclein in PS1 +/? neurons was ∼45 and 50 h, respectively, in PS1 −/− neurons the half-life of both α- and β-synuclein nearly doubled (Fig. 5 C). Thus, the increased levels of α-synuclein in PS1 −/− neurons appears to be the result of both increased production and decreased degradation, whereas the increase in β-synuclein is due primarily to decreased degradation. The half-life of synaptophysin was unchanged in PS1 −/− neurons relative to control cells.
Trafficking of other proteins is not impaired in PS1 −/− neurons
The accumulation of synucleins within the perinuclear region of PS1 −/− neurons suggests that the normal transport of these proteins to the synapse may be impaired. Altered transport of several transmembrane proteins has been demonstrated in PS1 −/− neurons (Naruse et al., 1998; Kaether et al., 2002). Furthermore, kinesin-directed vesicle transport is reduced in PS1 −/− neurons due to increased phosphorylation of kinesin (Pigino et al., 2003). α- and β-synuclein have been shown to be transported to the synapse via the fast rate component as well as by slow components a and b (Jensen et al., 1999; Li et al., 2004). The distribution of vesicular proteins transported by the kinesin-dependent fast rate component, such as synaptophysin and synapsin I, or the slow rate component protein NFL and β-tubulin was unaltered between PS1 −/− and PS1 +/? neurons (Figs. 1 and 6), and none of these proteins was found to colocalize with α- and β-synuclein within neuronal perikarya. Similar results were seen using antibodies to a large number of axonal and synaptic proteins (unpublished data). Thus, gross differences in axonal transport do not appear to account for the accumulation of synuclein in PS1 −/− neurons. Furthermore, overexpression of the PS1 substrates APP or Notch in PS1 −/− cells failed to demonstrate a colocalization of these proteins with the Lamp-positive organelles (unpublished data), demonstrating that these organelles are not simply a cellular repository for these uncleaved PS1 substrates.
Figure 6.
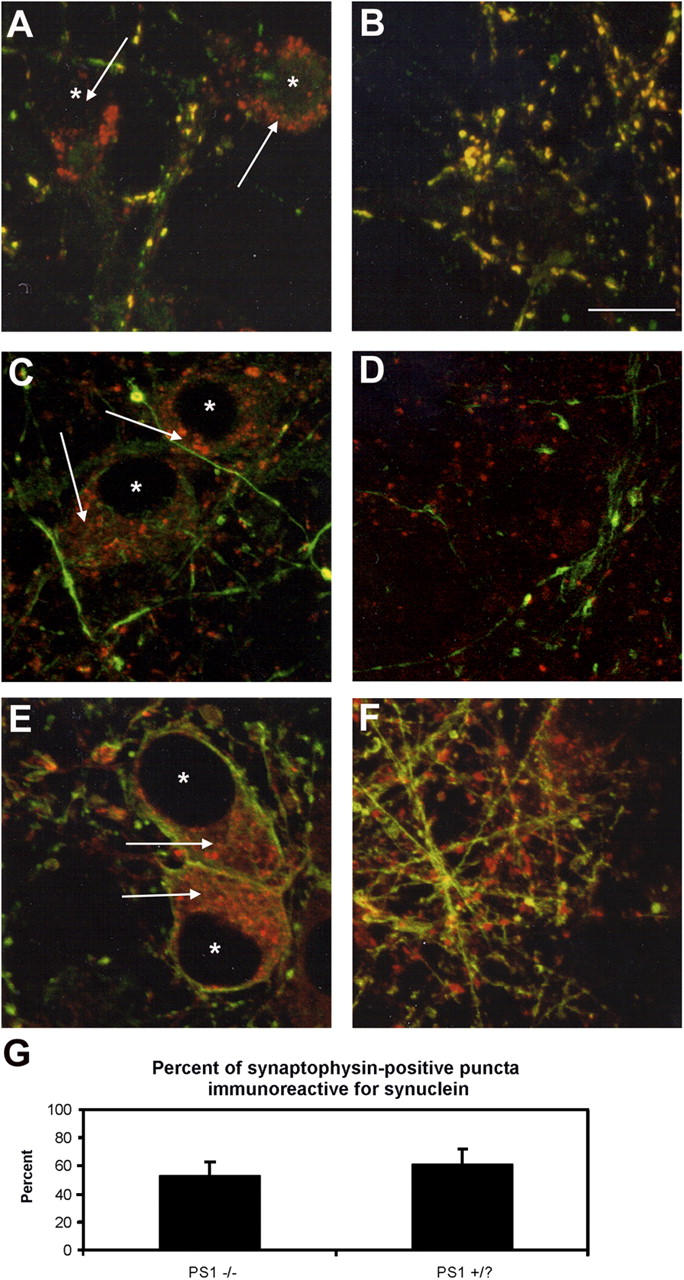
Axonal transport is not impaired in PS1 −/− neurons. The localization of proteins redistributed by the fast rate component of axonal transport (A and B, synaptophysin, green) as well as the slow rate component (C and D, NFL, green; E and F, β-tubulin, green) was compared with that of α- and β-synuclein (red, Syn 202) in (A, C, and E) PS1 −/− neurons and (B, D, and F) PS1 +/? neurons. None of the proteins examined appeared localized to the perinuclear synuclein accumulations. *, denotes nucleus. Bar, 5 μm. Arrows illustrate perinuclear synuclein accumulation. (G) The percentage of synaptophysin-positive putative synapses colabeled with synuclein immunoreactivity. Error bars represent SD.
Furthermore, although α- and β-synuclein accumulate in PS1 −/− neuronal perikarya, there does not appear to be a complete block in their transport, as some colocalization of α- and β-synuclein with other synaptic marker proteins could be visualized in punctate patterns characteristic of synapses even within PS1 −/− neurons (Figs. 1 and 6). Quantification of the colocalization of synucleins and synaptophysin in putative synaptic puncta demonstrated ∼50% colocalization in both PS1 −/− and control cells (Fig. 6 G). Furthermore, we found by EM that the distal pool of synaptic vesicles appeared to be similar between the two cell types (unpublished data), lending support to the view that synuclein transport to the synapses was unaltered in PS1 −/− neurons (unpublished data) because depletion of synuclein at the synapses results in a diminution of this particular pool of vesicles (Murphy et al., 2000; Cabin et al., 2002).
Pharmacological manipulation of autophagy impacts perikaryal α- and β-synuclein localization
To determine whether the aberrant translocation of α- and β-synuclein is due to the formation of autophagic vacuoles, we stimulated the formation of enlarged organelles by treating PS1 +/? neurons with staurosporine, which causes hyperaccumulation of large membrane-bound structures immunoreactive for lysosomal proteins (Adamec et al., 2000). After treatment with 100 nM staurosporine for 48 h, a significant number of cells displayed Lamp-2–positive intracellular organelles (Fig. 7, D–F) morphologically similar to those seen in PS1 −/− neurons. Control untreated PS1 +/? neurons did not develop such Lamp-2–positive organelles (Fig. 7, A–C). Immunolabeling with the antibody Syn 202 demonstrated that α- and β-synuclein colocalized with Lamp proteins along the outer membrane of these organelles. Thus, the formation of Lamp-positive organelles is sufficient to induce perikaryal colocalization of synucleins to these organelles.
Figure 7.

Induction of autophagy with staurosporine stimulates the association of synucleins with perinuclear lysosomal organelles. PS1 +/? neurons were treated with 100 nM staurosporine for 48 h to induce autophagy. Immunolabeling of (A–C) untreated and (D–F) treated neurons with antibodies to Lamp-2 (A and D, green) and synuclein (B and E, Syn 202, red). The overlap is shown in C and F. Nuclei were counterstained with DAPI (blue). Bar, 5 μm.
The formation of these organelles in PS1 −/− cells may be due to the loss of γ-secretase activity. To address this, PS1 +/? neurons were treated for 7 d with γ-secretase inhibitors, including transition-state analogs L-685,458 (Shearman et al., 2000) and III-21C (Esler et al., 2002), difluoroketone DFK-167 (Wolfe et al., 1998), dipeptide aldehyde 2-Napthyl-VF-CHO (Sinha and Lieberburg, 1999), and LiCl (Phiel et al., 2003). Cells were treated with doses shown previously to reduce the γ-secretase cleavage of APP overexpressed in PS1 +/? neurons (Wilson et al., 2002; and unpublished data). None of the compounds increased the percentage of neurons demonstrating Lamp-2–positive autophagic organelles, nor was there evidence of perikaryal localization of synuclein proteins (Fig. 8). Thus, the appearance of enlarged Lamp-2–positive organelles and the mislocalization of synuclein in PS1 −/− cells were not correlated with the absence of γ-secretase activity in these cells.
Figure 8.

Loss of PS1 γ-secretase activity does not result in the formation of enlarged perinuclear organelles. PS1 +/? neurons were treated for 7 d with the γ-secretase inhibitors L-685,458 (1 μM), DFK-167 (50 μM), III-31C (5 μM), 2-Napthyl-VF-CHO (20 μM), or LiCl (5 mM), and analyzed by light microscopy for Lamp-2 and synuclein immunoreactivity. None of these compounds increased the percentage of neurons containing Lamp-2–positive enlarged perinuclear organelles, nor did they induce the localization of synuclein proteins to this site. 100 nM staurosporine treatment for 48 h was included as a positive control. Asterisks indicate that the results are statistically significant, P < 0.005. Error bars represent SD.
To gain further insight into the possible mechanism leading to the formation of enlarged autophagosomes, we manipulated intracellular calcium levels in PS1 −/− neurons. Multiple intracellular signaling events have been demonstrated to influence degradative pathways, including dysregulation of intracellular calcium levels (for review see Codogno et al., 1997). Absence of PS1 has been linked to excessive activation of capacitative calcium channels in the plasma membrane responsible for refilling the ER calcium stores, which can be reversed by SKF 96365, a specific inhibitor of the channel (Leissring et al., 2000; Yoo et al., 2000). Thus, we sought to determine if SKF96365 affected the localization of synuclein proteins in enlarged lysosomal organelles. Treatment of 10-d-old PS1 −/− neuronal cultures with SKF 96365 for 48 h resulted in a dose-dependent decrease in both somatic synuclein accumulations (Fig. 9) and proliferation of Lamp-positive organelles (not depicted). In contrast, treatment with thapsigargin, an inhibitor of the ER calcium/ATPase responsible for maintaining the high concentrations of calcium within the ER, had no significant effect on the number of cells displaying either phenotype, nor did culturing the cells in medium containing a 10-fold reduction in calcium concentrations. Thus, the formation of enlarged autophagic vacuoles and the mislocalization of the synucleins are likely due to the deregulation of capacitative calcium in the absence of PS1.
Figure 9.

Inhibition of capacitative calcium channels eliminates the perinuclear distribution of synucleins in PS1 −/− neurons. PS1 −/− and PS1 +/? neurons cultured for 10 d in vitro were treated with SKF 96365 or thapsigargin, or incubated in low Ca2+ medium for 48 h. Representative images of (A) untreated PS1 +/? neurons, (B) untreated PS1 −/−neurons, or (C) PS1 −/− neurons treated with 500 nM SKF 96365 are shown. Nuclei are counterstained with DAPI (blue). Bar, 10 μm. (D) Quantification of the percentage of neurons displaying perinuclear synuclein immunoreactivity. Error bars represent SD.
Finally, we examined if somatic colocalization of synuclein and lysosomes is associated with disease pathology. Accumulations of enlarged lysosomes have been visualized early in the pathogenesis of AD (Cataldo et al., 1995), autophagy in the substantia nigra has been documented in PD (Anglade et al., 1997), and cell body α-synuclein accumulation in Lewy bodies is a classical feature of many α-synucleinopathies. In patients with LBVAD, irregular accumulations of α-synuclein that resemble those seen in PS1 −/− neurons were observed in neuronal cell bodies (Fig. 10 A, arrows) as well as close to the nuclei of nonneuronal cells (Fig. 10 A, arrowheads). Tissue from nondiseased controls (Fig. 10 B) and PD patients (not depicted) demonstrated only diffuse neuropil synuclein immunoreactivity. Likewise, Lamp-2 immunoreactivity was more pronounced in LBVAD dentate gyrus (Fig. 10 C) than controls (Fig. 10 D). Examination of serial sections showed similar cellular localization of α-synuclein and Lamp-2 in LBVAD hippocampi (compare Fig. 10 E with Fig. 10 F; and compare Fig. 10 G with 10 H). Thus, the perikaryal localization of α-synuclein to lysosomal structures in neuronal cell cultures may have counterparts in disease pathology.
Figure 10.
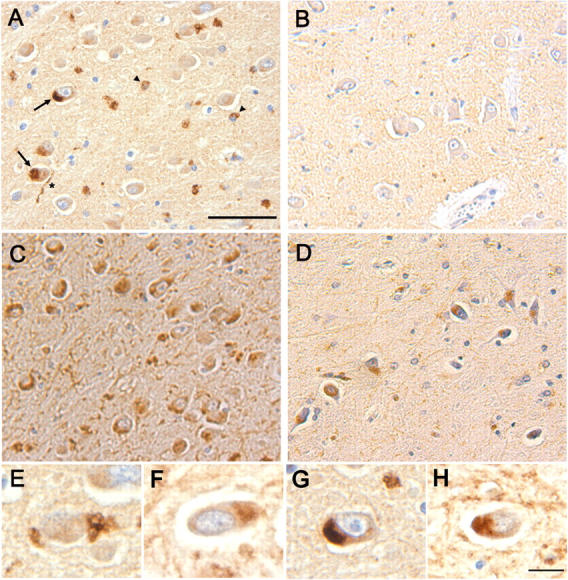
Perinuclear α-synuclein deposits colocalize with Lamp-immunoreactivity in dentate hilar neurons from patients with LBVAD. Hippocampal tissue from (A) LBVAD patients immunolabeled with Syn 303 revealed perinuclear synuclein immunoreactivity in neuronal cell bodies (arrows) and neurites (*) as well as near the nuclei of nonneuronal cells (arrowheads), which are not seen in tissue from (B) nondiseased controls. Similarly, Lamp-2 immunoreactivity is increased in (C) LBVAD dentate gyrus relative to (D) controls. A comparison of synuclein immunoreactivity (E and G, Syn 303) with Lamp-2 immunoreactivity in adjacent sections (F and H, Lamp-2 C20) demonstrated similar localization of the two proteins in cell bodies. Bars: (A–D) 30 μm; (E–H) 10 μm.
Discussion
We have demonstrated the formation of large vacuolar autophagic and/or lysosomal organelles in PS1 −/− cells and showed that α- and β-synuclein, normally presynaptic proteins, are found in association with these abnormal structures in PS1 −/− neuronal perikarya. Concomitantly, there was a large increase in the expression levels of both proteins. Similar perinuclear association with degradative organelles was demonstrated in cells treated with staurosporine to induce autophagy. Furthermore, the phenotype could be reversed by pharmacological blockade of capacitative calcium channels, suggesting that the role of PS1 in the maintenance of intracellular calcium stores may be involved.
The lysosomal Lamp-positive enlarged organelles described here in PS1 −/− neurons are morphologically similar to those seen in hereditary lysosomal storage diseases (Walkley, 1998) or aging (Cuervo and Dice, 2000), in neurons after injury (Hornung et al., 1989), or after treatment with lysosomal inhibitors (Bi et al., 1999) or cell stressor agents (Adamec et al., 2000). Organelles with morphological features similar to those described here and containing the novel protein telencephalin were reported previously in PS1 −/− neurons (Annaert et al., 2001). Interestingly, lysosomal and/or autophagic organelles are also increased in PC12 cells overexpressing α-synuclein containing the familial PD A53T mutation (Stefanis et al., 2001), as well as in hypothalamic cell lines transfected with WT α-synuclein (Hsu et al., 2000). An up-regulation of synuclein protein expression has also been documented in a variety of injury models, including ischemia (Ishimaru et al., 1998), administration of MPTP (Vila et al., 2000) or dopamine (Gomez-Santos et al., 2003), and targeted developmental injury to the striatum of rats (Kholodilov et al., 1999). Additionally, increased expression of both α- and β-synuclein was found in a mouse model of the lysosomal storage disorder GM2 gangliosidosis (Suzuki et al., 2003) and PD frequently occurs in patients with Gaucher's disease (Bembi et al., 2003). Somatic synuclein immunoreactivity can be induced by mitochondrial inhibitor pesticides such as rotenone (Betarbet et al., 2000), as well as physical injury such as nerve trans-section (Moran et al., 2001) and has been documented in the vicinity of lipofuscin granules, age-related tertiary lysosome derivatives, in MPTP-induced Parkinsonism in mice (Meredith et al., 2002).
The exact mechanism driving the accumulation of enlarged Lamp-positive organelles in PS1 −/− neurons remains unknown. Presenilins themselves were demonstrated recently within the lysosomal system (Pasternak et al., 2003) suggesting that they may play a critical role in the functions of these organelles. We have demonstrated that the formation of autophagic vacuoles in PS1 −/− cells is likely unrelated to PS1 γ-secretase activity, as treatment of control neurons with γ-secretase inhibitors failed to reproduce these effects. However, the decrease in enlarged lysosomal organelles in PS1 −/− neurons treated with a capacitative calcium channel inhibitor implicates an involvement of the ER calcium stores in the formation of these structures. ER Ca2+ stores are refilled via activation of capacitative calcium channels on the plasma surface. The activity of these cell surface refilling channels is significantly increased in PS1 −/− cells (Yoo et al., 2000). A role for ER calcium stores in the regulation of autophagic degradation has been described, although the exact mechanism remains elusive (Codogno et al., 1997). We have demonstrated that inhibition of calcium refilling with SKF 96365 was sufficient to block formation of autophagic/lysosomal organelles and concomitant localization of α- and β-synuclein in PS1 −/− cells. Surprisingly, we noted that thapsigargin, which inhibits the ER calcium/ATPase and thereby lowers ER calcium levels by passive diffusion, did not have the same effect as SKF 96365. This may suggest that the requirements for the formation of degradative organelles are more complex than simply the size of the ER pool, or alternatively may reflect the fact that capacitative calcium entry can be stimulated by thapsigargin-induced ER calcium release (Putney and McKay, 1999).
Synucleins may accumulate within Lamp-positive degradative organelles because they normally are degraded at this site. Inhibition of lysosomes with ammonium chloride reduces α-synuclein turnover (Paxinou et al., 2001), and α-synuclein can be degraded by autophagy when overexpressed in PC12 cells (Webb et al., 2003). The observation that the rate of α- and β-synuclein turnover is diminished in PS1 −/− neurons is consistent with this hypothesis. However, synucleins may be degraded normally by proteases such as calpain localized at the presynapse (Mishizen-Eberz et al., 2003), and thus the decrease in protein turnover in PS1 −/− neurons may simply reflect the spatial discrepancy between the somatic pool of synucleins and the presynaptic proteases.
Alternatively, α- and β-synuclein may be associated with the cytoplasmic surface of the enlarged organelles. High magnification confocal and immuno-EM demonstrated that synucleins are concentrated as ringlike immunoreactivity near the outer membrane of enlarged degradative organelles. Synucleins can bind to small synthetic vesicles in vitro (Davidson et al., 1998) and the perinuclear organelles may simply act as a nonspecific sink for synuclein binding. Alternatively, it is possible that synucleins play a more active role in the formation or maintenance of autophagic organelles in PS1 −/− neurons. Multiple presynaptic vesicle proteins either have homology with proteins regulating organelles in the degradative pathway or are themselves active at both sites, including isoforms of synaptotagmin (Martinez et al., 2000), synaptobrevin (Advani et al., 1999), and syntaxin (Darsow et al., 1997; Nakamura et al., 2000).
The aberrant α-, β-synuclein localization and expression in PS1 −/− neurons is not a direct model for the pathobiology in Lewy body diseases or other synucleinopathies. Several key characteristics distinguish the two phenomena, including the presence of β-synuclein and absence of neurofilaments, ubiquitinated proteins, and filamentous forms of α-synuclein in the PS1 −/− somatic accumulations. Nevertheless, the structures described herein may partially recapitulate early events in the development of α-synuclein pathology, as the accumulation of α-synuclein in the perinuclear region may lead to the formation of a nidus for fibrillization. Evidence for up-regulation of autophagy has been visualized in PD brain tissue (Anglade et al., 1997). We have demonstrated that α-, β-synuclein associate with degradative organelles structurally similar to these, both in cell culture and in human disease tissue, and that this association is accompanied by a significant increase in the intracellular expression of α-synuclein. Because the interactions of synucleins with lipid membranes are transient (Maroteaux and Scheller, 1991; George et al., 1995), a significant pool of synucleins could be dissociated from the organelles at any given time, perhaps reaching a high enough local concentration of α-synuclein to induce self-association and fibril formation. Interestingly, interaction of α-synuclein with synthetic lipid droplets in cells promotes the formation of oligomeric species (Cole et al., 2002). Further investigation is necessary to lend support to this model.
Materials and methods
Animals
The generation of PS1 knockout and rescue mice was described elsewhere (Wong et al., 1997; Qian et al., 1998). PS1 +/− animals were mated, and the pregnant dams were killed by cervical dislocation 15–16 d after conception. Embryos were visually classified as PS1 −/− or littermate control mice (designated PS1 +/? to include PS1 +/− and +/+ genotypes) by their distinct morphological characteristics (Wong et al., 1997). For experiments requiring either PS1 +/+ embryos or PS1 −/− embryos rescued with human wild-type PS1 (PS1WT rescue) or PS1 carrying the FAD A246E mutation (PS1A246E rescue), homozygous animals of each genotype were mated. PS1 −/−, PS1WT rescue, and PS1A246E rescue mice were provided by H. Zheng and S. Qian (Baylor College of Medicine, Houston, TX).
Cell culture
Cortices from mouse embryo brains were isolated and incubated in 0.1% trypsin/HBSS/0.5 mM EDTA without Ca2+ or Mg2+ (Invitrogen), and cells mechanically dissociated using a fire-polished pipette. Cells were plated in Dulbecco's minimum essential medium (DMEM) + 10% FBS in poly-d-lysine–coated 6-well plates at a density of 106 cells/well or on coated 10-mm coverslips (Bellco Glass, Inc.) at a density of 105cells/well. 24 h after plating, medium was replaced with DMEM plus B27 supplements (Invitrogen) to promote neuronal survival and inhibit growth of nonneuronal cells. Neurons were used for experimentation 10 d after plating unless stated otherwise. For pharmacological studies, staurosporine (Sigma-Aldrich), SKF 96365 (Sigma-Aldrich), thapsigargin (Sigma-Aldrich), or γ-secretase inhibitors MW-167, L-685,458, 2-Napthyl-VF-CHO, and WPE-III-21C (Calbiochem) were added to neurons in DMEM + B27 for the length of time indicated. Primary fibroblasts were generated from PS1 −/− and PS1 +/? mice, immortalized with v-myc, and maintained in DMEM + 10% FBS.
Antibodies
To assay synucleins, the following antibodies were used: Syn 202, Syn 205, and Syn 214 (recognizing both α- and β-synuclein), SNL-1 (specific for α-synuclein), Syn 207 (specific for β-synuclein), and Syn 303 (preferentially recognizing a pathological conformation of α-synuclein). Mapping and specificity of these antibodies has been documented previously (Giasson et al., 2000; Duda et al., 2002). Additionally, hu A is a rabbit polyclonal antibody raised to full-length recombinant α-synuclein that recognizes multiple epitopes on both α- and β-synuclein. Other antibodies used include those specific for synaptophysin (Boehringer), synapsin I (Molecular Probes, Inc.), Lamp-2 (ABL93; Iowa Developmental Hybridoma Studies Bank; and C20; Santa Cruz Biotechnology, Inc.), NFL (Tu et al., 1995), β-tubulin (Sigma-Aldrich), tau (14/46; Kosik et al., 1988), and neuron-specific enolase (NSE; Polysciences, Inc.).
Immunocytochemistry
Cultures were fixed in 4% PFA (Electron Microscopy Sciences) in PBS for 30 min, and blocked and permeabilized using 5% horse serum plus 0.1% saponin in PBS. Coverslips were incubated in primary antibody overnight followed by the appropriate secondary antibody conjugated to either Texas red or fluorescein (Jackson ImmunoResearch Laboratories). Coverslips were mounted in Vectashield (Vector Laboratories) and analyzed using either a confocal microscope (Leica) with software (Bio-Rad Laboratories), or an inverted microscope (Nikon) with CoolSnap software (Image Processing Solutions). To quantify synuclein-positive putative synapses, the number of puncta immunoreactive for both synuclein and synaptophysin was compared with the total number of synaptophysin-positive puncta.
Western blots
RIPA buffer (0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40, 5 mM EDTA, 150 mM NaCl, 50 mM Tris-HCl, pH 8.0) plus a cocktail of protease inhibitors (1 μg/ml each of leupeptin, pepstatin A, TPCK, TLCK, soybean trypsin inhibitor, and 0.5 mM PMSF) was used to extract proteins from cortical cultures and the resultant lysates were sonicated and spun at 100,000 g for 20 min. 25 μg of protein was resolved by SDS-PAGE on either 10% or 12% slab gels, electrophoretically transferred to nitrocellulose, and probed with primary antibody followed with HRP-conjugated mouse or rabbit secondary antibodies and visualized by chemiluminescence. For quantitative studies, blots were probed with an 125I-labeled secondary antibody and was analyzed by PhosphorImager (Molecular Dynamics, Inc.).
EM
Cortical neuronal cultures were fixed in 2% glutaraldehyde/PBS for 24 h, dehydrated in a graded series of ethanols, and embedded in Epon. For EM, blocks were cut on an UltracutE, stained with 1% uranyl acetate, and examined with a transmission electron microscope (model 1010; JEOL). For immuno-EM, coverslips were fixed briefly in 4% PFA/0.25% glutaraldehyde in PBS, permeabilized with ethanol, incubated with hu A antibody, followed by a goat anti–rabbit HRP-conjugated antibody (Santa Cruz Biotechnology, Inc.), and developed with 3,3′-DAB. DAB was enhanced for immuno-EM studies using a modification of the Rodriguez silver/gold enhancement method (Teclemariam-Mesbah et al., 1997). Coverslips were then fixed in 2% glutaraldehyde/PBS overnight, and prepared as described above.
Northern blots
Total RNA was extracted from cortical cultures using the RNeasy kit (QIAGEN). 5 μg RNA was denatured and loaded onto 2% agarose-formaldehyde gels, then transferred and cross-linked to nitrocellulose (Hybond N+; Amersham Biosciences). A synuclein-specific probe was prepared from full-length α-synuclein cDNA, labeled with [32P]-dCTP using DNA labeling beads (Amersham Biosciences) and purified using MicroSpin G-50 columns (Amersham Biosciences). Radioactive probe was hybridized to the membrane using QuickHyb solution (Stratagene), and the membrane was opposed to a PhosphorImager plate for analysis.
Translation assay
Cortical cultures were methionine-deprived for 30 min by incubation in methionine-free DMEM (Invitrogen) before adding 500 μCi [35S]methionine (NEN Life Sciences Products) per milliliter of DMEM with 5% dialyzed FBS (Invitrogen) for 1.5 h. Cells were rinsed twice with PBS and scraped into CSK buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 2 mM EGTA, 1% Triton X-100, 1 mM PMSF, protease inhibitor cocktail). Lysates were sonicated and spun at 100,000 g for 20 min. Cell lysates were immunoprecipitated with the antibody hu A using protein A/G agarose beads (Santa Cruz Biotechnologies), and proteins were resolved by electrophoresis on 12% SDS-PAGE gels. Gels were fixed in 50% methanol and 5% glycerol, dried, and exposed to a PhosphorImager screen.
Turnover assay
Cortical neurons cultured for 8 d in vitro were methionine deprived for 30 min in methionine-free DMEM before adding 500 μCi [35S]methionine per milliliter of DMEM with 5% dialyzed FBS for 2 h, after which the medium was replaced with DMEM plus B27 supplement. Cells were rinsed twice with PBS and scraped into CSK buffer at various times after the removal of radioactive media. Lysates were sonicated, spun at 100,000 g for 20 min, and immunoprecipitated with the antibody hu A using protein A/G beads. Proteins were resolved by electrophoresis on 12% SDS-PAGE gels, fixed in 50% methanol plus 5% glycerol, dried, and exposed to a PhosphorImager screen.
Immunohistochemistry
Postmortem hippocampal tissue from patients diagnosed with LBVAD or controls was obtained from the brain bank of the Center for Neurodegenerative Disease Research, University of Pennsylvania. 12 cases of LBVAD (9 males, 3 females; mean age 77.3 yr, range 65–87 yr; mean postmortem interval 13.2 h, range 3.5–46.5 h), 5 cases of PD (3 males, 2 females; mean age 76.0 yr, range 69–83 yr; mean postmortem interval 8.7 h, range 5–15 h), and 3 nondiseased control cases (2 males, 1 female; mean age 63.3 yr, range 56–72 yr; mean postmortem interval 3.5 h, range 2–8 h) were analyzed. Tissue was prepared as described previously (Trojanowski et al., 1989). In brief, tissue was fixed in 10% neutral-buffered formalin, rinsed in 50 mM Tris, and 150 mM NaCl, dehydrated through a graded series of ethanols into xylene at RT, infiltrated with paraffin, and cut into 6-μm sections. Sections were then rehydrated, treated with 88% formic acid, and quenched with 83% methanol/5% hydrogen peroxide before antibody addition. Sections were incubated with primary antibody in 0.1 M Tris, pH 7.6, plus 2% donor horse serum overnight at 4°C, then sequentially incubated with species-specific biotinylated secondary antibody and avidin–biotin complex (Vectastain ABC Elite kit; Vector Laboratories) for 1 h each at RT, and finally visualized with DAB and counterstained with hematoxylin.
Acknowledgments
The authors thank Drs. Hui Zheng and Su Qian for the generation of PS1 +/− and PS1 rescue animals, and Neelima Shah and the University of Pennsylvania Pathology Core Facility for assistance with electron microscopy studies.
This work was supported by grants AG11542 and AG09215 from the National Institute on Aging. C.A. Wilson was a Howard Hughes Predoctoral Fellow; B.I. Giasson was a recipient of a fellowship from the Canadian Institute of Health Research; J.Q. Trojanowski is the Measey-Schnabel professor of Geriatric Aging and Gerontology; and V.M.Y. Lee is the John H. Ware III professor of Alzheimer's Research.
D.D. Murphy's present address is National Institute of Neurological Diseases and Stroke, Neuroscience Center, Room 2228 6001 Executive Blvd., Rockville, MD 20852.
Abbreviations used in this paper: AD, Alzheimer's disease; APP, amyloid precursor protein; DMEM, Dulbecco's minimum essential medium; FAD, familial AD; LBVAD, Lewy body variant of AD; NFL, neurofilament-L; NSE, neuron-specific enolase; PD, Parkinson's disease; PS1, presenilin-1.
References
- Adamec, E., P.S. Mohan, A.M. Cataldo, A.N. Srinivasan, J.P. Vonsattel, and R.A. Nixon. 2000. Upregulation of the lysosomal system in experimental models of neuronal injury: Implications for Alzheimer's disease. Neuroscience. 100:663–675. [DOI] [PubMed] [Google Scholar]
- Advani, R.J., B. Yang, R. Prekeris, K.C. Lee, J. Klumperman, and R.H. Scheller. 1999. VAMP-7 mediates vesicular transport from endosomes to lysosomes. J. Cell Biol. 146:765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglade, P., S. Vyas, F. Javoy-Agid, M.T. Herrero, P.P. Michel, J. Marquez, A. Mouatt-Prigent, M. Ruberg, E.C. Hirsch, and Y. Agid. 1997. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol. Histopathol. 12:25–31. [PubMed] [Google Scholar]
- Annaert, W.G., C. Esselens, V. Baert, C. Boeve, G. Snellings, P. Cupers, K. Craessaerts, and B. De Strooper. 2001. Interaction with telencephalin and the amyloid precursor protein predicts a ring structure for presenilins. Neuron. 32:579–589. [DOI] [PubMed] [Google Scholar]
- Bembi, B., M.S. Zambito, E. Sidransky, G. Ciana, M. Carrozzi, C. Martini, M. Giolis, M.G. Pittis, and L. Capus. 2003. Gaucher's disease with Parkinson's disease: Clinical and pathogical aspects. Neurology. 61:99–101. [DOI] [PubMed] [Google Scholar]
- Betarbet, R., T.B. Sherer, G. MacKenzie, M. Garcia-Osuna, A.V. Panov, and J.T. Greenamyre. 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 3:1301–1306. [DOI] [PubMed] [Google Scholar]
- Bi, X., J. Zhou, and G. Lynch. 1999. Lysosomal protease inhibitors induce meganeurites and tangle-like structures in entorhinohippocampal regions vulnerable to Alzheimer's disease. Exp. Neurol. 158:312–327. [DOI] [PubMed] [Google Scholar]
- Cabin, D.E., K. Shimazu, D. Murphy, N.B. Cole, W. Gottschalk, K.L. McIlwain, B. Orrison, A. Chen, C.E. Ellis, R. Paylor, et al. 2002. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 22:8797–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo, A.M., J.L. Barnett, S.A. Berman, J. Li, S. Quarless, S. Bursztajn, C. Lippa, and R.A. Nixon. 1995. Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 14:671–680. [DOI] [PubMed] [Google Scholar]
- Codogno, P., E. Ogier-Denis, and J.J. Houri. 1997. Signal transduction pathways in macroautophagy. Cell. Signal. 9:125–130. [DOI] [PubMed] [Google Scholar]
- Cole, N.B., D.D. Murphy, T. Grider, S. Rueter, D. Brasaemle, and R.L. Nussbaum. 2002. Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha-synuclein. J. Biol. Chem. 277:6344–6352. [DOI] [PubMed] [Google Scholar]
- Cuervo, A.M., and J.F. Dice. 2000. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 275:31505–31513. [DOI] [PubMed] [Google Scholar]
- Darsow, T., S.E. Rieder, and S.D. Emr. 1997. A multispecificity syntaxin homologue, Vamp3p, essential for autophagic and biosynthetic protein transport to the vacuole. J. Cell Biol. 138:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson, W.S., A. Jonas, D.F. Clayton, and J.M. George. 1998. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273:9443–9449. [DOI] [PubMed] [Google Scholar]
- De Strooper, B., P. Saftig, K. Craessaerts, H. Vanderstichele, G. Guhde, W. Annaert, K. Von Figura, and F. Van Leuven. 1998. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 391:387–390. [DOI] [PubMed] [Google Scholar]
- De Strooper, B., W. Annaert, P. Cupers, P. Saftig, K. Craessaerts, J.S. Mumm, E.H. Schroeter, V. Schrijvers, M.S. Wolfe, W.J. Ray, et al. 1999. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 398:518–522. [DOI] [PubMed] [Google Scholar]
- Duda, J.E., V.M.-Y. Lee, and J.Q. Trojanowski. 2000. Neuropathology of synuclein aggregates. J. Neurosci. Res. 61:121–127. [DOI] [PubMed] [Google Scholar]
- Duda, J.E., B.I. Giasson, M.E. Mabon, V.M. Lee, and J.Q. Trojanowski. 2002. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann. Neurol. 52:205–210. [DOI] [PubMed] [Google Scholar]
- Esler, W.P., W.T. Kimberly, B.L. Ostaszewski, W. Ye, T.S. Diehl, D.J. Selkoe, and M.S. Wolfe. 2002. Activity-dependent isolation of the presenilin-gamma-secretase complex reveals nicastrin and a gamma substrate. Proc. Natl. Acad. Sci. USA. 99:2720–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin, J.E., K. Uryu, V.M.-Y. Lee, and J.Q. Trojanowski. 1999. Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc. Natl. Acad. Sci. USA. 96:13450–13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakopoulos, A., P. Marambaud, S. Efthimiopoulos, J. Shioi, W. Cui, H.C. Li, M. Schutte, R. Gordon, G.R. Holstein, G. Martinelli, et al. 1999. Presenilin-1 forms complexes with the cadherin/catenin cell-cell adhesion system and is recruited to intercellular and synaptic contacts. Mol. Cell. 4:893–902. [DOI] [PubMed] [Google Scholar]
- George, J.M. 2002. The synucleins. Genome Biol. 3:reviews3002.1–3002.6. [DOI] [PMC free article] [PubMed]
- George, J.M., H. Jin, W.S. Woods, and D.F. Clayton. 1995. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 15:361–372. [DOI] [PubMed] [Google Scholar]
- Giasson, B.I., R. Jakes, M. Goedert, J.E. Duda, S. Leight, J.Q. Trojanowski, and V.M.-Y. Lee. 2000. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson's disease. J. Neurosci. Res. 59:528–533. [DOI] [PubMed] [Google Scholar]
- Gomez-Santos, C., I. Ferrer, A.F. Santidrian, M. Barrachina, and J. Gil. 2003. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J. Neurosci. Res. 73:341–350. [DOI] [PubMed] [Google Scholar]
- Hornung, J.P., H. Koppel, and P.G. Clarke. 1989. Endocytosis and autophagy in dying neurons: an ultrastructural study in chick embryos. J. Comp. Neurol. 283:425–437. [DOI] [PubMed] [Google Scholar]
- Hsu, L.J., Y. Sagara, A. Arroyo, E. Rockenstein, A. Sisk, M. Mallory, J. Wong, T. Takenouchi, M. Hashimoto, and E. Masliah. 2000. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 157:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru, H., K. Ueda, A. Takahashi, and Y. Maruyama. 1998. Changes in presynaptic protein NACP/alpha-synuclein in an ischemic gerbil hippocampus. Brain Res. 788:311–314. [DOI] [PubMed] [Google Scholar]
- Iwai, A., E. Masliah, M. Yoshimoto, N. Ge, L. Flanagan, H. A.de Silva, A. Kittel, and T. Saitoh. 1995. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 14:467–475. [DOI] [PubMed] [Google Scholar]
- Iwata, A., S. Miura, I. Kanazawa, M. Sawada, and N. Nukina. 2001. alpha-synuclein forms a complex with transcription factor Elk-1. J. Neurochem. 77:239–252. [DOI] [PubMed] [Google Scholar]
- Jakes, R., M.G. Spillantini, and M. Goedert. 1994. Identification of two distinct synucleins from human brain. FEBS Lett. 345:27–32. [DOI] [PubMed] [Google Scholar]
- Jensen, P.H., J.Y. Li, A. Dahlstrom, and C.G. Dotti. 1999. Axonal transport of synucleins is mediated by all rate components. Eur. J. Neurosci. 11:3369–3376. [DOI] [PubMed] [Google Scholar]
- Kaether, C., S. Lammich, D. Edbauer, M. Ertl, J. Rietdorf, A. Capell, H. Steiner, and C. Haass. 2002. Presenilin-1 affects trafficking and processing of βAPP and is targeted in a complex with nicastrin to the plasma membrane. J. Cell Biol. 158:551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal, A., A. Almenar-Queralt, J.F. LeBlanc, E.A. Roberts, and L.S. Goldstein. 2001. Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature. 414:643–648. [DOI] [PubMed] [Google Scholar]
- Kang, D.E., S. Soriano, M.P. Frosch, T. Collins, S. Naruse, S.S. Sisodia, G. Leibowitz, F. Levine, and E.H. Koo. 1999. Presenilin 1 facilitates the constitutive turnover of beta-catenin: differential activity of Alzheimer's disease-linked PS1 mutants in the beta-catenin-signaling pathway. J. Neurosci. 19:4229–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodilov, N.G., M. Neystat, T.F. Oo, S.E. Lo, K.E. Larsen, D. Sulzer, and R.E. Burke. 1999. Increased expression of rat synuclein in the substantia nigra pars compacta identified by mRNA differential display in a model of developmental target injury. J. Neurochem. 73:2586–2599. [DOI] [PubMed] [Google Scholar]
- Kosik, K., L.D. Orecchio, L. Binder, J.Q. Trojanowski, V.M.-Y. Lee, and G. Lee. 1988. Epitopes that span the tau molecule are shared with paired helical filaments. Neuron. 1:817–825. [DOI] [PubMed] [Google Scholar]
- Leissring, M.A., Y. Akbari, C.M. Fanger, M.D. Cahalan, M.P. Mattson, and F.M. LaFerla. 2000. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell Biol. 149:793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W., P.N. Hoffman, W. Stirling, D.L. Price, and M.K. Lee. 2004. Axonal transport of human α-synuclein slows with aging but is not affected by familial Parkinson's disease-linked mutations. J. Neurochem. 88:401–410. [DOI] [PubMed] [Google Scholar]
- Lippa, C.F., H. Fujiwara, D.M. Mann, B. Giasson, M. Baba, M.L. Schmidt, L.E. Nee, B. O'Connell, D.A. Pollen, P. George-Hyslop, et al. 1998. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am. J. Pathol. 153:1365–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux, L., and R.H. Scheller. 1991. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res. Mol. Brain Res. 11:335–343. [DOI] [PubMed] [Google Scholar]
- Maroteaux, L., J.T. Campanelli, and R.H. Scheller. 1988. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8:2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, I., S. Chakrabarti, T. Hellevik, J. Morehead, K. Fowler, and N.W. Andrews. 2000. Synaptotagmin VII regulates calcium-dependent exocytosis of lysosomes in fibroblasts. J. Cell Biol. 148:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith, G.E., S. Totterdell, E. Petroske, K. Santa Cruz, R.C.J. Callison, and Y.S. Lau. 2002. Lysosomal malfunction accompanies alpha-synuclein aggregation in a progressive mouse model of Parkinson's disease. Brain Res. 956:156–165. [DOI] [PubMed] [Google Scholar]
- Mishizen-Eberz, A.J., R.P. Guttmann, B.I. Giasson, G.A. Day, R. Hodara, H. Ischiropoulos, V.M.-Y. Lee, J.Q. Trojanowski, and D.R. Lynch. 2003. Distinct cleavage patterns of normal and pathologic forms of α-synuclein by calpain I in vitro. J. Neurochem. 86:836–847. [DOI] [PubMed] [Google Scholar]
- Moran, L.B., S. Kosel, C. Spitzer, F.W. Schwaiger, O. Riess, G.W. Kreutzberg, and M.B. Graeber. 2001. Expression of alpha-synuclein in non-apoptotic, slowly degenerating facial motoneurones. J. Neurocytol. 30:515–521. [DOI] [PubMed] [Google Scholar]
- Murphy, D.D., S.M. Rueter, J.Q. Trojanowski, and V.M.-Y. Lee. 2000. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20:3214–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo, S., K. Tsukada, K. Omata, Y. Nakamura, and K. Nakaya. 1993. A new brain-specific 14-kDa protein is a phosphoprotein. Its complete amino acid sequence and evidence for phosphorylation. Eur. J. Biochem. 217:1057–1063. [DOI] [PubMed] [Google Scholar]
- Nakamura, N., A. Yamamoto, Y. Wada, and M. Futai. 2000. Syntaxin 7 mediates endocytic trafficking to late endosomes. J. Biol. Chem. 275:6523–6529. [DOI] [PubMed] [Google Scholar]
- Naruse, S., G. Thinakaran, J.J. Luo, J.W. Kusiak, T. Tomita, T. Iwatsubo, X. Qian, D.D. Ginty, D.L. Price, D.R. Borchelt, et al. 1998. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 21:1213–1221. [DOI] [PubMed] [Google Scholar]
- Okochi, M., J. Walter, A. Koyama, S. Nakajo, M. Baba, T. Iwatsubo, L. Meijer, P.J. Kahle, and C. Haass. 2000. Constitutive phosphorylation of the Parkinson's disease associated α-synuclein. J. Biol. Chem. 275:390–397. [DOI] [PubMed] [Google Scholar]
- Ostrerova, N., L. Petrucelli, M. Farrer, N. Mehta, P. Choi, J. Hardy, and B. Wolozin. 1999. alpha-Synuclein shares physical and functional homology with 14-3-3 proteins. J. Neurosci. 19:5782–5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak, S.H., R.D. Bagshaw, M. Guiral, S. Zhang, C.A. Ackerley, B.J. Pak, J.W. Callahan, and D.J. Mahuran. 2003. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J. Biol. Chem. 278:26687–26694. [DOI] [PubMed] [Google Scholar]
- Paxinou, E., Q. Chen, M. Weisse, B.I. Giasson, E.H. Norris, S.M. Rueter, J.Q. Trojanowski, V.M.-Y. Lee, and H. Ischiropoulos. 2001. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 21:8053–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, K., O.F. Olesen, and J.D. Mikkelsen. 1999. Developmental expression of alpha-synuclein in rat hippocampus and cerebral cortex. Neuroscience. 91:651–659. [DOI] [PubMed] [Google Scholar]
- Phiel, C.J., C.A. Wilson, V.M.-Y. Lee, and P.S. Klein. 2003. GSK-3 α regulates production of Alzheimer's disease amyloid-β peptides. Nature. 423:435–439. [DOI] [PubMed] [Google Scholar]
- Pigino, G., G. Morfino, A. Pelsman, M.P. Mattson, S.T. Brady, and J. Busciglio. 2003. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J. Neurosci. 23:4499–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney, J.W., and R.R. McKay. 1999. Capacitative calcium entry channels. Bioessays. 21:38–46. [DOI] [PubMed] [Google Scholar]
- Qian, S., P. Jiang, X.M. Guan, G. Singh, M.E. Trumbauer, H. Yu, H.Y. Chen, L.H. Van de Ploeg, and H. Zheng. 1998. Mutant human presenilin 1 protects presenilin 1 null mouse against embryonic lethality and elevates Abeta1-42/43 expression. Neuron. 20:611–617. [DOI] [PubMed] [Google Scholar]
- Selkoe, D.J., and R. Kopan. 2003. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu. Rev. Neurosci. 26:565–597. [DOI] [PubMed] [Google Scholar]
- Shearman, M.S., D. Beher, E.E. Clarke, H.D. Lewis, T. Harrison, P. Hunt, A. Nadin, A.L. Smith, G. Stevenson, and J.L. Castro. 2000. L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid beta-protein precursor gamma-secretase activity. Biochemistry. 39:8698–8704. [DOI] [PubMed] [Google Scholar]
- Shibayama-Imazu, T., I. Okahashi, K. Omata, S. Nakajo, H. Ochiai, Y. Nakai, T. Hama, Y. Nakamura, and K. Nakaya. 1993. Cell and tissue distribution and developmental change of neuron specific 14 kDa protein (phosphoneuroprotein 14). Brain Res. 622:17–25. [DOI] [PubMed] [Google Scholar]
- Sinha, S., and I. Lieberburg. 1999. Cellular mechanisms of beta-amyloid production and secretion. Proc. Natl. Acad. Sci. USA. 96:11049–11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza, J.M., B.I. Giasson, V.M.-Y. Lee, and H. Ischiropoulos. 2000. Chaperone-like activity of synucleins. FEBS Lett. 474:116–119. [DOI] [PubMed] [Google Scholar]
- Stefanis, L., K.E. Larsen, H.J. Rideout, D. Sulzer, and L.A. Greene. 2001. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 21:9549–9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K., E. Iseki, O. Katsuse, A. Yamaguchi, K. Katsuyama, I. Aoki, S. Yamanaka, and K. Kosaka. 2003. Neuronal accumulation of alpha- and beta-synucleins in the brain of a GM2 gangliosidosis mouse model. Neuroreport. 14:551–554. [DOI] [PubMed] [Google Scholar]
- Teclemariam-Mesbah, R., J. Wortel, H.J. Romijn, and R.M. Buijs. 1997. A simple silver-gold intensification procedure for double DAB labeling studies in electron microscopy. J. Histochem. Cytochem. 45:619–621. [DOI] [PubMed] [Google Scholar]
- Trojanowski, J.Q., T. Schuck, M.L. Schmidt, and V.M.-Y. Lee. 1989. Distribution of phosphate-independent MAP2 epitopes revealed with monoclonal antibodies in microwave-denatured human nervous system tissues. J. Neurosci. Methods. 29:171–180. [DOI] [PubMed] [Google Scholar]
- Tu, P.H., G. Elder, R.A. Lazzarini, D. Nelson, J.Q. Trojanowski, and V.M.-Y. Lee. 1995. Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. J. Cell Biol. 129:1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda, K., H. Fukushima, E. Masliah, Y. Xia, A. Iwai, M. Yoshimoto, D.A. Otero, J. Kondo, Y. Ihara, and T. Saitoh. 1993. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA. 90:11282–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila, M., S. Vukosavic, V. Jackson-Lewis, M. Neystat, M. Jakowec, and S. Przedborski. 2000. Alpha-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 74:721–729. [DOI] [PubMed] [Google Scholar]
- Walkley, S.U. 1998. Cellular pathology of lysosomal storage disorders. Brain Pathol. 8:175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb, J.L., B. Ravikumar, J. Atkins, J.N. Skepper, and D.C. Rubinsztein. 2003. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278:25009–25013. [DOI] [PubMed] [Google Scholar]
- Wilson, C.A., R.W. Doms, H. Zheng, and V.M.-Y. Lee. 2002. Presenilins are not required for A beta 42 production in the early secretory pathway. Nat. Neurosci. 5:849–855. [DOI] [PubMed] [Google Scholar]
- Withers, G.S., J.M. George, G.A. Banker, and D.F. Clayton. 1997. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res. Dev. Brain Res. 99:87–94. [DOI] [PubMed] [Google Scholar]
- Wolfe, M.S., M. Citron, T. Diehl, W. Xia, I.O. Donkor, and D.J. Selkoe. 1998. Cellular mechanisms of beta-amyloid production and secretion. J. Med. Chem. 41:6–9. [DOI] [PubMed] [Google Scholar]
- Wong, P.C., H. Zheng, H. Chen, M.W. Becher, D.J. Sirinathsinghji, M.E. Trumbauer, H.Y. Chen, D.L. Price, L.H. Van der Ploeg, and S.S. Sisodia. 1997. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 387:288–292. [DOI] [PubMed] [Google Scholar]
- Yoo, A.S., I. Cheng, S. Chung, T.Z. Grenfell, H. Lee, E. Pack-Chung, M. Handler, J. Shen, W. Xia, G. Tesco, et al. 2000. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 27:561–572. [DOI] [PubMed] [Google Scholar]