

Abstract
Cells lacking vinculin are highly metastatic and motile. The reasons for this finding have remained unclear. Both enhanced survival and motility are critical to metastasis. Here, we show that vinculin null (vin−/−) cells and cells expressing a vinculin Y822F mutant have increased survival due to up-regulated activity of extracellular signal–regulated kinase (ERK). This increase is shown to result from vinculin's modulation of paxillin–FAK interactions. A vinculin fragment (amino acids 811–1066) containing the paxillin binding site restored apoptosis and suppressed ERK activity in vin−/− cells. Both vinY822F and vin−/− cells exhibit increased interaction between paxillin and focal adhesion kinase (FAK) and increased paxillin and FAK phosphorylation. Transfection with paxillin Y31FY118F dominant-negative mutant in these cells inhibits ERK activation and restores apoptosis. The enhanced motility of vin−/− and vinY822F cells is also shown to be due to a similar mechanism. Thus, vinculin regulates survival and motility via ERK by controlling the accessibility of paxillin for FAK interaction.
Keywords: apoptosis; extracellular signal–regulated kinase; anoikis; MAPK; migration
Introduction
Adhesion complexes (i.e., focal adhesions and focal complexes) and cell–cell contacts are specialized structures that harbor a large number of cytoskeletal proteins and one of the highest concentrations of signaling molecules in cells (Ruoslahti and Obrink, 1996). These multiprotein complexes are capable of capturing and integrating many signals from the extracellular as well as intracellular environments (Rosales and Juliano, 1995). Such signals are indispensable for the coordinated control of fundamental cellular processes including differentiation, cell cycle control, apoptosis, and motility (Ruoslahti and Obrink, 1996; Huang and Ingber, 2000). Coordinating these processes is critical for the maintenance of tissue homeostasis (Huang and Ingber, 2000). In fact, disruption in the regulation of motility and survival is a major step in the pathogenesis of cancer (Parise et al., 2000).
Disregulation in the control of multiple cellular processes is seen in cells with altered expression and/or activation of molecules in focal adhesions and cell–cell contacts. Such molecules include paxillin, FAK (Rodina et al., 1999), and vinculin (Raz and Geiger, 1982; Lifschitz-Mercer et al., 1997). Cancer cells devoid of vinculin (Rudiger, 1998) are highly metastatic (Raz and Geiger, 1982; Lifschitz-Mercer et al., 1997) and motile (Coll et al., 1995; Xu et al., 1998a,b). Transfecting vinculin back into vinculin null cells remarkably represses their metastatic capacity (Rodriguez-Fernandez et al., 1992) as well as their enhanced motility (Coll et al., 1995; Xu et al., 1998b). The mechanisms through which vinculin controls motility and represses metastatic capacity remain unclear.
Highly metastatic cells, such as those lacking vinculin (Rodriguez-Fernandez et al., 1992), are usually very motile (Parise et al., 2000). Enhanced cell motility has been associated with increased survival (Frisch and Francis, 1994). Recent evidence has shown the existence of signaling cascades that coordinate these two processes (Cho and Klemke, 2000). Thus, vinculin could affect signaling through multiple proteins that mediate both survival and motility and are found in focal adhesions and/or cell–cell contacts. These proteins include p130 Crk-associated substrate (Cas) and Crk-II adaptor proteins (Nakamoto et al., 1997; Cho and Klemke, 2000), extracellular signal–regulated kinase (ERK)1/2 (Klemke et al., 1997; Fincham et al., 2000), phosphatidylinositol-3 kinase (PI-3K; Watton and Downward, 1999; Sasaki et al., 2000), and FAK (Hauck et al., 2002).
FAK is a particularly important signaling molecule, as it controls multiple fundamental cellular processes by regulating the assembly of different multiprotein complexes (Tachibana et al., 1995; Schwartz, 2001). Interaction between FAK and the adaptor protein paxillin is critical for the activation of signaling cascades involved in the control of cell survival and motility (Turner, 2000). FAK binds to paxillin through its COOH-terminal focal adhesion targeting (FAT) region (Hayashi et al., 2002). This FAT region is also contained within FAK-related nonkinase (FRNK), a naturally occurring fragment of FAK (Richardson and Parsons, 1996). Transfection with FRNK blocks the interaction of endogenous FAK with paxillin at focal adhesions, decreasing survival and motility (Richardson and Parsons, 1996; Hauck et al., 2000, 2002). Mutagenesis studies have shown that this interaction requires the sequence of the FAT region in FRNK (Hauck et al., 2002). Interestingly, the structure of the FAT region resembles that of vinculin's COOH-terminal “tail domain” (Bakolitsa et al., 1999; Hayashi et al., 2002). Furthermore, the FAT region and vinculin tail bind to overlapping repeats of leucine-rich sequences in paxillin named LD motifs (Turner et al., 1999; Turner, 2000; Tumbarello et al., 2002), suggesting that vinculin too may inhibit important FAK interactions. These observations led us to examine whether or not the highly metastatic and motile vinculin null cells have enhanced survival, and whether or not enhanced survival and the enhanced motility previously reported were due to disregulation of paxillin–FAK interactions. In this paper, we describe a novel pathway whereby vinculin controls cell survival and motility through regulation of paxillin–FAK interactions to alter ERK1/2 activation.
Results
Vinculin null cells are resistant to apoptosis
To test whether or not vinculin is important in apoptosis, we first examined apoptosis induced by serum withdrawal. Apoptosis was compared in wild-type mouse F9 embryonal carcinoma cells (WT F9), their vinculin null counterparts (F9 vin−/−), and F9 vin−/− rescue cells (stable cell lines in which vinculin had been reintroduced into the F9 vin−/− cells). Caspase-3 activation was assessed as an indicator of irreversible commitment to apoptosis (Nicholson et al., 1995). WT F9 cells exhibited caspase-3 activation reflected by the presence of cleaved caspase-3 after 10 h of serum withdrawal (Fig. 1 A). In contrast, F9 vin−/− cells showed a remarkable inhibition of caspase-3 activation, and caspase-3 activity was restored in the F9 vin−/− rescue cells (Fig. 1 A). N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (DEVD-AMC) cleavage quantifies the activity of caspase-3 as well as caspase-7 (Nicholson et al., 1995; He et al., 1998). Activation of caspase-7 is also an indicator of irreversible commitment to apoptosis (He et al., 1998; Riedl et al., 2001). DEVD-AMC cleavage was inhibited by 70% in F9 vin−/− cells compared with WT F9 cells after 10 h of serum withdrawal and was fully restored to wild-type levels in the F9 vin−/− rescue cells (Fig. 1 B).
Figure 1.

Vinculin null F9 embryonal carcinoma cells are resistant to a variety of apoptotic stimuli. (A) Detergent lysates from WT F9, F9 vin−/−, and F9 vin−/− rescue cells were analyzed by immunoblotting using anti–caspase-3 Ab 10 h after serum deprivation. The result shown is representative of three independent experiments. (B) DEVD-AMC cleavage, indicative of caspase-3 and -7 activation, was compared for WT F9 cells, F9 vin−/−, and F9 vin−/− rescue cells 10 h after serum withdrawal. (C) Cell extracts from WT F9, F9 vin−/−, and F9 vin−/− rescue cells were analyzed by immunoblotting using anti–caspase-3 Ab 3 h after disruption of cell–matrix interaction. The result shown is a representative example from three independent experiments. (D) DEVD-AMC cleavage was compared for WT F9 cells, F9 vin−/−, and F9 vin−/− rescue cells 3 h after disruption of cell–matrix interaction. (E) Immunoblotting analysis of cell extracts from WT F9, F9 vin−/−, and F9 vin−/− rescue cells treated for 2 h with 2 μM camptothecin using anti–caspase-3 Ab. The result shown is representative of three independent experiments. (F) DEVD-AMC cleavage was compared for WT F9 cells, F9 vin−/−, and F9 vin−/− rescue cells 2 h after exposure to 2 μM camptothecin. (G) Camptothecin-induced apoptotic cell death was also assessed through cellular morphologic changes. In B, D, F, and G, results shown are means ± SEM from at least three independent experiments. DEVD-AMC cleavage is shown in arbitrary fluorescence intensity units.
The role of vinculin in anoikis, apoptosis induced by detachment from the cellular substrate (Frisch and Francis, 1994), was also examined. Caspase-3 activation and DEVD-AMC cleavage were remarkably inhibited in F9 vin−/− cells compared with WT F9 cells (Fig. 1, C and D). Here too, both caspase-3 activation and DEVD-AMC cleavage were restored to wild-type levels in F9 vin−/− rescue cells.
Finally, caspase-3 activity and DEVD-AMC cleavage induced by 2-h incubation with camptothecin (Hertzberg et al., 1989), a cytotoxic drug widely used in cancer therapy, was also inhibited in F9 vin−/− cells compared with WT F9 cells (Fig. 1, E and F). The effect of camptothecin on caspase-3 activity was similar in wild-type and F9 vin−/− rescue cells. The rate of camptothecin-induced apoptosis was also determined using morphological criteria (blebbing and cell fragmentation; Subauste et al., 2000). As shown in Fig. 1 G, this rate was considerably reduced in F9 vin−/− cells compared with WT F9 or F9 vin−/− rescue cells. We also found that vinculin null embryonal fibroblasts are resistant to various apoptotic stimuli (unpublished data). Together, these findings show that vinculin regulates apoptosis induced by multiple different stimuli.
Vinculin regulation of apoptosis requires its hinge-tail fragment
Vinculin has multiple binding sites for interaction with components of cell contact sites (Rudiger, 1998). It consists of a large amino-terminal head and a rodlike tail connected by a flexible hinge region (840–857 aa; Rudiger, 1998). To address which regions of vinculin are important for control of apoptosis, cell death was examined in a F9 vin−/− cell clone (γ229) stably transfected with vinculin mutants (see Fig. 3). Results were confirmed with a different F9 vin−/− cell clone (γ227; see Fig. 4). For this work, we used cell clones derived from stable cell lines transfected with each construct (Fig. 2). Except for the hinge-tail fragment, the expression of these constructs was corroborated through immunoblotting with an antivinculin antibody (Ab; Fig. 2 B). The hinge-tail fragment is Flag tagged, so its expression was confirmed using an anti-Flag M2 Ab (Fig. 2 C).
Figure 3.

Role of various regions of vinculin in the control of apoptosis induction. (A) Cell extracts from WT F9 and F9 vin−/− cells (γ229 clone) stably expressing each construct were analyzed by immunoblotting using anti–caspase-3 Ab 10 h after serum withdrawal. (B) DEVD-AMC cleavage was compared for WT F9 cells versus F9 vin−/− cells stably expressing each construct 10 h after serum withdrawal. (C) Immunoblotting analysis of cell extracts from WT F9 and F9 vin−/− cells stably expressing each construct 3 h after disruption of cell–matrix interaction using anti–caspase-3 Ab. (D) DEVD-AMC cleavage was compared for WT F9 cells versus F9 vin−/− cells stably expressing vinY822F mutant and the hinge-tail fragment 3 h after disruption of cell–matrix interaction. (B and D) Results shown are means ± SEM from at least three independent experiments, in arbitrary fluorescence intensity units.
Figure 4.
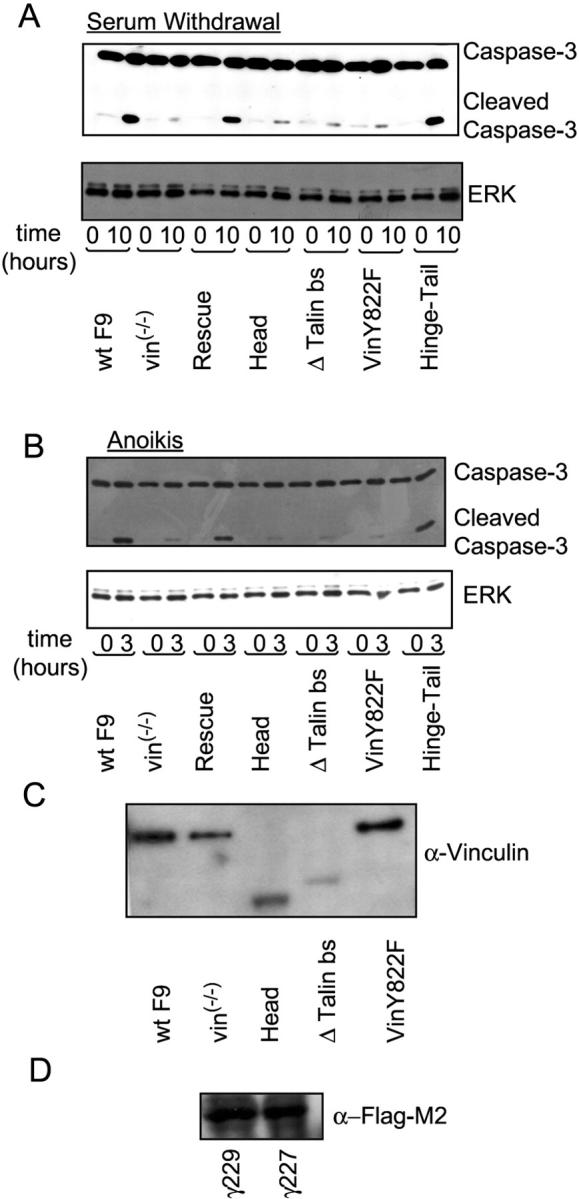
Role of various regions of vinculin in the control of apoptosis induction in a different F9 vin − / − clone (γ227). (A) Cell extracts from WT F9 and F9 vin−/− cells (γ227 clone) stably expressing each construct were analyzed by immunoblotting using anti–caspase-3 Ab 10 h after serum withdrawal. (B) Immunoblotting analysis of cell extracts from WT F9 and F9 vin−/− cells stably expressing each construct 3 h after disruption of cell–matrix interaction using anti–caspase-3 Ab. The results shown in A and B are representative examples from two independent experiments. (C) Expression of full-length vinculin, head, Δ talin bs, and vinY822F in γ227 clones. (D) Expression of the Flag M2–tagged hinge-tail fragment in γ229 and γ227 clones.
Figure 2.
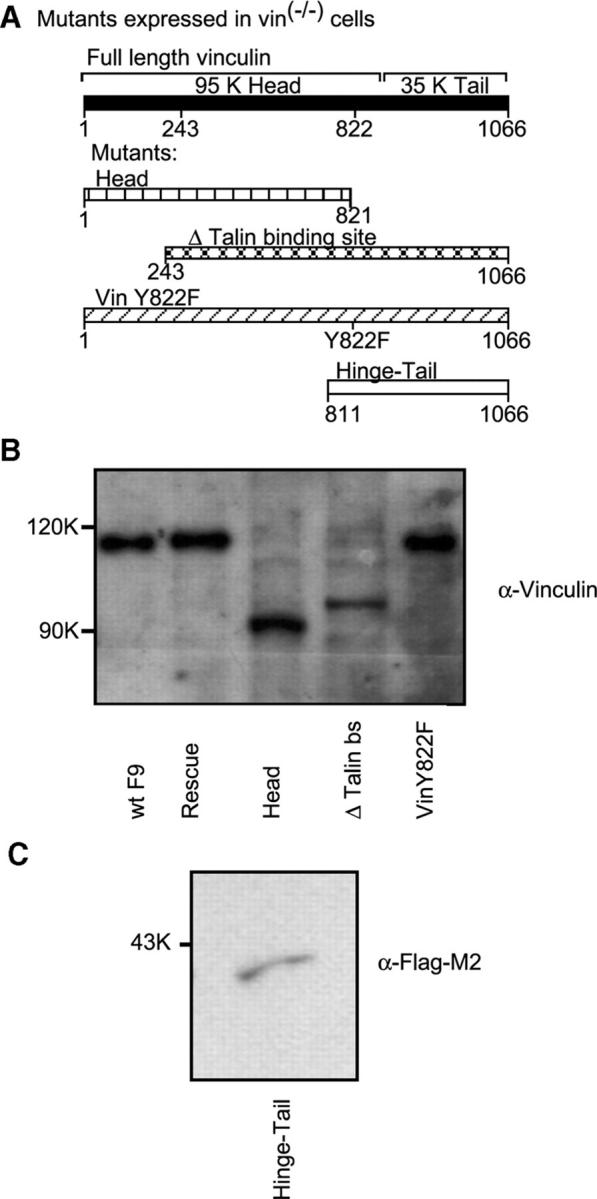
Role of various regions of vinculin in the control of apoptosis induction. (A) Vinculin mutants: the top line represents full-length vinculin. First mutant: head fragment (1–821). Second mutant: vinculin fragment with talin binding site deleted (243–1066). Third mutant: full-length vinculin containing a Y822F point mutation. Fourth mutant: hinge-tail fragment (811–1066). (B) Expression of these constructs, except for the hinge-tail fragment, was determined by immunoblotting with an antivinculin Ab. (C) Expression of the Flag M2-tagged hinge-tail fragment was determined by immunoblotting with an anti-Flag Ab.
The hinge-tail fragment (811–1066 aa) was able to restore apoptosis to wild-type levels, whereas the head fragment (1–821 aa) could not (Fig. 3, A and B), for apoptosis induced by serum withdrawal (Fig. 3, A and B) or anoikis (Fig. 3 C). This finding indicated that the critical portion of the vinculin molecule was within 811–1066 aa. Interestingly, the vinculin fragment 243–1066, in which there were deleted talin binding sites (Δ talin bs mutant), could not restore apoptosis (Fig. 3, A–C). This fragment cannot localize into focal adhesions (Goldmann et al., 1998) but can interact with α-catenin, a protein only present in cell–cell contacts (unpublished data), suggesting that focal adhesion localization is important for vinculin's regulation of apoptosis.
Vinculin with a Y822F point mutation was unable to restore apoptosis induced by serum withdrawal or cellular detachment, as demonstrated both by monitoring caspase-3 cleavage (Fig. 3, A–C) and DEVD-AMC cleavage (Fig. 3 D). Vinculin Y822F is able to localize into focal adhesions (Goldmann et al., 1998), suggesting that the mutation likely disrupts a specific interaction with vinculin's binding partners. The aforementioned data were also reproduced using a different F9 vin−/− cell clone (γ227) stably transfected with the various vinculin mutants (Fig. 4). Together, these data show that vinculin regulates apoptosis through its hinge-tail region via a process occurring within focal complexes.
Vinculin affects apoptosis by modulating ERK1/2 activity
Our results showed that vinculin controls apoptosis. Because vinculin also affects cellular motility (Coll et al., 1995), we searched for a signaling pathway through which vinculin might regulate both processes. ERK1/2 regulates motility and apoptosis and localizes in focal adhesions (Klemke et al., 1997; Fincham et al., 2000; Howe et al., 2002). Furthermore, ERK1/2 activity is elevated in cells that are highly metastatic and resist anoikis (Howe et al., 2002). Therefore, we assessed whether or not vinculin affected apoptosis by regulating ERK1/2 activity, concentrating on anoikis because of its significance in cancer biology (Frisch and Francis 1994; Howe et al., 2002).
As shown in Fig. 5 A, treatment of F9 vin−/− and F9 vinY822F with ERK antisense oligonucleotides restored anoikis-induced caspase-3 activation. Antisense treatment was shown to reduce ERK protein levels, scrambled antisense oligonucleotide controls had no effect, and blotting with anti-Erk Abs demonstrated that each cell line had similar Erk concentrations before antisense treatment (Fig. 5, A and B).
Figure 5.

Vinculin controls apoptosis induction by regulating ERK activity. (A) WT F9, F9 vin−/−, and F9 vin−/−Y822F cells were transfected with antisense ERK 1/2 and scrambled control oligonucleotides. DEVD-AMC cleavage was assayed 3 h after disruption of cell–matrix interaction. (B) Detergent lysates from these cells were analyzed by immunoblotting using anti-ERK1/2 and MEK Abs. The result shown are representative of three independent experiments. (C) WT F9, F9 vin−/−, and F9 vin−/−Y822F cells were left untreated or treated with 10 μM PD 98059 3 h before they were detached from gelatin-coated plates and during anoikis. DEVD-AMC cleavage was measured 3 h after disruption of cell–matrix interaction. Means ± SEM were calculated from at least three independent experiments. (D) WT F9, F9 vin−/−, and F9 vin−/−Y822F cells were left untreated or treated with 10 μM PD 98059 3 h before detachment from gelatin-coated plates and during anoikis. Detergent lysates of cells were analyzed by immunoblotting using anti-ERK1/2 and anti-phosphoERK1/2 Abs 30 min after disruption of cell–matrix interaction. The result shown is a representative example from two independent experiments. (E) A representative immunoblot of three showing phosphorylated ERK1/2 from WT F9, F9 vin−/−, F9 vin−/−Y822F, and F9 vin−/− hinge-tail cells after disruption of cell–matrix interactions. (F) A representative immunoblot of two showing phosphorylated ERK1/2 from WT F9, F9 vin−/−, and rescue cells. (G) Detergent lysates from WT F9, F9 vin−/−, rescue, F9 vin−/−Y822F, and F9 vin−/− hinge-tail cells were analyzed by immunoblotting using anti–caspase-9 Ab 30 min after disruption of cell–matrix interaction. The result is a representative example from two independent experiments. (H) WT F9, F9 vin−/−, and F9 vin−/−Y822F cells were left untreated or treated with 50 nM Wortmannin 1 h before they were detached from gelatin-coated plates and during anoikis. DEVD-AMC cleaving caspases activity was assayed 3 h after disruption of cell–matrix interactions. Means ± SEM were calculated from at least three independent experiments. A, C, and H show arbitrary fluorescence intensity units.
ERK1/2 is activated upon phosphorylation of its regulatory threonine and tyrosine residues by MAPK kinase (MEK; Ahn et al., 1991). The MEK inhibitor PD98059 restored caspase-3 activation to wild-type levels in F9 vin−/− and F9 vin−/−Y822F cells (Fig. 5 C), and this increase was largely eliminated by MEK inhibition (Fig. 5 D). This finding indicated that anoikis resistance was mediated by activation of the MEK–ERK pathway.
We also examined ERK1/2 activity in the apoptosis-resistant F9 vin−/− and F9 vin−/−Y822F cells, and the apoptosis-sensitive F9 vin−/− hinge-tail and WT F9 cells. Lysates from these cells were probed with Abs specific for the active, phosphorylated forms of ERK1/2 at different times after cellular detachment (Fig. 5 E). In both F9 vin−/− and F9 vin−/− Y822F cells, detachment led to ERK1/2 activation substantially above that in WT F9 cells. In contrast, F9 vin−/− hinge-tail cells showed ERK1/2 activation inhibited to levels seen in the wild-type cells. In F9 vin−/− cells stably transfected with vinculin, ERK1/2 activity after detachment was the same as in WT F9 cells (Fig. 5 F).
ERK1/2 was recently shown to down-regulate apoptosis by inhibiting activation of caspase-9 (Allan et al., 2003). Caspase-9 is an initiator protease that activates caspase-3 and other downstream caspases to trigger apoptosis, and it has been found to be particularly important in anoikis (Grossmann et al., 2001). Therefore, we examined caspase-9 activation after cellular detachment. WT F9, rescue, and F9 vin−/− hinge-tail cells showed cleaved caspase-9 30 min after cellular detachment (Fig. 5 G). In contrast, no cleaved caspase-9 was observed in F9 vin−/− and F9 vin−/−Y822F cells (Fig. 5 G). Together, these data indicate that vinculin affects apoptosis by regulating ERK1/2 activity through a mechanism requiring the hinge-tail region.
PI-3K is another signaling molecule that localizes into focal adhesions and affects both motility and apoptosis (Watton and Downward, 1999; Sasaki et al., 2000). We assessed whether or not vinculin affects apoptosis by regulating this pathway. As seen in Fig. 5 H, treatment of F9 vin−/− and F9 vin−/−Y822F cells with Wortmannin (50 μM), an inhibitor of PI-3K activity, was unable to restore apoptosis to wild-type levels. These results indicated that the survival pathway affected in vin−/− and vin−/−Y822F cells does not involve PI-3K. The WT F9 cells did exhibit more cell death upon inhibition of PI-3K, perhaps because they lack the enhanced survival mechanism produced by altering vinculin.
Vinculin regulates ERK1/2 activity through modulation of paxillin–FAK interaction
We found that vinculin regulates apoptosis via the ERK1/2 pathway, and that this regulation requires the vinculin hinge-tail region. The hinge-tail fragment is targeted to focal adhesions, where it colocalizes with the adaptor protein paxillin (Xu et al., 1998b). Furthermore, the vinculin tail domain and the FAT region of FAK bind to overlapping paxillin LD motifs (Turner et al., 1999; Turner, 2000; Tumbarello et al., 2002). FAK is an adaptor protein whose kinase activity is involved in the activation of ERK1/2, cell survival, and motility (Schwartz, 2001; Hauck et al., 2002). Therefore, we tested if the paxillin–FAK interaction is affected by vinculin.
To gauge the level of paxillin–FAK binding in different cell lysates, either paxillin or FAK were immunoprecipitated, and Western blotting was used to probe for the presence of the binding partner (Fig. 6, A and B). Paxillin–FAK interaction was enhanced relative to WT F9 cells in vin−/− and vinY822F cells. In contrast, paxillin–FAK binding is at wild-type levels in F9 vin−/− hinge-tail and rescue cells (Fig. 6, A and B).
Figure 6.

F9 vin −/− and F9 vin −/− Y822F cells have an increased paxillin–FAK interaction. (A) Cell extracts from WT F9, F9 vin−/−, rescue, F9 vin−/−Y822F, and F9 vin−/− hinge-tail cells were immunoprecipitated with a mouse antipaxillin Ab, followed by immunoblotting with Ab against FAK. (B) Cell extracts from WT F9, F9 vin−/−, rescue, F9 vin−/−Y822F, and F9 vin−/− hinge-tail cells were immunoprecipitated with a mouse anti-FAK Ab, followed by immunoblotting with Ab against paxillin. The results shown are representative examples from three independent experiments.
FAK interaction with paxillin, coupled with phosphorylation of both proteins, creates docking sites for signaling molecules involved in cell motility and survival (Turner, 2000; Schwartz, 2001; Hauck et al., 2002). Phosphorylation of FAK at tyrosine 397 (Y397) is critical for the induction of downstream effects (Hauck et al., 2000, 2002; Schwartz, 2001), including phosphorylation of paxillin at Tyrosine 118 (Y118; Turner, 2000). We assessed whether or not the enhanced paxillin–FAK interaction in F9 vin−/− and F9 vin−/−Y822F cells correlated with increased phosphorylation of FAK-Y 397 and paxillin-Y118. The data in Fig. 7 (A and B) show that these cells do in fact have constitutively increased FAK-Y397 and paxillin-Y118 phosphorylation. After cellular detachment, phosphorylation at these sites rapidly decreased (Fig. 7, A and B). In contrast, phosphorylation at both sites is at wild-type levels in F9 vin−/− hinge-tail cells as well as rescue cells (Fig. 7, A and B), both constitutively and during anoikis.
Figure 7.

F9 vin −/− and F9 vin −/− Y822F cells have increased FAK-Y397 and paxillin-Y118 phosphorylation. (A) A representative immunoblot showing FAK phosphorylated at Y397 in WT F9, F9 vin−/−, rescue, F9 vin−/−Y822F, and F9 vin−/− hinge-tail cells after disruption of cell–matrix interactions. (B) An immunoblot showing paxillin phosphorylated at Y118 in WT F9, F9 vin−/−, rescue, F9 vin−/−Y822F, and F9 vin−/− hinge-tail cells after disruption of cell–matrix interactions. The results shown are representative of three independent experiments.
Finally, we examined cells overexpressing paxillin mutated at the residues phosphorylated upon FAK interaction (paxillin Y31FY118F; Turner, 2000). This mutant disrupts important signaling events activated by the paxillin–FAK interaction (Petit et al., 2000). Overexpression of paxillin Y31FY118F restored anoikis in F9 vin−/− and F9 vin−/− Y822F cells to wild-type levels (Fig. 8 A). Importantly, this mutant also inhibited the enhanced ERK1/2 activity during cellular detachment (Fig. 8 B). Together, these data indicate that vinculin controls cell survival via ERK by regulating paxillin–FAK interaction.
Figure 8.

Expression of paxillin Y31F and Y118F in F9 vin −/− and F9 vin −/− Y822F cells restores apoptosis and reduces ERK activity to wild-type levels. (A) WT F9, F9 vin−/−, and F9 vin−/−Y822F cells were transfected with paxillin Y31FY118F-myc, wild-type paxillin-myc, or control mRFP-myc plasmids. DEVD-AMC cleavage was assayed 3 h after disruption of cell–matrix interaction. Means ± SEM were calculated from four independent experiments. (B) WT F9, F9 vin−/−, and F9 vin−/−Y822F were transfected with paxillin Y31FY118F-myc, wild-type paxillin-myc, or control mRFP-myc plasmids. Detergent lysates were analyzed by immunoblotting using ERK1/2 and anti-phosphoERK1/2 Abs 30 min after disruption of cell–matrix interaction. (C) Expression levels of control mRFP-myc, wild-type paxillin-myc, and paxillin Y31FY118F-myc proteins in WT F9, F9 vin−/−, and F9 vin−/−Y822F cells. DEVD-AMC cleavage is shown in arbitrary fluorescence intensity units. The results shown in B and C are representative of three independent experiments.
Vinculin regulates motility via paxillin and ERK1/2 activation
A previously published paper showed that the enhanced motility of F9 vin−/− cells was inhibited by expression of the vinculin hinge-tail fragment (Xu et al., 1998b). Because cellular motility and survival can be regulated by the same signaling pathways (Cho and Klemke, 2000), we assessed whether or not vinculin controls motility as well as apoptosis via ERK1/2 activation, and whether or not this is also dependent on paxillin–FAK interaction. In a Boyden chamber assay, WT F9 cells migrated significantly less than either F9 vin−/− cells or F9 vin−/−Y822F cells. Incubation with the MEK inhibitor PD 98059 led to a 50–55% inhibition of F9 vin−/− migration rates and decreased the enhanced migration of F9 vin−/−Y822F cells to wild-type levels (Fig. 9 A). The drug had little effect on the migration of WT F9 cells.
Figure 9.
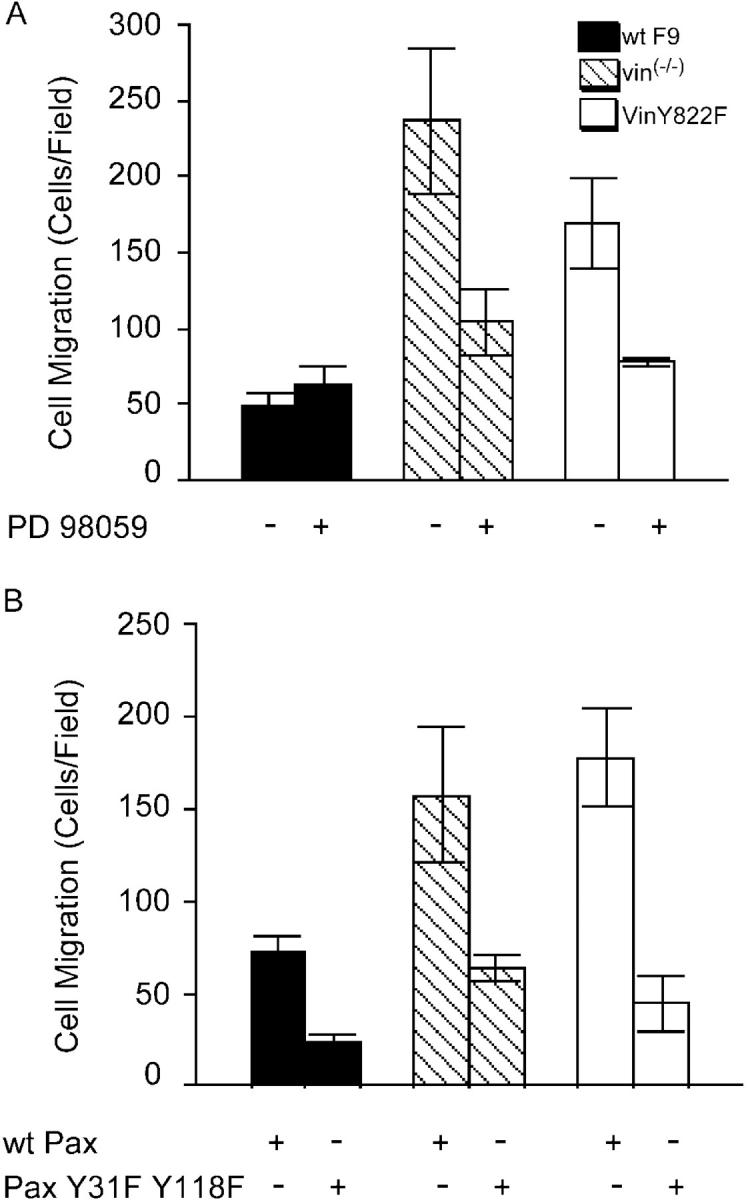
Enhanced motility in F9 vin −/− and F9 vin −/− Y822F cells is restored to wild-type levels upon inhibition of MEK and expression of paxillin Y31FY118F. (A) WT F9, F9 vin−/−, and F9 vin−/−Y822F cells were left untreated or treated with 10 μM PD 98059 3 h before they were detached from gelatin-coated plates for cell migration assays. Cells were allowed to migrate for 8 h using Boyden chambers coated with collagen. Mean number of cells per field ± SEM was calculated from at least three independent experiments. (B) WT F9, F9 vin−/−, and F9 vin−/−Y822F cells were transfected with paxillin Y31FY118F and wild-type paxillin. Cell migration was quantified as in A.
We had shown that expression of the paxillin Y31FY118F mutant led to restoration of anoikis in F9 vin−/− and F9 vin−/−Y882F cells. As seen in Fig. 9 B, overexpression of this mutant led to a remarkable inhibition of enhanced cell migration in F9 vin−/− and F9 vin−/−Y822F cells. These findings indicate that vinculin regulates both motility and survival through paxillin and ERK1/2 activation.
Discussion
Vinculin has a well characterized function as a cytoskeletal protein within adhesion complexes (Rudiger, 1998), but much of its role in signaling remains obscure. Tissue homeostasis is determined by the tightly regulated recruitment of signaling molecules (Huang and Ingber, 2000), such as FAK (Hauck et al., 2002) and paxillin (Turner, 2000; Schwartz, 2001), to specific subcellular locations. Our results demonstrate that vinculin controls both motility and survival by affecting ERK1/2 activity through modulation of paxillin–FAK interaction.
Vinculin null cells showed reduced susceptibility to apoptosis induced by either serum withdrawal, perturbation of cell attachment (anoikis), or camptothecin. Effects in these very different apoptosis induction pathways suggest that vinculin participates in one or more conserved and fundamental pathways. Reintroduction of vinculin was able to rescue normal apoptosis, proving that these results were not simply due to a compensatory mutation during the production of the vinculin null cells. By examining apoptosis after introduction of various vinculin mutants into vin−/− cells, we pinpointed the vinculin hinge-tail fragment (811–1066 aa) as essential for apoptosis regulation.
These experiments also suggested that vinculin's effect on apoptosis was dependent on its localization in focal adhesions. The hinge-tail fragment, which did restore apoptosis, is localized in focal adhesions and colocalizes with paxillin (Xu et al., 1998b). In contrast, Δ Talin bs mutant (243–1066 aa) is unable to localize in focal adhesions (Goldmann et al., 1998) and was unable to restore apoptosis in vin−/− cells. According to earlier works, the Δ talin bs (243–1066 aa) mutant would be expected to incorporate in focal adhesions through its tail domain; these works indicated that vinculin could target focal adhesions through both a talin binding site and another binding site in the tail (Bendori et al., 1989). These conclusions were based on the fact that a vinculin construct lacking 167–207 aa, thought to be vinculin's only talin binding site, was still able to incorporate into focal adhesions (Bendori et al., 1989). Later studies showed that vinculin's talin binding site actually extends from 1 to 253 aa (Gilmore et al., 1992). Thus, the vinculin construct lacking 167–207 aa still had residues involved in vinculin's interaction with talin, which could mediate localization into focal adhesions. In fact, a vinculin fragment consisting of 1–130 aa shows good talin binding (Bass et al., 2002), and a recent paper (Izard et al., 2004) has shown that talin's interaction with vinculin is critical for opening vinculin into its active conformation. Talin's interaction with vinculin causes vinculin's tail to dissociate from its head (Izard et al., 2004). Thus, the Δ talin bs mutant could not incorporate into the focal adhesion (Goldmann et al., 1998), as there was no talin binding to disrupt the high affinity interaction of its tail and residual head domain. The much smaller hinge-tail fragment is capable of incorporating into focal adhesions because it is completely devoid of vinculin's inhibitory head domain (Xu et al., 1998b).
Apoptosis resistance in vin−/− and vinY822F cells was ERK dependent. This conclusion is based on the fact that apoptosis of both F9 vin−/− and F9 vin−/−Y822F cells could be restored to wild-type levels using ERK antisense oligonucleotides or the MEK inhibitor PD98059. Moreover, recovery of apoptosis in F9 vin−/− hinge-tail cells was associated with a reduction of ERK1/2 activity to wild-type levels, and apoptosis-resistant F9 vin−/− and F9 vin−/−Y822F cells showed increased ERK1/2 activation after cellular detachment. Increased ERK1/2 phosphorylation after cellular detachment has been shown before in apoptosis-resistant lung adenocarcinoma cells (Wei et al., 2001). However, in contrast to our work, apoptosis resistance in these tumoral cells was ERK1/2-independent (Wei et al., 2001). Differences in the experimental design may account for this discrepancy. Specifically, in the present work, we inhibited ERK activity with PD98059 and ERK antisense oligonucleotides before cellular detachment, whereas in the previous work (Wei et al., 2001), ERK1/2 activity was inhibited with PD98059 after cellular detachment. This might be an important issue because basal activity levels might determine if a cell survives or dies upon apoptosis induction (Howe et al., 2002). However, consistent with our work, there was no difference in the constitutive ERK1/2 phosphorylation levels among apoptosis-resistant and apoptosis-sensitive cells (Wei et al., 2001). It is possible that only a small fraction of the total cellular ERK is involved in this regulation. ERK1/2 is known to be compartmentalized in various subcellular locations (Reffas and Schlegel, 2000; Pouyssegur et al., 2002; Smith et al., 2004).
Combining our own data with previous studies suggested that vinculin regulates motility and survival by affecting paxillin–FAK interaction. Expression of a vinculin hinge-tail fragment in F9 vin−/− cells reduced FAK-Y397 phosphorylation, cell motility (Xu et al., 1998b), and ERK1/2-dependent cell survival. These effects are very similar to those produced by transfection with FAK fragments containing the sequence of the FAT region, which also inhibits FAK-Y397 phosphorylation and downstream effects, including cell motility and survival (Richardson and Parsons, 1996; Hauck et al., 2000). Interestingly, the vinculin tail domain and the FAT region bind to overlapping paxillin LD motifs (Turner et al., 1999; Turner, 2000; Tumbarello et al., 2002). The FAT region contains two distinct paxillin binding subdomains (Turner et al., 1999; Tumbarello et al., 2002), which bind to paxillin LD2 and LD4 motifs (Hayashi et al., 2002). The tail domain of vinculin is capable of binding paxillin LD1, LD2, or LD4 motifs (Turner et al., 1999; Tumbarello et al., 2002). Vinculin oligomers (Rudiger, 1998) could bind simultaneously to paxillin LD2 and LD4 motifs from the same paxillin molecules, thus interfering with FAK binding. We showed that the highly motile and apoptosis-resistant F9 vin−/− cells did in fact exhibit enhanced paxillin–FAK interaction, and this was inhibited to wild-type levels upon expression of the vinculin hinge-tail region.
Paxillin–FAK interaction leads to paxillin phosphorylation at Y31 and Y118 (Turner, 2000). We addressed whether or not paxillin phosphorylation at these sites is mediating the increased motility and survival in F9 vin−/− and F9 vin−/−Y822F cells. Expression of a paxillin Y31FY118F mutant in these cells led to a substantial reduction in motility and survival mediated by ERK1/2. The link between paxillin–FAK binding and ERK activation may be through Crk, an important adaptor molecule involved in ERK1/2 activation (York et al., 1998). A previous paper demonstrated that paxillin Y31 and Y118 phosphorylation creates docking sites for Crk (Petit et al., 2000).
We also found that vinculin with a Y822F point mutation was unable to restore normal apoptosis induction. These profound effects from a point mutation strongly suggest that enhanced survival is not due to nonspecific cytoskeletal alterations, but rather from specific effects in survival signaling pathways (Judson et al., 1999; Rodina et al., 1999; Sattler et al., 2000; Turner, 2000; Hauck et al., 2002). Previous works indicate that the observed effects were not due simply to an inability of the mutant to localize in focal adhesions (Goldmann et al., 1998). Y822 is very close to the flexible hinge region of vinculin, so the mutation could affect the equilibrium between the open and closed form of the protein. When vinculin is in its open, active conformation it localizes into focal adhesions (Rudiger, 1998). Upon binding to phosphatidylinositol(4,5)-biphosphate (Steimle et al., 1999) at the cell membrane, vinculin unfolds, exposing its talin binding sites (Johnson and Craig, 1994). The talin binding sites are also critical for vinculin's focal adhesion localization in cells (Goldmann et al., 1998). In contrast, paxillin interactions are not sufficient to cause vinculin localization to focal adhesions in vivo (Goldmann et al., 1998), despite the fact that paxillin–vinculin interaction in vitro occurs for both open and closed conformations (Gilmore and Burridge, 1996). The Y822F mutation could decrease the stability of vinculin's open conformation, affecting an early stage of this complex process, before paxillin interaction.
Vinculin can be tyrosine phosphorylated (Sefton et al., 1981), and Y822 is a putative site for phosphorylation (Jockusch and Rudiger, 1996), but a physiological role for Y822 phosphorylation has not been demonstrated (Jockusch and Rudiger, 1996). Phosphorylation may be important for maintaining the open conformation, as for the cytoskeletal protein ezrin (Berryman et al., 1995). Thus, vinculin Y822F could be more prone than wild-type vinculin to adopt a closed conformation after its incorporation into focal adhesions. Elucidating the precise mechanism through which vinculin Y822F affects interactions with paxillin may be a valuable route to further understanding of cell motility and survival.
Understanding how cell motility and survival are regulated is an important fundamental goal in cell biology. Motility and survival are controlled by multimolecular signaling complexes created upon dynamic interaction between paxillin and FAK. Our results identify vinculin as a key molecule regulating these cellular processes and provides evidence of an important signaling mechanism tying vinculin to carcinogenesis.
Materials and methods
Cell culture
The following cell lines were used: WT F9 mouse embryonal carcinoma cells and vinculin-null F9 cells (F9 vin−/− clones γ229 and γ227; Coll et al., 1995); F9 vin−/− cells stably transfected with plasmid vectors expressing full-length vinculin (F9 vin−/− rescue); a fragment containing the sequences of the head portion of vinculin (F9 vin−/− head, 1 to 821 aa); a vinculin fragment 243–1066 in which the talin binding sites were deleted (F9 vin−/− Δ talin bs); a full-length vinculin containing a Y822F point mutation (F9 vin−/−Y822F); and cells expressing a vinculin hinge-tail fragment (F9 vin−/− hinge-tail, amino acid residues 811 to 1066), which was Flag M2-tagged (Xu et al., 1998b). These cells were cultured as described previously (Coll et al., 1995; Xu et al., 1998b).
Anoikis assay
Anoikis assays were performed according to a published protocol (Folkman and Moscona, 1978). In brief, 6-well plates (Costar) were coated with polyHEMA to create a nonadhesive surface. The wells were allowed to dry for 48 h and washed with PBS (GIBCO BRL) before experiments. Cells were added in each well at a concentration of 5 × 105 cells ml−1, and the extent of apoptotic cell death was determined 3 h later.
Live cell microscopy for apoptosis
Immediately before the experiment, the medium was changed to PBS supplemented with 10% FBS. Gelatin-coated coverslips with adherent F9, F9 vin−/−, and F9 vin −/− rescue cells were mounted in a sealed Dvorak chamber (Nicholson Instruments) held in a temperature-controlled stage (20/20 Technologies). Microscopy was performed using a microscope (model Axiovert 100 TV; Carl Zeiss MicroImaging, Inc.) modified with automated stage and filter wheels (LEP Ltd.) and a 40× NA 1.3 objective with differential interference contrast optics (Carl Zeiss MicroImaging, Inc.). Control of microscope automation and image analysis were performed using Inovision ISEE software. The extent of apoptotic cell death was assessed during the first 6 h after addition of 2 μM camptothecin (Sigma-Aldrich). As described previously (Subauste et al., 2000), individual cells were logged in using the automated microscope and scored for changes in morphology over time. Previously reported studies confirmed that the morphological features assayed (contraction and blebbing leading to cell fragmentation) were indicative of apoptosis (Subauste et al., 2000).
Caspase activity assay
The synthetic tetrapeptide fluorogenic substrate DEVD-AMC from Enzyme Systems Products was used as described previously (Subauste et al., 2000).
Preparation of protein extracts, immunoblotting, and immunoprecipitation
For immunoblotting analyses, cells were lysed in a buffer containing 20 mM Tris-HCl, pH 8, 100 mM NaCl, 10% glycerol, 1% Triton X-100, 50 mM NaF, 1 mM sodium vanadate, 1 mM benzamidine, 5 μg ml−1 leupeptin, and 1 mM PMSF. The cell lysates were clarified at 10,000 g for 10 min at 4°C. Samples were separated on SDS–polyacrylamide gels (Bio-Rad Laboratories). Proteins were transferred to Immobilon-P transfer membranes (Millipore) and blocked and probed with the following Abs: rabbit anti-ERK1/2 Ab, rabbit anti-phospho ERK1/2 (Thr202/Tyr204) Ab, rabbit anti-MEK Ab, rabbit antiphospho paxillin (Tyr118) Ab, and rabbit anti–Caspase-3 Ab from Cell Signaling Technology; rabbit anti-FAK Ab from Santa Cruz Biotechnology, Inc.; mouse monoclonal antipaxillin Ab; mouse monoclonal antiphospho FAK (Tyr 397) Ab from Becton Dickinson; rabbit anti–Caspase-9 Ab from BD Biosciences; and mouse monoclonal antivinculin Ab, mouse monoclonal anti-myc Ab, and mouse monoclonal anti-FLAG M2 Ab from Sigma-Aldrich. Bound Ab was detected by incubation with secondary Abs conjugated with HRP (rabbit and mouse HRP–derived Ab were obtained from Cell Signaling) and visualized by ECL (Pierce Chemical Co.). Equal loading in each lane was verified by assessing their ERK1/2 concentration.
Immunoprecipitation studies that assessed the interaction of FAK with paxillin were performed according to procedure described previously (Tachibana et al., 1995) with some minor modifications using the mouse monoclonal antipaxillin Ab as well as the rabbit anti-FAK Ab (the concentration used for both Abs was of 1/500).
Cell migration assays
Modified Boyden chambers (8 μm pore size; Transwell Costar Corporation) were used in cell migration assays. The filter inserts were coated with 10 μg ml−1 collagen type I (Upstate Biotechnology) for 2 h at 37°C, rinsed with PBS, and the inserts were placed into the lower chamber containing 0.5 ml of α-MEM media containing 10% FBS (GIBCO BRL). 2 × 105 cells that were previously serum starved (1% FBS) for 10 h were added to the top of each migration chamber and allowed to migrate to the underside of the top chamber for 8 h. Nonmigratory cells on the upper membrane surface were removed with a cotton swab, and the migratory cells attached to the bottom surface of the membrane were fixed with methanol and stained with 0.1% crystal violet in 0.1 M borate (Sigma-Aldrich), pH 9, and 2% ethanol for 20 min at RT. The number of migratory cells per membrane was counted with an inverted microscope (Nikon) using a 10× objective. Each determination represents the average of two individual wells. Background migration was assessed on BSA (1%)-coated membranes and subtracted from all data.
Inhibition of p42 and p44 MAPK (ERK1 and ERK2) with Erk antisense oligonucleotides
Depletion of MAPK (ERK1 and 2) from F9 embryonal carcinoma cells was performed using phosphorothioate-protected oligonucleotides according to the procedure of Sale et al. (1995) with some modifications. Erk antisense (5′-GCC GCC GCC GCC GCC AT-3′) and control oligonucleotides (5′-CGC GCG CTC GCG CAC CC-3′) were HPLC purified (Operon Biotechnology). Cells were grown to 30% confluency in 6-well tissue culture plates. Before transfection, lipofectin (GIBCO BRL) was diluted into 400 μl α-MEM at a concentration of 30 μg/ml for 40 min at RT. The diluted lipofectin was mixed with 400 μl α-MEM containing 6.0 μM ERK antisense or control phosphorothioate oligonucleotides for 15 min at RT. This mixture was added to the cells. The final concentrations of lipofectin and oligonucleotides were 15 μg/ml and 3 μM, respectively. Cells were incubated for 8 h at 37°C in the presence of 5% CO2. They were rinsed, incubated for another 40 h in fresh culture medium containing oligonucleotides, and tested as described in Anoikis assays. An aliquot of the aforementioned cells was analyzed in parallel for changes in ERK protein levels using ERK-specific Abs and immunoblotting as described in Preparation of protein extracts, immunoblotting, and immunoprecipitation. As a control for nonspecific depletion of proteins, MEK concentration of each sample was assessed by immunoblotting.
Plasmids and transfection
pcDNA3 wild-type and Y31FY118F paxillin constructs were previously described (Petit et al., 2000). To myc tag these constructs at their NH2 terminus, they were excised with BamHI and EcoRI enzymes and subcloned in a pTriExx4 His-myc vector. A monomeric red fluorescent protein (mRFP) subcloned in the same vector was used as a control. Transient transfections were done using lipofectamine and Plus Reagent (GIBCO BRL) following the manufacturer's instructions. In brief, F9 cell lines were transfected with 8 μl lipofectamine, 16 μl of Plus Reagent, and 7 μg of the plasmids. Cells were used 48 h after transfection. The plasmids expressions were confirmed with a mouse anti-myc Ab (Sigma-Aldrich).
Acknowledgments
We thank the National Institutes of Health for financial support (grant AI 01684-02 to M.C. Subauste, CA 67888 to E.D. Adamson, GM 47607 to C.E. Turner, Swiss National Science Foundation to O. Pertz, and AG 15430 to K.M. Hahn). This is Scripps manuscript number 14121-CB.
Abbreviations used in this paper: Δ talin bs, deleted talin binding sites; Ab, antibody; DEVD-AMC, N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin; ERK, extracellular signal–regulated kinase; FAT, focal adhesion targeting; FRNK, FAK-related nonkinase; MEK, MAPK kinase; mRFP, monomeric red fluorescent protein; PI-3K, phosphatidylinositol-3 kinase.
References
- Ahn, N.G., R. Seger, R.L. Bratlien, C.D. Diltz, N.K. Tonks, and E.G. Krebs. 1991. Multiple components in an epidermal growth factor-stimulated protein kinase cascade. In vitro activation of a myelin basic protein/microtubule-associated protein 2 kinase. J. Biol. Chem. 266:4220–4227. [PubMed] [Google Scholar]
- Allan, L.A., N. Morrice, S. Brady, G. Magee, S. Pathak, and P.R. Clarke. 2003. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat. Cell Biol. 5:647–654. [DOI] [PubMed] [Google Scholar]
- Bakolitsa, C., J.M. de Pereda, C.R. Bagshaw, D.R. Critchley, and R.C. Liddington. 1999. Crystal structure of the vinculin tail suggests a pathway for activation. Cell. 99:603–613. [DOI] [PubMed] [Google Scholar]
- Bass, M.D., B. Patel, I.G. Barsukov, I.J. Fillingham, R. Mason, B.J. Smith, C.R. Bagshaw, and D.R. Critchley. 2002. Further characterization of the interaction between the cytoskeletal proteins talin and vinculin. Biochem. J. 362:761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendori, R., D. Salomon, and B. Geiger. 1989. Identification of two distinct functional domains on vinculin involved in its association with focal contacts. J. Cell Biol. 108:2383–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryman, M., R. Gary, and A. Bretscher. 1995. Ezrin oligomers are major cytoskeletal components of placental microvilli: a proposal for their involvement in cortical morphogenesis. J. Cell Biol. 131:1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, S.Y., and R.L. Klemke. 2000. Extracellular-regulated kinase activation and CAS/Crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. J. Cell Biol. 149:223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll, J.-L., A. Ben-Ze'ev, R.M. Ezzell, J.L. Rodriguez-Fernandez, H. Baribault, R.G. Oshima, and E.D. Adamson. 1995. Targeted disruption of vinculin genes in F9 and embryonic stem cells changes cell morphology, adhesion, and locomotion. Proc. Natl. Acad. Sci. USA. 92:9161–9165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham, V.J., M. James, M.C. Frame, and S.J. Winder. 2000. Active ERK/MAP kinase is targeted to newly forming cell-matrix adhesions by integrin engagement and v-Src. EMBO J. 19:2911–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman, J., and A. Moscona. 1978. Role of cell shape in growth control. Nature. 273: 345–349. [DOI] [PubMed] [Google Scholar]
- Frisch, S.M., and H. Francis. 1994. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 124:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore, A.P., and K. Burridge. 1996. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-biphosphate. Nature. 381:531–535. [DOI] [PubMed] [Google Scholar]
- Gilmore, A.P., P. Jackson, G.T. Waites, and D.R. Critchley. 1992. Further characterization of the talin binding site in the cytoskeletal protein vinculin. J. Cell Sci. 103:719–731. [DOI] [PubMed] [Google Scholar]
- Goldmann, W.H., R. Galneder, M. Ludwig, W. Xu, E.D. Adamson, N. Wang, and R.M. Ezzell. 1998. Differences in elasticity of vinculin-deficient F9 cells measured by magnetometry and atomic force microscopy. Exp. Cell Res. 239:235–242. [DOI] [PubMed] [Google Scholar]
- Grossmann, J., K. Walther, M. Artinger, S. Kiessling, and J. Scholmerich. 2001. Apoptotic signaling during initiation of detachment-induced apoptosis (“anoikis”) of primary intestinal epithelial cells. Cell Growth Differ. 12:147–155. [PubMed] [Google Scholar]
- Hauck, C.R., D.A. Hsia, and D.D. Schlaepfer. 2000. Focal adhesion kinase facilitates platelet-derived growth factor-BB-stimulated ERK2 activation required for chemotaxis migration of vascular smooth muscle cells. J. Biol. Chem. 275:41092–41093. [DOI] [PubMed] [Google Scholar]
- Hauck, C.R., D.A. Hsia, and D.D. Schlaepfer. 2002. The focal adhesion kinase—a regulator of cell migration and invasion. Life. 53:115–119. [DOI] [PubMed] [Google Scholar]
- Hayashi, I., K. Vuori, and R.C. Liddington. 2002. The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat. Struct. Biol. 9:101–106. [DOI] [PubMed] [Google Scholar]
- He, J., C.M. Whitacre, L.Y. Xue, N.A. Berger, and N.L. Oleinick. 1998. Protease activation and cleavage of poly (ADP-ribose) polymerase: an integral part of apoptosis in response to photodynamic treatment. Cancer Res. 58:940–946. [PubMed] [Google Scholar]
- Hertzberg, R.P., M.J. Caranfa, and S.M. Hecht. 1989. On the mechanism of topoisomerase I inhibition by camptothecin: evidence for binding to an enzyme-DNA complex. Biochemistry. 28:4629–4638. [DOI] [PubMed] [Google Scholar]
- Howe, A.K., A.E. Aplin, and R.L. Juliano. 2002. Anchorage-dependent ERK signaling—mechanisms and consequences. Curr. Opin. Genet. Dev. 12:30–35. [DOI] [PubMed] [Google Scholar]
- Huang, S., and D.E. Ingber. 2000. Shape-dependent control, differentiation, and apoptosis: switching between attractors in cell regulatory networks. Exp. Cell Res. 261:91–103. [DOI] [PubMed] [Google Scholar]
- Izard, T., G. Evans, R.A. Borgon, C.L. Rush, G. Bricogne, and P.R.J. Bois. 2004. Vinculin activation by talin through helical bundle conversion. Nature. 427:171–175. [DOI] [PubMed] [Google Scholar]
- Jockusch, B.M., and M. Rudiger. 1996. Crosstalk between cell adhesion molecules: vinculin as a paradigm for regulation by conformation. Trends Cell Biol. 6:311–315. [DOI] [PubMed] [Google Scholar]
- Johnson, R.P., and S.W. Craig. 1994. An intramolecular association between the head and tail domains of vinculin modulates talin binding. J. Biol. Chem. 269:12611–12619. [PubMed] [Google Scholar]
- Judson, P.L., X. He, W.G. Cance, and L.Van Le. 1999. Overexpression of focal adhesion kinase, a protein tyrosine kinase, in ovarian carcinoma. Cancer. 86:1551–1556. [DOI] [PubMed] [Google Scholar]
- Klemke, R.L., S. Cai, A.L. Gianini, P.J. Gallagher, P. de Lanerolle, and D.A. Cheresh. 1997. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 137:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifschitz-Mercer, B., B. Czernobilsky, E. Feldberg, and B. Geiger. 1997. Expression of the adherens junction protein vinculin in human basal and squamous cell tumors: relationship to invasiveness and metastatic potential. Hum. Pathol. 28:1230–1236. [DOI] [PubMed] [Google Scholar]
- Nakamoto, T., R. Sakai, H. Honda, S. Ogawa, H. Ueno, T. Suzuki, S.-I. Aizawa, Y. Yazaki, and H. Hirai. 1997. Requirements for localization of p130cas to focal adhesions. Mol. Cell. Biol. 17:3884–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, D.W., A. Ali, N.A. Thornberry, J.P. Vaillancourt, C.K. Ding, M. Gallant, Y. Gareau, P.R. Griffin, M. Labelle, and M. Lazebnik. 1995. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 376:500–503. [DOI] [PubMed] [Google Scholar]
- Parise, L.V., J. Lee, and R.L. Juliano. 2000. New aspects of integrin signaling in cancer. Semin. Cancer Biol. 10:407–414. [DOI] [PubMed] [Google Scholar]
- Petit, V., B. Boyer, D. Lentz, C.E. Turner, J.P. Thiery, and A.M. Valles. 2000. Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. J. Cell Biol. 148:957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouyssegur, J., V. Volmat, and P. Lenormand. 2002. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem. Pharmacol. 64:755–763. [DOI] [PubMed] [Google Scholar]
- Raz, A., and B. Geiger. 1982. Altered organization of cell-substrate contacts and membrane-associated cytoskeleton in tumor cell variants exhibiting different metastatic capabilities. Cancer Res. 42:5183–5190. [PubMed] [Google Scholar]
- Reffas, S., and W. Schlegel. 2000. Compartment-specific regulation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinases (MAPKs) by ERK-dependent and non-ERK-dependent inductions of MAPK phosphatase (MKP)-3 and MKP-1 in differentiated P19 cells. Biochem. J. 352:701–708. [PMC free article] [PubMed] [Google Scholar]
- Richardson, A., and T. Parsons. 1996. A mechanism for regulation of the adhesion-associated protein tyrosine kinase pp125FAK. Nature. 380:538–540. [DOI] [PubMed] [Google Scholar]
- Riedl, S.J., P. Fuentes-Prior, M. Renatus, N. Kairies, S. Krapp, R. Huber, G.S. Salvesen, and W. Bode. 2001. Structural basis for the activation of human procaspase-7. Proc. Natl. Acad. Sci. USA. 98:14790–14795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodina, A., K. Schramm, E. Musatkina, E.-D. Kreuser, A. Tavitian, and A. Tatosyan. 1999. Phosphorylation of p125FAK and paxillin focal adhesion proteins in scr-transformed cells with different metastatic capacity. FEBS Lett. 455:145–148. [DOI] [PubMed] [Google Scholar]
- Rodriguez Fernandez, J.L., B. Geiger, D. Salomon, I. Sabanay, M. Zoller, and A. Ben-Ze'ev. 1992. Suppression of tumorigenicity in transformed cells after transfection with vinculin cDNA. J. Cell Biol. 119:427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales, C., and R.L. Juliano. 1995. Signal transduction by cell adhesion receptors in leukocytes. J. Leukoc. Biol. 57:189–198. [DOI] [PubMed] [Google Scholar]
- Rudiger, M. 1998. Vinculin and α-catenin: shared and unique functions in adherens junctions. Bioessays. 20:733–740. [DOI] [PubMed] [Google Scholar]
- Ruoslahti, E., and B. Obrink. 1996. Common principles in cell adhesion. Exp. Cell Res. 227:1–11. [DOI] [PubMed] [Google Scholar]
- Sale, E.M., P.G.P. Atkinson, and G.J. Sale. 1995. Requirement of MAP kinase for differentiation of fibroblasts to adipocytes, for insulin activation of p90 S6 kinase and for insulin or serum stimulation of DNA synthesis. EMBO J. 14:674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, T., J. Irie-Sasaki, R.G. Jones, A.J. Oliveira-dos-Santos, W.L. Stanford, B. Bolon, A. Wakeham, A. Itie, D. Bouchard, I. Kozieradzki, N. Joza, T.W. Mak, P.S. Ohashi, A. Suzuki, and J.M. Penninger. 2000. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science. 287:1040–1046. [DOI] [PubMed] [Google Scholar]
- Sattler, M., E. Pisick, P.T. Morrison, and R. Salgia. 2000. Role of the cytoskeletal protein paxillin in oncogenesis. Crit. Rev. Oncog. 11:63–74. [PubMed] [Google Scholar]
- Schwartz, M.A. 2001. Integrin signaling revisited. Trends Cell Biol. 11:466–470. [DOI] [PubMed] [Google Scholar]
- Sefton, B.M., T. Hunter, E.H. Ball, and S.J. Singer. 1981. Vinculin: a cytoskeletal target of the transforming protein Rous sarcoma virus. Cell. 24:165–174. [DOI] [PubMed] [Google Scholar]
- Smith, E.R., J.L. Smedberg, M.E. Rula, and X.-X. Xu. 2004. Regulation of Ras–MAPK pathway mitogenic activity by restricting nuclear entry of activated MAPK in endoderm differentiation of embryonic carcinoma and stem cells. J. Cell Biol. 164:689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steimle, P.A., J.D. Huffert, N.B. Adley, and S.W. Craig. 1999. Polyphosphoinositides inhibit the interaction of vinculin with actin filaments. J. Biol. Chem. 274:18414–18420. [DOI] [PubMed] [Google Scholar]
- Subauste, M.C., M. Von Herrath, V. Benard, C.E. Chamberlain, T.-H. Chuang, K. Chu, and K.M. Hahn. 2000. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J. Biol. Chem. 275:9725–9733. [DOI] [PubMed] [Google Scholar]
- Tachibana, K., T. Sato, N. D'Avirro, and C. Morimoto. 1995. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J. Exp. Med. 182:1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello, D.A., M.C. Brown, and C.E. Turner. 2002. The paxillin LD motifs. FEBS Lett. 513:114–118. [DOI] [PubMed] [Google Scholar]
- Turner, C.E. 2000. Paxillin and focal adhesion signalling. Nat. Cell Biol. 2:E231–E236. [DOI] [PubMed] [Google Scholar]
- Turner, C.E., M.C. Brown, J.A. Perrota, M.C. Riedy, S.N. Nikolopoulos, A.R. McDonald, S. Bagrodia, S. Thomas, and P.S. Leventhal. 1999. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: A role in cytoskeletal remodeling. J. Cell Biol. 145:851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watton, S.J., and J. Downward. 1999. Akt/PKB localization on 3′ phosphoinositide generation at sites of epithelial cell-matrix and cell-cell interaction. Curr. Biol. 9:433–436. [DOI] [PubMed] [Google Scholar]
- Wei, L., Y. Yang, and Q. Yu. 2001. Tyrosine kinase-dependent, phosphatidylinositol 3′-kinase, and mitogen-activated protein kinase-independent signaling pathways prevent lung adenocarcinoma cells from anoikis. Cancer Res. 61:2439–2444. [PubMed] [Google Scholar]
- Xu, W., H. Baribault, and E.D. Adamson. 1998. a. Vinculin knockout results in heart and brain defects during embryonic development. Development. 125:327–337. [DOI] [PubMed] [Google Scholar]
- Xu, W., J.-L. Coll, and E.D. Adamson. 1998. b. Rescue of the mutant phenotype by reexpression of full-length vinculin in null F9 cells; effects on cell locomotion by domain deleted vinculin. J. Cell Sci. 111:1535–1544. [DOI] [PubMed] [Google Scholar]
- York, R.D., H. Yao, T. Dillon, C.L. Ellig, S.P. Eckert, E.W. McCleskey, and P.J.S. Stork. 1998. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature. 392:622–626. [DOI] [PubMed] [Google Scholar]