

Abstract
Entry into mitosis in vertebrate cells is guarded by a checkpoint that can be activated by a variety of insults, including chromosomal damage and disrupting microtubules (Rieder, C.L., and R.W. Cole. 1998. J. Cell Biol. 142:1013–1022; Rieder, C.L., and R.W. Cole. 2000. Curr. Biol. 10:1067–1070). This checkpoint acts at the end of interphase to delay cells from entering mitosis, causing cells in prophase to decondense their chromosomes and return to G2 phase. Here, we show that in response to microtubule poisons this “antephase” checkpoint is primarily mediated by the p38 stress kinases and requires the Chfr protein that is absent or inactive in several transformed cell lines (Scolnick, D.M., and T.D. Halazonetis. 2000. Nature. 406:430–435) and lung tumors (Mizuno, K., H. Osada, H. Konishi, Y. Tatematsu, Y. Yatabe, T. Mitsudomi, Y. Fujii, and T. Takahashi. 2002. Oncogene. 21:2328–2333). Furthermore, in contrast to previous reports, we find that the checkpoint requires ubiquitylation but not proteasome activity, which is in agreement with the recent demonstration that Chfr conjugates ubiquitin through lysine 63 and not lysine 48 (Bothos, J., M.K. Summers, M. Venere, D.M. Scolnick, and T.D. Halazonetis. 2003. Oncogene. 22:7101–7107).
Keywords: checkpoint; ubiquitin; cyclin; phosphorylation; proteolysis
Introduction
The decision to enter mitosis is not lightly made. There are several distinct checkpoints that prevent mitosis when internal or external conditions are not favorable. The most well-characterized checkpoints are triggered by unreplicated or damaged DNA, which prevent mitosis by pathways dependent on the ATM and ATR protein kinases. These checkpoints block cells from entering mitosis by preventing the Cdc25 phosphatases from activating the cyclin B1-Cdk1 kinase. Cyclin B1-Cdk1 is fully activated at the end of prophase (Jackman et al., 2003), and this, along with nuclear envelope breakdown in metazoans, correlates with irreversible entry into mitosis. Thus, there are grounds to consider the bulk of prophase not as part of mitosis but as the final part of interphase. Moreover, a wide variety of insults in late G2 phase and early prophase will prevent cells from initiating mitosis. Indeed, Bullough and Johnson (1951) coined the term “antephase” to describe the period in late G2 phase, just before the first visible signs of chromosome condensation become evident, during which normal mouse epidermal cells are reversibly delayed when subjected to stresses including radiation damage, hypothermia, fluoride treatment, and lack of microtubule function. Rieder and Cole (1998)(2000) further showed that rat kangaroo, green monkey kidney, and pig kidney cells in early prophase will transiently return to antephase when they are exposed to cold, osmotic shock, or microtubule poisons such as colcemid (for review see Pines and Rieder, 2001; Mikhailov and Rieder, 2002). With the agreement of Conly Rieder we will refer to this as the antephase checkpoint to make clear that this acts at the end of interphase and not in mitosis. This checkpoint is distinct from the DNA damage checkpoint because it does not require ATM or ATR (Rieder and Cole, 2000). It is also weakened or absent in several tumors and transformed cell lines (Rieder and Cole, 1998). The checkpoint is most easily assayed in early prophase, during chromosome condensation, when colcemid treatment is able to cause PtK1 cells in prophase to return to antephase, but only before the nucleoli have broken down. After the nucleoli have disassembled, colcemid is unable to trigger the return to antephase and cells initiate mitosis (Rieder and Cole, 1998). Nucleolar breakdown correlates with the activation and nuclear translocation of cyclin B1-Cdk1 and, therefore, could be considered one of the first signs of entry into mitosis. At present, the mechanisms behind the antephase checkpoint are ill defined.
One protein that has been suggested to be part of the antephase checkpoint is Chfr (checkpoint protein with an FHA domain and ring finger; Scolnick and Halazonetis, 2000), a ubiquitin ligase that is down-regulated in several cell lines through methylation of its promoter (Mizuno et al., 2002). Chfr was originally reported to delay progress to prometaphase in the presence of colcemid (Scolnick and Halazonetis, 2000), and cells were surprisingly described as delaying with high cyclin B1-Cdk1 activity (Scolnick and Halazonetis, 2000), which conflicted with a role as part of the antephase checkpoint because cyclin B1-Cdk1 is fully activated only in late prophase. However, in Xenopus laevis extracts, Chfr is able to delay the activation of cyclin B-Cdk1, apparently by targeting the Polo-like kinase, Plx, for degradation by the proteasome (Kang et al., 2002), thereby preventing the activation of the Cdc25 phosphatase that activates Cdk1. Chfr has also been reported to affect Polo-like kinase levels in human cells in response to DNA damage (Shtivelman, 2003). But whether Chfr does target Polo for degradation or not is debatable because Chfr has been shown to conjugate ubiquitin via its lysine 63 residue (Bothos et al., 2003) that normally acts in signal transduction, especially for stress signals (Deng et al., 2000; Ulrich and Jentsch, 2000; Hofmann and Pickart, 2001; Pickart, 2001; Wang et al., 2001), rather than to target proteins to the proteasome.
The ability of a variety of stress stimuli to delay entry to prophase might implicate the p38 stress-activated kinases as components of the antephase checkpoint. Members of the family of p38 kinases can be activated by a variety of stresses (for review see Nebreda and Porras, 2000), and some have been shown to be able to delay the cell cycle in G1 and in G2 phase (for review see Bulavin et al., 2002). In animal cells, the p38α kinase has been reported to phosphorylate and inactivate the Cdc25B phosphatase in response to UV damage in G2 phase (Bulavin et al., 2001), thus delaying mitosis. p38α has also been reported to be part of the spindle assembly checkpoint that delays cells in mitosis when chromosomes are not properly attached to the spindle (Takenaka et al., 1998). However, until now the stress kinases have not been shown to have a role in the antephase checkpoint.
Here, we have investigated the mechanisms required for the antephase checkpoint in mammalian cells. We show that in response to microtubule poisons the checkpoint requires the Chfr ubiquitin ligase, but not the proteasome, and that Chfr does not delay mitosis by targeting Plk1 for degradation. Rather, we show that the microtubule-dependent antephase checkpoint acts through the p38 kinases.
Results
Several transformed cell lines had been shown to lack the antephase checkpoint (Rieder and Cole, 2000), and some of these had also been described as lacking Chfr (Scolnick and Halazonetis, 2000). Therefore, we were intrigued by the possibility that the antephase checkpoint might require Chfr. To investigate this possibility, we established two assays for the antephase checkpoint and compared PtK1 cells, where the checkpoint was originally defined (Rieder and Cole, 1998), with HeLa and U2OS cells that lack the checkpoint (Rieder and Cole, 2000). Chfr is not expressed in HeLa cells (Fig. 1 D) and is mutated in U2OS cells (Scolnick and Halazonetis, 2000). In our primary assay, we identified cells in early to mid-prophase by DIC microscopy using the criterion of discernible chromosome condensation with an intact nucleolus (Fig. 1 A and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1; Rieder and Cole, 1998) and continually monitored the response of the cells by time-lapse microscopy. When prophase PtK1 cells were treated with nocodazole or colcemid and followed by time-lapse DIC microscopy, they decondensed their chromosomes after a variable period of time and returned to antephase, which is illustrative of an intact antephase checkpoint (Fig. 1 A and Video 1). These cells remained in antephase for anywhere between 1 and 6 h (Fig. 1 E) before returning to mitosis (Video 1 and Fig. 1 F). In contrast, HeLa cells and U2OS cells did not return to interphase but continued on into mitosis (Fig. 1, B and C). In agreement with Rieder and Cole's (1998) original observations, PtK1 cells were unable to return to interphase once the nucleoli began to disassemble, which, from our previous studies on cyclin B1, is the time when cyclin B1 is activated and moves into the nucleus (Furuno et al., 1999; Jackman et al., 2003). To corroborate these results, we used a second assay according to Rieder and Cole (2000). We treated a population of PtK1 cells with nocodazole or colcemid and, at various times after treatment, stained the cells with Hoechst 33342 and determined the proportion of cells at each stage of mitosis by fluorescence microscopy. We confirmed that cells with an intact antephase checkpoint show a marked decrease in the number of cells in prophase after drug treatment (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1).
Figure 1.
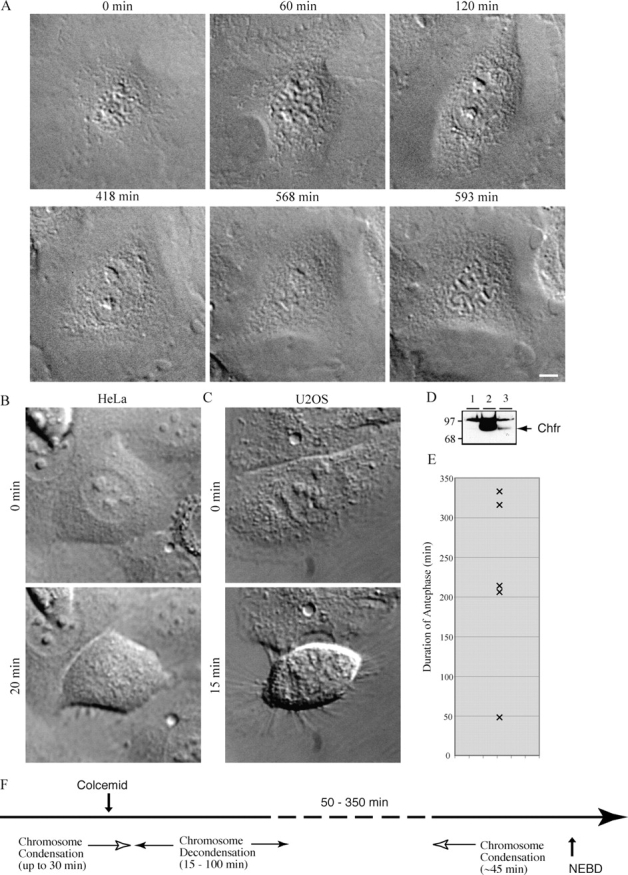
Assay for the antephase checkpoint. DIC images of a Ptk1 (A), HeLa (B), and U2OS (C) cell in early prophase, identified by partial chromosome condensation. Cells were treated with 15 μM colcemid at 0 min (top panels). Approximately 20 to 100 min after treatment, Ptk1 cells decondensed their chromosomes and returned to antephase, whereas Hela and U2OS cells continued into mitosis (bottom panels). After a variable time with decondensed chromosomes, the PtK1 cells returned to prophase and subsequently reentered mitosis (A; Video 1). Cells are representative of more than six Ptk1, eight HeLa, and nine U2OS cells in 12 different experiments. Bar, 10 μm. The complete series of images for the Ptk1 cell are presented in Quicktime format as Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1. (D) HeLa cells do not express Chfr. (lane 1) Anti-Chfr immunoblot of whole cell lysates from HeLa cells; (lane 2) HeLa cells transfected with Chfr under the CMV promoter; (lane 3) normal diploid fibroblasts. Equal amounts of protein were loaded per lane. Molecular mass markers are indicated on the left. (E) Duration of antephase delay. The time between initial chromosome decondensation and subsequent return to prophase (visible chromosome condensation) was measured for five PtK1 cells from the time-lapse videos. (F) Schematic timeline of the antephase checkpoint.
Chfr is required for the antephase checkpoint triggered by microtubule poisons
To determine if Chfr was required for the antephase checkpoint we generated a dominant-negative mutant by deleting the FHA domain from Chfr (Scolnick and Halazonetis, 2000). When injected into PtK1 cells, the mutant but not the wild-type protein abrogated the antephase checkpoint; early prophase cells no longer returned to interphase after treatment with nocodazole or colcemid but continued on to breakdown their nuclear envelope (Fig. 2 A, Table I, and Video 2, available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1). Some cells arrested in prophase and were separately scored (Table I). As a more direct test for whether or not Chfr was required for the antephase checkpoint, we generated cell lines in which Chfr was expressed under an inducible promoter (the tet-off system). We used U2OS cells as the parental cell line, as in U2OS cells the endogenous Chfr is inactive due to a point mutation in the carboxyl-terminus (Scolnick and Halazonetis, 2000). We isolated clones of cells that on an immunoblot expressed approximately equivalent levels of wild-type Chfr or Chfr with a mutation in the RING finger to disable its ubiquitylation activity (Fig. 2 B). Immunofluorescence analysis showed that most individual cells expressed similar levels of the proteins (Fig. 2 C), and that at least 50 and 80% of the cells in the population expressed wild-type or mutant Chfr, respectively. We found that cells expressing wild-type Chfr were able to return to interphase after challenging with colcemid in early prophase, whereas the uninduced cells and cells expressing the inactive Chfr mutant continued on to mitosis (Fig. 2 D and Videos 3 and 4, available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1). The cells were fixed and stained after the assays to show that they were expressing the ectopic Chfr protein (Fig. 2 D and Videos 3 and 4). We also synchronized cells expressing wild-type Chfr in S phase, and used time-lapse microscopy to compare their entry into mitosis with and without colcemid treatment in G2 phase. This comparison showed that colcemid delayed mitosis by an average of 3 h in a substantial proportion of the cells (Fig. 2 E). Thus, Chfr is able to restore the microtubule-dependent antephase checkpoint.
Figure 2.
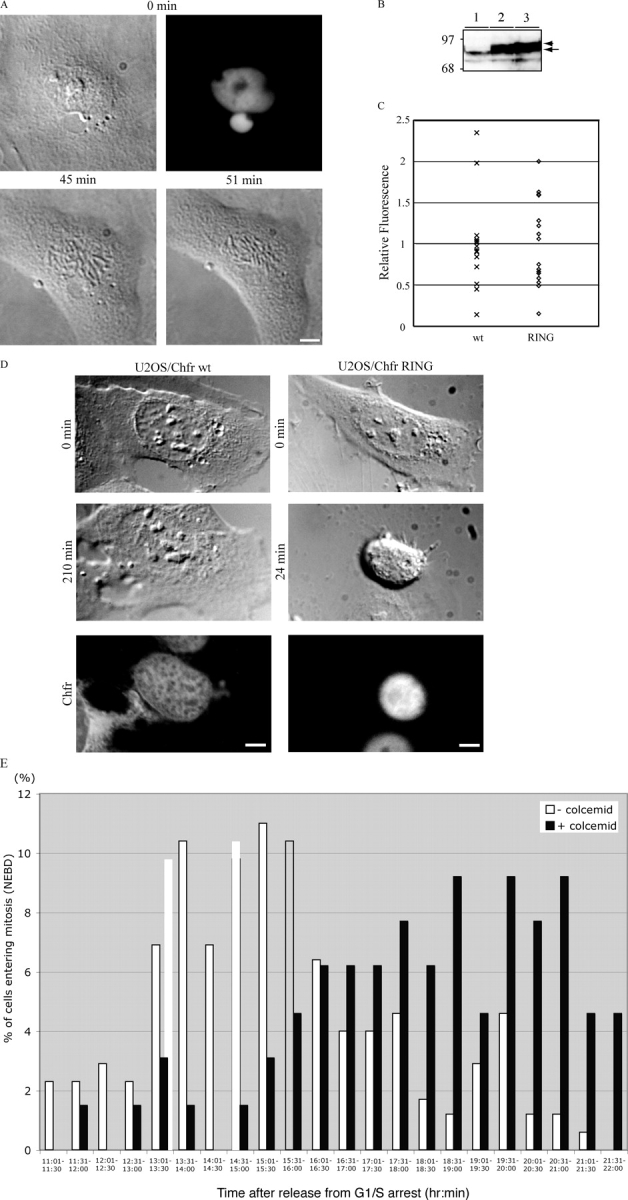
Chfr is required for the antephase checkpoint. (A) Fluorescence (top right) and DIC images of Ptk1 cell expressing a GFP tagged ΔFHA mutant of Chfr. The cell was treated with 15 μM colcemid at 0 min (top left) and continued on into mitosis (bottom panels). The complete series of DIC images are presented in Quicktime format as Video 2. (B) Anti-Chfr immunoblot of tetracycline-inducible Chfr cell lines. (lane 1) Uninduced cells; (lane 2) cells expressing epitope-tagged wild-type Chfr; (lane 3) cells expressing epitope-tagged RING-finger mutant of Chfr. Epitope-tagged Chfr is marked by the arrowhead, endogenous Chfr is marked by the arrow. Molecular mass markers are indicated on the left. (C) Box plot of the relative fluorescence intensities of individual U2OS cells of the stable cell lines expressing inducible Chfr proteins. Cells were stained with anti-Xpress epitope antibody to detect wild type (wt, n = 15) or the Ring finger mutant (RING, n = 16) of Chfr. (D) DIC images of U2OS cell lines expressing an inducible wild type (left) or a RING finger mutant (right) of Chfr were treated with 15 μM colcemid at 0 min (top). Cells expressing wild-type Chfr decondensed their chromosomes and returned to antephase for least 3.5 h (middle left). In contrast, cells expressing mutant Chfr continued on into mitosis (middle right). Cells were then fixed and stained with anti-Xpress tag antibody to assay for expression of the ectopic Chfr (bottom). Bar, 10 μm. The complete series of images are presented in Quicktime format for the cell expressing wild-type Chfr as Video 3 and for the RING finger mutant as Video 4. (E) Colcemid delays mitosis in U2OS cells expressing wild-type Chfr. Synchronized U2OS cells expressing wild-type Chfr from an inducible promoter were treated or not with 15 μM colcemid in G2 phase. Cells were monitored by time-lapse microscopy, and the time at which they broke down their nuclear envelopes was plotted as a frequency histogram. Cells are representative of two separate experiments. Videos are available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1.
Table I. Induction of the antephase checkpoint in PtK1 cells.
| Challenge | Return to interphase |
Continue to mitosis |
Arrest in prophase |
|||
|---|---|---|---|---|---|---|
| Cells expressing wild-type Chfr | ||||||
| Colcemid | 5 | 0 | 2 | |||
| Cells expressing ΔFHA-Chfr | ||||||
| Challenge | Return to interphase |
Continue to mitosis |
Arrest in prophase |
|||
| Colcemid | 0 | 7 | 5 | |||
| Anisomycin | 5 | 1 | 0 | |||
| Inject p38α | 4 | 1 | 2 | |||
PtK1 cells expressing wild type or ΔFHA-Chfr were treated as described and their behavior was assayed by time-lapse DIC microscopy. Cells that decondensed their chromosomes were scored as returning to interphase, and cells that broke down their nuclear envelopes were scored as continuing to mitosis.
The antephase checkpoint requires ubiquitylation but not proteasome activity
Chfr had been reported to ubiquitylate and target for destruction the Polo-like kinase in X. laevis cycling extracts, thereby delaying the activation of cyclin B-CDK1 (Kang et al., 2002). Therefore, we assayed whether or not Plk1 proteolysis was involved in the antephase checkpoint in mammalian somatic cells. However, we were unable to detect any loss of Plk1 upon activation of the antephase checkpoint, either by immunofluorescence analysis (not depicted) or by quantifying the level of a Plk1-GFP fusion protein in living cells (Fig. 3 A). (This Plk1-GFP fusion protein mimics the endogenous protein because it is degraded at the end of mitosis [Lindon and Pines, 2004].) Moreover, we found that the antephase checkpoint did not require proteasome-dependent degradation. Prophase PtK1 cells treated with the specific proteasome inhibitors MG132 or epoxomicin returned to interphase after treatment with nocodazole or colcemid, which is indicative of a functional checkpoint (Table II). In these experiments, MG132 and epoxomicin did block protein degradation because prophase cells treated with these compounds without nocodazole subsequently arrested in metaphase. Thus, mammalian cells appeared to differ from X. laevis extracts where proteasome inhibitors blocked the ability of Chfr to inhibit mitosis (Kang et al., 2002).
Figure 3.

Ubiquitylation but not proteasome-dependent degradation is required for the antephase checkpoint. (A) Plk1 is not degraded by the antephase checkpoint. Ptk1 cells were injected with an expression construct encoding YFP fused to Plk1 and followed by time-lapse fluorescence and DIC microscopy at 3-min intervals. In early prophase, the cell was treated with 15 μM colcemid and the cell returned to antephase. The total cell fluorescence minus background was quantified for each cell in successive images of a time series and plotted over time. Graph is from a cell representative of three cells in four independent experiments. (B) Ubiquitylation is required for the antephase checkpoint. DIC images of PtK1 cells injected with wild type (top) or methylated ubiquitin (bottom) in early prophase and then treated with 15 μM colcemid at 0 min. Cells with wild-type ubiquitin returned to antephase, whereas cells injected with methylated ubiquitin continued into mitosis. Cells are representative of 8 out of 10 wild type and 4 out of 8 methylated ubiquitin-injected cells in three independent experiments. Bar, 10 μm.
Table II. Effect of ubiquitin and the proteasome on the antephase checkpoint.
| Treatment | Return to interphase |
Continue to mitosis |
Arrest in prophase |
|---|---|---|---|
| Ubiquitin | 8 | 1 | 1 |
| Methyl-ubiquitin | 1 | 4 | 3 |
| MG132 | 12 | 2 | 4 |
| Epoxomicin | 4 | 1 | 1 |
PtK1 cells were treated as described and their behavior was assayed by time-lapse DIC microscopy. Cells that decondensed their chromosomes were scored as returning to interphase, and cells that broke down their nuclear envelopes were scored as continuing to mitosis.
Although polyubiquitinated proteins are often targeted for destruction by the proteasome, they have also been shown to act as signaling molecules, especially when conjugated via K63. Moreover, Chfr has been characterized as a ubiquitin ligase that requires Ubc13-Mms2 (Bothos et al., 2003), a dimeric ubiquitin-conjugating enzyme that preferentially conjugates ubiquitin through its K63 and not K48 residue. To test whether or not the antephase checkpoint required ubiquitylation, we injected methyl-ubiquitin into cells to block ubiquitylation. This treatment abrogated the antephase checkpoint in 4 out of 8 cells, which carried on into mitosis (3 other cells arrested in prophase), whereas injecting wild-type ubiquitin allowed 8 out of 10 cells to return to interphase when challenged with colcemid (Fig. 3 B and Table II). Thus, the antephase checkpoint appeared to require ubiquitylation but not proteasome-dependent proteolysis.
The p38 stress kinase is required for the antephase checkpoint
Ubiquitylation through K63 has been shown to activate the stress kinase pathway through the TAK1 kinase in response to interleukin-1 (Wang et al., 2001). In addition, cells treated with low doses of anisomycin have a reduced mitotic index (Lindon, C., personal communication). Anisomycin has been shown to be a potent activator of p38 stress kinases (Cano et al., 1994), and p38 kinases can also be activated by treating late G2/mitotic cells with nocodazole (Takenaka et al., 1998). Therefore, we analyzed if the p38 kinases could play a part in the antephase checkpoint. We found that anisomycin was as effective as nocodazole or colcemid in both reducing the number of prophase cells in the population (supplemental material) and causing mid-prophase cells to return to interphase (Fig. 4 A and Video 5, available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1). Treatment with other stimuli known to activate the p38 kinases such as 5 μM H2O2 or 10 j/m2 UV also caused a decline in the number of prophase cells (supplemental material). To support a role for the p38 stress kinases, we compared the effects of the p38α and β kinase inhibitors SB203580 and SB202190, with the inactive compound SB202474 as a control. In both of our assays, SB203580 and SB202190 abrogated the antephase checkpoint induced by colcemid, whereas SB202474 had no effect (Fig. 4 B and supplemental material). In these experiments, cells treated with colcemid subsequently arrested for several hours in prometaphase in the presence or absence of p38 inhibitors, which is indicative of an intact spindle assembly checkpoint.
Figure 4.

p38 stress kinases are required for the antephase checkpoint. (A) DIC images of Ptk1 cells in early prophase before (left) or after (right) treatment with 50 ng/ml anisomycin. The complete series of images for the cell treated with anisomycin are presented in Quicktime format as Video 5, available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1. (B) DIC images of Ptk1 cells in early prophase that were treated at 0 min with 15 μM colcemid plus 5 μM SB203580 (top), or 15 μM colcemid plus 5 μM SB202474 (bottom) and followed by time-lapse microscopy. Cells with the active p38 inhibitor (SB203580) continued into mitosis. Cells are representative of four cells in two experiments for each SB compound. (C) Ptk1 cells in early prophase were injected with active p38α (top) or active ERK2 (bottom) and followed by time-lapse DIC microscopy at 3-min intervals. The cell injected with p38 returned to antephase. Bars, 10 μm.
These results strongly indicated that the p38 α and/or β kinases were necessary for the antephase checkpoint. However, although SB203580 and SB202190 had been shown to be highly specific inhibitors of p38 in vitro, they could have acted by inhibiting another, related kinase in vivo. To eliminate this possibility, we injected active p38 kinases into early prophase PtK1 cells and found that either p38α or p38β forced the majority of cells to return to interphase, whereas p38δ did not (Fig. 4 C and Table III). Injecting the related MAP kinase family member, Erk2, had no effect on progression through prophase (Fig. 4 C and Table III). Thus, we concluded that the p38α and β stress kinases were important components of the antephase checkpoint.
Table III. Effect of stress kinases and cyclin A–Cdk kinases on the antephase checkpoint.
| Protein injected | Return to interphase |
Continue to mitosis |
Arrest in prophase |
|---|---|---|---|
| GST-p38α | 6 | 1 | 0 |
| p38α | 6 | 3 | 0 |
| p38β | 7 | 2 | 1 |
| p38δ | 1 | 5 | 4 |
| Cyclin A–CDK2AF | 0 | 9 | 4 |
| Cyclin A–CDK2 | 7 | 4 | 3 |
PtK1 cells were injected with the proteins as described and their behavior was assayed by time-lapse DIC microscopy. Cells that decondensed their chromosomes were scored as returning to interphase, and cells that broke down their nuclear envelopes were scored as continuing to mitosis.
To discriminate between whether the p38 kinases lay up or downstream of Chfr, we treated U2OS cells, which have a mutant Chfr, with 50 ng/ml anisomycin for 30 min to activate the p38 stress kinases and followed their entry into mitosis by time-lapse microscopy. Anisomycin blocked the appearance of prophase cells within 30 min and delayed entry to mitosis in the majority of the population by an average of 90 min, indicating that U2OS cells might have some of the components necessary for the antephase checkpoint (Fig. 5 A). However, the decline in mitotic cells might have been caused by cells arresting in late G2 phase, and we wished to determine whether or not anisomycin could cause cells to return from prophase to antephase. Therefore, we monitored the behavior of the centrosomes in HeLa cells expressing α-tubulin-YFP, which normally accumulated on centrosomes in early prophase (Khodjakov and Rieder, 1999; Fig. 5 B and Video 6, available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1), and found that anisomycin was able to reverse this recruitment (Fig. 5 C and Video 7, available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1). Thus, even in the absence of a functional Chfr protein, activating the p38 kinases appeared to be able to mediate the antephase checkpoint.
Figure 5.

p38 is downstream of Chfr. (A) Anisomycin treatment delays entry into mitosis. Synchronized U2OS cells were treated or not in G2 phase with anisomycin for 30 min, and then followed by time-lapse DIC microscopy at 3-min intervals, and the time when the cell completed nuclear envelope breakdown was assayed and plotted as a frequency histogram. Cells are representative of three different experiments. (B and C) HeLa cells stably expressing α-tubulin-EYFP were followed by time-lapse DIC and fluorescence microscopy at 3-min interval. 29 z-sections spaced 0.4 μm apart were taken for each time point, deconvolved, and used to generate a maximum intensity projection at each time point. In untreated cells (B), α-tubulin accumulated on centrosomes in prophase just before nuclear envelope breakdown (the complete series of images are presented in Quicktime format as Video 6). The accumulation of α-tubulin on centrosomes was reversed when the cell was treated with 1 μg/ml anisomycin. The complete series of images are presented in Quicktime format as Video 7. (D) Fluorescence (top) and DIC images of a Ptk1 cell expressing dominant-negative GFP-Chfr. The cell was injected in early prophase (time 0) with active p38α and followed by time-lapse DIC and fluorescence microscopy at 3-min intervals as it returned to antephase. The cell is representative of four cells in seven experiments. Bar, 10 μm. Videos are available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1.
One interpretation of these results was that Chfr was required to activate p38 when cells were treated with nocodazole. To test this interpretation, we abrogated the antephase checkpoint in PtK1 cells by expressing the ΔFHA mutant of Chfr. When challenged with anisomycin or injected with active p38 kinase the cells returned to interphase (Fig. 5 D and Table I), indicating that p38 was likely to be downstream of Chfr.
Active cyclin A–CDK2 kinase can overcome the antephase checkpoint
Lastly, we considered the possible mechanisms by which the p38 kinases might reverse progress through prophase. We had previously shown that cyclin A–dependent kinase activity was important for cells to enter and progress through prophase (Furuno et al., 1999) and that cyclin B1-Cdk1 kinase was significantly activated only towards the end of prophase (Jackman et al., 2003). Thus, it was possible that p38 kinases blocked or reversed cells in early prophase by inhibiting cyclin A–dependent kinases, perhaps by inactivating the Cdc25 phosphatases. If inactivation of cyclin A–Cdk complexes was required for the antephase checkpoint, we reasoned that cyclin A in a complex with a mutant form of Cdk that could not be inactivated by phosphorylation should overcome the checkpoint. In support of this reasoning, when we injected cells with cyclin A bound to a mutant form of CDK2 that could not be phosphorylated and inhibited (CDK2AF), 13 out of 13 injected cells were no longer able to return to antephase when challenged with colcemid in prophase (Fig. 6 and Table III). As a control, we injected cells with wild-type CDK2 and cyclin A. In this case, 7 out of 14 prophase cells still returned to antephase after being challenged with colcemid (Table III).
Figure 6.
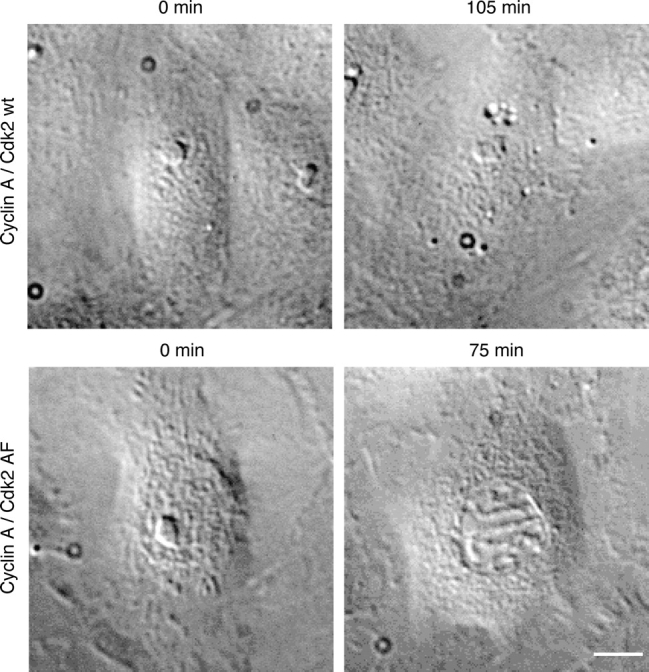
Active cyclin A–Cdk2 complexes override the antephase checkpoint. DIC images of Ptk1 cells. Cells in early prophase were injected with cyclin A bound to either wild-type Cdk2 (top) or a Cdk2 T14A/Y15F mutant (bottom) that cannot be inactivated by phosphorylation. 5 min after injection, the cells were treated with 15 μM colcemid (0 min) and followed by time-lapse microscopy. Some cells arrested in prophase and were scored separately. Cells are representative of 9 mutant and 7 wild-type Cdk-injected cells in 14 independent experiments. Bar, 10 μm.
Discussion
Here, we have shown that the Chfr protein is an important component of the antephase checkpoint in mammalian cells and that the p38 stress kinases appear to be downstream effectors of the checkpoint. As part of the checkpoint mechanism, Chfr requires its ubiquitin ligase activity, but its substrates do not need to be degraded by the proteasome. This is the first direct demonstration of a link between Chfr and the antephase checkpoint; originally Chfr was reported to be required for a checkpoint that prevented entry to metaphase after cyclin B1-Cdk1 is activated (Scolnick and Halazonetis, 2000). However, our evidence indicates that Chfr is required for the antephase checkpoint, and, in agreement with Rieder and Cole (1998), we find that cells no longer respond to the antephase checkpoint when the nucleoli begin to breakdown, which is the time when significant amounts of active cyclin B1 first appear in the cell and move into the nucleus (Jackman et al., 2003). We also find that active cyclin A–Cdk complexes can override the checkpoint, indicating that cyclin A–Cdks may be targets of the checkpoint.
At present, we are unable to explain exactly how Chfr acts in the checkpoint. Clearly, Chfr must be acting as an ubiquitin ligase because wild-type Chfr, but not a RING finger mutant, is able to restore the antephase checkpoint in U2OS cells. Moreover, methylated ubiquitin, which blocks polyubiquitylation, abolishes the checkpoint. In contrast to X. laevis extracts, in mammalian cells Chfr does not need to target its substrates for proteasome-dependent degradation to block progress into mitosis because the checkpoint is unaffected by proteasome inhibitors. Thus, we believe that Chfr acts in a signaling pathway by generating ubiquitin chains that are conjugated through K63. In support of this belief, we find that Chfr binds to Ubc13 in a yeast 2 hybrid assay, and in collaboration with J. Chen (personal communication), we have found that Chfr requires Ubc13 to autoubiquitylate (unpublished data). This finding is in agreement with the recent report that Chfr auto-ubiquitylates through K63 (Bothos et al., 2003). One intriguing possibility is that Chfr could activate the p38 stress kinase pathway by activating the TAK1 kinase. TAK1 is a MAP kinase kinase kinase in the p38 kinase pathway and can be activated by the TRAF6 ubiquitin ligase in response to interferon (Wang et al., 2001). Indeed, in a yeast 2 hybrid screen we have isolated the T6BP (TRAF6 binding protein) protein, which interacted with Chfr in a RING finger-dependent manner (unpublished data). T6BP is a protein that can interact with the TRAF6 protein (Ling and Goeddel, 2000). This finding raises the possibility that Chfr in combination with T6BP might be able to activate TAK1 in response to microtubule depolymerization. However, as yet, we have been unable biochemically to confirm this model.
Our evidence indicates that the p38 stress kinases are required to effect the antephase checkpoint and that they appear to act downstream or in parallel to Chfr. Chemical inhibitors of the p38α and β kinases abrogate the antephase checkpoint, and an active form of p38α, but not p38δ or the related ERK2 kinase, is able to return early prophase cells to interphase. In animal cells, the p38 stress kinases have previously been implicated in two different late cell cycle checkpoints: in G2 phase (Bulavin et al., 2001) and in mitosis itself (Takenaka et al., 1998). Bulavin et al. (2001) showed that, in vitro, the p38 stress kinases are able to phosphorylate the Cdc25B phosphatase that can play a role in the initiation of mitosis, and that this may underlie the G2 arrest in response to UV damage (Bulavin et al., 2001). This mechanism may also be the means by which the p38 stress kinases are able to return prophase cells to interphase, and we show that cyclin A in a complex with a form of CDK2 that is independent of Cdc25 is able to overcome the antephase checkpoint. However, as yet, we have been unable to detect biochemically a change in the phosphorylation state of CDK2 bound to cyclin A when we activate the antephase checkpoint, nor when we activate the p38 kinases directly with anisomycin (Koop, L., personal communication; unpublished data), although this may simply reflect the practical difficulty in obtaining sufficient numbers of cells in late G2/early prophase.
Takenaka et al. (1998) reported that the p38 stress kinases are required to arrest mammalian cells in prometaphase when microtubules are destabilized with nocodazole. Thus, they implicated the p38 stress kinases as important components of the spindle assembly checkpoint. In fission yeast, the p38 stress kinase has been reported to be a component of a checkpoint required to arrest cells in mitosis when the spindle is misoriented (Gachet et al., 2001). However, we were unable to detect any change in the ability of cells to arrest in mitosis in response to nocodazole in the presence or absence of p38 kinase inhibitors. At present, we are unable to explain the apparent discrepancy in our results from those of Takenaka et al. (1998), although one difference in experimental protocols is that we assayed mitotic arrest by counting cells stained with Hoechst 33342, whereas Takenaka et al. (1998) assayed arrest more indirectly by measuring H1 kinase activity in cell lysates.
Chfr is inactivated or absent from several cell lines (Scolnick and Halazonetis, 2000) and tumors (Mizuno et al., 2002), perhaps indicating that the loss of Chfr may confer a growth advantage, at least in cell culture. Our results could be interpreted as showing that cells lacking Chfr have a lesion in at least one stress response pathway, which might be an advantage in a tumor environment. However, we find that these cells still delay division in response to UV (unpublished data) or H2O2 treatment, making it less likely that the advantage conferred by the loss of Chfr is the ability to divide in low O2 tension environments. Nevertheless, Chfr may potentially link perturbations in the cytoskeleton to the stress kinase pathway and indicate that other ubiquitin ligases may function in a similar role to block entry into mitosis in response to other forms of stress (Fig. 7).
Figure 7.

Schematic for the antephase checkpoint. Several different insults are able to block cells in antephase, some or all of which may act through the p38 stress kinases. The Chfr protein appears to be required to activate the checkpoint in response to microtubule poisons but not to UV or H2O2 and probably signals through ubiquitin conjugation, perhaps through the TAK1 kinase. Note that this is only a tentative model.
Materials and methods
Cell culture, synchronization, and drug treatments
HeLa and U2OS cells were cultured and synchronized as described previously (Jackman et al., 2003). Cell lines expressing inducible Chfr were cultured in DME plus 5% FBS, 5% newborn calf serum, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 0.292 mg/ml glutamine, 1% (vol/vol) Fungizone (GIBCO BRL), 0.25 mg/ml hygromycin B, 1 μg/ml tetracycline, and 0.5 mg/ml geneticin at 37°C/5% CO2. Ptk1 cells were cultured in Ham's F-12 medium (GIBCO BRL), 10% FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 1 mM Na pyruvate, and 0.1% (vol/vol) Fungizone (GIBCO BRL) at 37°C/5% CO2. Colcemid was added to a final concentration of 15 μM, anisomycin to 50 ng/ml, SB203580 and SB202474 (Calbiochem) to 5 μM, H2O2 to 0.0 2 mM, MG-132 to 42 nM, MG-115 (Calbiochem) to 50 nM, and epoxomicin (Calbiochem) to 10 μM.
Protein expression, purification, and injection
Cyclin A–Cdk2 and cyclin A–Cdk2AF were expressed in and purified from Escherichia coli BL21 cells as described previously (Brown et al., 1999). Proteins were >90% pure on Coomassie blue R250–stained gels. Proteins were concentrated in injection buffer (12.5 mM Tris-HCl, pH 8.0, 200 mM NaCl, 2.5 mM DTT, and 1 mM EGTA) in a Vivaspin 5,000 MW cut-off microconcentrator (Vivascience). Approximately 5% of the cell volume was injected into cells using a semiautomatic microinjector (Eppendorf) attached to a microscope (model DMIRBE; Leica). p38α, p38β, and p38δ were gifts of C. Smythe (University of Sheffield, Sheffield, UK). Ubiquitin (50 mg/ml) and methylated ubiquitin were purchased from Calbiochem.
Antephase checkpoint assay
For live cell assay, cells were cultured on 0.15-mm ΔT dishes (Bioptechs) at 37°C, treated with the drugs colcemid (15 μM) or anisomycin (50 ng/ml), and followed by time-lapse DIC microscopy at 3-min intervals. Early prophase PtK1 cells were identified by the beginnings of chromosome condensation. U2OS cells were treated 11 h after release from a double thymidine block to increase the number of mitotic cells. When treated with 50 ng/ml anisomycin 30 min after the addition of the drug, the U2OS cultures were washed with fresh prewarmed medium six times and then followed by DIC microscopy. At least 200 cells were examined for each experiment, scored for nuclear envelope breakdown, and plotted on a graph.
Image acquisition
Images were acquired using a microscope and a 40× 1.0 NA or 63× 1.35 NA oil immersion objective as described previously (Karlsson and Pines, 1998) with the addition of a motorized XY stage (Prior Scientific) to visualize multiple fields of cells per time point. Images were taken with a cooled CCD camera (model Pentamax or Micromax; Roper Scientific) using IP Lab software (Scanalytics) and analyzed using ImageJ software (National Institutes of Health) before exporting to Adobe Photoshop for printing or Adobe Premiere to generate the Quicktime videos. The four-dimensional imaging of α-tubulin YFP cells were imaged at 37°C on a microscope (model DeltaVision Spectris; Applied Precision) equipped with a 40× 1.35 NA lens (Olympus) and a cooled CCD camera (model CoolSnap HQ; Roper Scientific). Stacks of 29 z-images 0.4 μm apart were taken every 3 min, and the maximum intensity projections were combined using SoftWorxTM software for the images and videos in Fig. 5.
Immunofluorescence
To detect epitope-tagged Chfr, cells were fixed and permeabilized with 50:50 vol/vol MeOH/Acetone and stained with anti-Xpress antibody (Invitrogen) at 1:2,000 dilution followed by an Alexa Fluor 488–labeled anti–mouse secondary antibody (Molecular Probes).
Fixed cell assay for prophase checkpoint
15 μM colcemid, 1 μg/ml anisomycin, 15 μM colcemid plus 5 μM SB203580, 15 μM colcemid plus 5 μM SB202474, or their carrier DMSO were directly added to the medium of cells growing on glass coverslips. At 0, 60, and 120 min after drug treatment, coverslips were removed from medium and fixed with 3% PFA in 1× PBS. Cultures were stained with Hoechst 33342. Coverslips were examined and mitotic cells were scored by epifluorescence using a microscope (model Optiphot; Nikon) equipped with a 20× 0.75 NA and a 40× 1.3 NA lens (Nikon).
Online supplemental material
A fixed cell assay showing that p38 kinases are required for the antephase checkpoint is shown in Fig. S1. Video 1 shows live cell imaging of the antephase checkpoint. Video 2 shows that ΔFHA Chfr abrogates the antephase checkpoint. Video 3 shows that wild-type Chfr rescues the antephase checkpoint in U2OS cells. Video 4 shows that the Ring finger mutant of Chfr fails to rescue an antephase checkpoint defect. Video 5 shows that anisomycin can activate the antephase checkpoint. Video 6 shows live cell imaging of HeLa cells stably expressing α-tubulin-EYFP. Video 7 shows that anisomycin reverses the accumulation of α-tubulin on centrosomes. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200401139/DC1.
Acknowledgments
We are very grateful to Catherine Lindon who made the observation that the mitotic index decreased in anisomycin-treated cells and to Conly Rieder for advice, discussions, and communicating unpublished results. Many thanks to Cecile Pickert, Carl Smythe, and Angel Nebreda for help and advice, and to James Chen for ongoing collaborative efforts.
This work was supported by a Japan Science Promotion Society fellowship to T. Matsusaka, the Association for International Cancer Research, and programme grant C29/A1782 to J. Pines from Cancer Research UK.
References
- Bothos, J., M.K. Summers, M. Venere, D.M. Scolnick, and T.D. Halazonetis. 2003. The Chfr mitotic checkpoint protein functions with Ubc13-Mms2 to form Lys63-linked polyubiquitin chains. Oncogene. 22:7101–7107. [DOI] [PubMed] [Google Scholar]
- Brown, N.R., M.E. Noble, J.A. Endicott, and L.N. Johnson. 1999. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1:438–443. [DOI] [PubMed] [Google Scholar]
- Bulavin, D.V., Y. Higashimoto, I.J. Popoff, W.A. Gaarde, V. Basrur, O. Potapova, E. Appella, and A.J. Fornace, Jr. 2001. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature. 411:102–107. [DOI] [PubMed] [Google Scholar]
- Bulavin, D.V., S.A. Amundson, and A.J. Fornace. 2002. p38 and Chk1 kinases: different conductors for the G(2)/M checkpoint symphony. Curr. Opin. Genet. Dev. 12:92–97. [DOI] [PubMed] [Google Scholar]
- Bullough, W.S., and M. Johnson. 1951. The energy relations of mitotic activity in adult mouse epidermis. Proc. R. Soc. Lond. B. Biol. Sci. 138:562–575. [DOI] [PubMed] [Google Scholar]
- Cano, E., C.A. Hazzalin, and L.C. Mahadevan. 1994. Anisomycin-activated protein kinases p45 and p55 but not mitogen-activated protein kinases ERK-1 and -2 are implicated in the induction of c-fos and c-jun. Mol. Cell. Biol. 14:7352–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, L., C. Wang, E. Spencer, L. Yang, A. Braun, J. You, C. Slaughter, C. Pickart, and Z.J. Chen. 2000. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 103:351–361. [DOI] [PubMed] [Google Scholar]
- Furuno, N., N. den Elzen, and J. Pines. 1999. Human cyclin A is required for mitosis until mid prophase. J. Cell Biol. 147:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gachet, Y., S. Tournier, J.B. Millar, and J.S. Hyams. 2001. A MAP kinase-dependent actin checkpoint ensures proper spindle orientation in fission yeast. Nature. 412:352–355. [DOI] [PubMed] [Google Scholar]
- Hofmann, R.M., and C.M. Pickart. 2001. In vitro assembly and recognition of Lys-63 polyubiquitin chains. J. Biol. Chem. 276:27936–27943. [DOI] [PubMed] [Google Scholar]
- Jackman, M., C. Lindon, E.A. Nigg, and J. Pines. 2003. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 5:143–148. [DOI] [PubMed] [Google Scholar]
- Kang, D., J. Chen, J. Wong, and G. Fang. 2002. The checkpoint protein Chfr is a ligase that ubiquitinates Plk1 and inhibits Cdc2 at the G2 to M transition. J. Cell Biol. 156:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson, C., and J. Pines. 1998. Green fluorescent protein. Cell Biology: A Laboratory Handbook. Vol. 4. J. Celis, editor. Academic Press, San Diego, CA. 246–252.
- Khodjakov, A., and C.L. Rieder. 1999. The sudden recruitment of γ-tubulin to the centrosome at the onset of mitosis and its dynamic exchange throughout the cell cycle, do not require microtubules. J. Cell Biol. 146:585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindon, C., and J. Pines. 2004. Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. J. Cell Biol. 164:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, L., and D.V. Goeddel. 2000. T6BP, a TRAF6-interacting protein involved in IL-1 signaling. Proc. Natl. Acad. Sci. USA. 97:9567–9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov, A., and C.L. Rieder. 2002. Cell cycle: stressed out of mitosis. Curr. Biol. 12:R331–R333. [DOI] [PubMed] [Google Scholar]
- Mizuno, K., H. Osada, H. Konishi, Y. Tatematsu, Y. Yatabe, T. Mitsudomi, Y. Fujii, and T. Takahashi. 2002. Aberrant hypermethylation of the CHFR prophase checkpoint gene in human lung cancers. Oncogene. 21:2328–2333. [DOI] [PubMed] [Google Scholar]
- Nebreda, A.R., and A. Porras. 2000. p38 MAP kinases: beyond the stress response. Trends Biochem. Sci. 25:257–260. [DOI] [PubMed] [Google Scholar]
- Pickart, C.M. 2001. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70:503–533. [DOI] [PubMed] [Google Scholar]
- Pines, J., and C.L. Rieder. 2001. Re-staging mitosis: a contemporary view of mitotic progression. Nat. Cell Biol. 3:E3–E6. [DOI] [PubMed] [Google Scholar]
- Rieder, C.L., and R.W. Cole. 1998. Entry into mitosis in vertebrate somatic cells is guarded by a chromosome damage checkpoint that reverses the cell cycle when triggered during early but not late prophase. J. Cell Biol. 142:1013–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder, C.L., and R. Cole. 2000. Microtubule disassembly delays the G2-M transition in vertebrates. Curr. Biol. 10:1067–1070. [DOI] [PubMed] [Google Scholar]
- Scolnick, D.M., and T.D. Halazonetis. 2000. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature. 406:430–435. [DOI] [PubMed] [Google Scholar]
- Shtivelman, E. 2003. Promotion of mitosis by activated protein kinase B after DNA damage involves polo-like kinase 1 and checkpoint protein CHFR. Mol. Cancer Res. 1:959–969. [PubMed] [Google Scholar]
- Takenaka, K., T. Moriguchi, and E. Nishida. 1998. Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science. 280:599–602. [DOI] [PubMed] [Google Scholar]
- Ulrich, H.D., and S. Jentsch. 2000. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 19:3388–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C., L. Deng, M. Hong, G.R. Akkaraju, J. Inoue, and Z.J. Chen. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 412:346–351. [DOI] [PubMed] [Google Scholar]