

Abstract
Although the importance of mitochondria in patho-physiology has become increasingly evident, it remains unclear whether these organelles play a role in Ca2+ handling by skeletal muscle. This undefined situation is mainly due to technical limitations in measuring Ca2+ transients reliably during the contraction–relaxation cycle. Using two-photon microscopy and genetically expressed “cameleon” Ca2+ sensors, we developed a robust system that enables the measurement of both cytoplasmic and mitochondrial Ca2+ transients in vivo. We show here for the first time that, in vivo and under highly physiological conditions, mitochondria in mammalian skeletal muscle take up Ca2+ during contraction induced by motor nerve stimulation and rapidly release it during relaxation. The mitochondrial Ca2+ increase is delayed by a few milliseconds compared with the cytosolic Ca2+ rise and occurs both during a single twitch and upon tetanic contraction.
Keywords: calcium; cameleon; in vivo; Na+/Ca2+ exchange; two-photon microscopy
Introduction
Up until now it has been unclear, and often the subject of heated debates, whether or not mitochondria in skeletal muscle take up Ca2+ during increase in the cytoplasmic concentration of Ca2+ ([Ca2+]c) accompanying the contraction process. Although at one stage this was intensely discussed (for review see Carafoli, 2003), the functional role of mitochondria in [Ca2+]c variations in striated muscle under physiological conditions has been generally neglected; this is mainly due to the early discovery of the ER–SR function (Hasselbach and Makinose, 1961; Ebashi and Lipman, 1962) and the prejudice that the mitochondrial Ca2+ uniporter, as studied in vitro (Gunter and Pfeiffer, 1990), is slow and has too low affinity for Ca2+ to comply with the extremely high speed [Ca2+]c changes that occur in striated muscle (for review see Bernardi, 1999) . With the advent of targeted chemical and protein probes capable of monitoring levels of mitochondrial Ca2+ ([Ca2+]m) in living cells, the role of mitochondria in Ca2+ signaling, both in general and specifically in muscle tissue, began to be reevaluated (Hajnoczky et al., 2000; Pozzan et al., 2000; Rizzuto et al., 2000). In heart muscle cells, beat-to-beat rises of [Ca2+]m have been observed using targeted aequorins and pericams (Robert et al., 2001a) and [Ca2+] increases in individual mitochondria loaded with rhod-2 have been observed in cardiac myotubes during localized release of Ca2+ from ryanodine receptors (Pacher et al., 2002). Uptake of Ca2+ into mitochondria has also been reported in single mature cardiac muscle fibers upon depolarization (Trollinger et al., 2000).
As to skeletal muscle, using targeted aequorins, mitochondria in myotubes were shown to accumulate Ca2+ in response to different stimuli (Brini et al., 1997; Challet et al., 2001), and myotubes from dystrophic mdx mice showed an elevated uptake of Ca2+ into mitochondria during potassium-induced cell depolarization (Robert et al., 2001b). There is also indirect evidence that [Ca2+]m increases may serve to adjust ATP production to the physiological demands of muscle and other tissues (McCormack and Denton, 1990; Robb-Gaspers et al., 1998; Jouaville et al., 1999). Furthermore, with using rhod-2, rises of [Ca2+]m after but not during tetanic stimulation were observed in mature isolated skeletal muscle fibers (Bruton et al., 2003a,c); but see Lannergren et al. (2001) for contrasting results. All these studies, valuable as they are, present well-known shortcomings. First, rhod-2 (used generally as an AM-ester) appears to distribute to mitochondria only, or mainly, when applied at low temperature (Trollinger et al., 1997); this limits the use of rhod-2 to in vitro systems in which the incubation temperature can be easily influenced. Second, rhod-2 measures [Ca2+] solely by changes in fluorescence intensity and is therefore prone to the general limitations inherent to nonratiometric dyes, including major artifacts due to movement; this is obviously critical for observations during muscle contraction. Third, the amount of photons emitted by recombinant aequorin is quite low and thus makes the analysis of single cells difficult and with relatively low time resolution. Fourth, all measurements of [Ca2+]m in skeletal muscle during contraction have been performed in in vitro systems using myocytes (Trollinger et al., 1997) or myotubes (Brini et al., 1997; Challet et al., 2001; Robert et al., 2001b); i.e., muscle fibers at a largely immature stage where the kinetics of the Ca2+ changes are far slower than those occurring in vivo (Koopman et al., 2003).
Genetically encoded fluorescent ratiometric Ca2+ sensors (called “yellow cameleon” [YC]) were introduced in 1997 (Miyawaki et al., 1997). Upon binding of Ca2+, they respond with an increase in fluorescence resonance energy transfer (FRET; Miyawaki et al., 1997) and are therefore optimal for experiments with monochromatic excitation and ratiometric read-out, as used in two-photon confocal microscopy. Genetic engineering enables YCs to be specifically targeted to different subcellular locations (such as the nucleus [Miyawaki et al., 1997], ER ER [Miyawaki et al., 1997], and mitochondria [Arnaudeau et al., 2001]) and thus permits the measurement of [Ca2+] therein. Recently, cytoplasmic YCs have been used in living invertebrates (Kerr et al., 2000; Diegelmann et al., 2002; Fiala et al., 2002; Reiff et al., 2002; Ikeda et al., 2003; Shyn et al., 2003; Suzuki et al., 2003), and the zebrafish (Higashijima et al., 2003); surprisingly, YCs have yet to be shown to function correctly in live mammals.
We transfected mouse hind-limb muscle in situ with a second generation YC (YC2; Miyawaki et al., 1997) targeted to the cytoplasm or mitochondria, and monitored [Ca2+]c and [Ca2+]m by two-photon microscopy. The initial part of our study sought to validate YC2 as a reliable Ca2+ indicator and the data obtained show that cytoplasmic YC2 is capable of recording short-lived rises in [Ca2+]c in the millisecond range, and correcting for gross movement artifacts. Our subsequent studies to address [Ca2+]m homeostasis demonstrated that the rise in [Ca2+]m observed during single twitches and tetanic stimulation in live mammalian skeletal muscle is synchronized with the physiological induction of muscle contraction.
Results
Expression rate and distribution of transfected fibers
Tibialis anterior muscle was transfected with YC2 cDNA as described previously (Dona et al., 2003; see also Materials and methods section). 3 wk later it was detached, snap-frozen, and transversal (Fig. 1, A1 and A2) or longitudinal sections (Fig. 1, B1 and B2) prepared. Fluorescence microscopy revealed that ∼50% of all fibers exhibited a strong diffuse YC2 signal; interestingly, there was an uneven distribution of transfected fibers, the majority being in the peripheral regions (Fig. 1 A2). Conveniently, the highest concentration was found in the anterior periphery (Fig. 1, A1 and A2, upper right), which is the area exposed for microscopic analysis. The unequal distribution of positive fibers is most likely due to the polarized injection of cDNA and/or to the efficacy of the electroporation in the fibers closest to the electrodes.
Figure 1.

Genetic expression of YC2 and its visualization in mouse skeletal muscle. Tibialis anterior muscle was transfected as described in the Materials and methods section. (A and B) 3 wk after transfection, the muscle was detached, snap-frozen, sectioned transversally (10-μm slices; A) or longitudinally (30-μm slices; B), and examined by either brightfield (A1 and B1) or fluorescence (A2 and B2) microscopy. Fibers expressing YC2 are visible as bright circular areas (A2) or horizontal stripes (B2). (C) Sketch illustrating the setup for in vivo observation of YC2. The tibialis anterior muscle is exposed to microscopic examination after detaching the distal tendon. Muscle contraction is induced by electric stimulation of the sciatic nerve with a custom-made silver electrode. A Tsunami pulsed femto-second laser beam (830 nm wavelength) is used to excite YC2 expressed in the muscle. CFP- and YFP-fluorescence emissions are simultaneously recorded by two photomultiplier tubes equipped with HQ450/80 and HQ535/50 emission filters, respectively. (D) Representative two-photon micrograph of four fibers expressing YC2 in vivo. The apparent decrease in the width of the fibers is due to the curvature of the muscle, causing the optical section to pass through adjacent fibers at slightly different heights.
Observation of calcium spikes during muscle contraction in situ
To test whether YC2 was able to report [Ca2+]c changes in relaxed and contracted muscle in vivo, tibialis anterior muscle expressing cytoplasmic YC2 was used for two-photon analyses, as described in the Materials and methods section and depicted in Fig. 1 C. As expected, the distribution of the Ca2+ probe was uniform throughout the whole myoplasm (Fig. 1 D). In general, the nuclei appeared negative or weakly positive (unpublished data). A simple protocol alternating periods of stimulation of the sciatic nerve with an extracellular electrode (2.5 s at 50 Hz stimulation frequency) and relaxation (10 s) was applied. A representative trace of such an experiment is shown in Fig. 2, A and B, where four aspects are noteworthy. First, the decrease (∼20%) in YFP/CFP ratio (Fig. 2 B) seen in the first few frames (without stimulation), is due to the Ca2+-independent photo-conversion of the YFP moiety (Fig. 2 A; Miyawaki et al., 1997; Filippin et al., 2003). Second, during prolonged phases without stimulation, the basal YFP/CFP ratio showed minimal variation (Fig. 2 B, see region covering 2–10 s). This demonstrates that slight lateral and vertical movements (which occur spontaneously in the living animal) have a negligible influence on the ratio value, sustaining the crucial notion that ratiometric probes effectively correct for movement artifacts (Silver, 1998; Kerr et al., 2000; Rudolf et al., 2003). Third, as a response to the tetanic stimulation, the ratio rises to a maximum value of ∼1.4 (Fig. 2 B); in the experiment presented, the increase in ratio is the result of the anti-parallel behavior of the CFP and YFP moieties (Fig. 2 A), as expected from the Ca2+-dependent FRET signal of this probe. It should be noted, however, that in some experiments this anti-parallelism can be masked by marked changes in total fluorescence intensity (due to fiber movement out of focal plane during contraction); importantly, however, the resulting ratio values are always similar to the one shown in Fig. 2 B. Finally, upon suspension of stimulation, the YFP/CFP ratio returns to basal values (Fig. 2, B and C). A representative trace of a single episode of contraction–relaxation is shown in Fig. 2 C, together with pseudo-colored ratio images at specific time points; the images show two horizontally oriented fibers, and the central frame depicts both fibers in full contraction. It is interesting to note in detail the second frame, captured during stimulation onset: image acquisition is performed as line scans from top to bottom, with each line taking ∼0.5 ms; the upper half of the image still shows no signal change but in the bottom half, a clear increase in [Ca2+]c is already seen, nicely corresponding with the deflection of the lower fiber.
Figure 2.
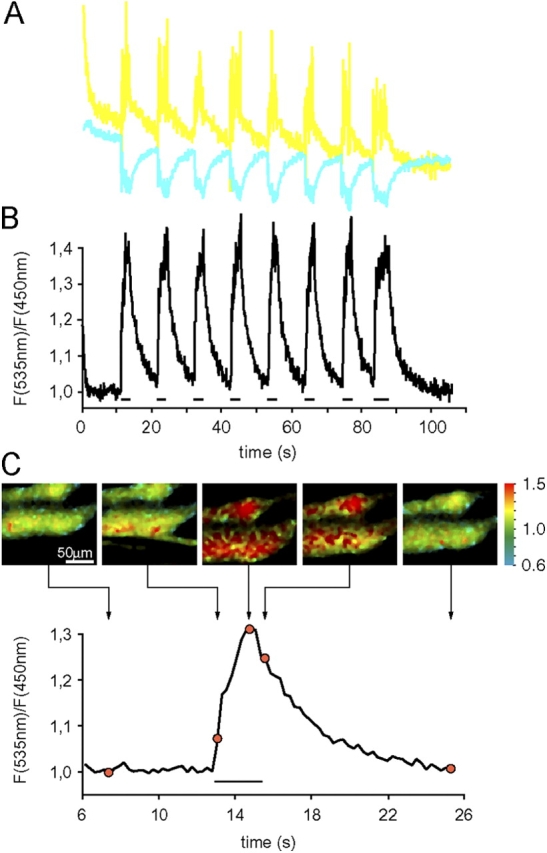
Observation of cytosolic [Ca 2+ ] spikes during muscle contraction in vivo. Tibialis anterior muscle expressing YC2 was monitored in situ using two-photon microscopy.(A) Individual traces of the fluorescence intensity at 450 ± 40 nm (CFP signal, cyan line) and 535 ± 25 nm (YFP signal, yellow line); the acquisition rate was 9.35 Hz, and the image size 64 × 64 pixels. (B) Corresponding ratio values (YFP/CFP) of the traces shown in A. (C) Expanded trace of a single contraction–relaxation cycle, with representative pseudo-colored ratio images; the exact location of each image in the time course is indicated. The horizontal bars below the traces in B and C represent trains of stimulation with a frequency of 50 Hz; note that the last stimulation in B is longer than the preceding ones, giving rise to a more sustained [Ca2+]c rise.
Kinetics of the YC2 response
The frequency of the motor neuron impulses modulates the nature (fast or slow) of the target muscle fibers (for review see Pette and Staron, 2001). The anterior tibialis muscle is primarily, but not exclusively, composed of fast fibers (Sartorius et al., 1998) and the typical frequency of nerve impulses is around 50–100 Hz for fast fibers and 10–20 Hz for slow ones (Hennig and Lomo, 1985). We therefore applied a protocol of nerve stimulation with increasing frequencies (from 1 to 50 Hz) to muscle expressing YC2 and recorded the fluorescence changes in vivo as described in the Materials and methods section. The average ratio values over the whole fiber length from a typical experiment are depicted in Fig. 3 A, with stimulation periods and their frequencies indicated by horizontal bars; the increase in mean ratio intensity that accompanies increased stimulation frequency is evident. It should be stressed that this must not be interpreted as an indication that the peak of [Ca2+]c at each single episode of contraction is different depending on the stimulation frequency; rather the values are an indication of the average [Ca2+]c in that muscle during the whole stimulation period. In other words, at a stimulation rate of 1 Hz for 10 s, [Ca2+]c is almost maximally elevated at each peak, but due to the very short duration of each twitch and its concomitant Ca2+ transient, for most of the time the [Ca2+]c is close to basal. Similar experiments were conducted on muscle expressing split YC2 (Miyawaki et al., 1997) or, as control, a combination of ECFP–CaM and EYFP, i.e., a construct very similar to split YC2, but devoid of M13 and therefore insensitive to [Ca2+]c. The ratio–frequency plot of all variants is shown in Fig. 3 B, where the values represent mean ratios of the analyzed data points for a given stimulation frequency; although both YC2 and its split variant yield similar results, the control shows no variation in ratio. These data confirm that the increase in FRET signal observed upon muscle contraction is not due to movement artifacts or changes in cytosolic pH, but reflect the increase in [Ca2+]c.
Figure 3.

Calibration of the YC2 response to stimulation frequency. Trains with different stimulation frequencies ranging from 1 to 50 Hz were applied as indicated to tibialis anterior muscle expressing either YC2 (A and B), split YC2 (B), or a combination of ECFP-CaM and EYFP (B, control), and the response was monitored in situ by two-photon microscopy. (A) Time course of the YFP/CFP ratio values of a representative calibration experiment. (B) Graph showing the mean YFP/CFP ratio as a function of stimulation frequency. Shown are mean YFP/CFP ratio values of the analyzed images for each stimulation frequency. Error bars show ± SD. All data points are derived from at least two different mice and four different fibers.
Ratio images obtained at different stimulation frequencies were then analyzed in more detail, exploiting an inherent characteristic of confocal laser scanning microscopy: image acquisition is performed as a series of line scans (usually from top to bottom) that require a discrete time period to be completed; given the total number of lines that comprise the whole image, it is possible to calculate the length of time that separates specific image regions. Given that the Ca2+ transient during a single twitch in a mouse muscle fiber lasts ∼50 to100 ms (Baylor and Hollingworth, 2003), and that the complete image acquisition lasts ∼500 ms (as exemplified in Fig. 4 A), a single twitch will be captured in a limited number of lines (Fig. 4 A, 1 Hz panel). The contraction of the fiber and its kinetics are conveniently revealed by small shifts of the fiber body (Fig. 4 A, 1–10 Hz panels); as expected for a fast muscle fiber, each event lasted ∼100 ms. In each shift, it is also clearly evident that the YC2 ratio increases (as indicated by the change in pseudo-color). It is important to note that the increase in [Ca2+]c, as revealed by the pseudo-color scale, should not be misinterpreted to indicate that the rise occurred only in a single region of the fiber; rather, due to time constraints, only that part of the fiber was imaged during the [Ca2+]c rise. This is again demonstrated when the stimulation frequency is increased to 5 Hz (Fig. 4 A), where two episodes of contraction, separated by ∼200 ms, were captured during the complete scan. Fig. 4 B shows a statistical analysis of the ratio intensities measured along the fiber length (and the corresponding time scale) for the different stimulation frequencies (0–50 Hz), together with matched control values (using ECFP-CaM and EYFP). Transient rises in [Ca2+]c corresponding to individual twitches could be distinguished up to a stimulation frequency of 10 Hz. Beyond this, an almost continuous high ratio was observed. Most confocal microscopes also allow what is generally called single-line scans (i.e., instead of generating complete images, the intensity is collected by scanning repeatedly along a single line); this method is often used to improve the time resolution at the expense of two dimensional information. We tried this procedure but results obtained were not informative: indeed, the ratio increases during contraction were systematically very small or even absent (unpublished data), presumably reflecting a damage of the probe by the continuous illumination with the laser beam. This protocol was therefore not further applied.
Figure 4.
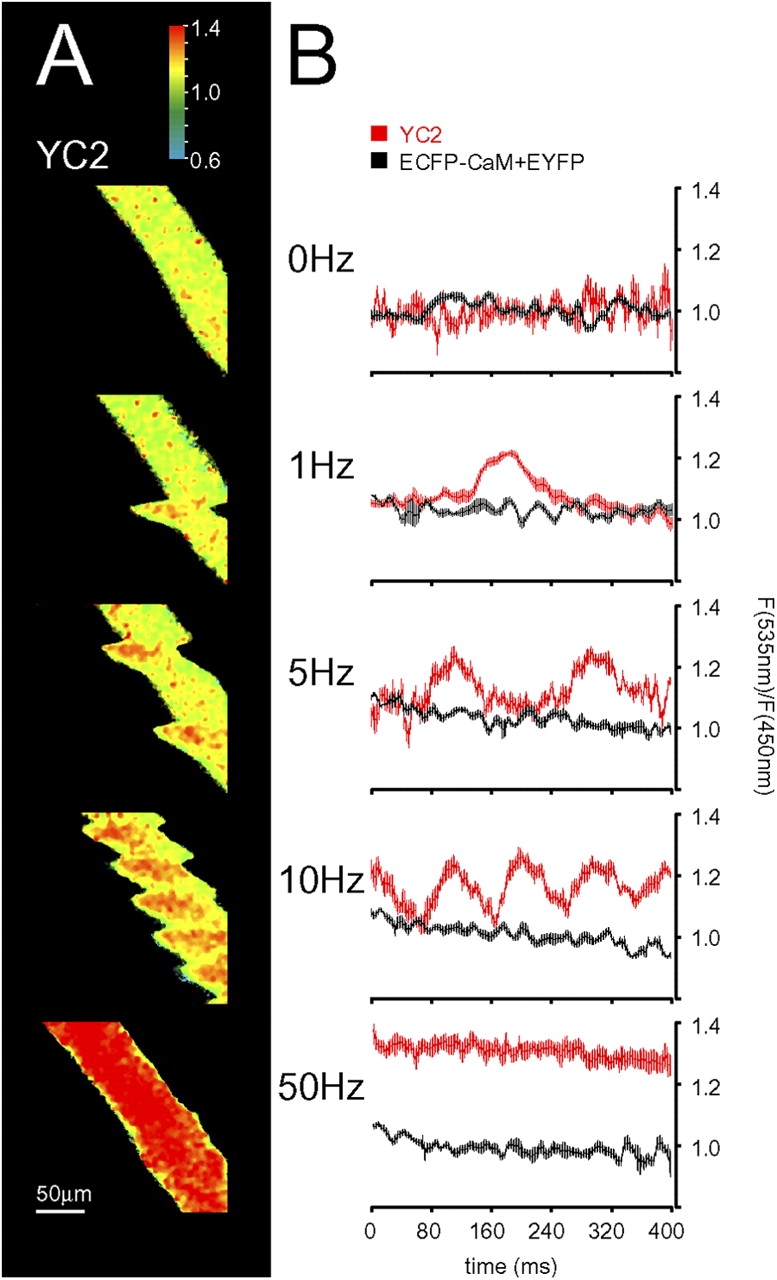
Kinetic analysis of single twitches with YC2. Tibialis anterior muscle expressing ECFP-CaM and EYFP (B) or YC2 alone (A and B) was observed in situ by two-photon microscopy as described. Muscle contraction was stimulated with different frequencies (1 to 50 Hz) as indicated. (A) Representative ratio images. (B) Graphs showing the distribution of ratio intensities along the fiber length. The ratio along parallel lines within the fiber was measured, and the average calculated; for each stimulation frequency, the same procedure was performed on 10 images. The average intensities along the fiber were “aligned” to the beginning of the first twitch observed in the respective image, and a pooled average was calculated and plotted. Error bars show ± SEM.
Calcium spikes in mitochondria in situ
The role played by mitochondria in the complex Ca2+ choreography of a living cell has become increasingly important in recent years. As such, we next addressed the question of whether changes in [Ca2+] do occur in this organelle during muscle contraction. For this we used YC with mitochondrial targeting signal (2mtYC2), modified to improve the efficiency of targeting (unpublished data). This probe showed a markedly striated localization (Fig. 5, A, F, I, and J), being in clear contrast to nontargeted YC2 (Fig. 5 E) and in accordance with the expected mitochondrial pattern (Ogawa et al., 1995). To minimize artifacts due to specimen thickness and movement, we analyzed fixed fibers using conventional confocal microscopy, obtaining very low levels of mislocalization in the cytoplasm (below 10% of that found in the mitochondria). Furthermore, the fluorescence signal from the mitochondrial marker TMRM (Fig. 5, C and H) showed a high degree of colocalization (Fig. 5, B and G) with that of 2mtYC2, providing further evidence for the efficient mitochondrial targeting of the Ca2+ probe; as expected, addition of the uncoupler FCCP (Farkas et al., 1989) abolished the striated signal of TMRM (Fig. 5, C and D). Having established the correct localization of the probe, tibialis anterior muscle expressing 2mtYC2 in vivo was analyzed by two-photon microscopy, as described in the Materials and methods section. Muscle contraction was triggered by pulses of 2.5 s (Fig. 5 K) at 50 Hz. As shown by the trace in Fig. 5 K, the ratio values increased during contraction (indicating a Ca2+ uptake by mitochondria) and subsequently returned to prestimulation levels, with a time course that was on average similar to that observed for the cytoplasm. After injection of FCCP (with or without oligomycin), such rises in [Ca2+]m were abolished (unpublished data). The dynamics of this phenomenon are illustrated in Fig. 5 K, where a series of ratio images depict the fiber at different time points: (a) at rest, (b) with basal Ca2+ levels; at the peak of ratio increase, and (c and d) during the decay phase. In the experiment presented in Fig. 5 L, the stimulation to the sciatic nerve was applied at 50 Hz for only 500 ms, but approximately every 2 s. It is evident that under these conditions, although a clear rise in [Ca2+]m is obtained at each stimulation, the concentration of Ca2+ within the mitochondrial matrix did not return to the basal level between two consecutive stimulations (Fig. 5 L, top trace). Such a behavior of [Ca2+]m was found for most of the tested fibers (5 out of 7), whereas in the cytoplasm [Ca2+] returned to baseline values much more rapidly (Fig. 5 L, bottom trace).
Figure 5.
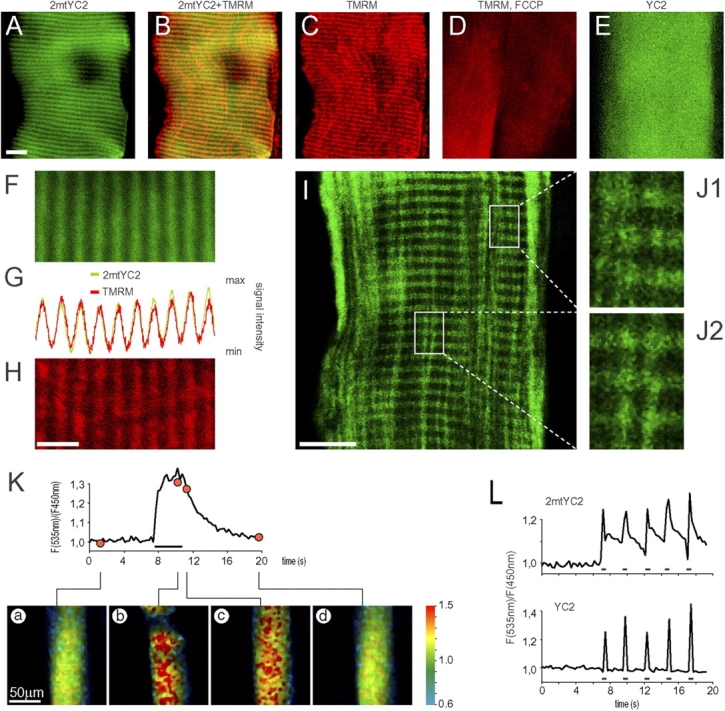
Co-localization of 2mtYC2 with TMRM and observation of mitochondrial [Ca 2+ ] spikes during muscle contraction in vivo. Tibialis anterior muscles expressing 2mtYC2 (A–D, F–K, and L [top trace]) or YC2 (E and L [bottom trace]) were observed in vivo with two-photon microscopy (A–H, K, and L) or after longitudinal slicing with confocal microscopy (I and J) as described in the Materials and methods section. (A–D) The transfected muscles were injected with TMRM. Fluorescence signals were visualized in vivo. (A) Fluorescence signal of 2mtYC2 pseudo-colored green). Bar, 10 μm. (B) Overlay of 2mtYC2 (green) and TMRM (red) signals; colocalization is shown in yellow. (C) Fluorescence signal of TMRM (red). (D) Fluorescence signal of TMRM (red) in two adjacent fibers after injection of the mitochondrial uncoupler FCCP: the striated localization of TMRM seen in C vanishes almost completely. (E) Fluorescence signal of YC2 (green). (F–H) Co-localization of 2mtYC2 (F; green) and TMRM fluorescence (H; red) shown by a signal intensity profile plot (G). Bar, 5 μm. (I) Confocal section of a longitudinal muscle slice showing 2mtYC2 fluorescence (green). Bar, 10 μm. (J1 and J2) Enlargements of the framed areas in I. (K and L) Trains of 2.5 s (horizontal bar below the trace in K) or 0.5 s (horizontal bars below the traces in L), with a stimulation frequency of 50 Hz, were applied to the muscle. The traces show the YFP/CFP ratio values of 2mtYC2 (K and L, upper trace) and YC2 (L, lower trace) as a function of time. Below K are shown representative ratio images: (a) at rest, (b) during contraction, (c) immediately after the suspension of the stimulus, and (d) back to basal conditions.
Uptake of Ca2+ into mitochondria during single twitches
The experiments presented above clearly demonstrate that during a tetanic stimulation mitochondria are able to accumulate Ca2+. The key question then arises as to whether the speed of mitochondrial Ca2+ uptake and release are compatible with the very rapid kinetics of [Ca2+]c rise and decay occurring during a single twitch. This is particularly important in a fast skeletal muscle where the peak rise in Ca2+ in the cytosol is obtained within 3 to 4 ms from the beginning of depolarization and the whole cycle lasts ∼100 ms (Baylor and Hollingworth, 2003). Muscles transfected with either YC2 or 2mtYC2 were observed by two-photon microscopy as described in the Materials and methods and single twitches were elicited by applying 5-Hz stimulation to the sciatic nerve through the extracellular electrode. As depicted in Fig. 6 B, mitochondria take up Ca2+ during each twitch in a manner that reflects the changes in the cytoplasm (Fig. 6 A), generally with a slight temporal delay. The traces show that the mitochondrial signal begins to rise within 10 ms of that observed in the cytoplasm and reaches its maximum with a delay of ∼10 to 20 ms with respect to the peak of cytosolic YC2 (see also the Discussion section). We next addressed the issue of the mechanism of the the fast decay of [Ca2+]m; to this end, we analyzed the kinetics of [Ca2+]m in the presence of CGP37157, an inhibitor of the mitochondrial Na+/Ca2+ exchanger. As shown in Fig. 6 C, under these conditions the decrease in [Ca2+]m was markedly slower. Our data show clearly that mitochondria take up Ca2+ during single twitches under highly physiological conditions, supporting the notion that these organelles are directly involved in mammalian skeletal muscle calcium handling. The fast decay of [Ca2+]m seems to be facilitated largely by the Na+/Ca2+ exchanger.
Figure 6.

Mitochondria take up Ca2 + during single twitches. Tibialis anterior muscles expressing (A) YC2 or (B and C) 2mtYC2 were observed with two-photon microscopy as described in the Materials and methods section. In C, CGP37157 was applied. Contractions were elicited by nervous stimulation at 5 Hz. Average YFP/CFP ratio values were determined in 10 ms windows, individual contractions (24, 64, and 24 for A–C, respectively) were synchronized, and the mean ratio values (normalized to the maximum) determined; the degree of contraction is indicated in gray. The maximal ratios reached for the analyzed twitches were on average 1.14 and 1.12 for YC2 and 2mtYC2, respectively. Error bars show ± SEM.
Discussion
Over the last decade, the capacity of mitochondria to accumulate Ca2+ under physiological conditions has been at the center of renewed interest (Duchen, 1999; Hajnoczky et al., 2000; Pozzan and Rizzuto, 2000; Rizzuto et al., 2000; Bootman et al., 2001). Plenty of evidence indicates that in many cell types, from leukocytes to neurons, these organelles are involved in Ca2+ homeostasis during cell activation. Much more debatable, on the other hand, is the involvement of mitochondria during the contraction–relaxation cycle of skeletal muscle. Although it is true that mitochondria in skeletal muscle myotubes can take up Ca2+ during single episodes of contraction (Brini et al., 1997; Challet et al., 2001; Robert et al., 2001b), the question still remains whether these are features of very immature or poorly differentiated muscle cells. Indeed, in these cultured cells, the kinetics of the Ca2+ changes are somewhat slower than those occurring in mature, differentiated muscle (Brini et al., 1997; Koopman et al., 2003). The objective of this study was thus to address the involvement of mitochondria in Ca2+ handling in skeletal muscle under conditions as close as possible to the physiological ones: in a live animal where the contraction of the muscle is induced by the acetylcholine released from the stimulated nerve terminals. Unraveling whether mitochondria in skeletal muscle can accumulate Ca2+ (and under which specific conditions) may be of key relevance for the understanding of a variety of patho-physiological processes, such as fatigue, apoptosis, or adaptation of oxidative metabolism to physiological demand. To this end, we developed a model system in which the anterior tibialis muscle of a mouse transiently expresses protein-based Ca2+ probes (targeted to the mitochondrial matrix or to the cytosol) and the fluorescence changes are monitored in the live, anesthetized animal by two-photon confocal microscopy. Our initial steps, sought to validate the approach, were thus to transfect the cells with a GFP-based probe particularly suited for two-photon microscopy, YC2. Initially, a cytosolic YC2 was used given that the behavior of [Ca2+]c is very well established. Having determined the robustness of our experimental model, we then proceeded with mitochondrial analyses by targeting the probe to this organelle. We show that under highly physiological conditions skeletal muscle mitochondria take up and release Ca2+ during the contraction–relaxation cycle.
For the transient expression of YC2 in mouse tibialis anterior muscle, we adopted an established electroporation-based protocol (Dona et al., 2003), with minor modifications. This technique permits the observation of YC2 fluorescence up to 3 wk after transfection (Fig. 1, A and B); i.e., long after cessation of any inflammatory responses and muscular repair mechanisms due to the operation. Indeed, the transfected muscles exhibited no obvious signs of damage or inflammation when prepared for microscopic visualization. This type of gene transfer provides an elegant and powerful way to study muscle physiology with genetically encoded probes, circumventing typical problems of other genetic manipulation techniques. First, it is fast, easy to perform, and shows a high transfection efficiency. Second, it is absolutely specific for the electroporated region and therefore is not expected to cause systemic interferences. Third, because it is applied only few days before microscopic examination, also ontogenetic disturbances are absent.
Using stimulation frequencies ranging from 1 to 50 Hz, we show that the mean [Ca2+]c in mammalian skeletal muscle in situ (as measured both by YC2 and split YC2) is in direct correlation to the intensity of stimulation (Fig. 3, A and B). Regarding the correlation shown in Fig. 3 B between stimulation frequency and YFP/CFP ratio, this is in agreement with work previously published for isolated mouse muscle fibers loaded with indo-1 (Helander et al., 2002). It is worth stressing that these results primarily reflect the number of twitches captured at the different stimulation frequencies (Fig. 3 A), although they do not provide any information about the ratio amplitude of each individual twitch. These measurements are of interest, nonetheless, because they provide an estimate of the mean cytosolic Ca2+ level in a period of several seconds, a potentially important parameter regarding the regulation of other Ca2+-dependent processes, such as the control of gene expression (Dolmetsch et al., 1998; Li et al., 1998). Evidence for the robustness of the method used in our studies is the fact that a Ca2+-insensitive construct similar to YC2 did not report any changes in ratio signal (Fig. 3 B). This clearly indicates the reliability of the YC2 read-out under conditions of strong movement of the observed object.
In isolated mouse extensor digitorum longus (EDL) muscle and soleus fibers, using furaptra, the onset of [Ca2+]c rise and the beginning of contraction are almost concomitant, whereas the maximum of contraction takes place typically several tens of milliseconds after the Ca2+ peak (Baylor and Hollingworth, 2003). The latter is reached at nearly physiological temperatures in 3 to 5 ms. Taking advantage of the scanning process of the confocal microscope, we could obtain good time resolution of the kinetics of both contraction and [Ca2+]c rise, as monitored by YC2. As demonstrated by Figs. 4 B and 6, the peak of the YC2 ratio change is reached with a significant delay with respect to that revealed by a fast Ca2+ dye such as furaptra (Baylor and Hollingworth, 2003); indeed, the peak of [Ca2+]c as revealed by YC2 and maximum contraction seem to coincide. We have not attempted to analyze in detail the reasons for this relatively slow response of YC2 (a clear limitation for the detailed kinetic analysis of cytoplasmic Ca2+ handling in muscle tissue), because this study does not delve into the kinetics of [Ca2+]c versus that of contraction; rather, our main concern was to establish that relatively fast [Ca2+] transients (within 10 to 50 ms) are capable of being detected by YC2, even in conditions of substantial specimen movement. Regarding the decay phase, at low frequency stimulation (Fig. 4 B, 1–10 Hz), the YFP/CFP ratio returned to baseline values within milliseconds, practically in synchrony with contraction. In contrast, at 50 Hz, the recovery to basal [Ca2+]c lasted some seconds (Fig. 2). As judged from the concise response of YC2 upon single twitches (Fig. 4 A, see 1 Hz and 5 Hz ratio images), this long-lasting recovery seems to reflect a true process, though at present some artifact due to an intrisic delay of the probe cannot be completely excluded (for example see Kerr et al., 2000). Regarding the peak maxima shown in Fig. 4 B, ratio values reach 1.27 ± 0.02 (mean ± SEM, n = 10) at the point of maximum contraction when using stimulation frequencies of 1–10 Hz. At 50 Hz, a slightly higher peak value of 1.32 ± 0.02 (mean ± SEM, n = 10) was observed, correlating with what is known about muscle physiology.
As to mitochondrial Ca2+ accumulation, to our knowledge, there is no data until now concerning what happens in the organelles of mature muscle fibers during contraction itself; furthermore, data obtained seconds after suspension of stimulus are contradictory (Lannergren et al., 2001; Bruton et al., 2003c). It should also be noted that the in vitro studies on mature fibers presented so far are based on rhod-2 fluorescence measurements, well-known to be prone to movement artifacts (Lannergren et al., 2001; Bruton et al., 2003b,c). At present, the only data available concerning the behavior of [Ca2+]m during skeletal muscle contraction are those obtained in myocytes (Trollinger et al., 1997, 2000) and myotubes (Brini et al., 1997; Challet et al., 2001; Robert et al., 2001b), but their relevance with respect to the behavior of the mature fibers is debatable. Using mitochondrially targeted YC2, we show here for the first time that mitochondria in mouse skeletal muscle observed in situ and under highly physiological conditions do indeed take up Ca2+ both during tetanic contraction (Fig. 5, K and L) and single twitches (Fig. 6 B). In terms of amplitude, assuming that the affinity of mitochondrially located YC2 is the same as that observed in the cytosol, the peaks in [Ca2+] are somewhat smaller in mitochondria (mean 1.14 ± 0.02 SEM, n = 21 videos, 150 contractions, six experiments) compared with the cytosol (mean 1.26 ± 0.03 SEM, n = 18 videos, 97 contractions, three experiments). During tetanic stimulation no obvious major differences were noticed in the kinetics of rise and decay of the mitochondrial Ca2+ signal compared with those in the cytosol, although when the phenomenon was analyzed at higher temporal resolution (as in the single twitch experiments) mitochondria generally showed some delay, in the order of 10 ms, both in the beginning of the Ca2+ rise and in reaching the peak when compared with the cytosol (Fig. 6). Similarly, the decay phase is slightly slower in mitochondria. We would like to stress that whereas the fast Ca2+ uptake rate could have been predicted (particularly now that the kinetic properties of the mitochondrial uptake system have been unraveled by direct electrophysiological measurements by Kirichok et al., 2004), the speed of the decay phase, completed in a period of ∼100 ms, is much faster than it could have been anticipated from in vitro measurements. To get some insight into how this fast expulsion of Ca2+ may occur, we tested its kinetics in the presence of an inhibitor of the Na+/Ca2+ exchanger. Our data show that under these conditions the decay in [Ca2+]m is slower than in the control (Fig. 6 , B and C), suggesting an important role of the Na+/Ca2+ exchanger for this process.
A final important observation is obtained by the experimental protocol of Fig. 5 L. Here, we used a stimulation protocol that is similar to that physiologically delivered by fast motor neurons to their target fibers, i.e., bursts of high frequency (50 Hz) and short temporal extension (500 ms; Hennig and Lomo, 1985). From the data presented in Fig. 5 L (upper panel), it seems that under these conditions [Ca2+]m remains elevated within the mitochondrial matrix far longer than the stimulus itself and if another train of stimuli is delivered at short distances, i.e., within 2 to 3 s, [Ca2+]m remains continuously elevated above basal level. In contrast, in the cytoplasm under the same experimental regime the basal value is achieved almost instantaneously after cessation of the stimulus (Fig. 5 L, lower panel). Although the degree of elevation varied between the different fibers, and we cannot exclude an inherent slow of rate of the YC2 probe, our data suggest that under conditions similar to a strong physiological stimulation of the muscle, the full power of the Ca2+-dependent mitochondrial dehydrogenases should be available, thus permitting the most efficient use of oxidative substrates and a maximal speed of ATP synthesis.
In summary, the data obtained with mammalian skeletal muscle indicate that (a) in vivo transfection using electroporation is a powerful method for expressing second messenger–sensitive probes in the muscle of a living mammal, (b) cytoplasmic YC2, in spite of some kinetic limitations, is a useful reporter of changes in [Ca2+]c during contraction and relaxation, and (c) the mitochondrially targeted YC2 variant reveals a rise in [Ca2+]m during contraction, not only during tetanic stimulation, but also in single twitches.
Materials and methods
Expression plasmids and chemicals
Transfection experiments used the following constructs, all expressed in pcDNA3 (Invitrogen) or pMITO (Clontech Laboratories, Inc.): YC2, CFP-CaM, M13-YFP, YFP, mtCFP, and mtM13-YFP in the former, and 2mtYC2 in the latter. The first three are gifts from R.Y. Tsien (University of California, San Diego, CA) (Miyawaki et al., 1997); mitochondrial targeting used the presequence of COX VIII (Rizzuto et al., 1992; Filippin et al., 2003); 2mtYC2 possesses a tandem repeat of the mitochondrial-targeting sequence of COX VIII for enhanced specificity (Galanis et al., 1991). The mitochondrial marker TMRM (tetramethylrhodamine methyl ester perchlorate) was acquired from Molecular Probes. The mitochondrial uncoupler FCCP (carbonyl cyanide 4-[trifluoromethoxy] phenylhydrazone) was from Sigma-Aldrich and the inhibitor of the mitochondrial Na+/Ca2+ exchanger CGP37157 was from Tocris. All drugs and cDNAs injected into muscle were diluted in sterile physiological solution (0.9% NaCl). All chemicals used were of the highest available grade and, unless otherwise indicated, obtained from Sigma-Aldrich.
Animals
For most of the shown experiments, C57BL/10 mice with an age of 6–12 mo were used; for calibration experiments, SV159 mice were also used; all animals were obtained from Charles River Italia. Use and care of animals was approved by the local authority for veterinary services and followed the guidelines of the Italian Health Ministry (Law 116/92 and Art. nr. 8/94). An i.p. injection containing a combination of Rompun® (Bayer) and Zoletil 100® (Laboratoires Virbac) was used for anesthesia.
Transfection method
For the heterologous expression of YCs, an electroporation-based transfection method of the tibialis anterior muscle was used very similar to the one described recently (Dona et al., 2003). In brief, the shaved skin of the left lower hind limb of the anesthetized animal was opened at the border between anterior and posterior hind limb muscles with a longitudinal cut of ∼1 cm length. The fascia was cut and an electrode was inserted between muscle and bone. With a modified 100-μl microsyringe (Exmire Microsyringe; ITO Corp.), 30 μg of expression plasmid were injected into the exposed tibialis anterior muscle before placing the second electrode on the muscle. With an Electro Square Porator (model ECM 830; BTX Genetronics), five pulses of 20 ms duration were applied with a voltage of 25 V. The electrodes were removed, and the wound was closed and sterilized. The antibiotic Terramicina® (Pfizer Italia) was injected as antiinflammatory prevention.
In vivo two-photon microscopy
Preparation of muscle and nerve.
2–3 wk after transfection the operated animal was anesthetized and the area around the original incision shaved. The sciatic nerve was isolated and a custom-made silver-microelectrode was inserted for subsequent stimulation of muscle contraction. The distal tendon of the transfected tibialis anterior muscle was severed and the fascia removed. The mouse was then transferred onto a custom-made plastic table, the muscle was fixed with a surgical clamp attached to the severed tendons, and the complete assembly mounted for observation.
Microscopy.
A multi-photon system (Radiance 2100 MP; Bio-Rad Laboratories), equipped with a Tsunami mode-locked, tunable, femto-second–pulsed Ti/sapphire laser, optically pumped by a Millennia VsS 5W green laser (both from Spectraphysics Lasers), was used for two-photon microscopy. The laser output is capable of generating 100-femto-second pulse trains at a rate of 82 MHz. The excitation wavelength was 830 nm, controlled with a spectrometer (Ocean Optics USB2000; getSpec). The microscope (Eclipse E600FN; Nikon) was equipped with a Leica objective (PL APO 63×/1.2W CORR; Leica); an optical gel (0.2% polyacryl; Viscotirs Gel®; Ciba Vision) was used as immersion medium. A Direct Detection System (Bio-Rad Laboratories), fitted with a 500LP DC dichroic mirror and HQ450/80 and HQ535/50 emission filters (Chroma Technology Corp.), was used for the simultaneous detection of CFP and YFP emission signals, respectively. Typically, gain and offset of the photomultipliers were kept constant while laser intensity was adjusted according to the depth of the observed fiber; only fibers from the first to the second outer layer (i.e., up to 100 μm in depth) were imaged. The LaserSharp2000 software package (Bio-Rad) was used for data acquisition. Unless otherwise indicated, video microscopy was performed at 128 × 128 pixels per image, with a 3.95-Hz acquisition frequency. For colocalization studies, images were acquired at 1,024 × 1,024 pixels, using a 560DCLPXR dichroic mirror; emission filters E560SP and HQ600/50 were used for the detection of YC2 and TMRM, respectively. Calcium levels were monitored in the cytoplasm with YC2 or split YC2, and in mitochondria with 2mtYC2. Co-expressed CFP-CaM and YFP in the cytoplasm, or mtCFP and mtM13-YFP in mitochondria, served as controls, and did not show an increase in YFP/CFP fluorescence intensity ratio during muscle contraction; in the controls for mitochondria, a decrease in ratio was sometimes observed during contraction. The latter effect is most likely due to the following: in kinetic experiments, the high image acquisition rate imposes a limited spatial resolution; individual mitochondria are thus not resolved and background correction becomes defective; because the background level is higher in CFP images, there is an artificial enhancement of the CFP signal during contraction.
Induction of muscle contraction.
For the stimulation of muscle contraction, a custom-made electrode was attached to the sciatic nerve and connected to an adjustable stimulator (Scientific Research Instruments). The typical voltage applied was in the 0.1–0.2-V range, with variable stimulation frequencies, as indicated in the text; each pulse had a duration of 5 ms. The stimulation intensity was adapted to the lateral movement of the fiber during contraction in the microscopic field.
Injection of probes.
500 nM TMRM, 10 μM FCCP, and 200 μM CGP37157 were injected locally into the observed muscle with a 30-gauge needle (Artsana) with a typical volume of 50–60 μl. To allow the muscle to recover from the injection-induced swelling, microscopic observation was interrupted for 2–5 min.
Cryopreservation and sectioning of muscle
After imaging, the complete anterior tibialis muscle was removed, washed in physiological solution, incubated for 10 min in relaxation buffer containing (in mM): 100 KCl, 5 EGTA, 5 MgCl2, 3 2,3-butanedione monoxime, 0.25 dithiothreitol, 10 histidine, pH 7.8, and then fixed for 2 h at 4°C in PFA solution (4% wt/vol paraformaldehyde, 4% wt/vol sucrose in PBS, pH 7.4). After washing in distilled water, the fixed muscles were then snap-frozen in isopentane cooled in liquid nitrogen (Wu et al., 1985), and stored at −80°C. For sectioning, muscles were embedded according to standard procedures in Jung Tissue Freezing Medium (Leica) at −20°C. A microtome (model CM1850; Leica) was used to prepare 10- and 30-μm-thick sections (for transversal and longitudinal sections, respectively), which were placed on electrostatically charged microscope slides (Superfrost Plus; BDH Laboratory Supplies). Transmission and fluorescence images were acquired using either a microscope (model BX60 equipped with a 50W HBO mercury arc lamp, a PLAN2×/0.05 objective, and a DP50 camera; Olympus), using Photoshop 6.0 (Adobe Systems), or a Bio-Rad Laboratories confocal microscope (Radiance 2100 MP equipped with an argon laser for 488 nm excitation of cameleon fluorescence, a Nikon 60×/1.4 Plan Apo objective, a 500 DCLPXR beamsplitter, and a HQ515/30 emission filter; all filters were from Chroma Technology Corp.); confocal images were taken at 1,024 × 1,024 pixel resolution with LaserSharp 2000 software (BioRad).
Data analysis and ratiometric videos
Analysis of the two-photon in vivo imaging data was performed using the public domain ImageJ program (developed at the United States National Institutes of Health by Wayne Rasband and available freely on the Internet at http://rsb.info.nih.gov/ij/). For the calculation of YFP/CFP fluorescence intensity ratios, the following procedure was used: the fluorescent fibers were outlined as regions of interest (ROI) by thresholding the YFP image sets; the mean greyscale values of the ROIs were calculated in both YFP and CFP image sets; the obtained values were transferred to Excel 2002 (Microsoft), where all subsequent calculations and graphs were performed. Ratio plots were normalized using as reference the average value of the images before the first stimulation (because laser illumination causes an initial photo-isomerization of YFP; Miyawaki et al., 1997; Filippin et al., 2003), the average used for normalization considered only images after a plateau of the YFP/CFP ratio was reached). The kinetics of the YFP/CFP ratio during twitches in the cytosol (as shown in Fig. 4 B) was determined by measuring ratio values along six parallel lines within the fiber, thus generating average ratio intensities along the fiber; the average ratio intensities along the fiber were “aligned” to the beginning of the first twitch observed in the respective image. For each stimulation frequency, the same procedure was performed on different images (n = 10). The kinetics of the YFP/CFP ratio during twitches in the cytosol and mitochondria (as shown in Fig. 6) were determined by the fluorescence intensities of rectangular ROIs on the respective ratiometric videos: these ROIs had a width of five lines (corresponding to 10 ms in real time), the height of the fiber diameter, and scanned the entire fiber length; alignment was performed as described above. Ratiometric videos were generated in ImageJ using the following scheme: the original 8-bit videos were median filtered (1-pixel kernel), and a pixel-by-pixel ratio image generated using a custom-made plugin (RatioPlus; available freely at the ImageJ web site). The resulting 32-bit greyscale images were pseudo-colored using a rainbow-like look-up table where low and high values are represented by dark blue and intense red, respectively. For visualization only, the pseudo-color ratio images were intensity calibrated: the brightness of each color (representing a specific ratio value) was adjusted based on the fluorescence intensity of the original YFP and CFP images. Unless otherwise indicated, ratio values refer to YFP fluorescence intensity divided by CFP fluorescence intensity.
Acknowledgments
We are grateful to R.Y. Tsien for the original cameleon constructs and to L. Filippin for help in molecular cloning. We acknowledge the technical and scientific help of E. Bigon, R. Betto, T. Cesetti, D. Danieli, M. Sandri, S. Schiaffino, and M. Zaccolo.
The work of R. Rudolf has been funded by a Marie-Curie fellowship (HPMF CT200201543.01), a grant from the University of Padua, and from the Deutsche Forschungsgemeinschaft (RU 923/1-1). This work was also partially supported by Italian Telethon (grant 1226), from the Italian Association for Cancer Research, from the Italian Ministry of University (Special Projects Prin RBNE01ERXR-001 and 20033054449-001 and FIRB 2002053318-005), from the Italian Ministry of Health, and from the CARIPARO foundation to T. Pozzan.
Abbreviations used in this paper: 2mtYC2, YC with mitochondrial targeting signal; [Ca2+], concentration of Ca2+; [Ca2+]c, cytoplasmic concentration of Ca2+; [Ca2+]m, mitochondrial Ca2+; FRET, fluorescence resonance energy transfer; ROI, region of interest; YC, yellow cameleon.
References
- Arnaudeau, S., W.L. Kelley, J.V. Walsh Jr., and N. Demaurex. 2001. Mitochondria recycle Ca(2+) to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J. Biol. Chem. 276:29430–29439. [DOI] [PubMed] [Google Scholar]
- Baylor, S.M., and S. Hollingworth. 2003. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. J. Physiol. 551:125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi, P. 1999. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 79:1127–1155. [DOI] [PubMed] [Google Scholar]
- Bootman, M.D., P. Lipp, and M.J. Berridge. 2001. The organisation and functions of local Ca(2+) signals. J. Cell Sci. 114:2213–2222. [DOI] [PubMed] [Google Scholar]
- Brini, M., F. De Giorgi, M. Murgia, R. Marsault, M.L. Massimino, M. Cantini, R. Rizzuto, and T. Pozzan. 1997. Subcellular analysis of Ca2+ homeostasis in primary cultures of skeletal muscle myotubes. Mol. Biol. Cell. 8:129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton, J.D., A.J. Dahlstedt, F. Abbate, and H. Westerblad. 2003. a. Mitochondrial function in intact skeletal muscle fibres of creatine kinase deficient mice. J. Physiol. 552:393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton, J.D., R. Lemmens, C.L. Shi, S. Persson-Sjogren, H. Westerblad, M. Ahmed, N.J. Pyne, M. Frame, B.L. Furman, and M.S. Islam. 2003. b. Ryanodine receptors of pancreatic beta-cells mediate a distinct context-dependent signal for insulin secretion. FASEB J. 17:301–303. [DOI] [PubMed] [Google Scholar]
- Bruton, J., P. Tavi, J. Aydin, H. Westerblad, and J. Lannergren. 2003. c. Mitochondrial and myoplasmic Ca2+ in single fibres from mouse limb muscles during repeated tetanic contractions. J. Physiol. 551: 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli, E. 2003. Historical review: mitochondria and calcium: ups and downs of an unusual relationship. Trends Biochem. Sci. 28:175–181. [DOI] [PubMed] [Google Scholar]
- Challet, C., P. Maechler, C.B. Wollheim, and U.T. Ruegg. 2001. Mitochondrial calcium oscillations in C2C12 myotubes. J. Biol. Chem. 276:3791–3797. [DOI] [PubMed] [Google Scholar]
- Diegelmann, S., A. Fiala, C. Leibold, T. Spall, and E. Buchner. 2002. Transgenic flies expressing the fluorescence calcium sensor Cameleon 2.1 under UAS control. Genesis. 34:95–98. [DOI] [PubMed] [Google Scholar]
- Dolmetsch, R.E., K. Xu, and R.S. Lewis. 1998. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 392:933–936. [DOI] [PubMed] [Google Scholar]
- Dona, M., M. Sandri, K. Rossini, I. Dell'Aica, M. Podhorska-Okolow, and U. Carraro. 2003. Functional in vivo gene transfer into the myofibers of adult skeletal muscle. Biochem. Biophys. Res. Commun. 312:1132–1138. [DOI] [PubMed] [Google Scholar]
- Duchen, M.R. 1999. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. J. Physiol. 516:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebashi, S., and F. Lipman. 1962. Adenosine triphosphate-linked concentration of calcium ions in a particulate fraction of rabbit muscle. J. Cell Biol. 14:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas, D.L., M.D. Wei, P. Febbroriello, J.H. Carson, and L.M. Loew. 1989. Simultaneous imaging of cell and mitochondrial membrane potentials. Biophys. J. 56:1053–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala, A., T. Spall, S. Diegelmann, B. Eisermann, S. Sachse, J.M. Devaud, E. Buchner, and C.G. Galizia. 2002. Genetically expressed cameleon in Drosophila melanogaster is used to visualize olfactory information in projection neurons. Curr. Biol. 12:1877–1884. [DOI] [PubMed] [Google Scholar]
- Filippin, L., P.J. Magalhaes, G. Di Benedetto, M. Colella, and T. Pozzan. 2003. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J. Biol. Chem. 278:39224–39234. [DOI] [PubMed] [Google Scholar]
- Galanis, M., R.J. Devenish, and P. Nagley. 1991. Duplication of leader sequence for protein targeting to mitochondria leads to increased import efficiency. FEBS Lett. 282:425–430. [DOI] [PubMed] [Google Scholar]
- Gunter, T.E., and D.R. Pfeiffer. 1990. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 258:C755–C786. [DOI] [PubMed] [Google Scholar]
- Hajnoczky, G., G. Csordas, M. Madesh, and P. Pacher. 2000. The machinery of local Ca2+ signalling between sarco-endoplasmic reticulum and mitochondria. J. Physiol. 529:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselbach, W., and M. Makinose. 1961. Die Calcium Pumpe der “Erschlaffungsgrana” des Muskels und ihre Abhangigkeit von der ATP-Spaltung. Biochem. Z. 333:518–528. [PubMed] [Google Scholar]
- Helander, I., H. Westerblad, and A. Katz. 2002. Effects of glucose on contractile function, [Ca2+]i, and glycogen in isolated mouse skeletal muscle. Am. J. Physiol. Cell Physiol. 282:C1306–C1312. [DOI] [PubMed] [Google Scholar]
- Hennig, R., and T. Lomo. 1985. Firing patterns of motor units in normal rats. Nature. 314:164–166. [DOI] [PubMed] [Google Scholar]
- Higashijima, S., M.A. Masino, G. Mandel, and J.R. Fetcho. 2003. Imaging neuronal activity during zebrafish behavior with a genetically encoded calcium indicator. J. Neurophysiol. 90:3986–3997. [DOI] [PubMed] [Google Scholar]
- Ikeda, M., T. Sugiyama, C.S. Wallace, H.S. Gompf, T. Yoshioka, A. Miyawaki, and C.N. Allen. 2003. Circadian dynamics of cytosolic and nuclear Ca2+ in single suprachiasmatic nucleus neurons. Neuron. 38:253–263. [DOI] [PubMed] [Google Scholar]
- Jouaville, L.S., P. Pinton, C. Bastianutto, G.A. Rutter, and R. Rizzuto. 1999. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA. 96:13807–13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr, R., V. Lev-Ram, G. Baird, P. Vincent, R.Y. Tsien, and W.R. Schafer. 2000. Optical imaging of calcium transients in neurons and pharyngeal muscle of C. elegans. Neuron. 26:583–594. [DOI] [PubMed] [Google Scholar]
- Kirichok, Y., G. Krapivinsky, and D.E. Clapham. 2004. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 427:360–364. [DOI] [PubMed] [Google Scholar]
- Koopman, W.J., M. Renders, A. Oosterhof, T.H. van Kuppevelt, B.G. van Engelen, and P.H. Willems. 2003. Upregulation of Ca2+ removal in human skeletal muscle: a possible role for Ca2+-dependent priming of mitochondrial ATP synthesis. Am. J. Physiol. Cell Physiol. 285:C1263–C1269. [DOI] [PubMed] [Google Scholar]
- Lannergren, J., H. Westerblad, and J.D. Bruton. 2001. Changes in mitochondrial Ca2+ detected with Rhod-2 in single frog and mouse skeletal muscle fibres during and after repeated tetanic contractions. J. Muscle Res. Cell Motil. 22:265–275. [DOI] [PubMed] [Google Scholar]
- Li, W., J. Llopis, M. Whitney, G. Zlokarnik, and R.Y. Tsien. 1998. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature. 392:936–941. [DOI] [PubMed] [Google Scholar]
- McCormack, J.G., and R.M. Denton. 1990. The role of mitochondrial Ca2+ transport and matrix Ca2+ in signal transduction in mammalian tissues. Biochim. Biophys. Acta. 1018:287–291. [DOI] [PubMed] [Google Scholar]
- Miyawaki, A., J. Llopis, R. Heim, J.M. McCaffery, J.A. Adams, M. Ikura, and R.Y. Tsien. 1997. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 388:882–887. [DOI] [PubMed] [Google Scholar]
- Ogawa, H., S. Inouye, F.I. Tsuji, K. Yasuda, and K. Umesono. 1995. Localization, trafficking, and temperature-dependence of the Aequorea green fluorescent protein in cultured vertebrate cells. Proc. Natl. Acad. Sci. USA. 92:11899–11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher, P., A.P. Thomas, and G. Hajnoczky. 2002. Ca2+ marks: miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc. Natl. Acad. Sci. USA. 99:2380–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pette, D., and R.S. Staron. 2001. Transitions of muscle fiber phenotypic profiles. Histochem. Cell Biol. 115:359–372. [DOI] [PubMed] [Google Scholar]
- Pozzan, T., P. Magalhaes, and R. Rizzuto. 2000. The comeback of mitochondria to calcium signalling. Cell Calcium. 28:279–283. [DOI] [PubMed] [Google Scholar]
- Pozzan, T., and R. Rizzuto. 2000. High tide of calcium in mitochondria. Nat. Cell Biol. 2:E25–E27. [DOI] [PubMed] [Google Scholar]
- Reiff, D.F., P.R. Thiel, and C.M. Schuster. 2002. Differential regulation of active zone density during long-term strengthening of Drosophila neuromuscular junctions. J. Neurosci. 22:9399–9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto, R., P. Bernardi, and T. Pozzan. 2000. Mitochondria as all-round players of the calcium game. J. Physiol. 529:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto, R., A.W. Simpson, M. Brini, and T. Pozzan. 1992. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 358:325–327. (published erratum appears in Nature. 1992. 360:768) [DOI] [PubMed] [Google Scholar]
- Robb-Gaspers, L.D., P. Burnett, G.A. Rutter, R.M. Denton, R. Rizzuto, and A.P. Thomas. 1998. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 17:4987–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert, V., P. Gurlini, V. Tosello, T. Nagai, A. Miyawaki, F. Di Lisa, and T. Pozzan. 2001. a. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 20:4998–5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert, V., M.L. Massimino, V. Tosello, R. Marsault, M. Cantini, V. Sorrentino, and T. Pozzan. 2001. b. Alteration in calcium handling at the subcellular level in mdx myotubes. J. Biol. Chem. 276:4647–4651. [DOI] [PubMed] [Google Scholar]
- Rudolf, R., M. Mongillo, R. Rizzuto, and T. Pozzan. 2003. Looking forward to seeing calcium. Nat. Rev. Mol. Cell Biol. 4:579–586. [DOI] [PubMed] [Google Scholar]
- Sartorius, C.A., B.D. Lu, L. Acakpo-Satchivi, R.P. Jacobsen, W.C. Byrnes, and L.A. Leinwand. 1998. Myosin heavy chains IIa and IId are functionally distinct in the mouse. J. Cell Biol. 141:943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyn, S.I., R. Kerr, and W.R. Schafer. 2003. Serotonin and Go modulate functional states of neurons and muscles controlling C. elegans egg-laying behavior. Curr. Biol. 13:1910–1915. [DOI] [PubMed] [Google Scholar]
- Silver, R.B. 1998. Ratio imaging: practical considerations for measuring intracellular calcium and pH in living tissue. In Video Microscopy. Vol. 56. G. Sluder and D.E. Wolf, editors. Academic Press, San Diego, CA. 237–251. [DOI] [PubMed]
- Suzuki, H., R. Kerr, L. Bianchi, C. Frokjaer-Jensen, D. Slone, J. Xue, B. Gerstbrein, M. Driscoll, and W.R. Schafer. 2003. In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron. 39:1005–1017. [DOI] [PubMed] [Google Scholar]
- Trollinger, D.R., W.E. Cascio, and J.J. Lemasters. 1997. Selective loading of Rhod 2 into mitochondria shows mitochondrial Ca2+ transients during the contractile cycle in adult rabbit cardiac myocytes. Biochem. Biophys. Res. Commun. 236:738–742. [DOI] [PubMed] [Google Scholar]
- Trollinger, D.R., W.E. Cascio, and J.J. Lemasters. 2000. Mitochondrial calcium transients in adult rabbit cardiac myocytes: inhibition by ruthenium red and artifacts caused by lysosomal loading of Ca(2+)-indicating fluorophores. Biophys. J. 79:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J.S., G.R. Hogan, and J.D. Morris. 1985. Modified methods for preparation of cryostat sections of skeletal muscle. Muscle Nerve. 8:664–666. [DOI] [PubMed] [Google Scholar]