

Abstract
The axonal initial segment is a unique subdomain of the neuron that maintains cellular polarization and contributes to electrogenesis. To obtain new insights into the mechanisms that determine protein segregation in this subdomain, we analyzed the trafficking of a reporter protein containing the cytoplasmic II–III linker sequence involved in sodium channel targeting and clustering (Garrido, J.J., P. Giraud, E. Carlier, F. Fernandes, A. Moussif, M.P. Fache, D. Debanne, and B. Dargent. 2003. Science. 300:2091–2094). Here, we show that this reporter protein is preferentially inserted in the somatodendritic domain and is trapped at the axonal initial segment by tethering to the cytoskeleton, before its insertion in the axonal tips. The nontethered population in dendrites, soma, and the distal part of axons is subsequently eliminated by endocytosis. We provide evidence for the involvement of two independent determinants in the II–III linker of sodium channels. These findings indicate that endocytotic elimination and domain-selective tethering constitute a potential mechanism of protein segregation at the axonal initial segment of hippocampal neurons.
Keywords: axonal initial segment; ankyrin G tethering; sorting; neuronal polarity; sodium channels
Introduction
The asymmetrical architecture of the neuron defines two domains: the somatodendritic domain and the axonal domain. The boundary between the soma and the axon comprises a unique and highly specialized subdomain of the neuron, the axonal initial segment (AIS). The molecular architecture of the AIS shares some homology with the organization of the nodes of Ranvier (Salzer, 2003). It is characterized by segregation of the cytoskeletal adaptor complex ankyrin G/β IV spectrin, linked to a dense network of actin, and by a high density of the voltage-gated sodium channels Nav1.2 and Nav1.6 and of members of the L1 cell adhesion molecule (CAM) family (Peles and Salzer, 2000; Boiko et al., 2003; Salzer, 2003). The AIS constitutes a barrier that restricts the lateral mobility of membrane proteins through differential tethering to cytoskeletal components (Kobayashi et al., 1992; Winckler et al., 1999) and blocks the diffusion of phospholipids (Nakada et al., 2003). The formation of this barrier is linked to the presence of the actin network (Winckler et al., 1999) and correlates with the accumulation of membrane proteins like sodium channels (Nakada et al., 2003). The high density of sodium channels ensures generation of the action potential, a fundamental signal in neuronal communication (Catterall, 2000). Thus, the AIS maintains neuronal polarization and contributes to electrogenesis.
Little is known about the biogenesis of the AIS, but information has recently emerged indicating the pivotal role of the cytoskeletal adaptor complex ankyrin G/β IV spectrin (Bennett and Baines, 2001; Salzer, 2003). The expression of either ankyrin G (Zhou et al., 1998; Jenkins and Bennett, 2001) or β IV spectrin (Komada and Soriano, 2002) is mandatory for the assembly of molecules of the L1 CAM family and sodium channels. At the molecular level, sodium channels from rat brain are composed of an α subunit, the pore-forming protein Nav1, and the auxiliary subunits, β2 or β4 and β1 or β3 (Catterall, 2000; Isom, 2001; Salzer, 2003; Yu et al., 2003). This complex molecular organization has hampered dissection of targeting and/or clustering motifs of sodium channels. Therefore, we developed an approach based on CD4 chimera expression in cultured hippocampal neurons to assess whether any of the large intracellular regions of Nav1.2 contained sufficient information for sorting and specific membrane organization. Using this approach, we have recently shown that sodium channel targeting and clustering at the AIS is specified by a motif within the cytoplasmic loop linking homologous domains II–III (the II–III linker) of the Nav1 proteins (Garrido et al., 2003). This signal was sufficient to relocalize the somatodendritic potassium channel Kv2.1 at the AIS of hippocampal neurons (Garrido et al., 2003). When the ankyrin binding motif of neurofascin (Davis and Bennett, 1994), a member of L1 CAMs, was replaced by the II–III linker of sodium channel Nav1.2, the resulting neurofascin–sodium channel chimera was concentrated at the AIS of hippocampal neurons (Lemaillet et al., 2003). This process involves a direct association with the ankyrin repeat domain of ankyrin G (Lemaillet et al., 2003).
The upstream events leading to membrane protein segregation within the AIS have yet to be dissected (Winckler and Mellman, 1999). Taking into account very recent studies on axonal sorting, several models are conceivable, that may not be mutually exclusive. One scenario is that an AIS protein is nonselectively inserted in the plasma membrane of both somatodendritic and axonal domains and subsequently eliminated in the dendrites, the soma, and in the distal part of the axon by endocytosis where it is tethered by ankyrin G at the AIS. Such a multistep process would be coordinated by independent molecular determinants, i.e., internalization and tethering motifs. It is significant that somatodendritic endocytosis has been shown to be involved in the axonal compartmentalization of a CD4 chimera bearing the COOH terminus of Nav1.2 (Garrido et al., 2001) and more recently, of the synaptic protein VAMP2 (Sampo et al., 2003). In each case, the abrogation of the internalization signal impaired axonal polarization (Garrido et al., 2001; Sampo et al., 2003). A second possibility is that an AIS protein is selectively sorted and inserted in the axonal domain, as observed in the case of NgCAM, the avian homologue of L1 (Sampo et al., 2003). After lateral diffusion, a fraction is tethered at the AIS whereas the distal population is eliminated by endocytosis. Alternatively, membrane proteins preassembled with ankyrin G can be selectively sorted to the AIS, as a consequence of polarized transport along microtubules involving KIF5, a member of the kinesin super family (Nakata and Hirokawa, 2003). Finally, the possibility that segregation of a given protein at the AIS involves both direct routing and transcytosis cannot be excluded. For example, Sampo et al. (2003) have shown that NgCAM is selectively routed to axons via a targeting motif localized in its extracellular domain. Wisco et al. (2003) have shown that NgCAM is preferentially inserted in the somatodendritic domain and subsequently sorted to the axons by transcytosis, a process mediated by an internalization motif located in the cytoplasmic COOH terminus of NgCAM.
The present study was aimed at analyzing the trafficking of a CD4 chimera bearing the II–III linker of Nav1.2 (CD4-Nav1.2 II–III) in hippocampal neurons, to obtain new insights into the mechanisms that determine protein segregation in the AIS. At the steady-state, the surface distribution of CD4-Nav1.2 II–III is restricted to the AIS of transfected hippocampal neurons (Garrido et al., 2003). We show here that CD4-Nav1.2 II–III is preferentially inserted in the somatodendritic domain but is subsequently eliminated by endocytosis, whereas it is accumulated at the AIS by a diffusion trap due to the high concentration of ankyrin G. Ankyrin G tethering involves a conserved glutamate residue within the sodium channel clustering motif whereas endocytosis is governed by a segment located in the NH2 terminus of linker II–III of Nav1.2.
Results
Involvement of a glutamate residue in sodium channel clustering at the AIS
The addition of the II–III linker of Nav1.2 to the human CD4 receptor deleted of its cytoplasmic tail, resulted in a surface distribution of the chimera (CD4-Nav1.2 II–III) markedly restricted to the AIS when expressed by transfection in hippocampal neurons (Fig. 1 A, left). The AIS was identified by the absence of the somatodendritic marker MAP2, and by staining for ankyrin G. In addition, CD4-Nav1.2 II–III was resistant to Triton X-100 extraction before cell fixation (Fig. 1 A, right), a feature reflecting tethering to the cytoskeleton, presumably via ankyrin G (Winckler et al., 1999). These features are governed by a 27-residue AIS motif (Garrido et al., 2003) that contains two potential phosphorylation sites for casein kinase II and clusters of acidic residues (Fig.1 C). To identify the critical residue(s) in this motif we decided to evaluate the contribution of acidic and serine residues by site-directed mutagenesis (Fig. 1, B–E). The substitution of both E1111 and D1113 with alanine residues drastically altered the distribution of the chimera. CD4-Nav1.2 II–III E1111A-D1113A was localized in both somatodendritic and axonal domains with a loss of concentration at the AIS. In contrast, replacing E1115, EE1120-1121, E1125, and D1127A by alanine did not affect the steady-state distribution of the chimeras. When only E1111 was substituted by either an alanine (Fig. 1 B, left) or a glutamine residue, chimeras were no longer concentrated at the AIS whereas the D1113A mutation did not modify the distribution of CD4-Nav1.2 II–III (Fig. 1 B, right). Consistent with these observations, mutants E1111A-D1113A and E1111A lost resistance to detergent extraction, an index of cytoskeletal tethering (Fig. 1 E). Finally mutation of each of the serine residues (S1112 and S1126) did not alter the steady-state distribution of CD4-Nav1.2 II–III (unpublished data).
Figure 1.

A point mutation inhibits sodium channel clustering at the AIS. (A, left) Cell surface distribution of CD4-Nav1.2 II–III in transfected hippocampal neurons. CD4-Nav1.2 II–III was immunodetected with an anti-CD4 antibody (green) before permeabilization and the somatodendritic domain was subsequently identified by MAP2 staining (red). CD4-Nav1.2 II–III was restricted at the AIS (arrow). (A, right) CD4-Nav1.2 II–III was resistant to Triton X-100 extraction before cell fixation and colocalized with ankyrin G. (B) Cell surface distribution of the indicated CD4-Nav1.2 II–III mutants. (C) Schematic representation of mutations within the AIS motif of Nav1.2. (D) Histogram of the cell surface distribution of CD4-Nav1.2 II–III mutants. For each mutant, the percentage of transfected CD4-positive hippocampal neurons were classified into three categories: CD4 staining concentrated at the AIS (hatched), localized on axons (gray shading), and uniformly distributed at the cell surface of somatodendritc and axonal domains (black) taking as 100% the total population of transfected neurons. Data are means ±SD from three to five different experiments. n denotes the total number of cells analyzed for each mutation. (E) In parallel, the resistance of mutated proteins to Triton X-100 extraction before cell fixation was determined. Bars, 20 μm.
The II–III linker of Nav1.2 is recognized by an endocytic pathway
Having demonstrated that a single mutation within the AIS motif impaired CD4-Nav1.2 II–III polarization, we examined whether this motif is sufficient for segregation of the chimera at the AIS. We replaced the COOH terminus of CD4 by a segment of 47 amino acids encompassing the AIS motif (CD4-Nav1.2 1082–1128). At the steady-state, the cell surface distribution of CD4-Nav1.2 1082–1128 differed from that of CD4-Nav1.2 II–III, displaying expression in dendrites, soma, and axons but with notably brighter staining at the AIS (Fig. 2 A). Nevertheless, CD4-Nav1.2 1082–1128 located at the AIS was still resistant to Triton X-100 extraction (Fig. 2 B). Thus, the AIS motif of Nav1.2 is necessary for tethering to the cytoskeleton but is not sufficient to restrict a CD4 chimera to the AIS. In the light of our previous study (Garrido et al., 2001) and because of the differences observed in the steady-state distribution of CD4-Nav1.2 II–III and CD4-Nav1.2 1082–1128, we evaluated the possibility that endocytosis contributes to compartmentalization at the AIS. We first applied an immunoendocytosis assay to COS-7 cells expressing CD4-Nav1.2 II–III. When the surface population of CD4-Nav1.2 II–III was prelabeled with an anti-CD4 antibody at 4°C, followed by a 20-min incubation at 37°C, it was found to be located in intracellular vesicles visualized by confocal microscopy (Fig. 3 A). The typical endocytic pattern was resistant to an acid-stripping treatment (Fig. 3 A, top). In COS-7 cells expressing CD4-Nav1.2 I-II (amino acids 428–753), antibody-labeled protein was confined at the cell surface and removed by acid stripping treatment (Fig. 3 A, bottom). We next applied a similar endocytosis assay to transfected hippocampal neurons. Antibody-labeled CD4-Nav1.2 II–III vesicles were visualized in the soma and throughout the dendrites after 30 min of endocytosis (Fig. 3 B). The presence of internalized CD4-Nav1.2 II–III in endosomes throughout the somatodendritic domain was further confirmed by colocalization with EEA1, a marker of somatodendritic early endosomes (Wilson et al., 2000; Fig. 3 B, bottom).
Figure 2.

Cell surface distribution of a CD4 chimera bearing the clustering motif of sodium channels. (A) CD4-Nav1.2 1082–1128 was localized at the cell surface of soma, dendrites, and axons but with a higher density at the AIS (arrow). (B) CD4-Nav1.2 1082–1128 localized at the AIS was resistant to Triton X-100 extraction before cell fixation. Somatodendritic domains and AIS were identified by staining for MAP2 and ankyrin G. Bar, 20 μm.
Figure 3.
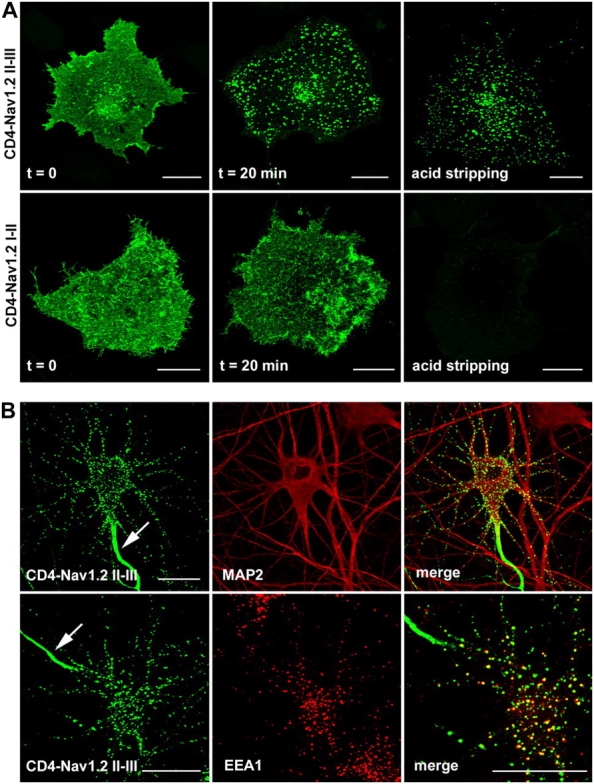
The cytoplasmic II–III linker of Nav1.2 is recognized by an endocytotic pathway. (A) COS-7 cells transfected with either CD4-Nav1.2 II–III or CD4-Nav1.2 I-II constructs were processed for an immunoendocytosis assay. After the antibody prelabeling step at 4°C, CD4-Nav1.2 II–III and CD4-Nav1.2 I–II showed a diffuse cell surface staining. After 20 min of incubation at 37°C, antibody-prelabeled CD4-Nav1.2 II–III displayed a punctate intracellular distribution resistant to an acidic stripping, whereas CD4-Nav1.2 I–II was confined to the cell surface and stripped by an acidic wash. (B) Endocytosis of CD4-Nav1.2 II–III in the somatodendritic domain of hippocampal neurons. Cells expressing CD4-Nav1.2 II–III were submitted to an immunoendocytosis assay for 30 min. CD4-Nav1.2 II–III containing vesicles were distributed throughout soma and dendrites identified by MAP2 costaining (top) and colocalized with EEA1 (right enlarged view). Note the concentration of CD4-Nav1.2 II–III at the AIS (arrow). Bars, 20 μm.
Involvement of an elimination-retention mechanism in CD4-Nav1.2 II–III segregation at the AIS
We next identified at the molecular level the internalization signal in Nav1.2 II–III. We tested the internalization of several mutants in COS-7 cells. The sequential truncation of Nav1.2 II–III up to amino acid 1030 did not impair internalization. For each mutant, the internalized antibody-prelabeled population located in intracellular vesicles was visualized by confocal microscopy. In contrast, mutants Δ1020, Δ1010, and Δ993 were confined at the cell surface (Fig. 4) and were not resistant to acid-stripping treatment (unpublished data). Two internal deletions (Δ1010-1030 and Δ1002-1011) were further generated; Δ1010-1030 mutant impaired internalization unlike Δ1002-1011. These data indicated that 19 amino acids located in the NH2-terminal region of Nav1.2 II–III are critical for endocytosis in COS-7 cells. We next compared endocytosis of CD4-Nav1.2 II–III and CD4-Nav1.2 II–III Δ1010-1030 in hippocampal neurons (Fig. 5 A). Differential staining of the surface and internalized populations was performed. Positive staining for the internalized population was detected in 80% of cells expressing CD4-Nav1.2 II–III (n = 184). This type of staining was only detected in 18% of cells expressing Δ1010-1030 (n = 295), indicating that the mutation impaired endocytosis. An internalized population was observed in neurons expressing Δ1098-1111, a mutant lacking the AIS motif (Fig. 5 A). The steady-state cell surface distribution of Δ1010-1030 differed from that of CD4-Nav1.2 II–III, displaying expression in soma, dendrites, and axons (Fig. 5 B, top). An accumulation at the AIS was however clearly observed, that was resistant to detergent extraction (Fig. 5 B, left bottom). The distribution of the Δ1010-1030 mutant was strongly reminiscent of CD4-Nav1.2 1082–1128 (Fig. 2 A). Thus, when internalization of CD4-Nav1.2 II–III was impaired, its steady-state distribution was drastically altered in spite of the presence of a cytoskeletal tethering motif.
Figure 4.

A determinant of the linker II–III of Nav1.2 is involved in endocytosis of CD4-Nav1.2 II–III. (A) Schematic representation of mutations generated in Nav1.2 II–III. The position of the AIS motif is delineated between amino acid 1102 and 1128. Internalization in COS-7 cells is indicated (+ or −). (B) COS-7 cells expressing the indicated mutants were submitted to an immunoendocytosis assay for 20 min at 37°C. Mutants Δ1030 and Δ1002-1011 underwent endocytosis whereas Δ1010 and Δ1010-1030 were confined to the plasma membrane. Bars, 20 μm.
Figure 5.

Involvement of somatodendritic endocytosis in the segregation of CD4-Nav1.2 II–III at the AIS. (A) Δ1010-1030 failed to be endocytosed in hippocampal neurons. After an immunoendocytosis assay, neurons expressing the indicated constructs were submitted to differential staining of the surface population (green) and of the internalized one (red). Note the absence of internalized Δ1010-1030, unlike CD4-Nav1.2 II–III and Δ1098-1111. (B) Cell surface distribution of Δ1010-1030 and CD4-Nav1.8 II–III in hippocampal neurons. Δ1010-1030 displayed a cell surface distribution in MAP2-positive domains as well as axons, with an enrichment at the AIS (top). Δ1010-1030 was resistant to detergent extraction (bottom left). Cell surface distribution of CD4-Nav1.8 II–III (bottom middle). CD4-Nav1.8 II–III localized at the AIS was resistant to Triton X-100 extraction (bottom right). (C) Histogram of the cell surface distribution of the indicated constructs. The percentage of transfected CD4-positive hippocampal neurons were classified into four categories: CD4 staining concentrated at the AIS ([AIS]), distributed at the cell surface of somatodendritc and axonal domains with a enrichment at the AIS (SD-[AIS]-DA), distributed in distal axon with no concentration at the AIS (axonal), and uniformly distributed at the cell surface (nonpolarized) taking as 100% the total population of transfected neurons. Data are means ±SD from three to five different experiments. n denotes the total number of cells analyzed for each construct. Arrows indicate the AIS. Bars, 20 μm.
A comparison of the sequences of the II–III linker region in different sodium channel types revealed that the segment involved in endocytosis is less conserved in Nav1.8 and 1.9 (see Fig. S1). In view of these differences, we generated a CD4 chimera containing the II–III linker of Nav1.8. In COS-7 cells, CD4-Nav1.8 II–III was not endocytosed, unlike CD4-Nav1.2 II–III (see Fig. S2). When expressed in hippocampal neurons, CD4-Nav1.8 II–III was distributed at the surface of the somatodendritic and axonal domains with an accumulation at the AIS, like that observed with CD4-Nav1.2 Δ1010-1030 (Fig. 5 B). It was also resistant to Triton X-100 extraction (Fig. 5 B). A histogram of the cell surface distribution of the constructs is shown in Fig. 5 C. All together, these findings strongly suggest that an elimination-retention mechanism accounts for CD4-II–III segregation at the AIS.
Kinetics of insertion of CD4-Nav1.2 II–III in the plasma membrane of hippocampal neurons
We next examined whether the insertion of newly synthesized CD4-Nav1.2 II–III occurs preferentially in the somatodendritic domain or in the axonal domain. With this aim, we looked at the time course of cell surface insertion of CD4-Nav1.2 II–III in hippocampal neurons by using brefeldin A (BFA) to block Golgi apparatus trafficking (Cid-Arregui et al., 1995; Jareb and Banker, 1997). After a 4-h posttransfection period, hippocampal neurons were treated overnight with BFA, resulting in protein accumulation in the Golgi apparatus. After removal of BFA, the cell surface distribution of CD4-Nav1.2 II–III was visualized by immunostaining at different time intervals (Fig. 6 A). At t = 0, upon BFA removal and after cell permeabilization, an intracellular accumulation was observed in the perinuclear region (unpublished data) and staining that was consistent with BFA block. After 30 min of recovery, CD4 surface staining was visualized on the somatodendritic membrane with an equivalent signal at the AIS in the majority of cells (53%). This cell population was designated somatodendritic (SD)-AIS. A second population SD-[AIS], with brighter CD4 staining at the AIS was also observed in 24% of the cells. At this stage of recovery, no signal was visualized on the distal part of axons (DA; Fig. 6 A). These two cell populations were also predominant 60 min after BFA removal but decreased markedly afterwards. Concomitantly, CD4-Nav1.2 II–III appeared in the distal region of axons. A surface distribution in both domains with an enrichment at the AIS (SD-[AIS]-DA) was predominant after 4 and 8 h of recovery (82 and 88%, respectively). Restriction at the AIS, as observed in the absence of BFA treatment, began to be preponderant 24 h after removal (Fig. 6 A). In parallel, we determined the kinetics of the insertion of CD4-Nav1.2 II–III E1111A (Fig. 6 B), a mutant that is no longer segregated at the AIS (Fig. 1). After 30 min of recovery, the E1111A mutant was visualized on the somatodendritic membrane with an equivalent signal at the AIS in 81% of cells (cell population designated SD-AIS). At this stage of recovery, no signal was visualized on the distal part of axons, similar to that observed with CD4-Nav1.2 II–III (Fig. 6 A). A surface distribution in both domains (SD-A), but with no enrichment at the AIS was largely predominant after 2 h of recovery (77%). These observations indicated that CD4-Nav1.2 II–III is preferentially inserted in the somatodendritic membrane and in the AIS, where it is enriched, and subsequently in the distal part of the axons.
Figure 6.

Time course of the insertion of CD4-Nav1.2 II–III and CD4-Nav1.2 II–III E1111A in the neuronal plasma membrane. Hippocampal neurons were transfected with either CD4-Nav1.2 II–III or CD4-Nav1.2 II–III E1111A. After a 4-h posttransfection step, neurons were treated overnight either in the presence of BFA or in the absence (−BFA). The cell surface distribution of the CD4-Nav1.2 II–III and E1111A mutant was detected by CD4 immunostaining at different time intervals after BFA removal. (A) Time course of the insertion of CD4-Nav1.2 II–III. For each indicated time, CD4-Nav1.2 II–III staining was classified into four categories: SD-AIS (uniform labeling on somatodendritic and the AIS surface), SD-[AIS] (as in SD-AIS but with a brighter signal on the AIS), SD-[AIS]-DA (uniform labeling in somatodendritic domain and the distal axon with a brighter signal on the AIS), and [AIS] (staining restricted at the AIS) taking as 100% the number of CD4-positive cells. (B) Time course of the insertion of CD4-Nav1.2 II–III E1111A mutant. CD4 staining was classified into two categories: SD-AIS as described above, and SD-A (uniform labeling in somatodendritic and axonal surface with no concentration at the AIS). Data are means ±SD from three to four experiments (the number of cells counted ranged from 200 to 800 cells for each indicated time). A representative example of each staining category is shown in the top part of the figure; arrows indicate the AIS and the asterisk the axon. Bars, 20 μm.
Discussion
In the present study, we analyzed the trafficking of CD4-Nav1.2 II–III, a chimera bearing the cytoplasmic region that determines sodium channel targeting and clustering at the AIS (Garrido et al., 2003), to explore potential mechanisms involved in protein accumulation in this membrane subdomain. Evidence has been obtained for the following scenario. CD4-Nav1.2 II–III is first preferentially inserted in the plasma membrane of the soma and dendrites, and is concentrated at the AIS, before its insertion in axonal tips. The high density of ankyrin G-β IV spectrin complex presumably acts as a diffusion trap at the AIS. The untrapped population in soma, dendrites, and in the distal part of axons is subsequently eliminated by endocytosis. However, our findings do not exclude the possibility that additional mechanisms contribute to the accumulation of proteins at the AIS.
The time course of the appearance of CD4-Nav1.2 II–III at the cell surface revealed that the neo-synthesized protein is preferentially inserted in the somatodendritic domain rather than in distal regions of axons. This finding is unlikely to result from a side effect of BFA block because it has been shown recently that the initial insertion of membrane proteins in axonal tips is not perturbed by this type of treatment (Wisco et al., 2003). We also observed that an uniform somatodendritic distribution preceded enrichment at the AIS. Previous studies have indicated that the accumulation of membrane proteins like sodium channels and neurofascin at the AIS is coordinated by ankyrin G (Jenkins and Bennett, 2001) and is impaired by inactivation of either ankyrin G or β IV spectrin gene expression (Zhou et al., 1998; Komada and Soriano, 2002). When the II–III linker of Nav1.2 was fused to a cytosolic protein like the green fluorescent protein, the resulting chimera was trapped within the AIS (Garrido et al., 2003). Moreover, the AIS motif of Nav1.2 contains a 9-amino acid ankyrin binding site, highly conserved within the sodium channel Nav1 family (Lemaillet et al., 2003). A single glutamate residue (Nav1.2 E 1111) within this motif plays a critical role in CD4-Nav1.2 II–III tethering to cytoskeleton at the AIS. Substitution of alanine or glutamine for this residue resulted in a loss of chimera segregation at the AIS, a finding underlining the functional impact of removing the charge. Consistently, CD4 chimeras bearing the II–III linkers of Nav 1.6 (Garrido et al., 2003) and 1.8 were concentrated at the AIS and resistant to Triton X-100 extraction. Their accumulation at the AIS was abolished by mutation of the equivalent glutamate residue to alanine (unpublished data). Besides, in cultured hippocampal neurons, ankyrin B, which contains the conserved ankyrin repeat domain that binds to sodium channels (Bennett and Lambert, 1999; Lemaillet et al., 2003), is distributed in distal regions of axons, a localization distinct from that of ankyrin G. However, at the steady-state, CD4-Nav1.2 II–III was restricted at the AIS. For all these reasons, it is tempting to propose that CD4-Nav1.2 II–III diffuses laterally in the somatic plasma membrane and is subsequently trapped by the cystokeleton adaptor complex ankyrin G-β IV spectrin, at the AIS. A similar mechanism could be involved in NgCAM accumulation. This axonal protein, that possesses the highly conserved ankyrin binding site of the L1 family (Garver et al., 1997), is initially incorporated in the somatodendritic domain of hippocampal neurons (Wisco et al., 2003). This interpretation is consistent with the fact that perturbation of the diffusion barrier formed by the AIS results in a somatic localization of NgCAM and sodium channels (Winckler et al., 1999; Nakada et al., 2003).
Despite the importance of anchoring, we show here that cytoskeleton tethering, presumably by ankyrin G-β IV spectrin, is not sufficient for CD4-Nav1.2 II–III segregation at the AIS, endocytosis is also required. The internalized population of CD4-Nav1.2 II–III was observed throughout the soma and dendrites and to a lesser extent, in the distal part of axons. It was never seen within the AIS, in all observed cells. The presence of endocytotic vesicles in these regions is consistent with the steady-state distribution of CD4-Nav1.2 II–III. The requirement for endocytosis was further demonstrated by fact that a mutation (Δ1010-1030) that abolished internalization of CD4-Nav1.2 II–III altered its steady-state distribution without inhibiting accumulation at the AIS. Consistent with this conclusion is the observation that CD4-Nav1.8 II–III, which was not internalized either in COS-7 cells or in hippocampal neurons, was distributed at the steady-state in soma, dendrites, and axons, and was enriched at the AIS. These findings also indicate that the internalization of CD4-Nav1.2 II–III is not involved in its accumulation at the AIS but rather in its elimination in the somatodendritic domain and in the distal part of axons. They imply that transcytosis is unlikely to be involved in the accumulation of CD4-Nav1.2 II–III at the AIS. However, we cannot exclude the possibility that the presence of internalized CD4-Nav1.2 II–III in the distal part of axons may reflect transcytosis from the somatodendritic domain to the axonal tips, as described for NgCAM (Wisco et al., 2003). The sequence that governs endocytosis of CD4-Nav1.2 II–III does not encompass a canonic internalization signal such as the di-leucine or tyrosine-based motifs (YxxΦ) recognized by the clathrin-mediated endocytotic pathway (Bonifacino and Traub, 2003). Hence, further investigation will be required to analyze the endocytotic pathway recognized by the II–III linker of Nav1.2. A differential regulation of endocytosis in the somatodendritic versus the axonal domain cannot be excluded.
In conclusion, our present study shows that endocytosis and domain-selective tethering confers CD4-Nav1.2 II–III segregation at the AIS. However, whether the Nav1.2 sodium channel follows a similar trafficking pathway remains to be explored. Multiple mechanisms probably play a role in establishing polarized sorting to the AIS. For instance, it is conceivable that ankyrin G-β IV spectrin may also act during sorting, involving preassembly with sodium channels and the CAM family. Consistent with this hypothesis is the recent observation that when RNAi was used to eliminate expression of Nav1.2 in cultured neurons, it resulted not only in a loss of sodium channels but also perturbed ankyrin G compartmentalization at the AIS (unpublished data). Hence further investigations are required for a better understanding of the molecular and cellular processes involved in the construction of the AIS.
Materials and methods
DNA constructs
CD4-Nav1.2 II–III was constructed and cloned into PCB6 as described previously (Garrido et al., 2001). Nav1.8 cDNA was a gift from John Wood (University College London, London, UK). CD4 chimeras composed of the II–III linker of Nav1.8 (amino acids 890–1149) were generated by sequential PCR amplification using Expand High Fidelity Taq DNA polymerase (Roche Molecular Biochemicals). The constructs were subcloned in pCRII-TOPO plasmid (Invitrogen) before cloning into PCB6. CD4-Nav1.2 II–III mutants were also generated by sequential PCR amplification. Site-directed mutants were obtained using Quick change X-L site-directed mutagenesis kit (Stratagene). All constructs were verified by DNA sequencing.
Cell culture and transfection
Primary hippocampal neurons were prepared from embryonic day 18 rats according to Goslin and Banker (1989) with slight modifications (Garrido et al., 2001). Neurons were transfected after 7 to 9 d in vitro using Lipofectamine 2000 (Invitrogen) (Garrido et al., 2003). COS-7 cells were cultured as described previously (Garrido et al., 2001) and transfected at a 80% confluence with Lipofectamine 2000, according to the manufacturer's instructions. Transfected cells were processed for immunofluorescence 24 h after transfection.
BFA treatment
Neurons (9 d in vitro) were transfected as described above. After 4 h, brefeldin A (Sigma-Aldrich) was added to yield 0.75 μg/ml final concentration in conditioned medium. After 14 h, coverslips were washed three times in conditioned medium and returned to the air/CO2 incubator at 37°C. Coverslips were removed, fixed at the indicated times, and processed for immunostaining. At t = 0, before washing, 10% of the transfected neurons displayed CD4 staining at the cell surface (n = 1,600, four independent experiments).
Immunofluorescence
To immunodetect the steady-state surface distribution of the chimeras in neurons, cells were fixed in 4% paraformaldehyde for 20 min. Nonspecific binding was blocked with 0.22% gelatin in 0.1 M phosphate buffer (PB). Cells were incubated for 1 h with anti-CD4 antibodies (monoclonal, 1:1,000; a gift from Jean Mérot, INSERM U533, Nantes, France); polyclonal T4-4, 1:1,500, National Institutes of Health AIDS and Reference Reagent Program). After a permeabilization step (0.066% saponin, 0.22% gelatin in PB), endogenous proteins were immunodetected either with anti-ankyrin G antibodies (monoclonal, 1:30; Zymed Laboratories; polyclonal 1:60, a gift from Gisèle Alcaraz, INSERM UMR 641, Marseille, France) or with an anti-MAP2 monoclonal, (1:400; Sigma-Aldrich) The secondary antibodies were goat anti–mouse or goat anti–rabbit conjugated to Alexa 563 or Alexa 488 (1:800; Molecular Probes). Coverslips were mounted in Mowiol.
Detergent extraction
Neurons were preincubated with anti-CD4 antibody at 16°C for 30 min, washed, and incubated in extraction buffer (30 mM Pipes, 1 mM MgCl2, 5 mM EDTA, 0.5% Triton X-100) for 10 min at 37°C as described previously (Garrido et al., 2003). Then, neurons were rinsed in PB, fixed, and processed by immunofluorescence.
Immunoendocytosis assay
COS-7 cells were exposed to the primary antibody diluted in PB with 0.22% gelatin for 45 min at 4°C, extensively washed, and then transferred to preheated DME medium and returned to the air/CO2 incubator at 37°C for 20 min. Cells were then fixed, permeabilized, and subjected to secondary antibody binding. For double immunostaining, a monoclonal anti-EEA1 antibody (1:400; Transduction Laboratories) was added to permeabilized cells followed by secondary antibodies. To dissociate membrane bound antibody before fixation, cells were incubated 10 min at 4°C in 0.2 M acetic acid and 0.5 M NaCl.
Neurons were incubated with primary antibody in glial-conditioned medium supplemented with 0.1% BSA for 30 min at 37°C, washed, and fixed. After the permeabilization step, cells were processed as described above. For differential staining of cell surface and internalized populations, endocytosis was stopped by transferring neurons to DME-Hepes at 16°C. Living cells were then exposed to Alexa 488–conjugated secondary antibody for 40 min at 16°C, washed, fixed, and further incubated 30 min with an excess of unlabeled secondary antibody (0.2 mg/ml diluted in PB with 0.22% gelatin). After a 15-min postfixation step, neurons were permeabilized and incubated with Alexa 563–conjugated secondary antibody.
Confocal microscopy
Labeling was viewed with a confocal laser scanning microscope (model TCS; Leica). Optical sections taken in the x–y plane from consecutive z positions every 0.45–0.5 μm, using the standard microscope software (Scanware; Leica). Images were processed with Adobe Photoshop software.
Online supplemental material
Fig. S1 shows the alignment of the NH2-terminal region of the linker II–III sequences of voltage-dependent sodium channels. Fig. S2 shows that the cytoplasmic II–III linker of Nav1.8 is not recognized by an endocytotic pathway in transfected COS-7 cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200312155/DC1.
Acknowledgments
We are grateful to colleagues for the gifts of cDNA. We thank L. Fronzaroli-Molinieres, F. Jullien, and S. Hutter for excellent technical assistance, and F. Castets and M. Seagar for critical reading of the manuscript.
This work was supported by INSERM and the “Association Francaise contre les Myopathies” grants and the “Fondation pour la Recherche Médicale” (M.-P. Fache and J.J. Garrido).
M.-P. Fache and A. Moussif contributed equally to this study.
J. J. Garrido's present address is Centro de Biología Molecular “Severo Ochoa,” Universidad Autónoma de Madrid, 28049 Cantoblanco, Spain.
Abbreviations used in this paper: AIS, axonal initial segment; [AIS], AIS concentration; BFA, brefeldin A; CAM, cell adhesion molecules; DA, distal axon; MAP2, microtubule associated protein 2; PB, phosphate buffer; SD, somatodendritic.
References
- Bennett, V., and A.J. Baines. 2001. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol. Rev. 81:1353–1392. [DOI] [PubMed] [Google Scholar]
- Bennett, V., and S. Lambert. 1999. Physiological roles of axonal ankyrins in survival of premyelinated axons and localization of voltage-gated sodium channels. J. Neurocytol. 28:303–318. [DOI] [PubMed] [Google Scholar]
- Boiko, T., A. Van Wart, J.H. Caldwell, S.R. Levinson, J.S. Trimmer, and G. Matthews. 2003. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J. Neurosci. 23:2306–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino, J.S., and L.M. Traub. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72:395–447. [DOI] [PubMed] [Google Scholar]
- Catterall, W.A. 2000. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 26:13–25. [DOI] [PubMed] [Google Scholar]
- Cid-Arregui, A., R.G. Parton, K. Simons, and C.G. Dotti. 1995. Nocodazole-dependent transport, and brefeldin A–sensitive processing and sorting, of newly synthesized membrane proteins in cultured neurons. J. Neurosci. 15:4259–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, J.Q., and V. Bennett. 1994. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J. Biol. Chem. 269:27163–27166. [PubMed] [Google Scholar]
- Garrido, J.J., F. Fernandes, P. Giraud, I. Mouret, E. Pasqualini, M.P. Fache, F. Jullien, and B. Dargent. 2001. Identification of an axonal determinant in the C-terminus of the sodium channel Na(v)1.2. EMBO J. 20:5950–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido, J.J., P. Giraud, E. Carlier, F. Fernandes, A. Moussif, M.P. Fache, D. Debanne, and B. Dargent. 2003. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 300:2091–2094. [DOI] [PubMed] [Google Scholar]
- Garver, T.D., Q. Ren, S. Tuvia, and V. Bennett. 1997. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J. Cell Biol. 137:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin, K., and G. Banker. 1989. Experimental observations on the development of polarity by hippocampal neurons in culture. J. Cell Biol. 108:1507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom, L.L. 2001. Sodium channel beta subunits: anything but auxiliary. Neuroscientist. 7:42–54. [DOI] [PubMed] [Google Scholar]
- Jareb, M., and G. Banker. 1997. Inhibition of axonal growth by brefeldin A in hippocampal neurons in culture. J. Neurosci. 17:8955–8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins, S.M., and V. Bennett. 2001. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J. Cell Biol. 155:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, T., B. Storrie, K. Simons, and C.G. Dotti. 1992. A functional barrier to movement of lipids in polarized neurons. Nature. 359:647–650. [DOI] [PubMed] [Google Scholar]
- Komada, M., and P. Soriano. 2002. βIV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J. Cell Biol. 156:337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaillet, G., B. Walker, and S. Lambert. 2003. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J. Biol. Chem. 278:27333–27339. [DOI] [PubMed] [Google Scholar]
- Nakada, C., K. Ritchie, Y. Oba, M. Nakamura, Y. Hotta, R. Iino, R.S. Kasai, K. Yamaguchi, T. Fujiwara, and A. Kusumi. 2003. Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat. Cell Biol. 5:626–632. [DOI] [PubMed] [Google Scholar]
- Nakata, T., and N. Hirokawa. 2003. Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J. Cell Biol. 162:1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peles, E., and J.L. Salzer. 2000. Molecular domains of myelinated axons. Curr. Opin. Neurobiol. 10:558–565. [DOI] [PubMed] [Google Scholar]
- Salzer, J.L. 2003. Polarized domains of myelinated axons. Neuron. 40:297–318. [DOI] [PubMed] [Google Scholar]
- Sampo, B., S. Kaech, S. Kunz, and G. Banker. 2003. Two distinct mechanisms target membrane proteins to the axonal surface. Neuron. 37:611–624. [DOI] [PubMed] [Google Scholar]
- Wilson, J.M., M. de Hoop, N. Zorzi, B.H. Toh, C.G. Dotti, and R.G. Parton. 2000. EEA1, a tethering protein of the early sorting endosome, shows a polarized distribution in hippocampal neurons, epithelial cells, and fibroblasts. Mol. Biol. Cell. 11:2657–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winckler, B., and I. Mellman. 1999. Neuronal polarity: controlling the sorting and diffusion of membrane components. Neuron. 23:637–640. [DOI] [PubMed] [Google Scholar]
- Winckler, B., P. Forscher, and I. Mellman. 1999. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 397:698–701. [DOI] [PubMed] [Google Scholar]
- Wisco, D., E.D. Anderson, M.C. Chang, C. Norden, T. Boiko, H. Folsch, and B. Winckler. 2003. Uncovering multiple axonal targeting pathways in hippocampal neurons. J. Cell Biol. 162:1317–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, F.H., R.E. Westenbroek, I. Silos-Santiago, K.A. McCormick, D. Lawson, P. Ge, H. Ferriera, J. Lilly, P.S. DiStefano, W.A. Catterall, et al. 2003. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J. Neurosci. 23:7577–7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, D., S. Lambert, P.L. Malen, S. Carpenter, L.M. Boland, and V. Bennett. 1998. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 143:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]