

Abstract
High mobility group box 1 (HMGB1) is an abundant chromatin protein that acts as a cytokine when released in the extracellular milieu by necrotic and inflammatory cells. Here, we show that extracellular HMGB1 and its receptor for advanced glycation end products (RAGE) induce both migration and proliferation of vessel-associated stem cells (mesoangioblasts), and thus may play a role in muscle tissue regeneration. In vitro, HMGB1 induces migration and proliferation of both adult and embryonic mesoangioblasts, and disrupts the barrier function of endothelial monolayers. In living mice, mesoangioblasts injected into the femoral artery migrate close to HMGB1-loaded heparin-Sepharose beads implanted in healthy muscle, but are unresponsive to control beads. Interestingly, α-sarcoglycan null dystrophic muscle contains elevated levels of HMGB1; however, mesoangioblasts migrate into dystrophic muscle even if their RAGE receptor is disabled. This implies that the HMGB1–RAGE interaction is sufficient, but not necessary, for mesoangioblast homing; a different pathway might coexist. Although the role of endogenous HMGB1 in the reconstruction of dystrophic muscle remains to be clarified, injected HMGB1 may be used to promote tissue regeneration.
Keywords: cell migration; cytokine; inflammation; stem cell; tissue damage
Introduction
High mobility group box 1 protein (HMGB1; also called amphoterin) is a very abundant chromatin-binding protein residing in the eukaryotic cell nucleus and acting in the assembly of nucleoprotein complexes (Bustin, 1999; Agresti and Bianchi, 2003). There are three HMGB proteins, with >80% amino acid identity among them; all are composed of two basic DNA-binding domains (HMG boxes A and B) and a long acidic COOH-terminal tail. HMGB1 is almost ubiquitous (Guazzi et al., 2003), and HMGB2 and HMGB3 are expressed during embryogenesis and have a very restricted expression pattern in adults (Vaccari et al., 1998; Ronfani et al., 2001). Further to binding to DNA, HMGB1 interacts with several transcription factors, viral replication proteins, the RAG1 recombinase, and steroid receptors, playing a crucial role in transcription (for review see Agresti and Bianchi, 2003). The phenotype of Hmgb1 knockout mice revealed that the gene is essential; mice die soon after birth, and have defects in the function of steroid receptors (Calogero et al., 1999).
Recently, several groups showed that HMGB1 has an extracellular role as a cytokine: (1) HMGB1 is secreted by macrophages and monocytes activated by IL-1β, TNF, or lipopolysaccharide (LPS; Wang et al., 1999; Andersson et al., 2000); (2) acts as a chemoattractant for myeloid cells (Abraham et al., 2000; Andersson et al., 2000) and smooth muscle cells (Degryse et al., 2001); (3) enhances the expression of vascular adhesion molecules in endothelial cells (Fiuza et al., 2003); and (4) impairs the barrier function of intestinal epithelia (Sappington et al., 2002). Most or all of these actions are initiated by binding of HMGB1 to the receptor for advanced glycation end products (RAGE), a multiligand receptor of the immunoglobulin superfamily (Hori et al., 1995).
Recently, we showed that necrotic cells release HMGB1 by simple diffusion, and thereby trigger inflammation; in contrast, apoptotic cells avidly retain HMGB1 bound to chromatin remnants even after their eventual lysis (Scaffidi et al., 2002). Monocytic cells actively secrete HMGB1 in a process that is independent from the ER and the Golgi apparatus, but depends on HMGB1 relocalization from the nucleus to special organelles, the secretory lysosomes (Gardella et al., 2002). Importantly, the secretion process requires the acetylation of HMGB1 on several specific lysine residues (Bonaldi et al., 2003). Thus, HMGB1 passively released from necrotic cells and HMGB1 actively secreted by inflammatory cells are molecularly different.
We have argued that extracellular HMGB1 is primarily a signal of tissue damage, and monocytes and macrophages have “learned” to mimic an ancient alarm signal (Scaffidi et al., 2002; Bonaldi et al., 2003). Presently, we build over this hypothesis. A signal of tissue damage is expected to promote the migration of stem cells to the damaged tissue, and to promote their proliferation. Stem cells migrate during normal and pathological conditions and do proliferate during tissue repair, but little is known about the mechanisms regulating these processes (Ferrari et al., 1998; Orlic et al., 2001; LaBarge and Blau, 2002; Vicario-Abejon et al., 2003). Here, we show that HMGB1 acts as a mitogenic and chemoattractant factor for embryonic and adult mesoangioblasts, both in vitro and in vivo.
Mesoangioblasts are a specific population of mesodermal stem cells that are associated to the wall of fetal and postnatal vessels (Minasi et al., 2002), and may derive from a primitive angioblast (Cossu and Bianco, 2003). Mesoangioblasts express angioblast markers such as Sca-1, Flk-1, and CD34, as well as genes typical of the mesoderm, including receptors and signaling molecules for classical mesoderm inducers, such as BMP, Wnt, and Notch (unpublished data). Mesoangioblasts can be grown extensively in culture (>50 passages) and are able to differentiate into most mesodermal cell types; most notably, they are able to home to damaged tissue muscle, and participate in muscle regeneration (Sampaolesi et al., 2003). Thus, they are ideal to study stem cell homing in vivo.
When injected into arteries of α-sarcoglycan (α-SG) null mice, a model for limb-girdle muscular dystrophy, mesoangioblasts accumulate in the first capillary filter they encounter, migrate outside of the vessel into the areas of muscle wastage and inflammation, and eventually correct morphologically and functionally the dystrophic phenotype (Sampaolesi et al., 2003). The nature of the molecules that chemoattract mesoangioblasts where they are required was unknown.
Here, we show that unmodified, bacterially made HMGB1 is sufficient to attract mesoangioblasts, both in vitro and when injected into healthy muscle. However, mesoangioblasts expressing a dominant-negative mutant of RAGE (dnRAGE), the only receptor for HMGB1 identified so far, still find their way into dystrophic muscle. These data indicate that mesoangioblasts can use receptors other than RAGE to navigate to dystrophic muscle. In turn, this may mean that dystrophic muscle recruits mesoangioblasts via as yet unidentified chemoattractants, or that acetylated HMGB1 secreted by inflammatory cells is indeed the chemoattractant released by dystrophic muscle, but is recognized by a receptor different from RAGE. Bacterially made HMGB1 may be used experimentally or therapeutically to direct mesoangioblast migration into nondystrophic muscle, or to enhance it in dystrophic muscle.
Results
HMGB1 stimulates the proliferation of vessel-associated embryonic stem cells
Stem cells isolated from mouse dorsal aorta of E9.5 C57Bl6 mouse embryos (mesoangioblasts) were cultured in vitro and tested for the presence of the CD34, Sca1, Flk1, and MEF2D cellular markers (Minasi et al., 2002). We used one of these clones (called D16; Sampaolesi et al., 2003) to assess whether HMGB1 can act as a mitogen.
D16 cells were seeded in RPMI medium with 20% FCS and then starved for 16 h in the absence of serum to synchronize the cell population. Increasing concentrations of HMGB1 were then added to the medium without serum. Fig. 1 A shows that there is a significant increase in the number of D16 mesoangioblasts after stimulation with HMGB1 up to d 2, whereas only slight proliferation occurs between d 2 and 3. All concentrations tested had similar effects. HMGB1-stimulated D16 cells had a normal morphology and excluded trypan blue up to the end of the experiment, whereas cells in control cultures without HMGB1 were dying (unpublished data). HMGB1 has no mitogenic effect on 3T3 fibroblasts (Fig. 1 C).
Figure 1.
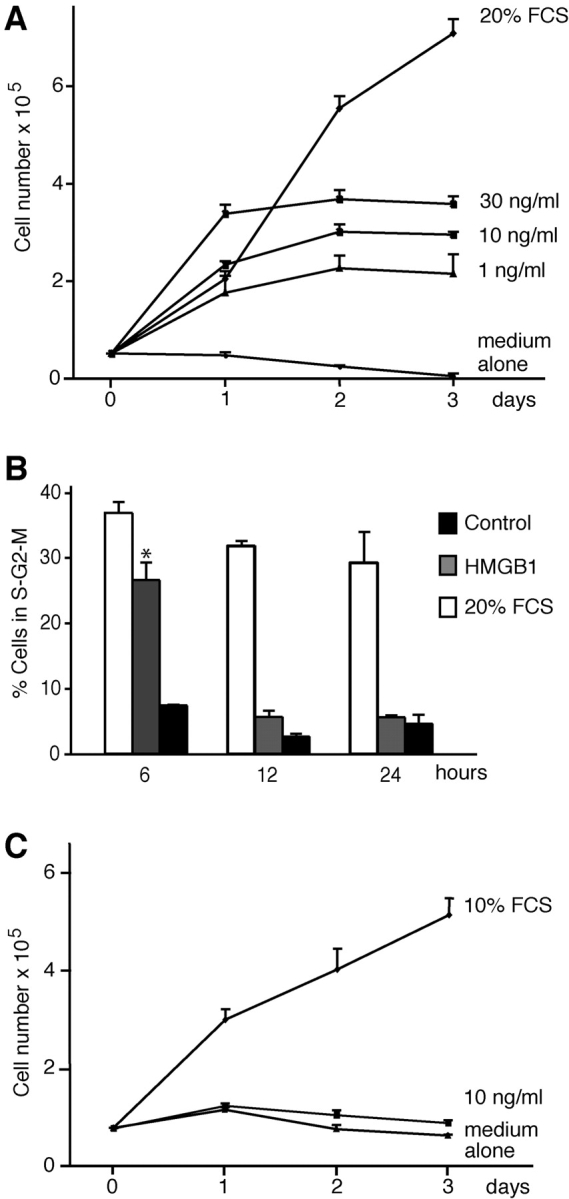
Effect of HMGB1 on embryonic mesoangioblast proliferation. (A) D16 cells were grown in RPMI medium containing no addition, HMGB1 at the indicated concentrations, or 20% FCS. HMGB1 induced cell proliferation at all concentrations tested, but the cell number reached a plateau after 48 h. Each point represents the mean ± SD (n = 3). The experiment was repeated three times. (B) D16 cell division was analyzed by FACS®. After 6 h in the presence of 30 ng/ml HMGB1 the DNA content increases, but returns to the normal diploid content after 24 h. Asterisk indicates statistical significance (P < 0.001). (C) 3T3 fibroblasts (treated as the D16 cells in A) do not divide in the presence of HMGB1.
We investigated in more detail the proliferative response of D16 cells to HMGB1. Cells were exposed for 6, 12, and 24 h to RPMI medium alone (negative control) or medium containing 30 ng/ml HMGB1 or 20% FCS, and were analyzed for DNA content by FACS® after propidium iodide staining. After 6 h of stimulation with HMGB1, the majority of mesoangioblasts had entered the cell cycle; after 24 h, most cells appeared to have a diploid DNA content and thus to be in G1 or G0 (Fig. 1 B). We evaluated the number of cell cycles triggered by HMGB1 by staining the cell membranes at time 0 with the fluorescent dye carboxyfluorescein diacetate succinimidyl ester (Lyons and Parish, 1994); after 48 h most cells had one-half of the initial quantity of dye (and so had undergone one division), and a minority had one-quarter (and had undergone two divisions). By comparison, all cells cultured in 20% FCS had divided at least twice (unpublished data).
These data indicate that HMGB1 induces a limited number of cell divisions. This might be due to a specific program of mesoangioblasts, or to the depletion of HMGB1 in the medium. Therefore, we added 30 ng/ml HMGB1 at time 0, and additional HMGB1 at 12, 36, and 60 h. Mesoangioblasts continually exposed to HMGB1 continued to divide (Fig. 2). After 48 h, no HMGB1 was detectable by Western blot in the medium of cells stimulated once, whereas HMGB1 equivalent to ∼40 ng/ml remained in the medium of multiply exposed cells (Fig. 2, inset). Together, these results indicate that HMGB1 acts as a growth factor for D16 cells, but is rapidly depleted.
Figure 2.

Continued HMGB1 stimulation sustains mesoangioblast proliferation. D16 cells were placed at time 0 in RPMI medium containing 30 ng/ml HMGB1; a similar amount of HMGB1 was also added at the times indicated with a triangle. Multiply stimulated D16 cells kept growing. Each point represents the average ± SD of two experiments performed in duplicate. Inset: Western blot of HMGB1 in the medium bathing D16 cells 48 h after the beginning of the experiment. HMGB1 was still present in the medium of restimulated cells, but not in the medium of cells stimulated once at time 0. This experiment was repeated two times with similar results.
HMGB1 induces mesoangioblast migration
Previously, we have shown that HMGB1 is a chemoattractant for rat smooth muscle cells (Degryse et al., 2001). We then investigated whether HMGB1 is also a chemoattractant for D16 cells. In a chemotaxis assay using modified Boyden chambers, HMGB1 stimulated migration of D16 cells in a concentration-dependent manner (Fig. 3 A). pAbs raised against aa 166–181 of HMGB1 (Fig. 3 A, anti 166–181) significantly reduced the migratory response. However, the migratory response was unaffected by an anti-HMGB1 mAb that recognizes box A.
Figure 3.

HMGB1 has chemotactic activity on embryonic mesoangioblasts. (A) D16 cells were subjected to chemotaxis assays with 10, 50, or 100 ng/ml HMGB1. Data represent the average ± SD of four experiments performed in duplicate; the effect of increasing HMGB1 concentrations is highly significant (P < 0.001 in ANOVA analysis). Addition of anti-HMGB1 antibodies recognizing the peptide 166–181 significantly reduced the chemotactic response (P < 0.05 in comparison to the sample without antibody), whereas the addition of monoclonal anti-box A antibodies had no effect. (B) Chemotactic activity on D16 cells of various HMGB1 fragments (all at 10 ng/ml). ABbt has a chemotactic effect comparable with full-length HMGB1 (P < 0.05 of proteins vs. medium alone). In contrast, boxes A and B and the AB didomain have no significant chemotactic activity. Bars represent the average ± SD of three experiments performed in duplicate. Asterisks indicate statistical significance (P< 0.01). (C) Schematic representation of full-length HMGB1, boxes A and B, the didomain AB, and tailless HMGB1 (ABbt). (D) Western blot with anti-RAGE antibodies on total D16 cell extract. (E) D16 cells transfected with dnRAGE-expressing plasmid or pCDNA3 empty vector were assayed for chemotaxis in response to medium with or without HMGB1. Cells transfected with dnRAGE exhibited a significant decrease in migration in comparison to cells transfected with control plasmid. Asterisks indicate statistical significance (P < 0.05).
We also tested the individual domains of HMGB1 for the ability to induce D16 cell migration (Fig. 3 B). Neither box A nor box B alone induced appreciable migration. The didomain fragment (comprising boxes A and B) had a nonsignificant effect, whereas the ABbt fragment, which only lacks the acidic tail (Fig. 3 C), was as potent as the full-length protein.
Residues 150–183 were identified as the HMGB1 segment that interacts with RAGE (Huttunen et al., 2002). Remarkably, the anti-HMGB1 antibodies that block D16 migration (Fig. 3 A) specifically interact with the amino acid stretch between residues 166 and 181 (Fig. 3 C), and therefore occlude the RAGE-interacting surface. The mAb that recognizes box A cannot prevent the interaction with RAGE.
These data suggested that RAGE is the receptor that mediates the HMGB1 chemoattractant function. In D16 cells, RNA profiling (unpublished data) indicates that they express RAGE, and RAGE protein is detectable by Western blot (Fig. 3 D). We then cotransfected D16 cells with plasmids expressing nuclear YFP and dnRAGE (lacking the intracytoplasmic domain) (Hofmann et al., 1999), or with the control empty vector. The entire cell population was subjected to a chemotaxis assay with Boyden chambers; however, only yellow fluorescent cells were scored. Fig. 3 E shows that mesoangioblasts transfected with dnRAGE were severely impaired in their ability to respond to extracellular HMGB1, supporting the conclusion that HMGB1 signals prevalently through RAGE.
HMGB1 induces mesoangioblast migration across endothelial monolayers
Mesoangioblasts are vessel-associated stem cells that can migrate to damaged tissues through the general circulation (Sampaolesi et al., 2003), and have the ability to transit through the endothelial barrier. We tested whether HMGB1 could also promote the transmigration of mesoangioblasts across an endothelial monolayer grown on the septum between the chambers of a Boyden apparatus. When we added 100 ng/ml HMGB1 to the medium in the lower chamber, the number of D16 cells crossing the monolayer increased eightfold when compared with medium without any addition (Fig. 4). HMGB1 has higher potency than VEGF, a signaling molecule known to promote cell migration across endothelial barriers. VEGF induces profound cytoskeletal reorganization of endothelial cells, characterized by the formation of trans-cytoplasmic stress fibers and the disassembly of adherens junctions, which are important to maintain the endothelial barrier function (Esser et al., 1998; Rousseau et al., 2000). Stimulation with HMGB1 caused a similar response in endothelial cells grown in vitro as monolayers (unpublished data).
Figure 4.

HMGB1 induces the transit of mesoangioblasts through an endothelial monolayer. D16 cells were placed in the upper compartment of Boyden apparatuses. The lower chambers contained RPMI alone (medium), RPMI plus 100 ng/ml HMGB1, or RPMI plus 10 ng/ml VEGF; chambers were separated by a confluent endothelial cell monolayer grown on polycarbonate filters. HMGB1 significantly stimulated D16 transmigration (P < 0.01). Bars represent the aver- age ± SD of three experiments performed in duplicate. Panels beside the bar graph show D16 cells stained with Giemsa after migration, toward medium alone or containing HMGB1. Asterisk indicates statistical significance (P < 0.05).
HMGB1 directs mesoangioblast homing in vivo
Next, we assessed HMGB1's ability to control mesoangioblast migration in vivo. We loaded heparin-Sepharose beads with HMGB1 at the concentration of 3 μg/μl and injected them with a fine needle into the tibialis anterior muscle of mice. D16 cells transduced by a lentiviral vector causing the expression of nuclear LacZ were injected after 30 min through the proximal femoral artery (see Materials and methods). The mice were killed after 24 h and the tibialis anterior muscle was removed, sectioned, and analyzed by immunohistochemistry. Muscles injected with HMGB1-loaded beads showed a considerable swelling compared with both sham-injected muscles (unpublished data) and muscles injected with unloaded heparin-Sepharose beads (Fig. 5 A), suggesting that HMGB1 caused considerable muscle inflammation. This is consistent with HMGB1's role as a proinflammatory cytokine.
Figure 5.

HMGB1 attracts mesoangioblasts in vivo. D16 cells were first transduced with a lentiviral vector encoding nuclear LacZ, and were then injected through the femoral artery of mice where heparin-Sepharose beads (either unloaded or loaded with HMGB1) had been injected in the tibialis anterior muscle. Mice were killed after 24 h. (A) Tibialis anterior muscles injected with HMGB1-loaded and control beads. (B–D) Cryosections of muscles treated with control heparin-Sepharose beads (control) or HMGB1-coated heparin-Sepharose beads (HMGB1). Arrows indicate the beads. Sections were stained with X-gal, and mesoangioblasts (arrowheads) appear blue. Mesoangioblasts were found in large clusters (C) or as isolated cells (D) only in muscles injected with HMGB1-coated beads. (E) Number of migrating D16 cells in tibialis anterior muscles of wild-type mice treated with HMGB1-loaded beads (gray bar; n = 2) or control beads coinjected with 1 μg LPS (black bar; n = 3). White bar represents the number of D16 cells found in tibialis anterior muscles of α-SG−/− dystrophic mice (n = 2).
Muscle sections were stained with X-gal, and LacZ+ (blue) cells were scored using computer-assisted imaging techniques. Large groups of blue cells were found in the vicinity of HMGB1-loaded beads (Fig. 5 C); a minority of sections displayed individual blue cells dispersed throughout the muscle (Fig. 5 D). The sections from muscles injected with unloaded beads had no blue cells at all (Fig. 5 B).
The number of mesoangioblasts colonizing healthy muscle implanted with HMGB1-loaded beads compares well with the number of mesoangioblasts homing into dystrophic muscle (Fig. 5 E). Inflammation was present in both HMGB1-treated wild-type and α-SG null muscle, and may be necessary to promote mesoangioblast invasion. However, inflammation by itself is not sufficient. Heparin-Sepharose beads injected together with 1 μg LPS in healthy muscle did cause inflammation and tissue swelling (unpublished data), but did not attract any mesoangioblast (Fig. 5 E). These observations indicate that HMGB1 is able to recruit mesoangioblasts in vivo.
The biological action of HMGB1 on adult mesoangioblasts and lin− hematopoietic stem cells
The previous experiments identified a role for extracellular HMGB1 on the migration and proliferation of embryonic mesoangioblasts. Next, we asked whether HMGB1 had an effect on adult mesoangioblasts as well. We used adult mesoangioblasts isolated from bone marrow (G1 clone; see Materials and methods); these cells express the same markers (CD34, Flk-1, Sca-1, and MEF2D) and show similar differentiation potency and comparable growth rate as mesoangioblasts isolated from embryos (unpublished data). Fig. 6 summarizes our findings. HMGB1 causes adult mesoangioblast proliferation (Fig. 6 A), chemotaxis, and transmigration (Fig. 6 B); like embryonic mesoangioblasts, adult ones can be recruited by HMGB1 into the tibialis anterior muscle (Fig. 6 C). Similar effects were also observed in additional experiments with different adult mesoangioblast lines derived from the aorta of 8-wk-old mice (unpublished data).
Figure 6.

Effect of HMGB1 on adult mesoangioblasts. (A) Mesoangioblasts of the G1 clone, obtained from mouse bone marrow, were grown in RPMI medium containing 1, 10, or 30 ng/ml HMGB1. For comparison, G1 cells were also grown in RPMI medium alone or in RPMI plus 20% FCS. (B) Migration of G1 cells toward the lower chamber of Boyden apparatuses containing RPMI (medium) or RPMI plus 10 ng/ml HMGB1 (HMGB1). In the migration experiment, the chambers were separated by a filter; in the transmigration experiment, the chambers were separated by a filter overgrown with a monolayer of endothelial cells. Each bar represents the average ± SD of three experiments, and the arrows indicate statistical significance (P < 0.05). (C) G1 cells were labeled with DiI and then injected through the femoral artery of mice where heparin-Sepharose beads (either loaded with HMGB1 or unloaded) had been implanted in the tibialis anterior muscle (arrows). Top, phase contrast; bottom, fluorescence. G1 cells (red fluorescence) migrate in the vicinity of HMGB1-loaded cells; no G1 cells are detected near control beads.
We then tested whether HMGB1 could also attract other types of stem cells. We injected a similar number of murine lineage-negative (lin−) hematopoietic stem cells in the femoral artery of mice that had been implanted with HMGB1-loaded beads. A few cells were indeed found around the beads (unpublished data), indicating that HMGB1 may have a general chemoattractant activity toward all or many types of stem cells. However, the response was quantitatively far less robust; ∼10-fold fewer hematopoietic cells migrated to the vicinity of beads compared with mesoangioblasts. This may be due to a quantitative difference of the chemoattractant properties of HMGB1 toward different stem cells, or to the different ability of stem cells to traverse the endothelium and invade muscle tissue. In fact, mesoangioblasts colonize dystrophic muscle in the mouse model of limb-girdle dystrophy much better than hematopoietic stem cells (unpublished data).
Mesoangioblasts expressing dnRAGE still home to dystrophic muscle
We chose mesoangioblasts as the object of our experiments precisely because they have the ability to navigate to dystrophic muscle; furthermore, the experiments shown above indicate that mesoangioblasts respond to extracellular HMGB1 in healthy muscle. Is then HMGB1 relevant for mesoangioblast homing to dystrophic muscle?
Both dystrophic and healthy muscles contain HMGB1, but HMGB1 is much more abundant in dystrophic muscle, as assayed by Western blot (Fig. 7 A). By immunohistochemistry (Fig. 7 B), it appears that both healthy and dystrophic muscle cells contain a similar amount of HMGB1 in their nuclei. However, infiltrating cells in dystrophic muscle contain a large amount of HMGB1, both in the cytoplasm and in the nucleus. The cytoplasmic location of HMGB1 indicates that inflammatory cells are activated, and that they have acetylated HMGB1 extensively (Gardella et al., 2002; Bonaldi et al., 2003).
Figure 7.

HMGB1 expression and mesoangioblast migration in α-SG −/− dystrophic muscles. (A) Western blot analysis of HMGB1 expression levels in tibialis anterior muscles from α-SG−/− dystrophic and wild-type mice. (B) Hematoxylin and eosin staining (H&E) of tibialis anterior muscle sections from α-SG−/− dystrophic and wild-type mice. Different sections from the same muscles were processed for immunofluorescence (right). A considerable number of foci containing inflammatory cells were evident in α-SG−/− dystrophic muscles, and these contained HMGB1 in the cytoplasm in addition to the nucleus (anti-HMGB1, green; DAPI, red pseudocolor). (C) Fraction of dnRAGE expressing D16 cells before and after injection and homing to tibialis anterior muscles of α-SG−/− dystrophic mice. About 500 cells were counted before and after injection in two mice. The difference before and after migration is not statistically different. (D) Number of migrating D16 cells in tibialis anterior muscles of α-SG−/− dystrophic mice injected with control beads (n = 2) or HMGB1-loaded beads (n = 2). Cells expressing dnRAGE and GFP are indicated in green; the nonexpressing cells are indicated in gray. The difference in total number of cells, and in cells not expressing dnRAGE, is significant (P < 0.05). The difference in cells expressing dnRAGE is not statistically significant.
Next, we injected into the femoral artery of α-SG−/− mice a mesoangioblast population that had been transfected with dnRAGE receptor and GFP. About 30% of cells expressed dnRAGE and GFP (green), and were unresponsive to bacterially made HMGB1 in vitro (Fig. 3 E) and in healthy muscle (unpublished data); all cells were also labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI; red). After homing to the dystrophic tissue, the ratio of green to red cells was again ∼30% (Fig. 7 C). This experiment indicates that mesoangioblasts do not need the RAGE receptor to home into dystrophic muscle. When we injected dystrophic muscles with HMGB1-loaded and control beads, many more mesoangioblasts homed to the HMGB1-loaded beads (Fig. 7 D). However, about the same number of dnRAGE-expressing green mesoangioblasts homed to control and HMGB1-loaded beads; the excess mesoangioblasts homing to HMGB1-loaded beads were red. Thus, mesoangioblasts can indeed use the RAGE receptor to home to the vicinity of injected unmodified HMGB1, even in dystrophic muscle.
Discussion
The discovery that necrotic cells release HMGB1 in the extracellular space, whereas apoptotic cells do not, led us to propose that extracellular HMGB1 is a signal of tissue damage (Scaffidi et al., 2002; Bianchi and Manfredi, 2004). Here, we have tested a stringent prediction: a signal of tissue damage should recruit stem cells, and should promote their proliferation. We have used mesoangioblasts because they can migrate inside damaged muscle and proliferate there to reconstruct the tissue (Sampaolesi et al., 2003).
Mesoangioblasts migrate in vitro in response to HMGB1 gradients, and proliferate in vitro in response to a continuing supply of HMGB1 as sole mitogenic factor in serum-free medium. RAGE appears to be the receptor involved: (1) the segment of HMGB1 protein required for mesoangioblast migration coincides with the segment required for RAGE signaling (Huttunen et al., 2002); (2) RAGE protein and mRNA are present in mesoangioblasts; and (3) dnRAGE significantly reduces the response of mesoangioblasts to HMGB1. In vitro, HMGB1 also induces the loss of cell–cell contacts in endothelial cell monolayers, and impairs their barrier function against the transit of cells.
The in vitro findings suggest that HMGB1 might have the characteristics required to attract mesoangioblasts in vivo. Thus, we performed an experiment where mesoangioblasts were injected in the femoral artery (and thus accumulated in the microvascular compartment of downstream skeletal muscles) while HMGB1 was released by beads implanted into the tibialis anterior muscle. Mesoangioblasts did migrate to the vicinity of HMGB1-loaded beads.
HMGB1 binds tightly to heparin (Bianchi, 1988), and it is difficult to measure or estimate the concentration of HMGB1 released locally in the muscle tissue. However, we can roughly estimate that each heparin-Sepharose bead contained ∼25 pg HMGB1, the equivalent of 50 nuclei, and that only a fraction of this would be released. Remarkably, the HMGB1 released in healthy muscle tissue under these conditions is able to promote both significant inflammation (as shown by fluid retention in the muscle and the recruitment of inflammatory cells around the HMGB1-loaded beads) and the recruitment of mesoangioblasts. Control beads recruited no mesoangioblasts, even when inflammation was elicited by coinjected LPS.
We also tested whether HMGB1 is able to attract hematopoietic stem cells. Lin− cells derived from bone marrow did home to the vicinity of HMGB1-loaded beads, but their quantitative response was far below that of mesoangioblasts. Hematopoietic stem cells also have a reduced ability to home to dystrophic muscle (unpublished data). We do not know with certainty whether this is due to a lower chemoattractant activity of HMGB1 toward lin− cells or to a reduced ability of lin− cells to home to the interior of muscle tissue. There is a clear possibility that other types of stem cells might also use extracellular HMGB1 as a cue to homing to damaged tissue.
The role of the HMGB1–RAGE interaction in the homing of mesoangioblasts to dystrophic muscle
Mesoangioblasts can home into dystrophic muscle (Sampaolesi et al., 2003), and our data identify extracellular HMGB1 and RAGE as a ligand/receptor pair sufficient for mesoangioblast homing in vivo. However, contrary to expectations, mesoangioblasts expressing dnRAGE also home into dystrophic muscle. The HMGB1–RAGE interaction is thus sufficient, but not necessary, in dystrophic muscle. This is indeed puzzling, and we discuss possible explanations.
The simplest explanation is that dystrophic muscle contains a lot of HMGB1, but such HMGB1 is entirely intracellular, either because inflammatory cells retain HMGB1 rather than secreting it, or because secreted HMGB1 is immediately destroyed. Infiltrating cells in dystrophic muscle cells contain HMGB1 in the cytoplasm, an indication that they are activated. However, HMGB1 secretion requires at least two signals: an inflammatory signal promotes HMGB1 acetylation and its relocation from the nucleus to the cytoplasm and into secretory vesicles (Bonaldi et al., 2003), and a secretion signal (extracellular ATP or lysophosphatidylcholine) promotes exocytosis (Gardella et al., 2002). The secretory signal may be absent in dystrophic muscle.
A second possibility is that HMGB1 is secreted by inflammatory cells, but acetylated HMGB1 attracts mesoangioblasts via an as yet unidentified receptor. In other words, RAGE would be activated by unmodified HMGB1, but not by acetylated HMGB1.
A third possibility is that dystrophic muscle uses an as yet unidentified chemoattractor (different from HMGB1 and binding to a receptor different from RAGE) to attract mesoangioblasts. The existence of a parallel signaling pathway that does not require HMGB1 as one of its signaling components would be required if no HMGB1 is released within dystrophic muscle, but is still very possible even if acetylated or unmodified HMGB1 is indeed released in dystrophic muscle.
None of these possible explanations is easily testable for the time being: (1) we have no way to determine whether HMGB1 is secreted by inflammatory cells in dystrophic muscle (assays for HMGB1 in blood have insufficient sensitivity); (2) the form of HMGB1 secreted by inflammatory cells and acetylated on specific lysines cannot be produced in meaningful amounts; and (3) alternative chemoattractors for mesoangioblasts have not been identified yet. However, the recognition that unmodified HMGB1 is sufficient to attract mesoangioblasts both in healthy and dystrophic muscle is highly valuable. Unmodified HMGB1 can be produced in bacteria in unlimited amounts, and can be used as a tool for research in stem cell mobilization, and potentially to improve the reconstruction of dystrophic muscle.
Materials and methods
HMGB1 and antibodies
Expression and purification of the full-length HMGB1 protein and fragments thereof was performed as described previously (Müller et al., 2001). Endotoxins were removed by passage through Detoxy-Gel columns (Pierce Chemical Co.).
Rabbit polyclonal anti-HMGB1 antibodies raised against peptide 166–181 were purchased from BD Biosciences, pAbs against box A were purchased from MBL International Corporation. The anti-RAGE goat antibody was purchased from CHEMICON International. Western blots were performed as described previously (Degryse et al., 2001), and films were scanned on a color scanner (StudioStar; Agfa).
Cells
Bovine aorta endothelial cells were isolated from a section of the thoracic aorta of a freshly slaughtered calf as described previously (Palumbo et al., 2002). Mesoangioblast cell lines were isolated from the dorsal aorta of mouse embryos (D16) or bone marrow (G1), cloned, and expanded as described previously (Minasi et al., 2002). Clones were checked for the presence of CD34, Kit, Flk1, and MEF2D markers. For the in vivo experiments, embryonic mesoangioblasts (D16) were transduced with a lentiviral vector encoding for nuclear LacZ, whereas adult mesoangioblasts (G1) were labeled with DiI (Molecular Probes, Inc.).
Lin− cells were isolated from CD-1 mice using the SpinSep™ kit (StemCell Technologies Inc.) according to the manufacturer's instructions. In brief, 2 × 107 bone marrow cells, obtained by flushing femurs and tibias with PBS containing 2% FCS, were subjected to centrifugation and were resuspended in 1 ml of the same flushing buffer; 10 μl SpinSep™ antibody cocktail (containing antibodies against CD5, CD45R, CD11b, TER 119, Gr-1, and neutrophils) was added to the cell suspension and incubated at 4°C for 30 min. After washing, 100 μl of the SpinSep™-dense particles was added to the cells and incubated on ice for 20 min. Cells were then diluted and layered on top of 4 ml SpinSep™ density medium and centrifuged for 10 min at 1,200 g at RT. The cell fraction enriched for hematopoietic progenitors (0.11–1.3 × 106 cells) was located in the interface between layers. After washing, cells were used for intra-arterial delivery.
Proliferation assay
Cells were seeded in 6-well plates (105 cells/well) and grown in RPMI supplemented with 20% FCS. After 24 h, the medium was replaced with serum-free RPMI for 16 h. Subsequently, the cells were grown with medium alone, or medium with the addition of 20% FCS or HMGB1 at the concentration of 1, 3, 10, and 30 ng/ml. Cells were counted after 1, 2, and 3 d, and trypan blue dye exclusion was used as an indicator of cell viability. All experiments were performed three times in duplicate.
Flow cytometry
Mesoangioblasts starved overnight were grown in RPMI medium alone, RPMI plus 20% FCS, or RPMI plus 100 ng/ml HMGB1 for 6, 12, 24, or 48 h. Cells were washed, fixed in 70% ethanol, stained with 50 μg/ml propidium iodide in PBS plus 50 μg/ml RNase A, and incubated for 30 min at RT. The DNA content was measured by flow cytometry (FACScan™; Becton Dickinson) using the standard CellQuest software.
Estimation of the number of cell divisions
One million embryonic or adult mesoangioblasts were seeded on 100-mm dishes in RPMI supplemented with 20% FCS. After 24 h, the cells were starved in RPMI alone overnight. Cells were then washed in PBS, and 2.5 μM carboxyfluorescein diacetate succinimidyl ester (Molecular Probes, Inc.) was added for 8 min at RT. Staining was quenched by the addition of 10% FCS, and cells were washed in RPMI. Fluorescently labeled cells were then grown in RPMI alone, RPMI plus 100 ng/ml HMGB1, or RPMI plus 20% FCS, and were harvested after 48 h. Cells were then analyzed on FACScan™.
Chemotaxis assay
Cell migration was assayed using Boyden chambers (Degryse et al., 2001). In brief, PVP-free polycarbonate filters with 8-μm pores (Costar) were coated with 5 μg/ml porcine skin gelatin (Sigma-Aldrich). Serum-free RPMI (negative control), RPMI containing 10, 50, or 100 ng/ml HMGB1, and RPMI with 20% serum (positive control) were placed in the lower chambers. D16 cells were grown in RPMI plus 10% FCS, starved overnight, washed twice with PBS to eliminate any floating cells, and harvested with trypsin. 50,000 cells resuspended in 200 μl RPMI were placed in the upper chambers and incubated at 37°C in 5% CO2 for 16 h. Cells remaining on the upper surface of the filters were mechanically removed, and those which had migrated to the lower surface were fixed with ethanol, stained with Giemsa stain (modified; Sigma-Aldrich), and counted at 400× in 10 random fields per filter. Assays were performed in triplicate and repeated three times in independent experiments.
Transmigration assay
Bovine aorta endothelial cells were grown in DME plus 10% FCS on polycarbonate transwell inserts (3-μm pores; Costar) for 5 d until they formed a monolayer. The inserts were then placed between chambers in Boyden apparatuses, and the tightness of monolayers was checked by measuring the diffusion of BSA between chambers. Mesoangioblasts (105 cells in 100 μl RPMI) were placed in the upper compartments, and RPMI containing HMGB1 or VEGF (mature 121 aa variant of human VEGF expressed in Escherichia coli, purchased from R&D Systems) was placed in the lower compartments (500 μl). After 8 h at 37°C, the filters were removed and the result was evaluated as described for the chemotaxis assay. These experiments were performed three times in duplicate.
Cell transfection
dnRAGE is a RAGE truncation lacking the entire intracytoplasmic domain (Hofmann et al., 1999). pNLS1-YFP was constructed like pNLS1-GFP (Bonaldi et al., 2003). One million D16 cells were cotransfected with 1 μg pNLS1-YFP and 3 μg plasmid encoding dnRAGE or pCDNA3 empty vector, using FuGENE™ reagent (Roche). After 24 h, the medium was replaced with RPMI plus 20% FCS; after an additional 24 h, the chemotaxis assay was performed as described above. Only YFP-positive cells were considered.
In the mesoangioblast homing experiment, cells were transfected in a similar way, but using pNLS1-GFP instead of pNLS1-YFP, and were then labeled with DiI. The ratio of green to red cells was evaluated by counting ∼500 cells before and after injection and homing.
Preparation of HMGB1-loaded beads
34-μm-diam heparin-Sepharose beads were recovered from a HiTrap™ heparin HP column (Amersham Biosciences) and were extensively washed in PBS. Beads (20 μl packed volume) were then incubated for 1 h at 4°C with 60 μg HMGB1, harvested by centrifugation, washed twice with PBS, and resuspended in PBS. SDS-PAGE was performed to check the amount of HMGB1 on the beads.
Intra-artery delivery of stem cells in mice
Heparin-Sepharose beads (a slurry containing 3 μg beads in 20 μl PBS), either loaded with HMGB1 or not, were injected with an insulin syringe into tibialis anterior muscles of 6-wk-old female CD-1 mice (three per group). After 1 h, mesoangioblasts or hematopoietic stem cells (4 × 105 cells/animal) were injected through the femoral artery as described previously (Torrente et al., 2001); animals were killed 24 h later. For histochemistry analysis, samples of tibialis anterior muscles were frozen in liquid nitrogen–cooled isopentane and were cryostat sectioned. 10-μm thick serial muscle sections were stained with X-gal for the experiment with LacZ-labeled cells (Totsugawa et al., 2002), or visualized directly under the fluorescence microscope for the experiment with DiI, and GFP was indicated.
Immunofluorescence and immunoblots to detect HMGB1 in muscle
Tibialis anterior muscles of α-SG−/− and C57Bl6 wild-type mice were removed after bead implantation and mesoangioblast injection, frozen in liquid nitrogen, and cryostat sectioned. Serial muscle sections were fixed in 4% PFA, permeabilized, saturated, and processed for immunofluorescence with rabbit anti-HMGB1 antibody (1:500 dilution) followed by Alexa Fluor® 488–conjugated goat anti–rabbit Ig (Molecular Probes, Inc.). Images were taken with a microscope (S100 TV; Carl Zeiss MicroImaging, Inc.) equipped with a 32×/0.4 Ph2 LD Achroplan objective (Carl Zeiss MicroImaging, Inc.).
For immunoblots, whole tibialis anterior muscles were removed and homogenized in PBS containing Protease Inhibitor Cocktail (Sigma-Aldrich). Protein concentrations were measured using a Bradford assay. Immunoblotting was performed using rabbit anti-HMGB1 (BD Biosciences) and anti-β tubulin (Sigma-Aldrich) antibodies.
Digital images
Digital images were elaborated using Adobe Photoshop®, and included in figures using Adobe Illustrator®.
Acknowledgments
A special thanks to the referees of this paper, who suggested a connection between dystrophic muscle invasion by mesoangioblasts and increased HMGB1 release. We thank Maria del Angels de Planell i Saguer and Elisa Latorre for constructing the dnRAGE plasmid, and Dr. Kevin P. Campbell (University of Iowa College of Medicine, Iowa City, IA) for providing the α-SG−/− dystrophic mice.
This work was supported by grants to G. Cossu from Telethon and the Parent Project Organization for Duchenne Muscular Dystrophy, and to M.E. Bianchi from the Ministries of Health and Education, University and Research of Italy, and the Associazione Italiana Ricerca sul Cancro. M.E. Bianchi and R. Palumbo have an indirect financial interest, being named as inventors on a patent application based partially on the data reported in this paper.
R. Palumbo and M. Sampaolesi contributed equally to this paper.
Abbreviations used in this paper: α-SG, α-sarcoglycan; DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; dnRAGE, dominant-negative mutant of RAGE; HMGB1, high mobility group box 1; lin−, murine lineage negative; LPS, lipopolysaccharide; RAGE, receptor for advanced glycation end products.
References
- Abraham, E., J. Arcaroli, A. Carmody, H. Wang, and K.J. Tracey. 2000. HMG-1 as a mediator of acute lung inflammation. J. Immunol. 165:2950–2954. [DOI] [PubMed] [Google Scholar]
- Agresti, A., and M.E. Bianchi. 2003. HMGB proteins and gene expression. Curr. Opin. Genet. Dev. 13:170–178. [DOI] [PubMed] [Google Scholar]
- Andersson, U., H. Wang, K. Palmblad, A.C. Aveberger, O. Bloom, H. Erlandsson-Harris, A. Janson, R. Kokkola, M. Zhang, H. Yang, and K.J. Tracey. 2000. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 192:565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi, M.E. 1988. Interaction of a protein from rat liver nuclei with cruciform DNA. EMBO J. 7:843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi, M.E., and A. Manfredi. 2004. Chromatin and cell death. Biochim. Biophys. Acta. In press. [DOI] [PubMed] [Google Scholar]
- Bonaldi, T., F. Talamo, P. Scaffidi, D. Ferrera, A. Porto, A. Bachi, A. Rubartelli, A. Agresti, and M.E. Bianchi. 2003. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 22:5551–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin, M. 1999. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol. Cell. Biol. 19:5237–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calogero, S., F. Grassi, A. Aguzzi, T. Voigtländer, P. Ferrier, S. Ferrari, and M.E. Bianchi. 1999. The lack of chromosomal protein HMG1 does not disrupt cell growth, but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 22:276–280. [DOI] [PubMed] [Google Scholar]
- Cossu, G., and P. Bianco. 2003. Mesoangioblasts—vascular progenitors for extravascular mesodermal tissues. Curr. Opin. Genet. Dev. 13:537–542. [DOI] [PubMed] [Google Scholar]
- Degryse, B., T. Bonaldi, P. Scaffidi, S. Müller, M. Resnati, F. Sanvito, G. Arrigoni, and M.E. Bianchi. 2001. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J. Cell Biol. 152:1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser, S., M.G. Lampugnani, M. Corada, E. Dejana, and W. Risau. 1998. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 111:1853–1865. [DOI] [PubMed] [Google Scholar]
- Ferrari, G., G. Cusella-De Angelis, M. Coletta, E. Paolucci, A. Stornaiuolo, G. Cossu, and F. Mavilio. 1998. Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 279:1528–1530. [DOI] [PubMed] [Google Scholar]
- Fiuza, C., M. Bustin, S. Talwar, M. Tropea, E. Gerstenberger, J.H. Shelhamer, and A.F. Suffredini. 2003. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 101:2652–2660. [DOI] [PubMed] [Google Scholar]
- Gardella, S., C. Andrei, D. Ferrera, L.V. Lotti, M.R. Torrisi, M.E. Bianchi, and A. Rubartelli. 2002. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 3:995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guazzi, S., A. Strangio, A.T. Franzi, and M.E. Bianchi. 2003. HMGB1, an architectural chromatin protein and extracellular signalling factor, has a spatially and temporally restricted expression pattern in mouse brain. Gene Expr. Patterns. 3:29–33. [DOI] [PubMed] [Google Scholar]
- Hofmann, M.A., S. Drury, C. Fu, W. Qu, A. Taguchi, Y. Lu, C. Avila, N. Kambham, A. Bierhaus, P. Nawroth, et al. 1999. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 97:889–901. [DOI] [PubMed] [Google Scholar]
- Hori, O., S.D. Yan, S. Ogawa, K. Kuwabara, M. Matsumoto, D. Stern, and A.M. Schmidt. 1995. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. J. Biol. Chem. 270:25752–25761. [DOI] [PubMed] [Google Scholar]
- Huttunen, H.J., C. Fages, J. Kuja-Panula, A.J. Ridley, and H. Rauvala. 2002. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 62:4805–4811. [PubMed] [Google Scholar]
- LaBarge, M.A., and H.M. Blau. 2002. Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell. 111:589–601. [DOI] [PubMed] [Google Scholar]
- Lyons, A.B., and C.R. Parish. 1994. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 171:131–137. [DOI] [PubMed] [Google Scholar]
- Minasi, M.G., M. Riminucci, L. De Angelis, U. Borello, B. Berarducci, A. Innocenzi, A. Caprioli, D. Sirabella, M. Baiocchi, R. De Maria, et al. 2002. The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development. 129:2773–2783. [DOI] [PubMed] [Google Scholar]
- Müller, S., M.E. Bianchi, and S. Knapp. 2001. Thermodynamics of HMG1 interaction with duplex DNA. Biochemistry. 40:10254–10261. [DOI] [PubMed] [Google Scholar]
- Orlic, D., J. Kajstura, S. Chimenti, I. Jakoniuk, S.M. Anderson, B. Li, J. Pickel, R. McKay, B. Nadal-Ginard, D.M. Bodine, et al. 2001. Bone marrow cells regenerate infarcted myocardium. Nature. 410:701–705. [DOI] [PubMed] [Google Scholar]
- Palumbo, R., C. Gaetano, A. Antonini, G. Pompilio, E. Bracco, L. Ronnstrand, C.H. Heldin, and M.C. Capogrossi. 2002. Different effects of high and low shear stress on platelet-derived growth factor isoform release by endothelial cells: consequences for smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 22:405–411. [DOI] [PubMed] [Google Scholar]
- Ronfani, L., M. Ferraguti, L. Croci, C.E. Ovitt, H.R. Schöler, G.G. Consalez, and M.E. Bianchi. 2001. Reduced fertility and spermatogenesis defects in mice lacking chromosomal protein Hmgb2. Development. 128:1265–1273. [DOI] [PubMed] [Google Scholar]
- Rousseau, S., F. Houle, H. Kotanides, L. Witte, J. Waltenberger, J. Landry, and J. Huot. 2000. Vascular endothelial growth factor (VEGF)-driven actin-based motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J. Biol. Chem. 275:10661–10672. [DOI] [PubMed] [Google Scholar]
- Sampaolesi, M., Y. Torrente, A. Innocenzi, R. Tonlorenzi, G. D'Antona, M.A. Pellegrino, R. Barresi, N. Bresolin, M.G. Cusella De Angelis, K.P. Campbell, et al. 2003. Cell therapy of α-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. 301:487–492. [DOI] [PubMed] [Google Scholar]
- Sappington, P.L., R. Yang, H. Yang, K.J. Tracey, R.L. Delude, and M.P. Fink. 2002. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology. 123:790–802. [DOI] [PubMed] [Google Scholar]
- Scaffidi, P., T. Misteli, and M.E. Bianchi. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 418:191–195. [DOI] [PubMed] [Google Scholar]
- Torrente, Y., J.P. Tremblay, F. Pisati, M. Belicchi, B. Rossi, M. Sironi, F. Fortunato, M. El Fahime, M.G. D'Angelo, N.J. Caron, et al. 2001. Intraarterial injection of muscle-derived CD34+Sca-1+ stem cells restores dystrophin in mdx mice. J. Cell Biol. 152:335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totsugawa, T., N. Kobayashi, T. Okitsu, H. Noguchi, T. Watanabe, T. Matsumura, M. Maruyama, T. Fujiwara, M. Sakaguchi, and N. Tanaka. 2002. Lentiviral transfer of the LacZ gene into human endothelial cells and human bone marrow mesenchymal stem cells. Cell Transplant. 11:481–488. [PubMed] [Google Scholar]
- Vaccari, T., M. Beltrame, S. Ferrari, and M.E. Bianchi. 1998. Hmg4, a new member of the Hmg1/2 gene family. Genomics. 49:247–252. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejon, C., M.J. Yusta-Boyo, C. Fernandez-Moreno, and F. de Pablo. 2003. Locally born olfactory bulb stem cells proliferate in response to insulin-related factors and require endogenous insulin-like growth factor-I for differentiation into neurons and glia. J. Neurosci. 23:895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., O. Bloom, M. Zhang, J.M. Vishnubhakat, M. Ombrellino, J. Che, A. Frazier, H. Yang, S. Ivanova, L. Borovikova, et al. 1999. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 285:248–251. [DOI] [PubMed] [Google Scholar]