

Abstract
CDC13 encodes a telomere-binding protein that prevents degradation of telomeres. cdc13-1 yeast grown at the nonpermissive temperature undergo G2/M arrest, progressive chromosome instability, and subsequent cell death. Recently, it has been suggested that cell death in the cdc13-1 mutant is an active process characterized by phenotypic hallmarks of apoptosis and caspase activation. In this work, we show that cell death triggered by cdc13-1 is independent of the yeast metacaspase Yca1p and reactive oxygen species but related to cell cycle arrest per se. Inactivating YCA1 or depleting reactive oxygen species does not increase viability of cdc13-1 cells. In turn, caspase activation does not precede cell death in the cdc13-1 mutant. Yca1p activity assayed by cell binding of mammalian caspase inhibitors is confounded by artifactual labeling of dead yeast cells, which nonspecifically bind fluorochromes. We speculate that during a prolonged cell cycle arrest, cdc13-1 cells reach a critical size and die by cell lysis.
Keywords: FITC-VAD-FMK; apoptosis; CDC13; YCA1; Saccharomyces cerevisiae
Introduction
Stability of chromosome ends in yeast cells is maintained by a telomere-specific DNA–protein complex (Lydall, 2003). A key component of this telomere cap is a single-stranded DNA-binding protein, Cdc13p (Nugent et al., 1996). In a cdc13-1 mutant incubated at the nonpermissive temperature, the uncapped telomeres are recognized as double strand breaks, leading to accumulation of single-stranded DNA at the ends of chromosomes (Garvik et al., 1995; Maringele and Lydall, 2002), inducing a DNA-damage checkpoint signal (Garvik et al., 1995) and promoting a terminal cell cycle arrest at G2/M (Weinert and Hartwell, 1993). When these cells are returned to permissive temperature after prolonged arrest, only a small proportion recover. This cell death has been attributed to loss of essential genes located in the subtelomeric regions (Garvik et al., 1995; Booth et al., 2001), conversion of single-stranded DNA at telomeres to unrepaired lethal lesions (Jia et al., 2004), and/or the formation of chromosome end-to-end fusions (DuBois et al., 2002).
Recently, Qi et al. (2003) have demonstrated that dying cdc13-1 cells display phenotypic markers of apoptosis such as exposure of phosphatidylserine on the outer leaflet of the plasma membrane, accumulation of reactive oxygen species (ROS), and induction of caspase activity based on retention of the caspase inhibitor FITC-VAD-FMK. Binding of the valyl-alanyl-aspartyl (VAD) sequence to an activated mammalian caspase allows the fluoromethyl ketone (FMK) moiety to react with the active site cysteine, resulting in inactivation and fluorescent labeling of the protein (Grabarek and Darzynkiewicz, 2002). Yeast express a single caspase-like protease, Yca1p. Like mammalian caspases, Yca1p undergoes cleavage, which activates its proteolytic activity toward several synthetic caspase substrates in vitro (Madeo et al., 2002a). Yca1p activity can be inhibited by Z-VAD-FMK (Madeo et al., 2002a). Uncleaved FITC-VAD-FMK can be washed out readily from viable cells, but dead cells retain FITC and can be detected by green fluorescence in flow cytometry. Thus, in vivo labeling of yeast with FITC-VAD-FMK has been attributed to bona fide caspase activation (Madeo et al., 2002a; Qi et al., 2003; Herker et al., 2004; Wadskog et al., 2004). Deletion of MEC1, a yeast ATM/ATR kinase homologue, prevented this cdc13-1 apoptosis, suggesting that unprotected telomeres generate a MEC1-dependent apoptotic death signal (Qi et al., 2003).
In this report, we show that cell death induced by the inactivation of Cdc13p is not dependent on the caspase-like protease Yca1 or increased ROS production. We further demonstrate that flow cytometric measurements of caspase activity and ROS production, using fluorochrome-conjugated caspase inhibitors and dihydrorhodamine 123 (DHR123), respectively, is confounded by binding to already-dead yeast cells. This work suggests that flow cytometric analysis of dying yeast cells is subject to significant artifacts and indicates a need to reinterpret prior results on mechanisms of yeast cell death.
Results and discussion
Yeast caspase-like protease is not involved in cell death triggered by cdc13-1 mutation
As previously reported, when the cdc13-1 mutant was transferred from vegetative growth in rich media at 24 to 37°C, cells arrested as large-budded dumbbells and gradually lost viability as measured by colony formation (Weinert and Hartwell, 1993). By 24 h, viability was decreased by at least 105 (unpublished data). By this time point, a majority of mutant cells appeared as weakly refractive “ghost” cells in phase microscopy, indicative of loss of plasma membrane integrity and subsequent leaking of cell contents. We considered that these cells might represent the postapoptotic population. To determine the degree to which the Yca1p caspase contributes to cell death in the cdc13-1 strain, we deleted the YCA1 gene in both wild-type and cdc13-1 backgrounds. Next, cdc13-1, yca1Δ, cdc13-1 yca1Δ, and wild-type cells were incubated at 24 or 37°C and caspase activity was measured by staining with the caspase inhibitor FITC-VAD-FMK (Fig. 1 A). We confirmed that arrested cdc13-1 cells label with FITC-VAD-FMK (Qi et al., 2003), suggesting activation of Yca1 metacaspase and induction of apoptosis (Fig. 1 A). Surprisingly, after 24 h at 37°C, cdc13-1 yca1Δ double mutants showed the same proportion of FITC-positive cells as cdc13-1, 80% (Fig. 1 A). In turn, deletion of YCA1 did not improve survival of cdc13-1 cells at the restrictive temperature (Fig. 1 B). FITC-VAD-FMK labeling of chronologically aged yca1 null mutant yeast has been reported (Herker et al., 2004), but attributed to other proteases that may recognize the caspase substrate. We considered an alternative hypothesis that FITC-VAD-FMK might also label dead or dying yeast cells unrelated to caspase activation or apoptosis.
Figure 1.
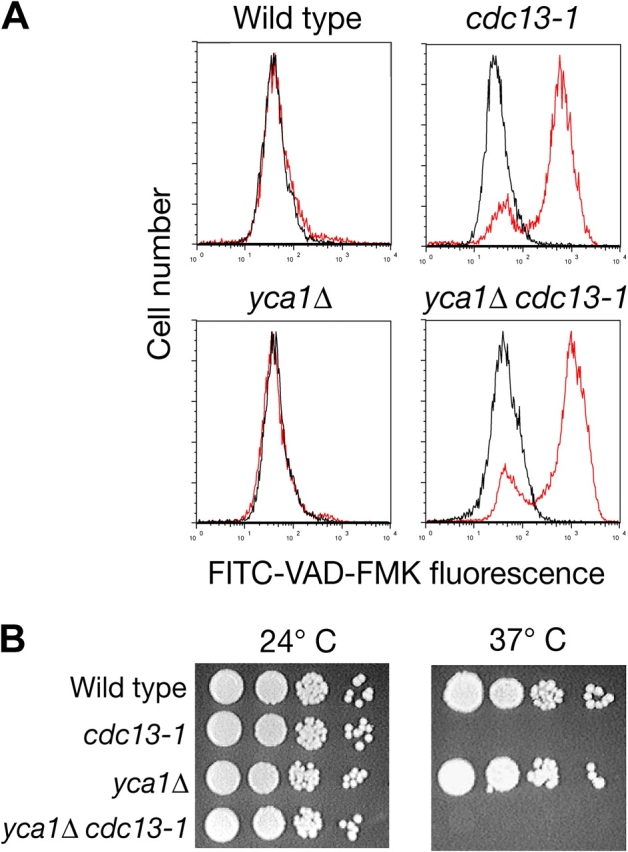
Deletion of YCA1 does not prevent accumulation of mammalian caspase inhibitor FITC-VAD-FMK or cell death in the cdc13-1 mutant. Wild-type, yca1Δ, cdc13-1, and yca1Δ cdc13-1 strains were incubated at 24°C or at nonpermissive 37°C for 24 h. (A) Caspase activation was assayed by staining with 10 μM FITC-VAD-FMK and measurement of fluorescence was done by flow cytometry. Black line, 24°C; red line, 37°C. (B) Viability was analyzed by serial 10-fold dilutions spotted onto YPD plates and incubated for 3 d at 24°C.
Thus, we performed dual staining of cdc13-1 cells with FITC-VAD-FMK and propidium iodide (PI), which is a reporter of plasma membrane integrity. Labeling with FITC-VAD-FMK and/or PI can distinguish four distinct cell states: (a) live nonapoptotic cells (FITC-negative/PI-negative), (b) live cells with activated caspases/early apoptotic cells (FITC-positive/PI-negative), (c) dead postapoptotic cells (FITC-positive/PI-positive), and (d) dead nonapoptotic cells (FITC-negative/PI-positive; Fig. 2 A). This assay can help establish the sequence of events leading to cell death (Jayaraman, 2003). First, we performed a series of control studies by comparing unstained, terminally arrested cdc13-1 cells with those stained only with PI and/or FITC-VAD-FMK (Fig. 2 A). Using bivariate flow cytometry analysis, cells detected by forward scatter are plotted by red fluorescence on the X-axis, reporting PI staining, and green fluorescence on the Y-axis, proportional to FITC-VAD-FMK binding. As expected, unstained cells were all found in the lower left quadrant (Fig. 2 A, a), FITC-VAD-FMK staining alone yielded cells in the upper left quadrant (Fig. 2 A, b), while PI stained cells appeared in the lower right quadrant (Fig. 2 A, d). Staining with PI or FITC-VAD-FMK alone revealed a similar number of labeled cells, 80% of the population. In turn, double staining revealed almost no FITC-positive/PI-negative (early apoptotic) cells, suggesting that only cells that had already lost plasma membrane integrity could be labeled with FITC-VAD-FMK (Fig. 2 A). To detect caspase activation and/or apoptosis at time points earlier than 24 h, we similarly stained cdc13-1 cells incubated at 37°C for 2, 4, 6, and 8 h. We were unable to detect FITC-positive cells not permeable to PI, and identical results at all time points were obtained for the cdc13-1 yca1Δ double mutant (unpublished data).
Figure 2.
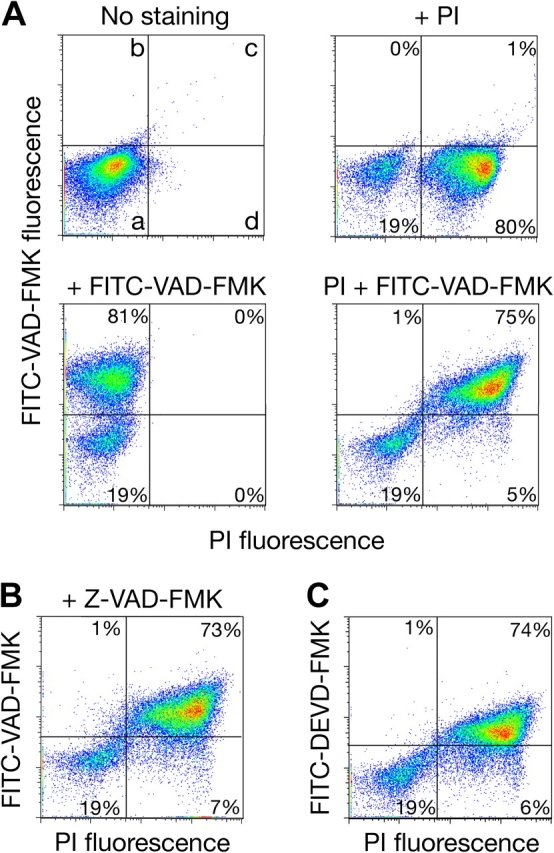
Cell death and binding of caspase inhibitors in the cdc13-1 mutant at nonpermissive temperature. (A) cdc13-1 cells in early logarithmic phase were shifted from 24 to 37°C for 24 h and then stained with 5 μg/ml propidium iodide (PI) and/or 10 μM FITC-VAD-FMK before analysis by flow cytometry. Four distinct subpopulations (a–d) can be identified on these scatter plots as described in the text. (B) Presence of caspase inhibitor Z-VAD-FMK does not block binding of FITC-VAD-FMK or cell death in the cdc13-1 mutant. (C) Dead cdc13-1 cells are unspecifically labeled with caspase inhibitor substrate FITC-DEVD-FMK, uncleavable by Yca1 protease (Madeo et al., 2002a).
To exclude a role for a caspase-like protease in cdc13-1 cell death and confirm the nonspecific binding of FITC-VAD-FMK to dead cells, we pretreated cdc13-1 cells for 2 h at 24°C with the nonfluorescent caspase inhibitor Z-VAD-FMK before shifting to 37°C (Fig. 2 B). Addition of Z-VAD-FMK to mammalian cells blocks the binding of FITC-VAD-FMK to mammalian caspases and promotes cell survival (Earnshaw et al., 1999). Yet, as shown in Fig. 2 B, the presence of Z-VAD-FMK throughout the 37°C incubation had no effect on subsequent FITC-VAD-FMK binding or cell death in cdc13-1 cells. Madeo et al. (2002a) used a fluorimetric assay to show that Yca1p cleaves several synthetic substrates, but does not cleave the colorimetric caspase-3/7 substrate DEVD-AMC (aspartyl-glutamyl-valyl-aspartyl-7-amino-4-methylcoumarin). Thus, cdc13-1 cells, live or dead, should not be labeled with FITC-DEVD-FMK, even if they express activated Yca1p caspase. However, most dead cdc13-1 cells were stained with this inhibitor, which is consistent with nonspecific binding of fluorescently tagged compounds to inviable cells (Fig. 2 C). These results suggest that death of cdc13-1 cells during cell cycle arrest is independent of caspase-like proteases including Yca1.
Dead yeast cells accumulate free fluorochromes
Having shown that dead yeast nonspecifically bind fluorescent-conjugated caspase inhibitors, we wondered how these cells retain FITC-VAD-FMK. Recently, Pozarowski et al. (2003) reported that human early apoptotic cells bind free fluorescein. To test this possibility in the cdc13-1 mutant, cells incubated 24 h at 37°C were stained with 10 μM of free FITC or fluorescein (Fig. 3 A). Flow cytometry analysis revealed that dead cdc13-1 cells preferentially bind free fluorochromes (Fig. 3 A). To further investigate this result, we examined whether or not the cause or rate of cell death might influence binding of FITC-VAD-FMK. An exponentially growing culture of wild-type yeast cells was split and one part was incubated for 5 min at 80°C to rapidly kill the cells and denature all proteins. Both the live and heat-killed cells were immediately stained with FITC-VAD-FMK and PI. All heat-killed cells accumulated both PI and FITC-VAD-FMK caspase inhibitor (Fig. 3 B). We conclude that nonspecific binding confounds the use of FITC-VAD-FMK and other fluorescent caspase inhibitors in detecting apoptosis in yeast.
Figure 3.
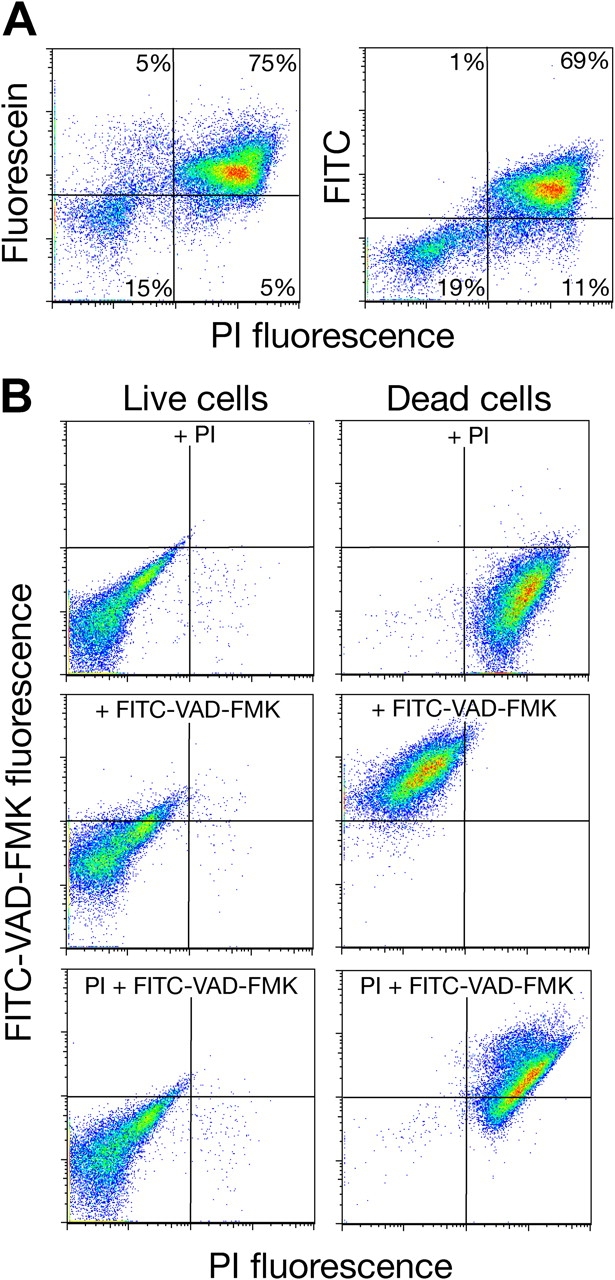
Dead yeast cells unspecifically bind fluorescein and FITC or FITC-conjugated caspase inhibitor FITC-VAD-FMK. (A) After 24-h incubation at 37°C, the cdc13-1 mutant cells were stained with 10 μM fluorescein or FITC and subjected to flow cytometry analysis. (B) Exponentially growing wild-type cells were either heated to 80°C for 5 min (Dead cells) or maintained at 24°C (Live cells). Cells from both cultures were subsequently labeled with PI and caspase inhibitor FITC-VAD-FMK. Green (FITC) and red (PI) fluorescence of stained cells was measured by flow cytometry.
Increased ROS production does not mediate cell death in cdc13-1 mutant
It is well established that ROS play a major role in signaling and/or effector functions in apoptosis (for review see Kannan and Jain, 2000). Indeed, antioxidants such as N-acetylcysteine (NAC) can block or delay animal cell death (Fleury et al., 2002). ROS may also have a critical role in programmed cell death in organisms such as yeast that lack homologues of key metazoan proapoptotic proteins (Madeo et al., 2002b). Consistent with this hypothesis, elevated ROS production was observed in Saccharomyces cerevisiae cells responding to death-inducing insults such as perturbations of the cell cycle, ectopic Bax expression, high levels of peptide mating pheromone, oxidative stress, and aging (Madeo et al., 2002b). Accumulation of ROS has been reported in cdc13-1 cells incubated at the restrictive temperature (Qi et al., 2003). In addition, apoptosis-like death in yeast can be suppressed by treatment with free-radical spin traps (Madeo et al., 1999) or by expression of the antiapoptotic proteins CED-9, Bcl-2, or Bcl-xL, which appear to protect yeast from oxidative stress (Chen et al., 2003). However, a pathway directly linking ROS to yeast cell death has yet to be elucidated.
To reexamine ROS production in cdc13-1 cells, the mutant was incubated at 37°C for 24 h and incubated with DHR123, a nonfluorescent precursor of green fluorescent rhodamine 123. In the presence of ROS, DHR123 can be oxidized so that rhodamine 123 accumulates in the mitochondria of live cells (Henderson and Chappell, 1993). Flow cytometry of cdc13-1 cells incubated 24 h at 37°C and then stained with DHR123 revealed that ∼85% showed high levels of green fluorescence distributed into “intermediate” and “high” fluorescence subpopulations (Fig. 4 A). Bivariate analysis of ROS content and PI permeability was used to explore if either subpopulation of fluorescent cells might be consistent with ROS accumulation in the absence of, or before, cell death. Strikingly, the high fluorescence population remained impermeable to PI, suggesting that ROS production indeed precedes cell death in the cdc13-1 mutant. However, depletion of ROS by addition of NAC (Fig. 4 A), or by expression of Bcl-xL (Fig. 4 B), did not abrogate cell death. Thus, production of ROS is not necessary for death commitment in cdc13-1 cells.
Figure 4.
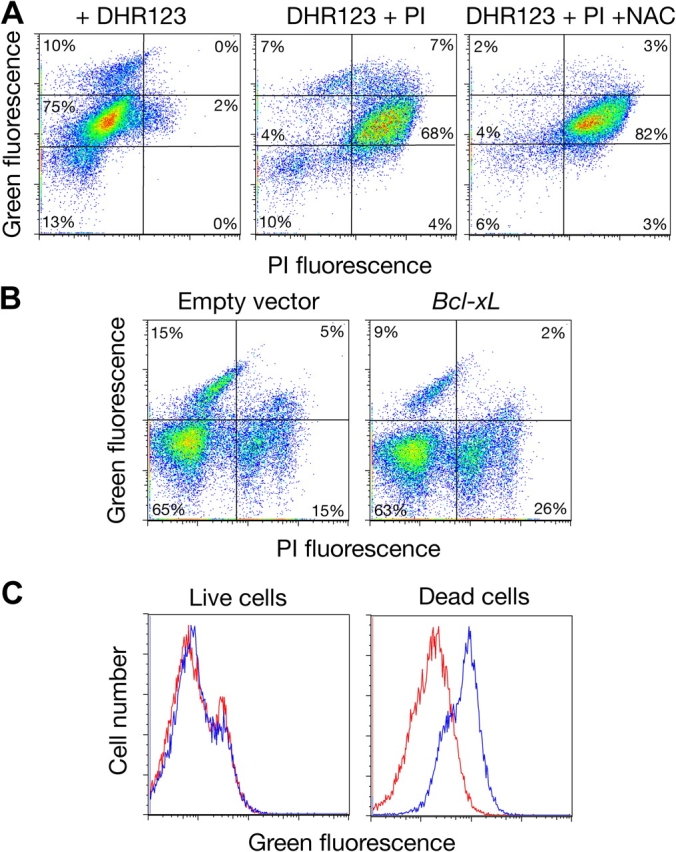
Staining of cdc13-1 after incubation at the restrictive temperature and heat-killed wild-type yeast cells with DHR123. (A) cdc13-1 cells were incubated at 37°C overnight in the presence or absence of the antioxidant N-acetylcysteine (NAC), stained with DHR123 and PI, and analyzed by flow cytometry. DHR123 staining reveals two populations of green fluorescence–positive cells: dead cells show intermediate green fluorescence, whereas a population with bright green fluorescence represents cells producing ROS. Control experiments with wild-type and cdc13-1 cells at 24°C revealed only a single nonfluorescent population restricted to the bottom left quadrant (not depicted). (B) Expression of the mammalian antiapoptotic protein Bcl-xL in cdc13-1 cells decreases ROS production but does not suppress death after 8-h incubation at 37°C. Functional expression of Bcl-xL in yeast cells was confirmed by rescuing toxicity of Bax expression. (C) Heat-killed wild-type cells show intermediate green fluorescence after staining with DHR123. Live and dead cells were prepared as described for Fig. 3 and stained with DHR123 (blue line) or left unstained (red line).
Because the intermediate fluorescence population was also permeable to PI and the antioxidant NAC could not reduce green fluorescence in these cells, we wondered if dead cells might stain with DHR123 without producing ROS. Notably, it was previously reported that dead cells of a Bax-expressing Kluyveromyces lactis strain nonspecifically labeled with DHR123 (Poliakova et al., 2002). Thus, we performed DHR123 staining on live and heat-killed wild-type yeast cells. Although live cells did not exhibit green fluorescence, fluorescent staining equivalent to that of the intermediate population (e.g., Fig. 4 A) was detected in dead cells (Fig. 4 C). Our data suggest that ROS measurements in yeast may require double staining with a membrane integrity reporter and antioxidant treatment as controls to exclude nonspecific binding to dead cells.
Cell cycle–dependent death in the cdc13-1 mutant
Our data argue against the hypothesis that cdc13-1 cells die by a MEC1 checkpoint signal-mediated pathway involving ROS signaling and caspase-dependent apoptosis (Qi et al., 2003). It is well known that cdc13-1 mec1 double mutants do not arrest at 37°C and rapidly lose viability as measured by colony formation by comparison to cdc13-1 alone. We were struck that such mutants, though replicatively dead, apparently retain plasma membrane integrity, indicating a direct link between cell cycle arrest and cell death.
Upon shift from 24 to 37°C, cdc13-1 cells arrest uniformly at G2/M and maintain high viability for several hours during which time cells continue to grow, increasing two- to threefold in diameter (unpublished data). A single unrepairable double-strand break in a gratuitous plasmid can induce a prolonged G2/M arrest. Cells that fail to adapt to the checkpoint signal eventually lyse (Lee et al., 1998). Two unrepairable double strand breaks are sufficient to prevent such adaptation (Lee et al., 1998), presumably fating cells to cell death. Unprotected telomeres are likely to sufficiently activate the checkpoint signaling pathway that adaptation is blocked. When such cells reach a critical size, they might simply rupture and lyse.
Because loss of cell wall integrity due to mutations in the protein kinase C pathway can be suppressed by addition of osmotic stabilizers (Heinisch et al., 1999), we tested the viability of cdc13-1 cells after 24-h incubation at 37°C in YPD supplemented with 1.2 M sorbitol. Sorbitol did not prevent cell cycle arrest or block cell death completely, but did markedly improve colony formation in the cdc13-1 mutant (Fig. 5). Perhaps, a significant cause of cell death in cdc13-1 is lysis due to the loss of cell integrity rather than DNA damage or apoptosis.
Figure 5.

Addition of sorbitol to media increases survival of cdc13-1 cells at the restrictive temperature. cdc13-1 and wild-type cells were grown in YPD medium in the presence or absence of 1.2 M sorbitol at 37°C for 24 h. Serial 10-fold dilutions were spotted on a YPD plate and incubated for 3 d at 24°C.
These data do not exclude caspase-independent programmed cell death in the cdc13-1 mutant. Autolysis is a long-studied form of cell death in S. cerevisiae stimulated by stress with features reminiscent of apoptosis but which may be mediated by a completely distinct pathway (Zhao and Fleet, 2003). Other data suggest that temperature-sensitive mutations leading to cell cycle arrest (and even expression of Bax) may induce autophagic death in yeast (Motizuki et al., 1995; Abudugupur et al., 2002; Camougrand et al., 2003). Autophagy is a highly conserved process from yeast to humans (for review see Bursch, 2001) characterized by formation of a large vacuole called an autophagosome in which cellular components are sequestered for degradation. Interestingly, yeast cells undergoing autophagy induced by cell cycle arrest or Bax expression also show some morphological changes similar to those observed in apoptosis (Abudugupur et al., 2002; Camougrand et al., 2003).
In conclusion, cell death during cell cycle arrest in the cdc13-1 mutant may well result from processes other than degradation of critical DNA sequences at chromosome ends. Instead, we infer that cdc13-1 cell death may arise as a result of loss of cell wall integrity in oversized, large-budded cells or via an active process such as induction of autolysis and/or autophagy. Nonetheless, our work appears to rule out reactive oxygen signaling and caspase activation as critical mediators of this death despite their established roles in the characteristic apoptotic response of metazoan cells confronted by DNA damage.
Materials and methods
Yeast strains and plasmids
S. cerevisiae strains used in this study are in the W303 background. A W303 cdc13-1 mutant was originally obtained from K. Nasmyth (Research Institute of Molecular Pathology, Vienna, Austria). Deletion of YCA1 in wild-type and cdc13-1 cells was performed by direct transformation with PCR product amplified from genomic DNA of the yca1Δ::kanMX strain from the BY4741 gene deletion library and selection on G418 media. The deletions were confirmed by PCR. The pOW4-Bcl-x L plasmid (CEN, URA3, ADH1 promoter) and corresponding control vector pOW4 have been described previously (Vander Heiden et al., 2002) and provided by J. Brace and C. Rudin (Johns Hopkins University, Baltimore, MD). Yeast cells were grown in standard rich YPD media or selective synthetic complete media. Plates were photographed using a Pixera Professional digital camera (Pixera), macro lens, and software.
Fluorochrome labeling
Yeast cells (5 × 106) were collected by centrifugation, washed once in 1 ml PBS, resuspended in 200 μl PBS containing 10 μM FITC-VAD-FMK (CaspACE™; Promega), FITC-DEVD-FMK (Caspase-3 Activity Detection kit; Oncogene Research Products), FITC (Fluka), or fluorescein (Sigma-Aldrich), and incubated for 20 min in the dark. Then, cells were rinsed twice with 1 ml PBS and resuspended in 1 ml PBS for flow cytometry analysis in an LSRII (Becton Dickinson) flow cytometer/FACS®. To detect loss of membrane integrity, 5 μg of PI was added to cell suspensions before analysis. All labeling experiments were repeated at least three times with similar results.
DHR123 staining
Yeast cells were labeled with DHR123 (Molecular Probes) as described previously (Madeo et al., 1999). After staining, cells were washed with PBS, resuspended in the same buffer or PBS containing 5 μg/ml of PI, and analyzed by flow cytometry.
Acknowledgments
We thank C. Rudin and A. Szallies for helpful suggestions and reagents and A. Javaheri for critical reading of the manuscript.
This work was generously supported by a National Institutes of Health grant (GM60443) and a Translational Grant from the Ludwig Fund for Cancer Research. S.J. Kron is a Scholar of the Leukemia & Lymphoma Society.
Abbreviations used in this paper: DHR123, dihydrorhodamine 123; FMK, fluoromethyl ketone; NAC, N-acetyl-cysteine; PI, propidium iodide; ROS, reactive oxygen species; VAD, valyl-alanyl-aspartyl.
References
- Abudugupur, A., K. Mitsui, S. Yokota, and K. Tsurugi. 2002. An ARL1 mutation affected autophagic cell death in yeast, causing a defect in central vacuole formation. Cell Death Differ. 9:158–168. [DOI] [PubMed] [Google Scholar]
- Bursch, W. 2001. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 8:569–581. [DOI] [PubMed] [Google Scholar]
- Camougrand, N., A. Grelaud-Coq, E. Marza, M. Priault, J.J. Bessoule, and S. Manon. 2003. The product of the UTH1 gene, required for Bax-induced cell death in yeast, is involved in the response to rapamycin. Mol. Microbiol. 47:495–506. [DOI] [PubMed] [Google Scholar]
- Chen, S.R., D.D. Dunigan, and M.B. Dickman. 2003. Bcl-2 family members inhibit oxidative stress-induced programmed cell death in Saccharomyces cerevisiae. Free Radic. Biol. Med. 34:1315–1325. [DOI] [PubMed] [Google Scholar]
- Booth, C., E. Griffith, G. Brady, and D. Lydall. 2001. Quantitative amplification of single-stranded DNA (QAOS) demonstrates that cdc13-1 mutants generate ssDNA in a telomere to centromere direction. Nucleic Acids Res. 29:4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBois, M.L., Z.W. Haimberger, M.W. McIntosh, and D.E. Gottschling. 2002. A quantitative assay for telomere protection in Saccharomyces cerevisiae. Genetics. 161:995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw, W.C., L.M. Martins, and S.H. Kaufmann. 1999. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68:383–424. [DOI] [PubMed] [Google Scholar]
- Garvik, B., M. Carson, and L. Hartwell. 1995. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol. 15:6128–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabarek, J., and Z. Darzynkiewicz. 2002. In situ activation of caspases and serine proteases during apoptosis detected by affinity labeling their enzyme active centers with fluorochrome-tagged inhibitors. Exp. Hematol. 30:982–989. [DOI] [PubMed] [Google Scholar]
- Heinisch, J.J., A. Lorberg, H.P. Schmitz, and J.J. Jacoby. 1999. The protein kinase C-mediated MAP kinase pathway involved in the maintenance of cellular integrity in Saccharomyces cerevisiae. Mol. Microbiol. 32:671–680. [DOI] [PubMed] [Google Scholar]
- Henderson, L.M., and J.B. Chappell. 1993. Dihydrorhodamine 123: a fluorescent probe for superoxide generation? Eur. J. Biochem. 217:973–980. [DOI] [PubMed] [Google Scholar]
- Herker, E., H. Jungwirth, K.A. Lehmann, C. Maldener, K.U. Fröhlich, S. Wissing, S. Büttner, M. Fehr, S. Sigrist, and F. Madeo. 2004. Chronological aging leads to apoptosis in yeast. J. Cell Biol. 164:501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury, C., B. Mignotte, and J.L. Vayssiere. 2002. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 84:131–141. [DOI] [PubMed] [Google Scholar]
- Jayaraman, S. 2003. Intracellular determination of activated caspases (IDAC) by flow cytometry using a pancaspase inhibitor labeled with FITC. Cytometry. 56A:104–112. [DOI] [PubMed] [Google Scholar]
- Jia, X., T. Weinert, and D. Lydall. 2004. Mec1 and Rad53 inhibit formation of single-stranded DNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics. 166:753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan, K., and S.K. Jain. 2000. Oxidative stress and apoptosis. Pathophysiology. 7:153–163. [DOI] [PubMed] [Google Scholar]
- Lee, S.E., J.K. Moore, A. Holmes, K. Umezu, R.D. Kolodner, and J.E. Haber. 1998. Saccharomyces Ku70, Mre11/Rad50, and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 94:399–409. [DOI] [PubMed] [Google Scholar]
- Lydall, D. 2003. Hiding at the ends of yeast chromosomes: telomeres, nucleases and checkpoint pathways. J. Cell Sci. 116:4057–4065. [DOI] [PubMed] [Google Scholar]
- Madeo, F., E. Fröhlich, M. Ligr, M. Grey, S.J. Sigrist, D.H. Wolf, and K.U. Fröhlich. 1999. Oxygen stress: a regulator of apoptosis in yeast. J. Cell Biol. 145:757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo, F., E. Herker, C. Maldener, S. Wissing, S. Lächelt, M. Herlan, M. Fehr, K. Lauber, S.J. Sigrist, S. Wesselborg, and K.U. Fröhlich. 2002. a. A caspase-related protease regulates apoptosis in yeast. Mol. Cell. 9:911–917. [DOI] [PubMed] [Google Scholar]
- Madeo, F., S. Engelhardt, E. Herker, N. Lehmann, C. Maldener, A. Proksch, S. Wissing, and K.U. Fröhlich. 2002. b. Apoptosis in yeast: a new model system with applications in cell biology and medicine. Curr. Genet. 41:208–216. [DOI] [PubMed] [Google Scholar]
- Maringele, L., and D. Lydall. 2002. EXO1-dependent single-stranded DNA at telomeres activates subsets of DNA damage and spindle checkpoint pathways in budding yeast yku70Δ mutants. Genes Dev. 16:1919–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motizuki, M., S. Yokota, and K. Tsurugi. 1995. Autophagic death after cell cycle arrest at the restrictive temperature in temperature-sensitive cell division cycle and secretory mutants of the yeast Saccharomyces cerevisiae. Eur. J. Cell Biol. 68:275–287. [PubMed] [Google Scholar]
- Nugent, C.I., T.R. Hughes, N.F. Lue, and V. Lundblad. 1996. Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science. 274:249–252. [DOI] [PubMed] [Google Scholar]
- Poliakova, D., B. Sokolikova, J. Kolarov, and L. Sabova. 2002. The antiapoptotic protein Bcl-xL prevents the cytotoxic effect of Bax, but not Bax-induced formation of reactive oxygen species, in Kluyveromyces lactis. Microbiology. 148:2789–2795. [DOI] [PubMed] [Google Scholar]
- Pozarowski, P., X. Huang, D.H. Halicka, B. Lee, G. Johnson, and Z. Darzynkiewicz. 2003. Interactions of fluorochrome-labeled caspase inhibitors with apoptotic cells: a caution in data interpretation. Cytometry. 55A:50–60. [DOI] [PubMed] [Google Scholar]
- Qi, H., T.S. Li, D. Kuo, A. Nur-E-Kamal, and L.F. Liu. 2003. Inactivation of Cdc13p triggers MEC1-dependent apoptotic signals in yeast. J. Biol. Chem. 278:15136–15141. [DOI] [PubMed] [Google Scholar]
- Vander Heiden, M.G., J.S. Choy, D.J. VanderWeele, J.L. Brace, M.H. Harris, D.E. Bauer, B. Prange, S.J. Kron, C.B. Thompson, and C.M. Rudin. 2002. Bcl-xL complements Saccharomyces cerevisiae genes that facilitate the switch from glycolytic to oxidative metabolism. J. Biol. Chem. 277:44870–44876. [DOI] [PubMed] [Google Scholar]
- Wadskog, M., C. Maldener, A. Proksch, F. Madeo, and L. Adler. 2004. Yeast lacking the SRO7/SOP1-encoded tumor suppressor homologue show increased susceptibility to apoptosis-like cell death on exposure to NaCl stress. Mol. Biol. Cell. 15:1436–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, T.A., and L.H. Hartwell. 1993. Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics. 134:63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J., and G.H. Fleet. 2003. Degradation of DNA during the autolysis of Saccharomyces cerevisiae. J. Ind. Microbiol. Biotechnol. 30:175–182. [DOI] [PubMed] [Google Scholar]