

Abstract
Fas (APO-1/CD95) is the prototypic death receptor, and the molecular mechanisms of Fas-induced apoptosis are comparably well understood. Here, we show that Fas activates NFκB via a pathway involving RIP, FADD, and caspase-8. Remarkably, the enzymatic activity of the latter was dispensable for Fas-induced NFκB signaling pointing to a scaffolding-related function of caspase-8 in nonapoptotic Fas signaling. NFκB was activated by overexpressed FLIPL and FLIPS in a cell type–specific manner. However, in the context of Fas signaling both isoforms blocked FasL-induced NFκB activation. Moreover, down-regulation of both endogenous FLIP isoforms or of endogenous FLIPL alone was sufficient to enhance FasL-induced expression of the NFκB target gene IL8. As NFκB signaling is inhibited during apoptosis, FasL-induced NFκB activation was most prominent in cells that were protected by Bcl2 expression or caspase inhibitors and expressed no or minute amounts of FLIP. Thus, protection against Fas-induced apoptosis in a FLIP-independent manner converted a proapoptotic Fas signal into an inflammatory NFκB-related response.
Keywords: apoptosis; Bcl2; CD95; FasL; IL8
Introduction
In susceptible cells, membrane FasL, agonistic Fas-specific antibodies, and secondarily aggregated soluble FasL induce reorganization of inactive, preassembled Fas complexes to supramolecular Fas clusters having the capacity to signal apoptosis. Upon formation of apoptosis-competent signaling clusters, Fas is able to recruit the cytoplasmic death domain–containing adaptor protein FADD (Fas-associated death domain protein), which in turn recruits procaspase-8. In context of this death-inducing signaling complex (DISC), procaspase-8 is activated by dimerization (Boatright et al., 2003; Donepudi et al., 2003), resulting in autoproteolytic processing and release of the mature and active heterotetrameric form of the enzyme. In so called type I cells, Fas-activated caspase-8 is sufficient to trigger efficient activation of effector caspases, especially caspase-3, resulting in execution of the final steps of apoptosis (Barnhart et al., 2003). In contrast, in type II cells, caspase-8 activation is less prominent and/or caspase-3 activation is counteracted by members of the inhibitor of apoptosis (IAP) protein family (Barnhart et al., 2003). In these cells, robust induction of apoptosis is therefore dependent on mitochondrial amplification mechanisms that can be triggered by caspase-8–mediated cleavage of Bid, a BH3-only protein. The resulting truncated Bid fragment induces Bax/Bak-dependent release of apoptogenic proteins from mitochondria including Smac/Diablo (the second mitochondria-derived activator of caspase/direct IAP binding protein with low PI), HtrA2/Omi, and cytochrome c (Barnhart et al., 2003). Although cytosolic cytochrome c assembles with ATP and the scaffold protein Apaf-1 (apoptosis promoting factor-1) to the apoptosome (Shi, 2002), which activates caspase-9, Smac/Diablo and HtrA2/Omi block caspase inhibition by members of the IAP protein family (Verhagen and Vaux, 2002). Both mechanisms enhance the effect of initially DISC-activated caspase-8. Due to cell type–specific relative contributions of these proapoptotic mitochondrial events to Fas-induced apoptosis, type I and type II cells have been experimentally defined in vitro by overexpression of Bcl2 or other proteins interfering with the Bax/Bak-mediated release of apoptogenic factors. In type I cells, death receptor–induced apoptosis was not affected by Bcl2 expression, whereas in type II cells Bcl2 expression inhibited or attenuated Fas-induced apoptosis. If and to which extent the release of mitochondrial proteins can contribute to the apoptotic effects of Fas in vivo is a matter of debate. Although some reports found a protective effect in hepatocytes of Bcl2 transgenic mice against Fas-mediated apoptosis induced by agonistic antibodies (Lacronique et al., 1996; Rodriguez et al., 1996), others found no protective effect by Bcl2 when Fas was challenged with aggregated soluble FasL (Huang et al., 1999). The latter study has shown in vitro that agonistic Fas-specific antibodies, but not cross-linked FasL, are much more active on type I cells than on type II cells. Therefore, these apparent discrepancies in various studies might be caused by analyzing Fas signals of different strengths. Embryonal fibroblasts of Apaf1-deficient mice (Cecconi et al., 1998) displayed somewhat lower Fas sensitivity, and Fas-mediated liver toxicity is also reduced in mice deficient for Bid (Yin et al., 1999) or Bak and Bax (Wei et al., 2001). In contrast, thymocytes of Bcl2 transgenic mice (Strasser et al., 1995; Huang et al., 1999), of caspase-9–deficient mice (Hakem et al., 1998), and of Bak/Bax double-deficient mice (Lindsten et al., 2000) as well as Bcl2-expressing granulocytes (Villunger et al., 2000) showed no significant decrease in Fas sensitivity, suggesting a cell type–specific nonessential contribution of the intrinsic mitochondrial apoptotic pathway to Fas-induced apoptosis.
Fas-induced apoptosis is inhibited by the long and short isoform of the cellular FLICE-inhibitory protein cFLIP. Similar to caspase-8, FLIPL (FLIP-long) consists of two amino-terminal death effector domains followed by an unfunctional caspase homology domain (Krueger et al., 2001; Thome and Tschopp, 2001). FLIPS (FLIP-short) has no caspase homology domain and mainly consists of the two death effector domains of the long isoform. Although FLIPS blocks autoproteolytical maturation of Fas-FADD–bound caspase-8 completely, FLIPL arrests this process at an intermediate state (Krueger et al., 2001; Thome and Tschopp, 2001).
Although Fas has been predominantly recognized as an apoptosis inducer, there is increasing evidence for additional apoptosis-independent functions of Fas, including induction of proliferation in T cells and fibroblasts, hepatocyte regeneration, chemokine production, DC regulation, and neurite outgrowth (for review see Desbarats et al., 2003; Wajant et al., 2003). However, the molecular mechanisms of Fas signaling in most of these processes are poorly understood. In this study, we identified FADD, caspase-8, and RIP as essential components of Fas-induced NFκB signaling. Moreover, we showed that FLIPS and especially FLIPL have an inhibitory role in Fas-induced NFκB activation.
Results
Bcl2 expression in HT1080 and KB cells confers resistance against Fas-induced apoptosis
Active caspases cleave components of the NFκB signaling cascade and efficiently inhibit activation of this pathway during apoptosis (for review see Wajant et al., 2003). Therefore, we decided to analyze FasL-induced NFκB signaling and gene induction in cells protected from the apoptotic action of FasL. This can be achieved in type I and type II cells by inhibition of caspases; e.g., by pharmacological inhibitors or by expression of FLIP and in vitro in type II cells, which show a strong contribution of the intrinsic apoptotic pathway to Fas-induced apoptosis, in addition by Bcl2 overexpression. We choose the latter possibility, in contrast to pharmacological caspase-8 inhibitors and FLIP, as Bcl2 does not target the Fas signaling complex and should therefore have no influence on receptor proximal events in Fas signaling. We used KB and HT1080 cells as well as transfectants derived thereof that have been stably transfected with a GFP fusion protein of Bcl2 (Fig. 1 A). FasL-induced apoptosis in parental KB cells required sensitization by cycloheximide (CHX), an inhibitor of protein synthesis. Parental HT1080 cells already underwent significant Fas-induced apoptosis in the absence of CHX, but nevertheless responded more strongly in the presence of CHX (unpublished data). Noteworthily, the CHX concentrations used (2.5 μg/ml or less) only reduced overall protein synthesis for 10 to 30% and resulted only in marginal cell death even after extended incubation times (unpublished data). Thus, in the time scale of our analysis, CHX treatment predominantly affected expression levels of short-lived proteins including antiapoptotic factors (e.g., FLIP). Parental KB and HT1080 cells both have an ED50 value for FasL-induced apoptosis of around 0.3–2 ng/ml. The KB-GFP-Bcl2 transfectants have an ED50 value of >1 μg/ml and HT1080-GFP-Bcl2 cells showed only marginal apoptosis (<10%) at FasL concentrations >1,000-fold higher than those necessary to kill the parental cells (Fig. 1 B). Next, we checked if KB and HT1080-GFP-Bcl2 cells are protected against apoptosis induction by cells expressing membrane FasL. For this purpose, we assayed cocultures of the various cells with Rapo cells and Rapo transfectants expressing membrane FasL (Fig. 1 C). Rapo cells are a Jurkat clone devoid of Fas expression, and these cells are therefore resistant against Fas-induced apoptosis. Rapo-FasL cells induced complete killing of KB and HT1080 cells in coculture assays, whereas the parental Rapo clone showed no effect. In contrast, KB-GFP-Bcl2 and HT1080-GFP-Bcl2 cells were completely (KB) or largely (HT1080) protected against the membrane FasL-expressing Rapo cells. Apoptosis induction by Rapo-FasL was blocked by the caspase inhibitor z-IETD-fmk and Fas-Comp, a pentameric fusion protein containing the extracellular domain of Fas (Fig. 1, D and E). Thus, in KB and HT1080 cells the intrinsic apoptotic pathway makes a significant contribution to Fas-induced apoptosis, even when Fas is stimulated with membrane FasL and allows analysis of nonapoptotic Fas signaling.
Figure 1.

Bcl2 expression protects HT1080 and KB cells from Fas-mediated apoptosis. (A) FACS® analysis of pools of HT1080 and KB cells stably transfected with GFP-Bcl2. (B) Indicated cells were seeded in 96-well plates (20 × 103 per well) and challenged the next day with the indicated concentrations of soluble Flag-tagged FasL complexed with 0.5 μg/ml of the Flag-specific mAb M2 in the presence of 2.5 μg/ml CHX. After an additional 18 h, cell viability was determined by crystal violet staining. (C) Cells were seeded in 96-well plates overnight, and the next day the indicated number of membrane FasL-expressing Rapo cells or parental Rapo cells were added to each well in the presence of 2.5 μg/ml CHX. After 18 h, Rapo, Rapo-FasL, and dead cells were removed by two washes with PBS, and finally, the remaining viable cells were quantified by crystal violet staining. HT1080 (D) and KB (E) cells were challenged with Rapo-FasL cells as described in C. However, this time either the HT1080 and KB cells were pretreated with 50 μM of the caspase-8 inhibitor z-IETD-fmk or the Rapo-FasL cells with 4 μg/ml of Fas-Comp. Viability data shown are averages and SDs of triplicates.
Fas-mediated NFκB activation and gene induction in Bcl2-protected cells
Next, we analyzed FasL-induced IL8 production, as this chemokine is a well established target gene of nonapoptotic Fas signaling and of pivotal importance for FasL-induced tumor cell rejection (for review see Wajant et al., 2003). Although in KB cells there was only a minor increase in IL8 production upon Fas stimulation, significant amounts of IL8 were induced in HT1080 cells (Fig. 2 A). Remarkably, in the presence of the caspase inhibitor z-VAD-fmk, which blocks apoptosis and the NFκB inhibitory effects of caspases, FasL-induced IL8 production in HT1080 cells was three to five times higher. Similarly, the low induction of IL8 in KB cells was also enhanced in the presence of z-VAD-fmk. In the presence of low doses of CHX (2.5 μg/ml), thus under circumstances where Fas induces activation of caspases and apoptosis, FasL-induced IL8 production was almost completely blocked in both cell lines. In the case of KB cells, there was a higher basal IL8 production in the presence of CHX (Fig. 2 A). However, when apoptosis induction was blocked by z-VAD-fmk, FasL-induced IL8 production was restored and even significantly higher than in the absence of CHX. These data not only illustrate that the low concentrations of CHX used had indeed no major effect on overall protein synthesis but also suggest that CHX facilitates Fas-induced IL8 production by bringing a short-lived inhibitory protein under a critical threshold. Bcl2 expression was sufficient to substitute z-VAD-fmk to allow enhanced FasL-mediated induction of IL8. Thus, in HT1080-GFP-Bcl2 cells, FasL-induced IL8 production reached comparable levels as in z-VAD-fmk–protected parental HT1080 cells (Fig. 2 A). Treatment of HT1080-GFP-Bcl2 cells with z-VAD-fmk did not lead to a further enhancement of IL8 production. In contrast to the parental cell lines, CHX treatment did not interfere or even sensitize the GFP-Bcl2 transfectants for FasL-induced IL8 production. In the presence of CHX and the absence of apoptosis (z-VAD-fmk and GFP-Bcl2 expression), FasL-induced IL8 levels almost reached values induced by TNF, which is a very potent inducer of IL8 production (unpublished data). IL8 production was similarly induced when membrane FasL-expressing cells were used instead of cross-linked soluble FasL (Fig. 2 B). Together, these data suggest that a CHX-sensitive, and therefore short-lived, protein exists that blocks both apoptosis and gene induction by FasL upstream of Bcl2. In accordance with the central role of NFκB in regulation of the IL8 gene (Hoffmann et al., 2002), FasL activated the NFκB pathway in z-VAD-fmk– and Bcl2-protected cells (Fig. 3, A and B). IκBα degradation was transient and rapid upon TNF stimulation (Fig. 3 B). In contrast, a delayed and sustained loss of IκBα protein was observed after FasL treatment in KB and HT1080 cells and in the corresponding Bcl2-transfectants in the presence of 2.5 μg/ml CHX (Fig. 3 B). To analyze the genes induced by FasL more comprehensively and, at the same time, compare them to TNF, we determined the expression of 110 genes relevant to inflammation using a customized DNA oligonucleotide microarray developed in our laboratory (Holzberg et al., 2003). In HT1080-GFP-Bcl2 cells, 40 genes were significantly expressed, 16 of which were induced by FasL by at least 1.5-fold on average and 27 by TNF (Table I). Inspection of the more strongly regulated genes revealed that FasL and TNF, despite their different kinetics of NFκB activation, induced an almost identical pattern of genes. Moreover, many of the induced genes are established targets of the NFκB pathway; e.g., such as ICAM-1, CCL2, IL8, JunB, or IκBα. Together, these studies demonstrated that at least in vitro Bcl2 expression can convert a proapoptotic Fas signal into an inflammatory NFκB-related response.
Figure 2.

FasL induces IL8 in Bcl2-protected HT1080 and KB cells. (A) Indicated cells were seeded in 96-well plates (20 × 103 per well). The next day, the medium was changed and cells were stimulated with 200 ng/ml of soluble Flag-tagged FasL complexed with 0.5 μg/ml of the Flag-specific mAb M2 or 20 ng/ml TNF in the presence of the indicated reagents (20 μM z-VAD-fmk; 2.5 μg/ml CHX). After 6 h, supernatants were removed, cleared, and analyzed for their IL8 content using an IL8-specific ELISA. (B) Cells were seeded in 96-well plates (20 × 103 per well). The next day, the medium was changed and Rapo or Rapo-FasL cells (40 × 103 per well) were added for 6 h in the presence or absence of 20 μM z-VAD-fmk and finally IL8 production was determined as in A. Data shown are averages and SDs of triplicates.
Figure 3.
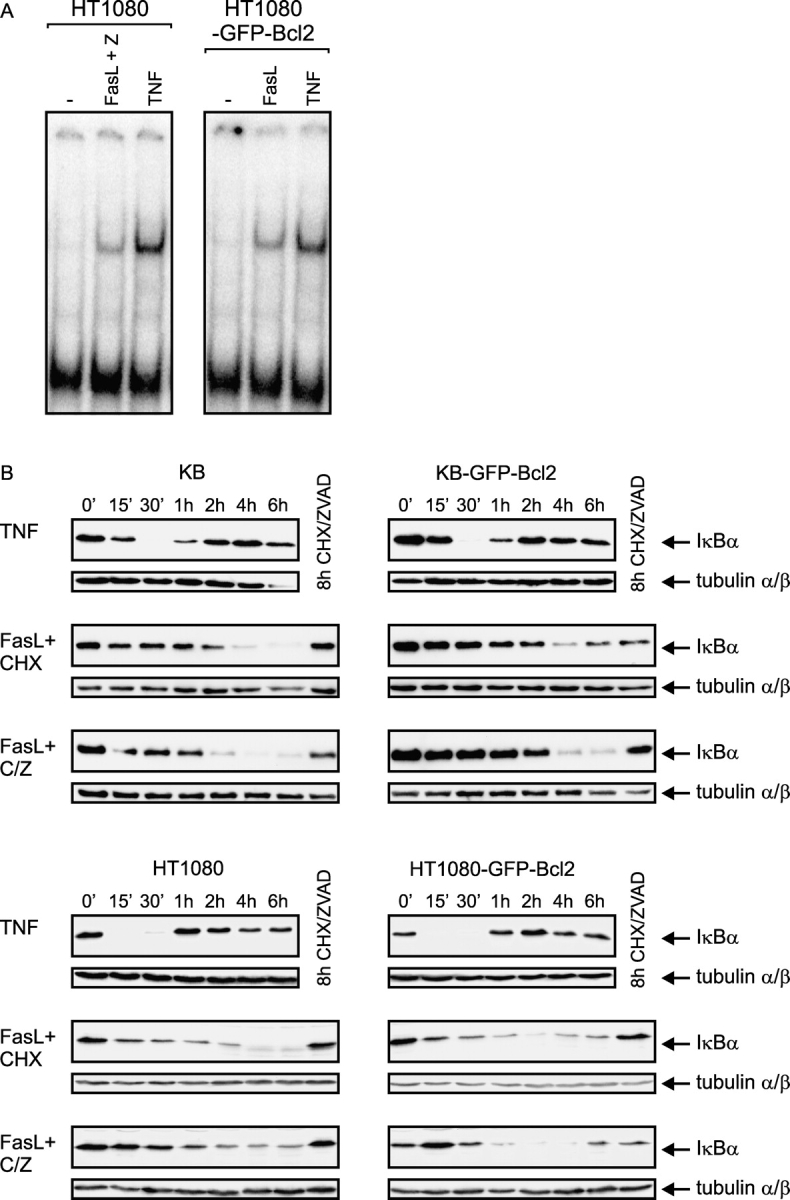
FasL induces NFκB activation in Bcl2-protected HT1080 and KB cells. (A) HT1080 and HT1080-GFP-Bcl2 cells were stimulated with M2–cross-linked Flag-FasL (200 ng/ml) or TNF (20 ng/ml), and in the case of the parental cells, the presence of 20 μM z-VAD-fmk. After 3 h, cells were analyzed for NFκB activation by electrophoretic mobility shift assay. (B) Indicated cells were stimulated for varying times with cross-linked Flag-FasL or TNF, and IκBα degradation was determined by Western blot analysis of cytosolic extracts with an IκBα-specific antibody. To control protein loads, filters were reprobed with antitubulin antibodies. Where indicated, 20 μM z-VAD-fmk and 2.5 μg/ml CHX were added 1 h before stimulation.
Table I. Comparison of the effects of FasL and TNF on inducible inflammatory gene expression of HT1080-Bcl2 cells.
| Relative induction
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FasL
|
TNF
|
|||||||||
| Accession number | Gene | Exp. 1 | Exp. 2 | Exp. 1 | Exp. 2 | |||||
| Signalling molecules | ||||||||||
| NM_001165 | hlAP-1 | 11.0 | 4.0 | 28.1 | 5.1 | |||||
| NM_020529 | IκB-α | 7.5 | 7.4 | 9.3 | 9.1 | |||||
| NM_001561 | ILA | 2.7 | 0.9 | 7.4 | 1.9 | |||||
| NM_002229 | JunB | 4.1 | 2.0 | 3.0 | 2.7 | |||||
| NM_001166 | hlAP-2 | 0.9 | 1.6 | 1.8 | 1.6 | |||||
| NM_031419 | 1.8 | 1.2 | 1.6 | 1.8 | ||||||
| NM_002228 | c-Jun | 1.3 | 1.4 | 0.9 | 0.8 | |||||
| Metabolic enzymes, miscellaneous | ||||||||||
| NM_002852 | PTX3 | 6.0 | 5.1 | 15.3 | 9.8 | |||||
| NM_000636 | MnSOD | 2.8 | 1.5 | 6.5 | 5.1 | |||||
| NM_002575 | PAI-2 | 3.2 | 1.4 | 5.0 | 3.2 | |||||
| NM_000161 | GTP cyclohydrolase I | 1.4 | 0.8 | 1.8 | 1.8 | |||||
| NM_001993 | Tissue factor | 1.5 | 1.0 | 1.1 | 0.9 | |||||
| NM_004385 | Versican | 1.3 | 0.5 | 1.0 | 1.1 | |||||
| NM_002032 | Ferritin heavy chain | 1.2 | 0.8 | 1.0 | 1.0 | |||||
| NM_000602 | PAI-1 | 0.9 | 1.2 | 0.9 | 0.9 | |||||
| Proteases | ||||||||||
| NM_002428 | MMP15 | 5.1 | 2.0 | 8.8 | 4.1 | |||||
| NM_004994 | MMP9 | 2.1 | 1.4 | 5.9 | 5.4 | |||||
| NM_002427 | MMP13 | 1.3 | 0.9 | 1.3 | 3.2 | |||||
| NM_002422 | MMP3 | 1.0 | 0.8 | 1.4 | 1.2 | |||||
| Cytokines–cytokine receptors | ||||||||||
| NM_000575 | IL-1 α | 4.9 | 2.3 | 9.1 | 3.9 | |||||
| NM_000758 | GM-CSF | 4.0 | 1.4 | 8.1 | 3.5 | |||||
| NM_000576 | IL-1 β | 3.1 | 2.2 | 7.6 | 6.2 | |||||
| NM_001066 | TNFR II | 1.7 | 0.6 | 3.0 | 1.4 | |||||
| NM_000882 | IL-12A (p35) | 1.4 | 1.1 | 1.7 | 1.9 | |||||
| NM_002341 | Lymphotoxin β | 1.3 | 0.9 | 1.6 | 1.5 | |||||
| NM_002006 | bFGF | 1.1 | 0.7 | 1.4 | 1.1 | |||||
| NM_005228 | EGFR | 0.9 | 0.9 | 1.1 | 0.9 | |||||
| NM_003266 | TLR 4 | 1.1 | 0.8 | 1.0 | 1.3 | |||||
| NM_000594 | TNF α | 1.4 | 1.9 | 0.9 | 1.6 | |||||
| NM_004513 | IL-16 | 0.9 | 0.5 | 0.8 | 1.1 | |||||
| NM_001901 | CTGF | 1.7 | 0.9 | 0.7 | 0.5 | |||||
| Chemokines–chemokine receptors | ||||||||||
| NM_002982 | CCL2 | 6.6 | 2.5 | 40.7 | 14.6 | |||||
| NM_000584 | CXCL8 | 14.8 | 8.1 | 38.4 | 13.5 | |||||
| NM_002985 | CCL5 | 1.2 | 0.9 | 2.3 | 1.5 | |||||
| NM_002986 | CCL11 | 1.2 | 0.7 | 1.8 | 1.6 | |||||
| NM_001295 | CCR1 | 1.6 | 0.8 | 1.5 | 2.1 | |||||
| NM_000648 | CCR2b | 1.1 | 0.8 | 0.9 | 1.1 | |||||
| Adhesion molecules | ||||||||||
| NM_000201 | ICAM1 | 2.2 | 1.8 | 4.1 | 4.3 | |||||
| Acute phase genes | ||||||||||
| NM_000331 | SAA 1 | 2.2 | 1.4 | 3.4 | 3.5 | |||||
| NM_001639 | SAP | 0.8 | 0.8 | 1.7 | 1.9 | |||||
HT1080-Bcl2 cells were treated with 200 ng/ml FasL for 6 h or left untreated. Parallel cells were treated with 20 ng/ml TNF for 6 h. Thereafter, total RNA was isolated from all samples and used to prepare double-stranded cDNA followed by cRNA synthesis. cRNA was labeled with Cy3 and hybridized independently to DNA microarrays containing amino-modified oligonucleotide probes, representing 110 genes relevant to inflammation as well as several housekeeping genes. Fluorescence intensities of bound cRNAs were recorded, normalized, and used to identify 40 inflammatory genes that were significantly expressed. Alterations imposed by the FasL or TNF on the inducible or basal expression of these genes were determined as a ratio of relative gene expression compared with unstimulated cells. The results from two independent experiments are shown (Exp. 1 and 2). Genes are arranged into functional groups and are ordered according to their relative induction by TNF in Exp. 1. GenBank accession numbers and gene names, respectively, are provided for identification. Genes induced for more than twofold by TNF or FasL are in bold.
Fas-induced up-regulation of IκBα occurs via FADD, caspase-8, and RIP
Next, we investigated Jurkat cells because for this cell type a variety of clones are available with defects in the expression of proteins related to NFκB and/or Fas signaling. In Jurkat cells, FasL did not induce IL8. We analyzed instead IκBα, which is a bona fide NFκB target gene (Karin and Ben-Neriah, 2000). Jurkat cells are highly sensitive toward FasL-induced apoptosis in the absence of CHX. To ensure analysis of nonapoptotic Fas signaling, we blocked caspase activity and apoptosis by adding z-VAD-fmk. Under these conditions, all parental Jurkat strains responded to FasL treatment with IκBα induction (Fig. 4, A and B, left). First, we analyzed a Jurkat clone deficient in expression of NEMO/IKKγ, a crucial component of the IKK complex (Karin and Ben-Neriah 2000), which therefore fails to activate the NFκB signaling pathway in response to a variety of inducers (Harhaj et al., 2000). In agreement with the idea that FasL-induced up-regulation of IκBα is due to the activation of NFκB, this effect was completely abolished in the NEMO/IKKγ-deficient Jurkat clone (Fig. 4 A, middle). FasL-induced up-regulation of IκBα also occurred in the presence of high concentrations of CHX (25–50 μg/ml), which fully block protein synthesis, indicating that IκBα induction is a direct consequence of Fas signaling (unpublished data). We also analyzed Jurkat cells deficient in the expression of the serine/threonine kinase RIP, which was originally identified as a death domain–containing Fas-interacting protein (Stanger et al., 1995) and which plays an essential role in NFκB activation by the death receptors TNF-R1 and TRAIL-R1 (Ting et al., 1996; Kelliher et al., 1998; Lin et al., 2000). FasL-induced up-regulation of IκBα was completely blocked in RIP-deficient Jurkat cells (Fig. 4 A, right), indicating that this kinase is also critically involved in Fas-induced NFκB activation. Remarkably, induction of cJun, which is regulated by AP1 and the JNK pathway, was only marginally affected (Fig. 4 A).
Figure 4.
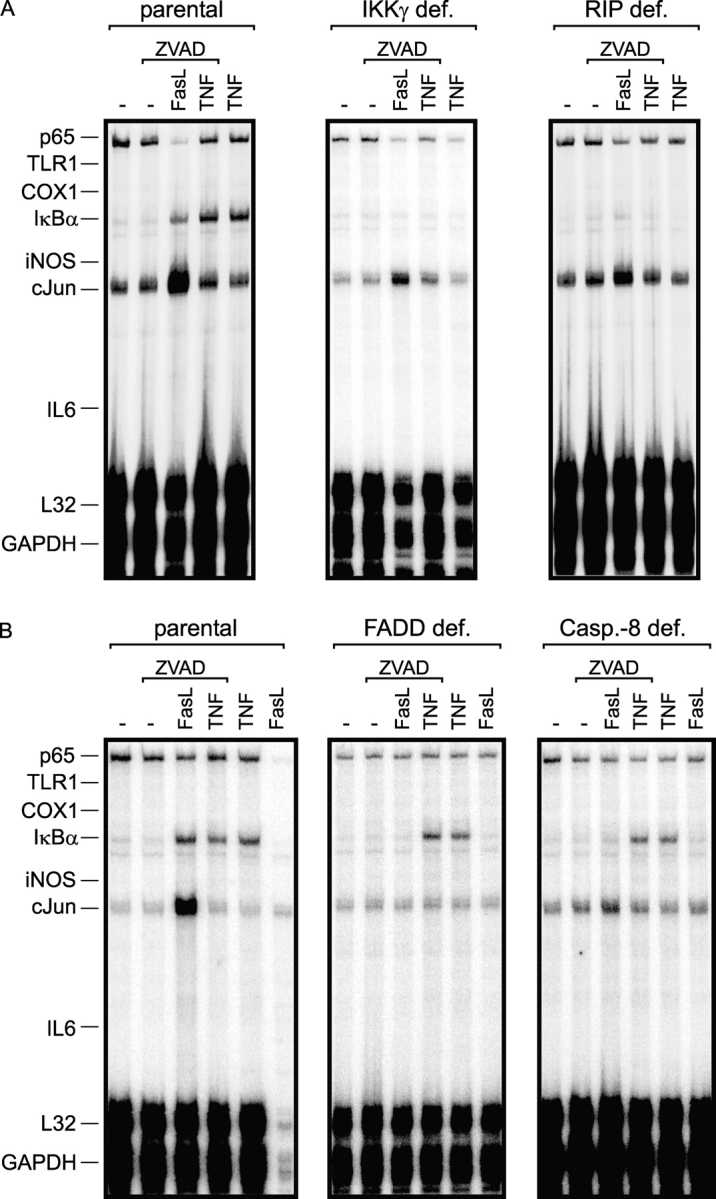
FasL-induced IκBα up-regulation occurs via a FADD, caspase-8, RIP-dependent pathway in Jurkat cells. (A and B) RIP- (A), NEMO/IKKγ- (A), FADD- (B), and caspase-8 (B)–deficient Jurkat clones and the corresponding parental cell lines were treated for 6 h with the indicated combinations of M2–cross-linked Flag-FasL (200 ng/ml), TNF (20 ng/ml), and z-VAD-fmk (20 μM). Total RNAs were isolated for RPA analyses and 10 μg of each RNA sample were analyzed with a Multi-Probe template set containing probes for the indicated mRNAs. Parental Jurkat cells were challenged with cross-linked FasL alone, but a reliable RPA analysis was not possible due to the massive induction of apoptosis leading to a strong reduction of the mRNA fraction in total RNA isolated.
FADD and caspase-8 are essential components of the DISC of Fas (Barnhart et al., 2003) and are also indispensable for FasL-induced up-regulation of cFos, which occurs in Jurkat cells under nonapoptotic circumstances (Siegmund et al., 2001). Analysis of FADD and caspase-8–deficient Jurkat clones (Juo et al., 1998, 1999) revealed that both proteins are also necessary for FasL-induced up-regulation of IκBα (Fig. 4 B). Fas signaling occurred in the presence of z-VAD-fmk, when caspase-8 activity was completely blocked. Thus, caspase-8 acts in FasL-induced NFκB signaling independent from its enzymatic activity. Moreover, FasL-induced cJun up-regulation was also completely blocked in FADD and caspase-8–deficient clones (Fig. 4 B). Together these data suggest that FADD and caspase-8 form a core signaling complex with Fas, whose structural integrity is necessary to allow activation of a diverse set of otherwise independent signaling pathways.
FLIP inhibits Fas-mediated up-regulation of NFκB target genes
Apoptosis induction and NFκB activation by Fas bifurcate at the level of FADD and caspase-8. Therefore, the potential short-lived inhibitor, which concomitantly blocks apoptotic and nonapoptotic Fas signaling, should target the Fas signaling complex. As discussed in an earlier work (Wajant et al., 2000), good candidates for such short-lived protein(s) are the FLIP isoforms as they (a) act on the Fas DISC and (b) exert the required high turnover (Fig. 5 A; Fulda et al., 2000; Leverkus et al., 2000a; Wajant et al., 2000; Kreuz et al., 2001). In further agreement with a negative-regulatory role of FLIP proteins in FasL-induced NFκB signaling, we found that FasL-induced up-regulation of IκBα is completely absent in a Jurkat clone stably transfected with FLIPL (Fig. 5 B). Notably, the expression level of FLIPL in this clone does not exceed endogenous FLIPL expression levels observed in other cell lines or mature dendritic cells (Fig. 5, A and C; Kreuz et al., 2001). A Jurkat clone transfected with FLIPS showed normal FasL-induced up-regulation of IκBα (unpublished data), but it cannot be ruled out that this was due to limited expression of FLIPS. Further, we observed that the FasL-induced response pattern in primary cells also supports an NFκB inhibitory role of FLIP. In mature dendritic cells having high endogenous levels of FLIP (Fig. 5 C; Leverkus et al., 2000b; Rescigno et al., 2000; Willems et al., 2000), FasL showed no effect on IκBα expression (Fig. 5 D, middle), whereas in primary T cells having barely detectable FLIP expression (Fig. 5 C; Irmler et al., 1997), IκBα mRNA was readily induced (Fig. 5 D, left and right). In the latter case, addition of z-VAD-fmk was required, as otherwise apoptosis interferes with the NFκB response. To analyze the role of FLIPL and FLIPS in Fas-mediated gene induction in KB and HT1080 cells, we produced pools of transfectants stably expressing GFP fusion proteins of FLIPL and FLIPS, respectively. All FLIPL/S expressing KB and HT1080 populations (KB-FLIPL/S-GFP and HT1080-FLIPL/S-GFP) showed unchanged Fas expression (not depicted) and were highly resistant against FasL-induced apoptosis also in the presence of CHX (Fig. 6, A and B). Although in z-VAD-fmk– and Bcl2-protected KB and HT1080 cells FasL strongly induced IL8 production, in FLIPL- and FLIPS-protected cells Fas-mediated IL8 up-regulation was blocked (Fig. 6, C and D). The inhibitory effect of FLIPL and FLIPS on IL8 production was specific for Fas, as TNF-induced IL8 up-regulation, which occurs via the Fas-related death receptor TNF-R1, only insignificantly varied for maximally 33% between the parental cells and the corresponding transfectants. Changes in basal IL8 production were negligible in the FLIPL/S-transfected KB populations (Fig. 6, C and D). However, the FLIPL and especially the FLIPS-expressing HT1080 cells showed significantly increased levels of constitutive IL8 production (Fig. 6, C and D). Similar effects were also found in respect to NFκB activation (unpublished data). Thus, dependent on the cell type, FLIPS and to a lesser extend FLIPL can have opposing effects, namely inhibition of Fas-inducible NFκB activation and up-regulation of NFκB by another yet unknown mechanism. FACS® analysis of the FLIP-GFP–expressing cells and EGFP FACS® calibration beads showed that 60–80% of the KB transfectants expressed <116,000 molecules of the GFP fusion proteins per cell, whereas the rest of the cells expressed >116,000 molecules per cell (Fig. 6 A). In TNF-stimulated KB cells (Fig. 7 B), mature DCs, and, as a positive control in the Hodgkin cell line L591 (Fig. 5), endogenous expression of FLIPL reached 0.25, 4, and 3 fg per cell; and FLIPS reached 0.5 and 6 fg per cell in KB and L591 cells. This corresponds to ∼3,000 to 50,000 FLIPL molecules and 15,000 to 180,000 FLIPS molecules per cell. As the inhibitory effects on FLIPL-GFP and FLIPS-GFP expression in KB cells were around 90% (Fig. 6 D), these data indicate that “physiological” levels of FLIPL and FLIPS are sufficient to block FasL-induced IL8 production. Next, we down-regulated expression of endogenous FLIP proteins by RNA interference–mediated gene silencing using the synthetic small interfering RNA (siRNA) F1, recognizing both FLIP isoforms (Fig. 7 A; Siegmund et al., 2002), and a new siRNA, F1490, which down-regulate FLIPL but not FLIPS (Fig. 7, A and B). In KB cells electroporated with the FLIP-specific siRNAs, FasL-induced IL8 production was substantially enhanced, as compared with cells mock electroporated or electroporated with control siRNA (Fig. 7 C). The increase of FasL-induced IL8 production reached by siRNA treatment was comparable, although FLIPS expression significantly exceeded FLIPL expression in KB cells (Fig. 5 A and 7 B). This finding suggests that FLIPL is more efficient than FLIPS in inhibition of Fas-induced NFκB activation. Basal IL8 production was also specifically increased in F1490- and F1-siRNA electroporated cells (Fig. 7 C). Thus, although in HT1080 cells that ectopically expressed FLIPS or FLIPL basal IL8 production was increased, endogenously expressed FLIP had an inhibitory role in KB cells. These opposing effects on Fas-independent basal IL8 production might reflect cell type–specific and/or FLIP isoform–specific functions of FLIP not related to Fas signaling (see Discussion).
Figure 5.


FLIPL inhibits FasL-induced IκBα up-regulation. (A) 106 L591 Hodgkin, KB, or Jurkat FLIPL cells were treated for the indicated times with 50 μg/ml CHX and were analyzed with respect to expression of FLIPL and FLIPS by Western blotting with a FLIP-specific antibody. To control protein loads, filters with L951 and KB cells were reprobed with an antitubulin antibody, and a FLIPL-GFP mass standard, which was normalized via its GFP part, was processed on the same filter to allow a quantitative estimation of FLIP expression. Based on this standard, FLIPL expression corresponds to 3,000 (KB) and 36,000 (L591) molecules per cell and FLIPS expression to 15,000 (KB) and 180,000 (L591) molecules per cell. (B) Parental Jurkat cells and a clone derived thereof, which stably overexpresses FLIPL, were challenged with the indicated combinations of soluble Flag-tagged FasL (200 ng/ml) cross-linked with the Flag-specific mAb M2 (0.5 μg/ml), TNF (20 ng/ml), and z-VAD-fmk (20 μM) for 6 h; and 10 μg of total RNA were used for RPA analyses with respect to the expression of the indicated target genes. (C) Cell lysates corresponding to 106 primary T cells, immature and mature DCs, and Jurkat-FLIPL transfectants were analyzed with respect to expression of FLIPL and FLIPS by Western blotting with a FLIP-specific antibody. Based on the FLIPL-GFP mass standard, FLIPL expression corresponds to 50,000 molecules per cell in mature DCs and Jurkat-FLIPL cells. (D) Human (left) and murine T cell blasts (right) as well as fully mature human dendritic cells (DCs; middle) were challenged for 6 h with M2–cross-linked Flag-FasL (200 ng/ml) in the presence of z-VAD-fmk. Total RNAs were finally analyzed with respect to expression of the indicated genes by RPA analysis.
Figure 6.



FLIPL and FLIPS protect HT1080 and KB cells from Fas-mediated apoptosis and block FasL-induced IL8 production. (A) FACS® analysis of GFP FACS® calibration beads with 0 (labeled “A”), 4,500 (labeled “B”), 15,000 (labeled “C”), 44,000 (labeled “D”), and 116,000 (labeled “E”) molecules of equivalent soluble fluorochrome as well as HT1080 and KB cell stably transfected with FLIPL-GFP or FLIPS-GFP. In the case of the KB cells, the localization of the calibration beads was corrected according to a minor difference in the autofluorescence of the parental cells and the nonfluorescent beads. (B) Indicated cells were seeded in 96-well plates (20 × 103 per well) and challenged with the indicated concentrations of soluble Flag-tagged FasL complexed with 0.5 μg/ml of the Flag-specific mAb M2 in the presence of 2.5 μg/ml CHX the next day. After an additional 18 h, cell viability was determined by crystal violet staining. (C) The indicated transfectants and cell lines were seeded in 96-well plates (20 × 103 per well). The next day, the medium was changed and cells were stimulated in triplicates with the indicated combinations of 200 ng/ml of cross-linked Flag-FasL, 20 ng/ml TNF, 20 μM z-VAD-fmk, and 2.5 μg/ml CHX. After 6 h, supernatants were removed and cleared, and IL8 concentrations were determined by ELISA analysis. (D) HT1080 and KB cells and FLIPL-GFP and FLIPS-GFP transfectants derived thereof were challenged for 6 h with 200 ng/ml of M2–cross-linked Flag-FasL or 20 ng/ml TNF in the presence of 20 μM z-VAD-fmk (HT1080) or a mixture of 2.5 μg/ml CHX and 20 μM Z-VAD-FMK (KB). Total RNAs were isolated and the expression of the indicated genes was determined by RPA analysis.
Figure 7.

Down-regulation of endogenous FLIPL is sufficient to enhance Fas-mediated IL8 production. (A) Expression plasmids for FLIPL-GFP, FLIPS-GFP, and DsRED were transiently transfected in triplicate with siRNAs (150 nM) specific for GFP (S15), FLIPL (F1490), or for both FLIP isoforms (F1). As a control, the various plasmids were cotransfected with a Bcl2-specific siRNA. After 48 h, cells were analyzed by FACS®, and relative expression levels were calculated from the product of the percentage of positive cells and the mean fluorescence intensity of positive cells. (B) The indicated siRNAs (150 nM) were introduced into KB cells by electroporation. The next day, expression of FLIPL and FLIPS was up-regulated by TNF stimulation (20 ng/ml) and determined by Western blot analysis of cell extracts with a FLIP-specific antibody. To control protein loads, filters were reprobed with antitubulin antibodies. (C) KB cells were seeded in 96-well plates (20 × 103 per well). 1 d later, cells were mock transfected (open bars) or transfected with control siRNA (hatched bars), a FLIPL/S-specific siRNA (F1; black bars), or a FLIPL-specific siRNA (F1490; gray bars). After an additional day, medium was changed and cells were stimulated for 6 h with 200 ng/ml of M2–cross-linked Flag-FasL or 20 ng/ml TNF. Finally, IL8 production was measured by ELISA analysis.
Discussion
The Fas-induced signaling pathways leading to up-regulation of IκBα and cJun were completely blocked in FADD- and caspase-8–deficient Jurkat cells in our study (Fig. 4 B). Moreover, induction of apoptosis and necrosis as well as up-regulation of cFos, which most likely occurs via the ERK pathway, have also been reported to be blocked in the FADD-deficient Jurkat clone used in our study (Juo et al., 1999; Holler et al., 2000; Siegmund et al., 2001). Further, with the exception of necrosis induction, all these Fas-induced processes are also abrogated in caspase-8–deficient Jurkat cells (Juo et al., 1998; Holler et al., 2000; Siegmund et al., 2001). Together this suggests that upon stimulation Fas forms a core signaling complex with FADD, which is essentially involved in all Fas-mediated signaling pathways (Fig. 8). With the exception of necrosis induction, caspase-8 is necessary for all other Fas signaling events. Noteworthily, only apoptosis induction appears to be dependent on the proteolytic activity of caspase-8 arguing for an additional scaffolding-related function of caspase-8 in context of the Fas signaling complex. Although RIP is necessary for Fas-induced NFκB activation (Fig. 4 A) and necrosis induction (Holler et al., 2000), this kinase seems dispensable for the pathway(s) leading to up-regulation of cJun and cFos (Fig. 4; Siegmund et al., 2001). For the reasons outlined in the next paragraph in detail, FLIPL/S appear to have an inhibitory role for all of these Fas-dependent signaling events by targeting the Fas/FADD–caspase-8 core complex. However, there is also evidence in the literature for Fas-independent signaling properties of FADD, caspase-8, and FLIP.
Figure 8.

Model of Fas signaling pathways. Relations 1–3, 7–9, and 10 are based on literature data (see Discussion for details). Relations 3–6 are evident from data shown in Fig. 5. Relations 11–13 are based on experiments shown in Figs. 1–4 and 7.
Transient overexpression experiments have revealed NFκB-inducing capacities of FADD, caspase-8, FLIPL, and FLIPS (Chaudhary et al., 2000; Hu et al., 2000; Wajant et al., 2000). Moreover, several studies have shown that stimulation of Fas can also result in NFκB activation, suggesting at first glance that a FADD/caspase-8/FLIPL (or FLIPS)–containing complex mediates this response (for review see Leverkus et al., 2003; Wajant et al., 2003). However, there are several concerns arguing against this simple concept. NFκB activation by overexpression of FADD, FLIPL, or FLIPS is blocked by proteins (CrmA and p35) and reagents (BD-fmk and z-VAD-fmk) that prevent caspase-8 activation (Chaudhary et al., 2000; Hu et al., 2000). In contrast, NFκB activation induced by overexpressed caspase-8 or stimulated Fas are rather enhanced by inhibition of caspase-8 activity (Chaudhary et al., 2000; Hu et al., 2000; Wajant et al., 2000). Nevertheless, as shown in Fig. 4 B, FasL-induced up-regulation of IκBα was completely absent in FADD and caspase-8–deficient cells. Thus, it seems possible that FADD and caspase-8 are not only necessary for caspase activity–independent Fas-induced NFκB activation but may also act in a Fas-independent but caspase activity–dependent NFκB-inducing pathway. We have further identified in this study RIP as an essential component of Fas-induced NFκB signaling. In accordance with this finding, it has been reported that a dominant-negative deletion mutant of RIP blocks NFκB activation induced by transiently overexpressed FADD and Fas (Hu et al., 2000). Notably, in this study, dominant-negative RIP failed to inhibit NFκB activation by overexpressed FLIP (Hu et al., 2000), arguing again for the existence of a Fas-independent role of FLIP in a yet unknown pathway leading to NFκB activation. Further, RIP is cleaved by caspase-8, resulting in an NFκB-inhibitory fragment (Lin et al., 1999). Recent studies suggest that enzymatically active heteromers of caspase-8 and FLIPL can be formed within the Fas signaling complex. Thus, the FLIPL-arrested DISC may also lead to the cleavage of RIP arguing against an NFκB-activating role of FLIPL in Fas signaling. It should be briefly mentioned here that Fas also induces necrosis in Jurkat cells under crucial involvement of RIP, but independent from NEMO/IKKγ (Holler et al., 2000; unpublished data). Thus, Fas-induced NFκB and necrosis signaling bifurcate at the level or downstream of RIP (Fig. 8). The Fas-independent NFκB-inducing capabilities of FLIPL/S might be related to the NFκB-activating properties of the viral FLIP homologue HHV8 vFLIP that interacts with the IKK complex (Liu et al., 2002; Field et al., 2003). Indeed, similarly to overexpressed FLIPL/S, HHV8 vFLIP activates NFκB independent of RIP (Matta et al., 2003). Moreover, HT1080 cells stably transfected with FLIPS, which is more closely related in its domain architecture to HHV8 vFLIP than FLIPL, showed a significantly higher basal NFκB activity than FLIPL-expressing HT1080 cells (Fig. 6). FLIP effects not related to Fas signaling might also be based on the recently described interaction of FLIPL with NFκB1/p105 (Li et al., 2003) and p38MAPK (Grambihler et al., 2003).
Opposing effects of FLIP have also been reported with respect to T cell activation. Although overexpressed FLIPL enhances TCR-induced ERK activation and IL2 production in Jurkat cells, it reduces these responses in DO11.10 T cells (Fang et al., 2004). Moreover, in one study, increased proliferation in FLIP-transgenic T cells upon stimulation with suboptimal concentrations of anti-CD3 has been observed (Lens et al., 2002), whereas in another study several transgenic lines with T cell–restricted FLIP expression exerted suppression of T cell activation over a wide range of anti-CD3 concentrations (Tai et al., 2004). However, the precise effects of FLIP on Fas signaling had not been addressed in these studies as no experiments with Fas stimulation have been performed. In KB and HT1080 cells, Bcl2 overexpression increased the ED50 value for FasL-induced apoptosis more than 500-fold (Fig. 1 B), suggesting an almost essential contribution of the intrinsic apoptotic pathway to Fas-induced apoptosis. However, in vivo, a significant contribution of the intrinsic pathway has so far only been observed in some studies when Fas was challenged with Fas-specific agonistic antibodies, making the physiological relevance of the intrinsic pathway for the in vivo effects of Fas a matter of debate. However, it should be taken into consideration that not only the specific cell type is of relevance for the question of whether a contribution of the intrinsic pathway to Fas-induced apoptosis gets apparent but also the strength of Fas activation and the time point after Fas triggering. Indeed, it has been shown in vitro that agonistic Fas antibodies alone are significantly less efficient than cross-linked soluble FasL or secondary aggregated Fas antibodies to kill type II cells (Huang et al., 1999). Noteworthily, in the studies showing protection against Fas-induced liver failure in Apaf1- (Cecconi et al., 1998), Bid- (Yin et al., 1999), or Bax- and Bak (Wei et al., 2001)-deficient mice, a suboptimal amount of agonistic antibodies was used to activate Fas. In contrast, in a study that fails to observe Bcl2-mediated protection against the hepatotoxic effects of Fas activation, cross-linked FasL was used (Huang et al., 1999). This suggests that there is no essential role of the intrinsic pathway in Fas-induced apoptosis in vivo but rather an accelerative and/or enhancing contribution, especially after a limited activation of Fas. Delayed apoptosis induction via Fas should already be sufficient to allow transient activation of the NFκB pathway by the mechanisms described in our study and might be of special relevance for the proinflammatory effects of FasL observed in various models of FasL-mediated tumor rejection. Although Fas-induced NFκB activation depends on caspase-8, the enzymatic activity of the latter appeared to be dispensable (Figs. 2–5). Thus, Fas-induced NFκB activation may attain importance under pathophysiological conditions where caspase-8 activity is blocked, e.g., by viral inhibitors such as CrmA or where caspase-8 is mutated.
Materials and methods
Reagents
Human recombinant Flag-tagged soluble FasL was purified from supernatants of Hek293 cells stably transfected with a corresponding expression plasmid by affinity chromatography with anti-FLAG M2 agarose beads (Sigma-Aldrich). Human recombinant TNF was obtained from Knoll AG. z-VAD-fmk was purchased from Bachem and z-IETD-fmk was obtained from Calbiochem. CHX, the anti-Flag mouse mAb M2, and alkaline phosphatase–conjugated goat anti–mouse IgG were purchased from Sigma-Aldrich. The anti-FLIP mouse mAb NF-6 was purchased from Qbiogene. The human epidermal cell line KB and the human fibrosarcoma cell line HT1080 were both obtained from the American Type Culture Collection. The Jurkat T cell lines deficient for FADD, caspase-8, NEMO/IKKγ, and RIP were supplied by J. Blenis (Harvard Medical School, Boston, MA), B. Seed (Massachusetts General Hospital, Boston, MA), and S.-C. Sun (Pennsylvania State University College of Medicine, Hershey, PA) and are described elsewhere (Ting et al., 1996; Juo et al., 1998, 1999; Harhaj et al., 2000). The Jurkat clones overexpressing FLIPL and FLIPS, respectively, were supplied by P. Schneider and J. Tschopp (University of Lausanne, Epalinges, Switzerland) and are described by Irmler et al. (1997). The F1 siRNA corresponds to positions 472–494 of U97074 (sense: 5′-atgtggttccacctaatgtca-3′; antisense: 5′-tgacattaggtggaaccacatct-3′) and F1490 to positions 1785 to 1805 whereby A1788 was substituted to G to match the mouse sequence (sense: 5′-agcacacucugaggaagaaac-3′; antisense: 5′-guuucuuccucagagugugcugc-3′).
Cell culture
KB, HT1080, and Jurkat cells were maintained in RPMI 1640 medium containing 10% heat-inactivated FCS in a humified 5.0% CO2 environment. For generation of polyclonal cell populations stably overexpressing GFP-fusion proteins, 10 × 106 cells were electroporated (4 mm cuvette; 250 V, 1800 μF, maximal resistance) with 10 μg of plasmid DNA in medium with 5% FCS. After 14 d of selection with 400–800 μg/ml G418 (GIBCO BRL), primary clones (n > 100) were pooled, expanded, and enriched for cells expressing the GFP-fusion proteins by two to three rounds of cell sorting using a FACStarPlus (Becton Dickinson). A Fas-negative, CD3-responsive Jurkat subclone (Janssen et al., 1996) was electroporated with a pEFBos-derived vector encoding full-length FasL and a hygromycin resistance vector (provided by B. Schraven, Otto-von-Guericke-University, Magdeburg, Germany). Resistant clones (Rapo-FasL) obtained were selected for high FasL membrane expression and subcloned under limiting dilution conditions. Human monocyte-derived dendritic cells were prepared and characterized as recently described (Leverkus et al., 2003). Human and murine T cells were prepared as described by Glauner et al. (2002) and Mack and Hacker (2002), respectively.
Cell death assays, Western blotting, and electrophoretic mobility shift assay analysis
These techniques were performed as described previously (Wajant et al., 2000; Kreuz et al., 2001).
IL8-ELISA
Cells were seeded in 96-well cell culture plates and cultivated overnight. The next day, the cell culture medium was exchanged and cells were stimulated with the reagents of interest for 6 h, and the IL8 concentrations reached in the supernatants were determined using the OptEIA™ human IL8 Set (BD Biosciences) according to the supplier's protocols. After subtraction of the extinction of the medium control, IL8 concentrations were calculated based on IL8 standards included in the analysis.
RNase protection assay (RPA)
Cells were treated as indicated, washed, and stored at −80°C until total RNAs were prepared with the peqGOLD RNAPure reagent (PeqLab Biotechnologie GmbH) according to the manufacturer's instructions. Total RNAs were analyzed using customer Multi-Probe template sets (BD Biosciences) with respect to the expression of the indicated genes. Probe synthesis, hybridization, and RNase treatment were performed with RiboQuant In vitro transcription and RiboQuant Multi-Probe RNase Protection Assay Systems (BD Biosciences) according to the manufacturer's recommendations. After RNase treatment, the protected transcripts were resolved by electrophoresis on a denaturing polyacrylamide gel (5%) and analyzed using a PhosphorImager operated by the ImageQuant software (Molecular Dynamics).
DNA oligonucleotide microarray analysis
The customized DNA microarray used in this study contains three oligonucleotide probes per gene for 110 genes relevant for inflammation. The specificity of the probes has been validated as described in detail previously (Holzberg et al., 2003). Fluorescent cRNA were prepared by reverse transcription of 3–5 μg of total RNA. Specifically, RNA was treated with DNase and double-stranded cDNAs followed by fluorophore-cRNAs that were synthesized using the cDNA synthesis system (Roche) and the MEGAscript T7 kit (Ambion) as directed by the manufacturers. 80 to 250 ng of double-stranded cDNA and 1.25 mM Cy3-UTP were used in each cRNA labeling reaction. Equal amounts (9 μg) of labeled cRNAs from each condition were hybridized individually to the DNA microarray in preprepared hybridization solution (MWG Biotech) at 42°C overnight and then washed sequentially in 2× SSC, 0.1% SDS, 1× SSC, and 0.5× SSC. Hybridized arrays were scanned at maximal resolution on a scanner (model 428; Affymetrix, Inc.). Fluorescence intensity values from TIFF images of Cy3 channels were integrated into one value per probe, normalized by the MAVI software (MWG Biotech), and further analyzed using Imagene 4.2 software (Biodiscovery). Genes whose expression changed by 1.5-fold on average and by at least 1.3-fold in each individual experiment were considered to be regulated by TNF or FasL, respectively. Genes whose expression ratio did not change were considered to be significantly expressed if the normalized signal intensity measured for this gene was two SDs higher than the average signal intensity obtained by comparing microarray results from five different cell lines.
Acknowledgments
We thank Svetla Chaneva for preparation of murine T-cells.
This work was supported by the Deutsche Forschungsgemeinschaft (grant Wa 1025/11-1, grant Le953/4-1, and Sonderforschungsbereich 495 project A5) and Deutsche Krebshilfe (grant 10-1751-Wa 3).
Abbreviations used in this paper: CHX, cycloheximide; DISC, death-inducing signaling complex; IAP, inhibitor of apoptosis; RPA, RNase protection assay; siRNA, small interfering RNA.
References
- Barnhart, B.C., E.C. Alappat, and M.E. Peter. 2003. The CD95 type I/type II model. Semin. Immunol. 15:185–193. [DOI] [PubMed] [Google Scholar]
- Boatright, K.M., M. Renatus, F.L. Scott, S. Sperandio, H. Shin, I.M. Pedersen, J.E. Ricci, W.A. Edris, D.P. Sutherlin, D.R. Green, and G.S. Salvesen. 2003. A unified model for apical caspase activation. Mol. Cell. 11:529–541. [DOI] [PubMed] [Google Scholar]
- Cecconi, F., G. Alvarez-Bolado, B.I. Meyer, K.A. Roth, and P. Gruss. 1998. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 94:727–737. [DOI] [PubMed] [Google Scholar]
- Chaudhary, P.M., M.T. Eby, A. Jasmin, A. Kumar, L. Liu, and L. Hood. 2000. Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene. 19:4451–4460. [DOI] [PubMed] [Google Scholar]
- Desbarats, J., R.B. Birge, M. Mimouni-Rongy, D.E. Weinstein, J.S. Palerme, and M.K. Newell. 2003. Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat. Cell Biol. 5:118–125. [DOI] [PubMed] [Google Scholar]
- Donepudi, M., A. Mac Sweeney, C. Briand, and M.G. Grutter. 2003. Insights into the regulatory mechanism for caspase-8 activation. Mol. Cell. 11:543–549. [DOI] [PubMed] [Google Scholar]
- Fang, L.W., T.S. Tai, W.N. Yu, F. Liao, and M.Z. Lai. 2004. Phosphatidylinositide 3-kinase priming couples c-FLIP to T cell activation. J. Biol. Chem. 279:13–18. [DOI] [PubMed] [Google Scholar]
- Field, N., W. Low, M. Daniels, S. Howell, L. Daviet, C. Boshoff, and M. Collins. 2003. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 116:3721–3728. [DOI] [PubMed] [Google Scholar]
- Fulda, S., E. Meyer, and K.M. Debatin. 2000. Metabolic inhibitors sensitize for CD95 (APO-1/Fas)-induced apoptosis by down-regulating Fas-associated death domain-like interleukin 1-converting enzyme inhibitory protein expression. Cancer Res. 60:3947–3956. [PubMed] [Google Scholar]
- Glauner, H., D. Siegmund, H. Motejadded, P. Scheurich, F. Henkler, O. Janssen, and H. Wajant. 2002. Intracellular localization and transcriptional regulation of tumor necrosis factor (TNF) receptor-associated factor 4 (TRAF4). Eur. J. Biochem. 269:4819–4829. [DOI] [PubMed] [Google Scholar]
- Grambihler, A., H. Higuchi, S.F. Bronk, and G.J. Gores. 2003. cFLIP-L inhibits p38 MAPK activation: an additional anti-apoptotic mechanism in bile acid-mediated apoptosis. J. Biol. Chem. 278:26831–26837. [DOI] [PubMed] [Google Scholar]
- Hakem, R., A. Hakem, G.S. Duncan, J.T. Henderson, M. Woo, M.S. Soengas, A. Elia, J.L. de la Pompa, D. Kagi, W. Khoo, et al. 1998. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 94:339–352. [DOI] [PubMed] [Google Scholar]
- Harhaj, E.W., L. Good, G. Xiao, M. Uhlik, M.E. Cvijic, I. Rivera-Walsh, and S.C. Sun. 2000. Somatic mutagenesis studies of NF-kappa B signaling in human T cells: evidence for an essential role of IKK gamma in NF-kappa B activation by T-cell costimulatory signals and HTLV-I Tax protein. Oncogene. 19:1448–1456. [DOI] [PubMed] [Google Scholar]
- Hoffmann, E., O. Dittrich-Breiholz, H. Holtmann, and M. Kracht. 2002. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 72:847–855. [PubMed] [Google Scholar]
- Holler, N., R. Zaru, O. Micheau, M. Thome, A. Attinger, S. Valitutti, J.L. Bodmer, P. Schneider, B. Seed, and J. Tschopp. 2000. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1:489–495. [DOI] [PubMed] [Google Scholar]
- Holzberg, D., C.G. Knight, O. Dittrich-Breiholz, H. Schneider, A. Dorrie, E. Hoffmann, K. Resch, and M. Kracht. 2003. Disruption of the c-JUN-JNK complex by a cell-permeable peptide containing the c-JUN delta domain induces apoptosis and affects a distinct set of interleukin-1-induced inflammatory genes. J. Biol. Chem. 278:40213–40223. [DOI] [PubMed] [Google Scholar]
- Hu, W.H., H. Johnson, and H.B. Shu. 2000. Activation of NF-kappaB by FADD, Casper, and caspase-8. J. Biol. Chem. 275:10838–10844. [DOI] [PubMed] [Google Scholar]
- Huang, D.C., M. Hahne, M. Schroeter, K. Frei, A. Fontana, A. Villunger, K. Newton, J. Tschopp, and A. Strasser. 1999. Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-x(L). Proc. Natl. Acad. Sci. USA. 96:14871–14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmler, M., M. Thome, M. Hahne, P. Schneider, K. Hofmann, V. Steiner, J.L. Bodmer, M. Schroter, K. Burns, C. Mattmann, et al. 1997. Inhibition of death receptor signals by cellular FLIP. Nature. 388:190–195. [DOI] [PubMed] [Google Scholar]
- Janssen, O., B. Lengl-Janssen, H.H. Oberg, M.J. Robertson, and D. Kabelitz. 1996. Induction of cell death via Fas (CD95, Apo-1) may be associated with but is not dependent on Fas-induced tyrosine phosphorylation. Immunol. Lett. 49:63–69. [DOI] [PubMed] [Google Scholar]
- Juo, P., C.J. Kuo, J. Yuan, and J. Blenis. 1998. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr. Biol. 8:1001–1008. [DOI] [PubMed] [Google Scholar]
- Juo, P., M.S. Woo, C.J. Kuo, P. Signorelli, H.P. Biemann, Y.A. Hannun, and J. Blenis. 1999. FADD is required for multiple signaling events downstream of the receptor Fas. Cell Growth Differ. 10:797–804. [PubMed] [Google Scholar]
- Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 18:621–663. [DOI] [PubMed] [Google Scholar]
- Kelliher, M.A., S. Grimm, Y. Ishida, F. Kuo, B.Z. Stanger, and P. Leder. 1998. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 8:297–303. [DOI] [PubMed] [Google Scholar]
- Kreuz, S., D. Siegmund, P. Scheurich, and H. Wajant. 2001. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell. Biol. 21:3964–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger, A., S. Baumann, P.H. Krammer, and S. Kirchhoff. 2001. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol. Cell. Biol. 21:8247–8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacronique, V., A. Mignon, M. Fabre, B. Viollet, N. Rouquet, T. Molina, A. Porteu, A. Henrion, D. Bouscary, P. Varlet, et al. 1996. Bcl-2 protects from lethal hepatic apoptosis induced by an anti-Fas antibody in mice. Nat. Med. 2:80–86. [DOI] [PubMed] [Google Scholar]
- Lens, S.M., T. Kataoka, K.A. Fortner, A. Tinel, I. Ferrero, R.H. MacDonald, M. Hahne, F. Beermann, A. Attinger, H.A. Orbea, et al. 2002. The caspase 8 inhibitor c-FLIP(L) modulates T-cell receptor-induced proliferation but not activation-induced cell death of lymphocytes. Mol. Cell. Biol. 22:5419–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverkus, M., M. Neumann, T. Mengling, C.T. Rauch, E.B. Brocker, P.H. Krammer, and H. Walczak. 2000. a. Regulation of tumor necrosis factor-related apoptosis-inducing ligand sensitivity in primary and transformed human keratinocytes. Cancer Res. 60:553–559. [PubMed] [Google Scholar]
- Leverkus, M., H. Walczak, A. McLellan, H.W. Fries, G. Terbeck, E.B. Brocker, and E. Kampgen. 2000. b. Maturation of dendritic cells leads to up-regulation of cellular FLICE-inhibitory protein and concomitant down-regulation of death ligand-mediated apoptosis. Blood. 96:2628–2631. [PubMed] [Google Scholar]
- Leverkus, M., A.D. McLellan, M. Heldmann, A.O. Eggert, E.B. Brocker, N. Koch, and E. Kampgen. 2003. MHC class II-mediated apoptosis in dendritic cells: a role for membrane-associated and mitochondrial signaling pathways. Int. Immunol. 15:993–1006. [DOI] [PubMed] [Google Scholar]
- Li, Z., J. Zhang, D. Chen, and H.B. Shu. 2003. Casper/c-FLIP is physically and functionally associated with NF-kappaB1 p105. Biochem. Biophys. Res. Commun. 309:980–985. [DOI] [PubMed] [Google Scholar]
- Lin, Y., A. Devin, Y. Rodriguez, and Z.G. Liu. 1999. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13:2514–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y., A. Devin, A. Cook, M.M. Keane, M. Kelliher, S. Lipkowitz, and Z.G. Liu. 2000. The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IkappaB kinase and c-Jun N-terminal kinase. Mol. Cell. Biol. 20:6638–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten, T., A.J. Ross, A. King, W.X. Zong, J.C. Rathmell, H.A. Shiels, E. Ulrich, K.G. Waymire, P. Mahar, K. Frauwirth, et al. 2000. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell. 6:1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L., M.T. Eby, N. Rathore, S.K. Sinha, A. Kumar, and P.M. Chaudhary. 2002. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J. Biol. Chem. 277:13745–13751. [DOI] [PubMed] [Google Scholar]
- Mack, A., and G. Hacker. 2002. Inhibition of caspase or FADD function blocks proliferation but not MAP kinase-activation and interleukin-2-production during primary stimulation of T cells. Eur. J. Immunol. 32:1986–1992. [DOI] [PubMed] [Google Scholar]
- Matta, H., Q. Sun, G. Moses, and P.M. Chaudhary. 2003. Molecular genetic analysis of human herpes virus 8-encoded viral FLICE inhibitory protein (vFLIP)-induced NF-kappa B activation. J. Biol. Chem. 278:52406–52411. [DOI] [PubMed] [Google Scholar]
- Rescigno, M., V. Piguet, B. Valzasina, S. Lens, R. Zubler, L. French, V. Kindler, J. Tschopp, and P. Ricciardi-Castagnoli. 2000. Fas engagement induces the maturation of dendritic cells (DCs), the release of interleukin (IL)-1beta, and the production of interferon gamma in the absence of IL-12 during DC-T cell cognate interaction: a new role for Fas ligand in inflammatory responses. J. Exp. Med. 192:1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, I., K. Matsuura, K. Khatib, J.C. Reed, S. Nagata, and P. Vassalli. 1996. A bcl-2 transgene expressed in hepatocytes protects mice from fulminant liver destruction but not from rapid death induced by anti-Fas antibody injection. J. Exp. Med. 183:1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Y. 2002. Apoptosome: the cellular engine for the activation of caspase-9. Structure (Camb.). 10:285–288. [DOI] [PubMed] [Google Scholar]
- Siegmund, D., D. Mauri, N. Peters, P. Juo, M. Thome, M. Reichwein, J. Blenis, P. Scheurich, J. Tschopp, and H. Wajant. 2001. Fas-associated death domain protein (FADD) and caspase-8 mediate up-regulation of c-Fos by Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) via a FLICE inhibitory protein (FLIP)-regulated pathway. J. Biol. Chem. 276:32585–32590. [DOI] [PubMed] [Google Scholar]
- Siegmund, D., P. Hadwiger, K. Pfizenmaier, H.P. Vornlocher, and H. Wajant. 2002. Selective inhibition of FLICE-like inhibitory protein expression with small interfering RNA oligonucleotides is sufficient to sensitize tumor cells for TRAIL-induced apoptosis. Mol. Med. 8:725–732. [PMC free article] [PubMed] [Google Scholar]
- Stanger, B.Z., P. Leder, T.H. Lee, E. Kim, and B. Seed. 1995. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 81:513–523. [DOI] [PubMed] [Google Scholar]
- Strasser, A., A.W. Harris, D.C. Huang, P.H. Krammer, and S. Cory. 1995. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 14:6136–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai, T.S., L.W. Fang, and M.Z. Lai. 2004. c-FLICE inhibitory protein expression inhibits T-cell activation. Cell Death Differ. 11:69–79. [DOI] [PubMed] [Google Scholar]
- Thome, M., and J. Tschopp. 2001. Regulation of lymphocyte proliferation and death by FLIP. Nat. Rev. Immunol. 1:50–58. [DOI] [PubMed] [Google Scholar]
- Ting, A.T., F.X. Pimentel-Muinos, and B. Seed. 1996. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 15:6189–6196. [PMC free article] [PubMed] [Google Scholar]
- Verhagen, A.M., and D.L. Vaux. 2002. Cell death regulation by the mammalian IAP antagonist Diablo/Smac. Apoptosis. 7:163–166. [DOI] [PubMed] [Google Scholar]
- Villunger, A., L.A. O'Reilly, N. Holler, J. Adams, and A. Strasser. 2000. Fas ligand, Bcl-2, granulocyte colony-stimulating factor, and p38 mitogen-activated protein kinase: Regulators of distinct cell death and survival pathways in granulocytes. J. Exp. Med. 192:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajant, H., E. Haas, R. Schwenzer, F. Muhlenbeck, S. Kreuz, G. Schubert, M. Grell, C. Smith, and P. Scheurich. 2000. Inhibition of death receptor-mediated gene induction by a cycloheximide-sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD). J. Biol. Chem. 275:24357–24366. [DOI] [PubMed] [Google Scholar]
- Wajant, H., K. Pfizenmaier, and P. Scheurich. 2003. Non-apoptotic Fas signaling. Cytokine Growth Factor Rev. 14:53–66. [DOI] [PubMed] [Google Scholar]
- Wei, M.C., W.X. Zong, E.H. Cheng, T. Lindsten, V. Panoutsakopoulou, A.J. Ross, K.A. Roth, G.R. MacGregor, C.B. Thompson, and S.J. Korsmeyer. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 292:727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems, F., Z. Amraoui, N. Vanderheyde, V. Verhasselt, E. Aksoy, C. Scaffidi, M.E. Peter, P.H. Krammer, and M. Goldman. 2000. Expression of c-FLIP(L) and resistance to CD95-mediated apoptosis of monocyte-derived dendritic cells: inhibition by bisindolylmaleimide. Blood. 95:3478–3482. [PubMed] [Google Scholar]
- Yin, X.M., K. Wang, A. Gross, Y. Zhao, S. Zinkel, B. Klocke, K.A. Roth, and S.J. Korsmeyer. 1999. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 400:886–891. [DOI] [PubMed] [Google Scholar]